

Abstract
Several diseases, including Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease (HD), are associated with specific proteins aggregating and depositing within tissues and/or cellular compartments. The aggregation of these proteins is characterized by the formation of extended, β-sheet rich fibrils, termed amyloid. In addition, a variety of other aggregate species also form, including oligomers and protofibrils. Specifically, HD is caused by the aggregation of the huntingtin (htt) protein that contains an expanded polyglutamine domain. Due to the link between protein aggregation and disease, small molecule aggregation inhibitors have been pursued as potential therapeutic agents. Two such small molecules are epigallocatechin 3-gallate (EGCG) and curcumin, both of which inhibit the fibril formation of several amyloid-forming proteins. However, amyloid formation is a complex process that is strongly influenced by the protein’s environment, leading to distinct aggregation pathways. Thus, changes in the protein’s environment may alter the effectiveness of aggregation inhibitors. A well-known modulator of amyloid formation is lipid membranes. Here, we investigated if the presence of lipid vesicles altered the ability of EGCG or curcumin to modulate htt aggregation and influence the interaction of htt with lipid membranes. The presence of 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine or total brain lipid extract vesicles prevented the curcumin from inhibiting htt fibril formation. In contrast, EGCG’s inhibition of htt fibril formation persisted in the presence of lipids. Collectively, these results highlight the complexity of htt aggregation and demonstrate that the presence of lipid membranes is a key modifier of the ability of small molecules to inhibit htt fibril formation.
Keywords: Curcumin, EGCG, polyglutamine, Huntington’s disease, amyloid
Graphical Abstract

INTRODUCTION
The polymerization of specific proteins into fibril aggregates with an underlying cross-β-structure, generically termed amyloid, is associated with a broad range of diseases. For example, aggregation of the amyloid-β peptide (Aβ) and tau are associated with Alzheimer’s disease; α-synuclein (α-syn, UniProtKB P37840) is implicated in Parkinson’s disease; aggregates of islet amyloid polypeptide (IAPP, UniProtKB P10997) form in diabetes mellitus type 2; and mutated forms of the huntingtin (htt, UniProtKB P42858) protein with an expanded polyglutamine (polyQ) stretch underlie Huntington’s disease (HD). Amyloid formation is a complex, heterogeneous process that involves a number of intermediate aggregate species, including a variety of oligomers, protofibrils, and amorphous aggregates.(1–3) In particular, oligomers have been implicated as prominent toxic species.(4–6) The aggregation process for any given amyloid-forming protein is highly dependent on its environment. For example, changes in pH,(7,8) metal ions,(9) and preparatory protocols(10) influence aggregate structure and morphology. In particular, HD is caused by an expanded polyglutamine domain that must surpass a critical threshold of ~35 repeat units,(11–13) and the extent of htt fibrillization correlates directly with an increase in polyQ length.(14–16) htt can form a heterogeneous mixture of oligomers(15,17–19) that have been observed in a variety of HD models.(20–24)
Due to the apparent role of protein aggregation in amyloid diseases, a number of small molecules that inhibit or modify amyloid formation have been identified.(25,26) Two specific polyphenols, (−)-epigallocatechin 3-gallate (EGCG) and curcumin, have been extensively studied due to their ability to generically inhibit amyloid formation. EGCG, a compound commonly found in green tea, reduces the level of aggregation and cytotoxicity of Aβ,(27–30) α-syn,(27,31,32) and tau.(33) For htt, EGCG decreases the level of formation of toxic htt aggregates in both yeast and Drosophila models.(34) Curcumin, an abundant molecule in the spice turmeric, inhibits the fibril formation of Aβ and α-Syn.(35–37) With regard to htt, the ability of curcumin to inhibit fibril formation is less clear, as it has been reported to both promote(38) and inhibit htt fibril formation.(39) This apparent contradiction may lie in the ability of other cellular factors to influence htt aggregation and the interaction between htt and curcumin.
With the notion that the cellular environment may influence the efficacy of small molecule inhibitors of amyloid formation, it becomes important to understand how specific cellular components influence the ability of small molecules to modify aggregation. Lipid membranes are known modulators of aggregation as altered aggregation in the presence of membranes, compared to bulk solution, has been observed for multiple amyloids, including htt,(40–43) Aβ,(44–47) and α-Syn.(48,49) The presence of cellular membranes comprised of lipid bilayers also alters amyloid aggregation by nucleating aggregation or stabilizing certain aggregate species and morphologies.(50) Specifically with regard to HD, mutant htt shows an increased binding affinity for phospholipids over nonmutated htt and can also disrupt the structural integrity of phospholipid bilayers.(51–53) EGCG has been shown to inhibit permeabilization of model membranes and mitochondrial membranes by Aβ and α-syn aggregates.(54–56) EGCG also interacts with and binds to membranes themselves but significantly weakens the membrane disruption capabilities of Aβ upon preincubation with the peptide.(57) To determine the effect these small molecules have on htt aggregation in membranous environments, we investigated if the presence of lipid altered the ability of either EGCG or curcumin to modulate aggregation and influence htt lipid binding. We found that the presence of either pure or physiological membrane systems interfered with the ability of curcumin to slow htt fibril formation, while EGCG’s inhibition of fibril formation persisted in the presence of lipids. Our studies suggest that the interaction between htt and curcumin is largely altered by cellular environment, while EGCG led to a propensity for the formation of amorphous aggregates regardless of the interaction with membrane systems.
MATERIALS AND METHODS
Purification of GST-htt-exon1 fusion protein.
Glutathione S-transferase (GST)-htt-exon1(46Q) fusion proteins were expressed and purified as previously described.(58) In short, the GST-htt fusion proteins were expressed by induction in Escherichia coli with isopropyl thiogalactopyranoside (IPTG) for 4 h at 30 °C. The cells were lysed using both lysozyme (0.5 mg/mL) and sonication, and the fusion proteins were purified by liquid chromatography (Bio-Rad LPLC) using a 5 mL GST affinity column. Fractions were analyzed using gel electrophoresis to verify the presence of htt and purity. Protein concentrations were determined using Coomassie Bradford reagent. Prior to any assay, fusion proteins were subjected to high-speed centrifugation at 20000g for 45 min at 4 °C to remove preexisting aggregates. To initiate aggregation for experimentation, the fusion proteins were incubated with Factor Xa (Promega, Madison, WI) to cleave the GST tag. All experiments were performed in buffer A [50 mM Tris-HCl (pH 7) and 150 mM NaCl].
Preparation of lipid vesicles.
Lipid vesicles of 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC) or total brain lipid extract (TBLE) were prepared by dissolving the lipid in buffer A. Lipid solutions were then subjected to 10 freeze/thaw cycles using liquid nitrogen followed by bath sonication for 30 min. The size and polydispersity of lipid vesicles were measured via dynamic light scattering (NanoBrook 90plus Particle Size Analyzer, Brookhaven Instruments) to verify the formation of large unilamellar vesicles (LUVs). The mean and standard deviation of vesicle sizes were determined by assuming a log-normal distribution of the DLS data.
ThioflavinT aggregation assays.
Purified htt-exon1(46Q) was diluted to a final concentration of 20 μM in the presence of 125 μM thioflavin T (ThT) (Sigma-Aldrich, St. Louis, MO) and the desired small molecule (20 or 100 μM). In experiments in which lipid was used, the lipid concentration was 400 μM, resulting in a 20:1 lipid:protein ratio. Reactions were run in a black Costar 96-well plate with a clear flat bottom, and the ThT fluorescence was monitored using a SpectraMax M2 microplate reader. Experiments were performed at 37 °C with 440 nm excitation and 484 nm emission, with readings every 5 min for 18 h. The initial rate of each ThT plot was calculated over a period of 3 h, starting at the point at which the intensity of the fluorescence signal was larger than the set threshold. The threshold was set at 10% of the maximum signal obtained for each condition; this ensures that the data have made a significant increase from the baseline and avoid rate calculations starting in the noise of the lag phase. These initial aggregation rates were normalized to the htt control (100%) for ease of comparison. The relative maximum fluorescence was determined by finding the maximum fluorescence intensity for each condition and normalizing them to the maximum fluorescence intensity of the htt control (100%).
Atomic Force Microscopy (AFM).
Incubations of htt-exon1(46Q) (20 μM) in the presence and absence of small molecule aggregation inhibitors (100 μM) were maintained at 37 °C and 1400 rpm using an orbital mixer. Control incubations of small molecule aggregation inhibitors (100 μM) alone were also used. In addition, incubations performed with lipid vesicles were performed at a lipid:peptide ratio of 20:1. At various time points, 2 μL aliquots of each condition were deposited on freshly cleaved mica (Ted Pella Inc., Redding, CA) for 1 min, washed with 200 μL of ultrapure water, and dried using a gentle stream of clean air. Images were collected using a Nanoscope V Multi-Mode scanning probe microscope (VEECO) equipped with a closed loop vertical engage J-scanner. Silicon oxide cantilevers with a nominal spring constant of 40 N/m and a resonance frequency of 300 kHz were used. Scan rates were set between 1 and 2 Hz with cantilever drive frequencies at 10% of resonance. All images were then analyzed using the MATLAB image processing toolbox (MathWorks) as previously described.(59,60)
Vesicle-Binding Assay.
To measure the interaction between the htt and lipid vesicles, a polydiacetylene (PDA) lipid binding assay was performed using previously reported protocols.(61,62) In short, diacetylene monomers of 10,12-tricosadiynoic acid (GFS Chemicals, Columbus, OH) and the lipid system of choice were mixed in a 2:3 molar ratio in a 4:1 chloroform/ethanol solution. The solution was evaporated under a gentle stream of nitrogen, and then the resulting dry films were reconstituted in buffer A at 70 °C. Lipid solutions were then sonicated at 100 W for 10 min using a sonic dismembrator (FisherSci) before being stored overnight at 4 °C to allow the self-assembly of vesicles. The next day, lipid mixtures were irradiated at 254 nm for 15 min with constant stirring to polymerize the 10,12-tricosadiynoic acid and result in a dark, royal blue solution. Experiments were performed in triplicate in a 96-well format, with the colorimetric response being recorded every 5 min for 18 h using a SpectraMax M2 microplate reader. The negative control included equal ratios of PDA/lipid solution and buffer, while the positive control included saturated NaOH (pH 12) to create a range of colorimetric responses for each lipid system. Polymerized vesicles were exposed to htt at a final concentration of 20 μM and small molecule concentrations of 100 μM. The PB, defined by Ablue/(Ablue + Ared), was calculated for the control (PB0) and each sample condition (PB). The percent colorimetric response (% CR) indicates the extent of insertion or disruption of the lipid membrane by htt and was calculated using the following equation:
| (1) |
Data were normalized to the maximum colorimetric response for each system, which was found by incubating the PDA/lipid solutions with 1:1 1 M NaOH.
RESULTS
Curcumin and EGCG inhibit htt fibrillization.
All experiments in this study were performed with a mutant htt fragment that expresses exon 1 with 46Q [htt-exon1(46Q)], which was purified from E. coli as a soluble fusion with GST. Cleavage of the GST moiety with factor Xa released the htt-exon1(46Q) fragment, initiating aggregation. To determine the relative ability of curcumin and EGCG to inhibit htt fibril formation, ThT aggregation assays were performed with htt-exon1(46Q) and varying ratios of the small molecules [1:1 and 5:1 small molecule:htt (Figure 1A,B)]. All relevant controls were measured and subtracted from the curves to show only the fluorescence signal from the htt aggregation (Figure S1), and multiple independent experiments were performed (Figure S2). For the initial relative aggregation rate, curcumin slowed aggregation to 50% and 35% of the control rate for 1:1 and 5:1 treatments, respectively (Figure 1C). At a 1:1 ratio, curcumin reduced the intensity of the overall ThT signal to 62% of the control signal, and increasing the dose of curcumin to a 5:1 ratio resulted in an additional reduction to 48% of the control signal (Figure 1D). EGCG was more effective in reducing the level of apparent fibril formation, as the relative initial aggregation rate was reduced to a larger extent [31% and 10% of the control rate for 1:1 and 5:1 treatments, receptively (Figure 1C)]. EGCG reduced the intensity of the maximum ThT fluorescence signal to 32% and 24% of the control singal for the 1:1 and 5:1 doses, respectively (Figure 1D). While ThT fluorescence is traditionally considered to track fibril formation, ThT signals are not always indicative of fibril formation. In addition, ThT does not provide information associated with the formation of other types of aggregate species. As a result, further experimentation is required to verify aggregate inhibition.
Figure 1.
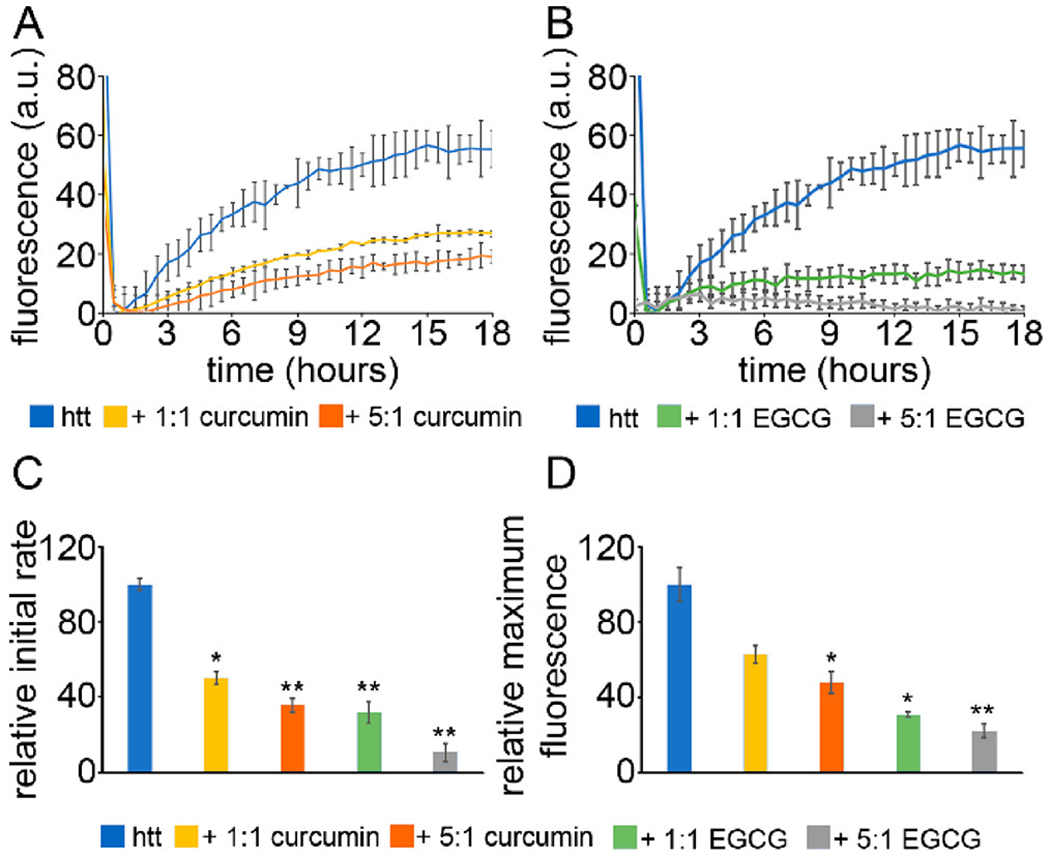
ThT aggregation assays for htt-exon1(46Q) in the presence of (A) curcumin or (B) EGCG. The htt-exon1(46Q) concentration was 20 μM. (C) The initial rate of aggregation and (D) the relative maximum fluorescence were determined with respect to the htt control. Normalization was performed with respect to the htt control. Analyses shown in panels C and D were determined as averages over all trials (shown in Figure S2). Error bars are provided for every sixth data point (30 min) and represent the standard error of the mean. One asterisk represents a p value of <0.05, and two asterisks represent a p value of <0.01.
To further establish the inhibition of htt-exon1(46Q) fibrillization by curcumin and EGCG and to determine the impact of these small molecules on aggregate morphology, AFM analysis was performed. This is particularly important to do with regard to EGCG, as it can compete with binding of ThT to fibrils for some amyloid-forming proteins.(29) Co-incubation experiments were performed with fresh preparations of htt-exon1(46Q) and with either curcumin or EGCG at a molar ratio of 5:1 (small molecule:protein). Control incubations of htt-exon1(46Q) without the small molecules were also analyzed. The htt-exon1(46Q) concentration was 20 μM. Representative AFM images of aliquots removed from solutions of htt-exon1(46Q) in the presence and absence of curcumin or EGCG after incubation for 1, 3, 5, and 8 h are shown in Figure 2. To quantify the effect of small molecules on aggregation, AFM images from all incubations were analyzed by counting the number of fibrils and oligomers per square micrometer (Figure 2B) using automated scripts written in Matlab. For this analysis, oligomers were defined as any feature more than 0.8 nm in height with an aspect ratio (longest distance across to shortest distance across) of <3.0, indicating a globular structure. Fibrils were defined as aggregates more than 0.8 nm in height with an aspect ratio of >3.0. These criteria were based on hand-measured characteristics of representative examples of each aggregate type. The height threshold of 0.8 nm was chosen on the basis of the background RMS roughness of mica that had been exposed to either curcumin or EGCG in the absence of htt (Figure S3).
Figure 2.
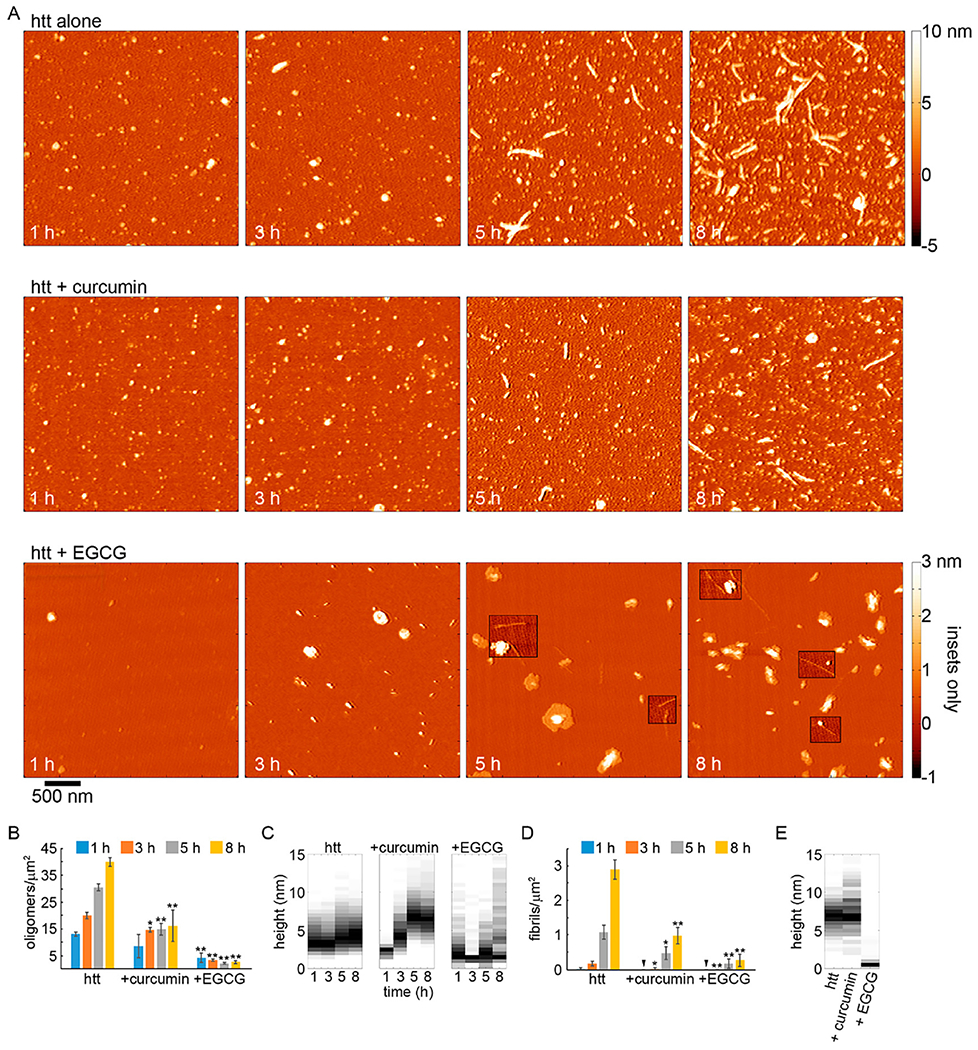
AFM analysis of the impact of curcumin or EGCG on htt-exon1(46Q) aggregation (5:1 small molecule:htt ratio). (A) Representative AFM images of 20 μM htt-exon1(46Q) incubated alone, with curcumin, or with EGCG as a function of time. The color map is the same for all images (see the top color bar). The insets in the AFM images of htt incubated with EGCG use a reduced color map (see bottom, labeled color bar) to make the shorter fibrils in these samples easier to see. (B) Image analysis of the number of oligomers per unit area as a function of time. (C) Height histograms of oligomers observed in AFM images as a function of time. (D) Image analysis of the number of fibrils per unit area as a function of time. (E) Height histograms of fibrils observed in AFM images as a function of time. For panels B and D, error bars represent the standard deviation, one asterisk represents a p value of <0.05, and two asterisks represent a p value of <0.01 using a Student’s t test.
Htt-exon1(46Q) incubated in the absence of small molecules exhibited extensive oligomer formation after incubation for 1 h with a steady increase in the number of oligomers with longer incubation times. After incubation for 1 and 3 h, oligomers were predominately 3–3.5 nm in height (Figure 2A,B). Oligomers became slightly larger with time [modes of 4–4.5 and 4.5–5 nm for 5 and 8 h time points, respectively (Figure 2C)]. Fibrils appeared after incubation for 3 h in htt-exon1(46Q) alone, and the number of fibrils steadily increased with time (Figure 2A,D). While the number of fibrils increased, the average height along the fibril contours did not change with time [mode of 6.5–7.5 nm (Figure 2E)]
When curcumin was co-incubated with htt-exon1(46Q), the formation of aggregates was inhibited. Oligomers appeared in similar numbers as the control after 1 h of co-incubation with curcumin; however, the number of oligomers per unit area was significantly reduced from the 3 h time point for the duration of the experiment (Fig 2 A and B). Fibrils were not detected until after 3 h of incubation, and they were significantly reduced in number compared to control (p < 0.05, Fig. 2 A and D). The fibril population continued to be significantly smaller in comparison with control for the duration of the experiment (p < 0.05 and p < 0.01 after 5 and 8 h respectively). While the number of oligomers and fibrils was reduced in the presence of curcumin, the size of the htt oligomers observed with curcumin was different compared to control experiments (Fig. 2 C). At 1 h, the mode height of oligomers formed in the presence of curcumin was 2.5 nm. With longer incubation times, the oligomers became progressively larger with a mode height of 4.0-4.5 nm after 3 h and 6.5-7.0 nm after 5 and 8 h of co-incubation with curcumin. This was consistent with a previously reported tendency of conditions that slow htt fibrillization promoting larger oligomeric aggregates.15, 63 Fibrils formed in the presence of curcumin were similar in morphology to control. The average height along the contour of the fibril did not change with time with an overall mode of 6.5–7.0 nm (Fig. 2E).
When EGCG was co-incubated with htt-exon1(46Q), the formation of aggregates was robustly inhibited. Both oligomer and fibril formation were significantly (p < 0.01) decreased at all time points (Fig. 2 A, B, and D). While the number of aggregates was reduced, the morphologies of the observed aggregates formed in the presence of EGCG were very different compared with those observed in control and + curcumin experiments (Fig. 2 C and E). Aggregates distinguished as oligomers from our automated scripts were often large and conglomerate-looking in morphology. That is, these aggregates were granular in appearance, adopted a variety of shapes, and often had a flat (~ 2-2.5 nm thick) region around its periphery. These aggregates were larger than the typical oligomers formed by htt, occupying a much larger area of the surface, and more heterogeneous in morphology (Fig. S2).The propensity to form these large aggregates of htt in the presence of EGCG has previously been reported.34 While a small population of fibrils were detected, the morphological characteristics of these fibrils were very different compared to control as they had a much smaller average height along their contours (mode of 1-1.5 nm) compared to fibrils formed by htt alone or with curcumin (Fig. 2E and 3). These thin, fibrillar species often (although not always) appeared to be growing from a larger, conglomerate aggregate (Fig. 3). This unique structure could represent a single protofilament that are incorporated into larger amyloid fibrils.
Figure 3.
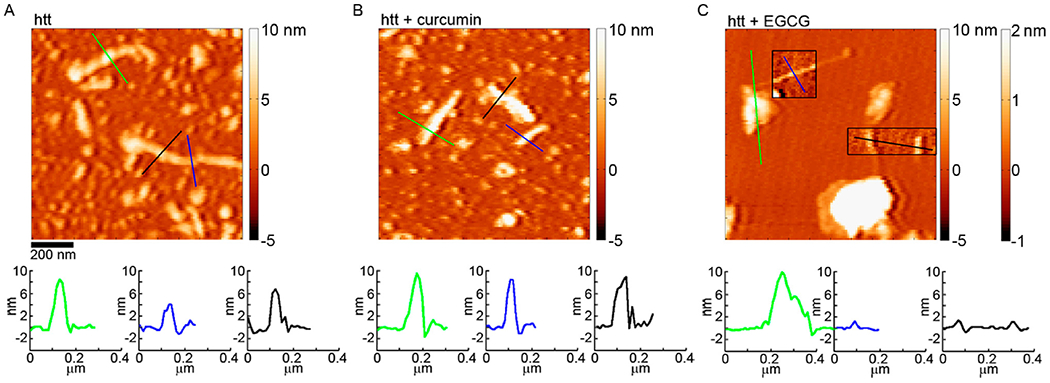
AFM images comparing the morphology of htt-exon1(46Q) fibrils formed (A) in the absence of small molecules, (B) with curcumin, or (C) with EGCG. In panel C, the boxed insets correspond to the second color bar to better visualize the fibril structures associated with htt incubated with EGCG. The color lines in each image correspond to the height profiles directly below each image.
Lipid membranes alter the ability of curcumin and EGCG to inhibit fibrillization.
To determine if the presence of lipid membranes impacts the ability of curcumin or EGCG to alter htt aggregation, we used two lipid systems. POPC was selected as it was previously shown to promote a distinct htt aggregation process.(43) The other system was total brain lipid extract (TBLE) as it also alters htt aggregation(40,64,65) and represents a more physiologically relevant mixture of lipids. Both lipid systems were introduced as vesicles, and the lipid:protein ratio was 20:1. Initially, the relative ability of curcumin and EGCG to inhibit htt fibril formation in the presence of POPC or TBLE vesicles was assessed by ThT aggregation assays (Figure 4; all independent trials are shown in Figure S2). As determined by DLS, the preparation protocol used produced POPC vesicles with an average diameter ~130–145 nm with a polydispersity index (PDI) of ~0.34–0.4 and TBLE vesicles with an average diameter of ~180–195 nm with a PDI of ~0.25–0.3 (representative DLS distributions are presented in Figure S5). The concentration of htt-exon1(46Q) was 20 μM, and the small molecule:protein ratio was 5:1.
Figure 4.
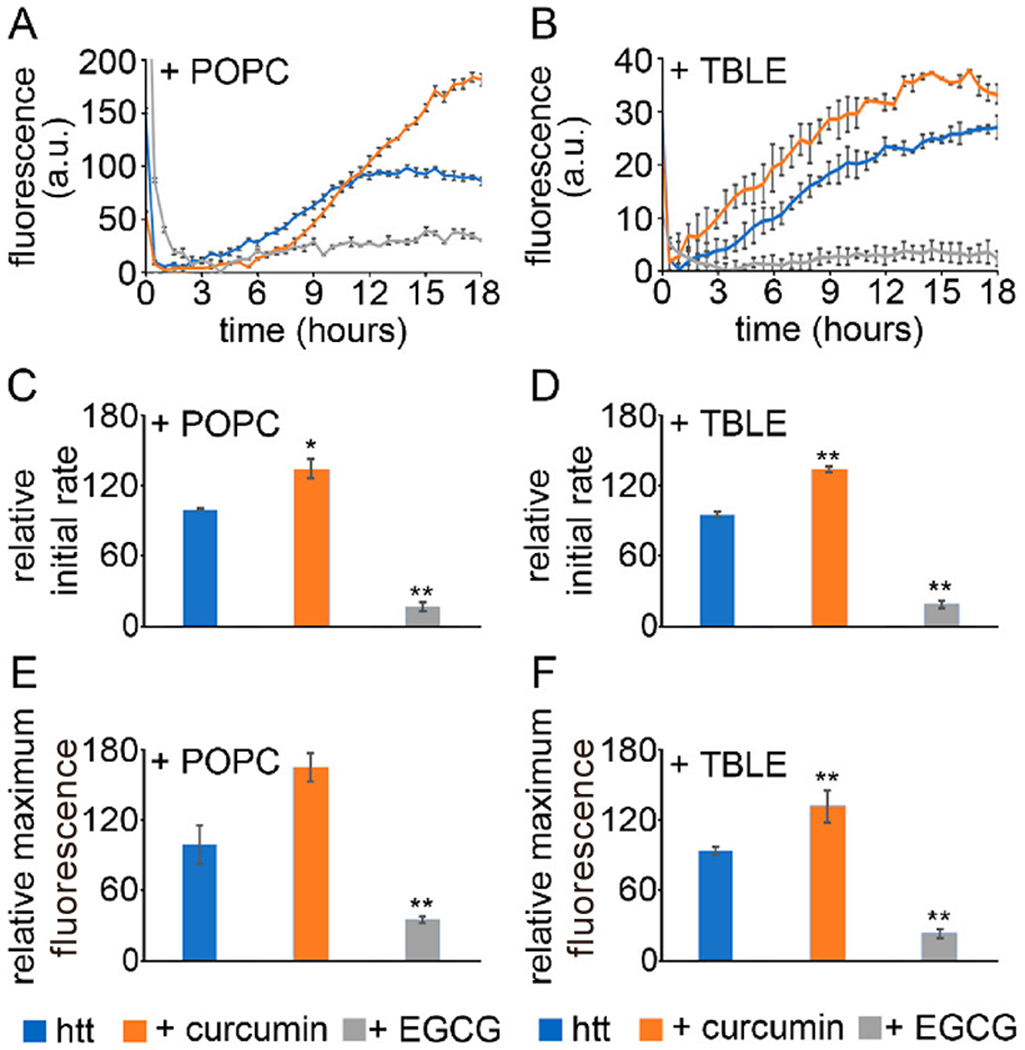
ThT aggregation assays for htt-exon1(46Q) aggregated in the presence of (A) POPC or (B) TBLE lipid vesicles. The htt-exon1(46Q) concentration was 20 μM. Curcumin and EGCG were added at a 5:1 small molecule:htt molar ratio. The initial rate of aggregation in the presence of (C) POPC or (D) TBLE vesicles was made relative to the aggregation rate of htt with the lipid vesicles in the absence of the small molecules. Finally, the relative maximum fluorescence values of all conditions in the presence of (E) POPC or (F) TBLE lipid vesicles at the end of the 18 h kinetic run are compared. Analyses shown in panels C–F were determined as averages over all trials (shown in Figure S2). Error bars are provided for every sixth data point (30 min) and represent the standard error of the mean. One asterisk represents a p value of <0.05, and two asterisks represent a p value of <0.01.
The impact of exposing htt-exon1(46Q) to these two lipid systems (in the absence of small molecules) was varied. The addition of POPC accelerated htt aggregation compared to the absence of lipids, whereas TBLE slowed htt aggregation (compare the fluorescence intensity of the htt alone experiments in Figures 1 and 4). In the presence of POPC, curcumin could delay the onset of fibrillization, as a lag phase was clearly observed. However, fibril growth occurred rapidly once initiated, as the initial aggregation rate exceeded that of htt alone in the presence of POPC (35% larger). The overall ability of curcumin to inhibit fibrillization was weakened in the presence of POPC, with the intensity of the maximum ThT signal being 68% larger than the control. In the presence of TBLE vesicles, htt aggregation proceeded faster without a discernible lag phase in the presence of curcumin compared to control (35% increase in the initial aggregation rate), and the intensity of the relative maximum ThT signal was increased by 40% compared to that of the control. htt-exon1(46Q) aggregation in the presence of POPC and TBLE vesicles was inhibited by EGCG based on the ThT assay (intensities of relative maximum ThT signals that were reduced to 41% and 22% compared to the control signal for incubations with POPC or TBLE, respectively).
To further establish that POPC or TBLE vesicles altered the ability of curcumin and EGCG to inhibit htt-exon1(46Q) fibrillization, AFM analysis was performed. As before, co-incubation experiments were performed with fresh preparations of htt-exon1(46Q) (20 μM) and with either curcumin or EGCG at a molar ratio of 5:1 (small molecule:protein); however, lipid vesicles of either POPC or TBLE were also added to the co-incubations at a 20:1 lipid:protein ratio. Control incubations of htt-exon1(46Q) with POPC and TBLE vesicles but without the small molecules were also performed (Figure S3). Representative AFM images of aliquots removed from solutions of htt-exon1(46Q) in the presence and absence of curcumin or EGCG after incubation for 1, 3, 5, and 8 h in the presence of POPC or TBLE vesicles are shown in Figures 5 and 6, respectively. To quantify the effect of the small molecules on aggregation in the presence of lipids, AFM images from all incubations were analyzed by counting the number of fibrils and oligomers per square micrometer (Figures 5 and 6) using automated scripts written in Matlab. As the addition of lipids to these incubations resulted in taller backgrounds once samples had been deposited on mica (Figure S3), the height threshold used for image analysis was increased to 1.5 nm. As a result, oligomers were defined as any feature more than 1.5 nm in height with an aspect ratio of <3.0, indicating a globular structure. Fibrils were defined as aggregates taller than 1.5 nm in height with an aspect ratio of >3.0.
Figure 5.
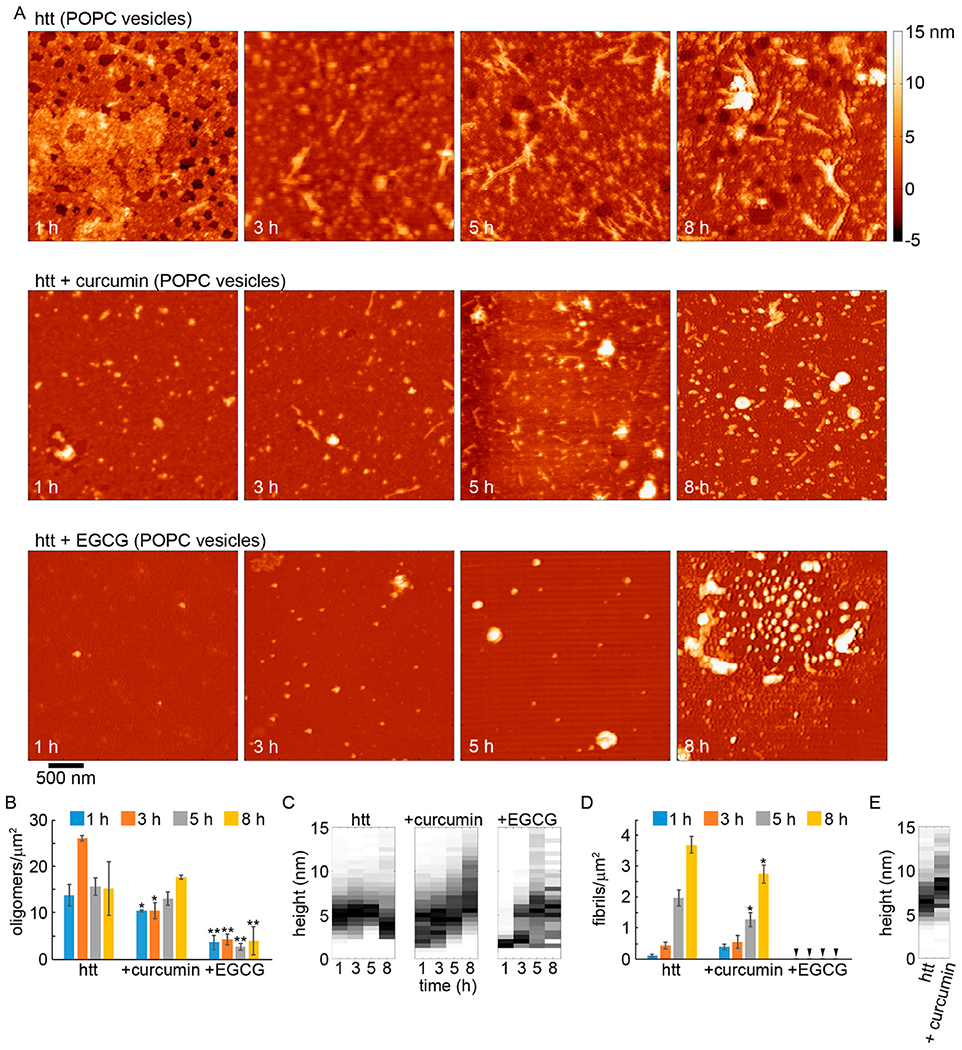
AFM analysis of the impact of curcumin or EGCG on htt-exon1(46Q) aggregation (5:1 small molecule:htt ratio) in the presence of POPC lipid vesicles (20:1 lipid:protein ratio). (A) Representative AFM images of 20 μM htt-exon1(46Q) incubated with POPC with no small molecules, with curcumin, or with EGCG as a function of time. The color map is the same for all images. (B) Image analysis of the number of oligomers per unit area as a function of time. (C) Height histograms of oligomers observed in AFM images as a function of time. (D) Image analysis of the number of fibrils per unit area as a function of time. Arrows indicate that no fibrils were observed. (E) Height histograms of fibrils observed in AFM images as a function of time. As fibrils were not observed for htt-exon1(46Q) incubations with POPC and EGCG, this condition is not presented in the fibril histograms. For panels B and D, error bars represent the standard deviation, one asterisk represents a p value of <0.05, and two asterisks represent a p value of <0.01 using a Student’s t test.
Figure 6.
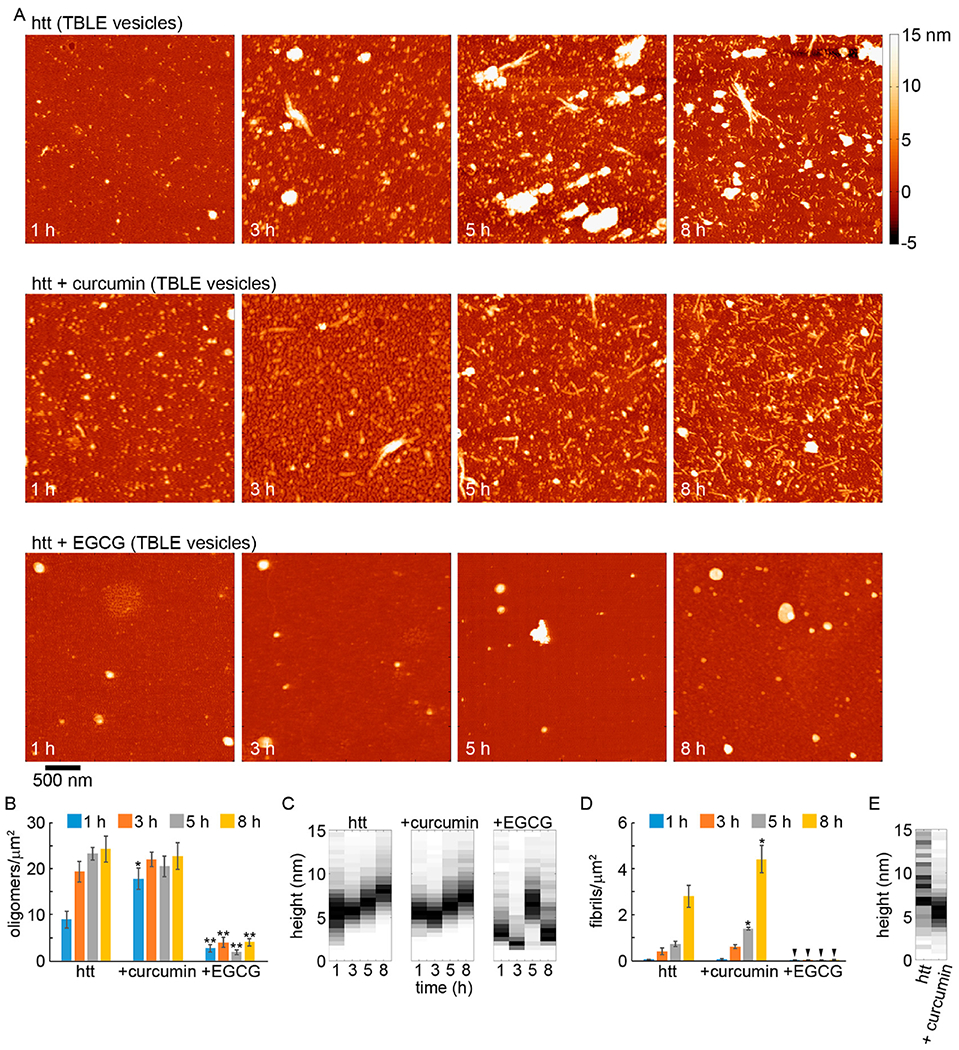
AFM analysis of the impact of curcumin or EGCG on htt-exon1(46Q) aggregation (5:1 small molecule:htt ratio) in the presence of TBLE vesicles (20:1 lipid:protein ratio). (A) Representative AFM images of 20 μM htt-exon1(46Q) incubated with TBLE with no small molecules, with curcumin, or with EGCG as a function of time. The color map is the same for all images. (B) Image analysis of the number of oligomers per unit area as a function of time. (C) Height histograms of oligomers observed in AFM images as a function of time. (D) Image analysis of the number of fibrils per unit area as a function of time. Arrows indicate that no fibrils were observed. (E) Height histograms of fibrils observed in AFM images as a function of time. As fibrils were not observed for incubations of htt-exon1(46Q) with TBLE and EGCG, this condition is not presented in the fibril histograms. For panels B and D, error bars represent the standard deviation, one asterisk represents a p value of <0.05, and two asterisks represent a p value <0.01 using a Student’s t test.
Htt-exon1(46Q) incubated in the presence of POPC exhibited extensive oligomer formation after 1 h of incubation (Fig. 5). While the number of detectible oligomers increased after 3 h, the oligomer population remained relatively stable during the time course of the experiment (Fig. 5B). After 1 h of incubation the mode height of oligomers was 4–5.5 nm (Fig. 5C). Oligomers were slightly larger at 3 and 5 h of incubation as the mode height was 5–6 nm at these time points. At 8 h of incubation, oligomers decreased in size with a mode height of ~2.5-3.5 nm. These oligomer species may contain lipid components; however, it is unlikely that they are comprised of lipids alone, as features observed from pure lipid incubations were all smaller than 1.5 nm in height and were not globular in appearance (Fig. S1). Fibrils appeared after 1 h of incubation in the htt-exon1(46Q) with POPC incubations, which was earlier than without any lipids (Fig 5A and D). The number of fibrils steadily increased with time, reaching a larger population compared with htt incubations without lipids, which is consistent with results from the ThT assays. While the number of fibrils increased, the average height along the fibril contours (mode of 6–7 nm) did not change with time and was comparable to fibrils formed in the absence of lipids (Figs. 5E and 7A).
Figure 7.
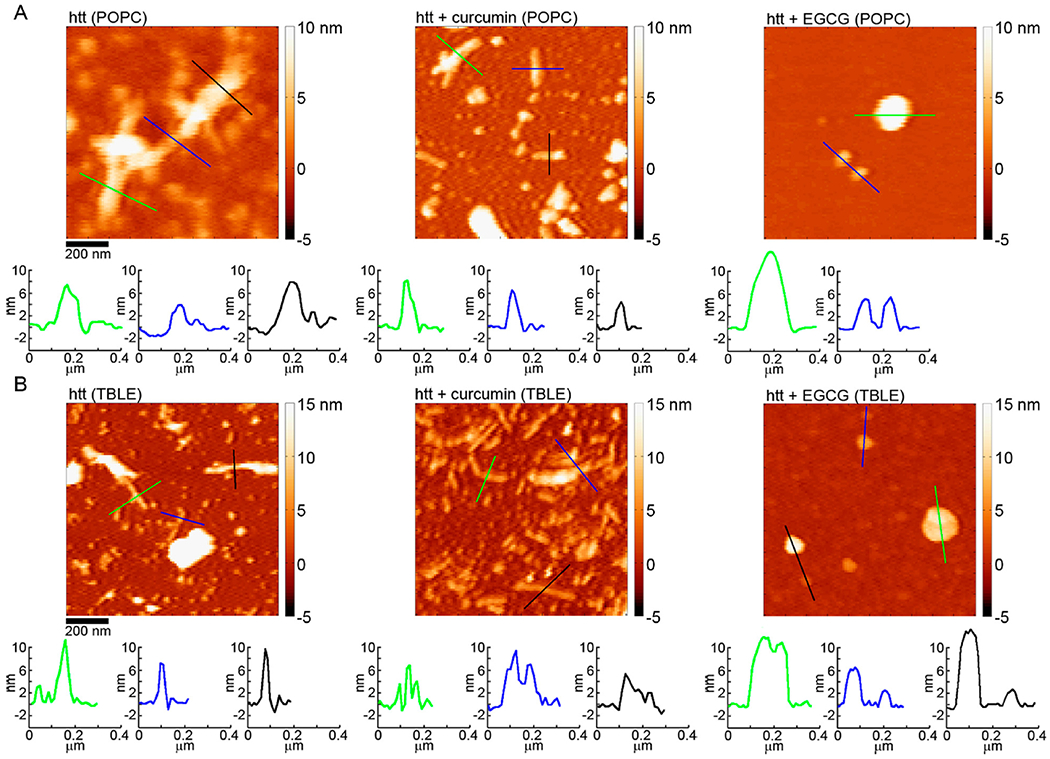
AFM images comparing the morphology of htt-exon1(46Q) fibrils formed in the absence of small molecules, with curcumin, or with EGCG for incubations performed in the presence of (A) POPC or (B) TBLE vesicles. The color lines in each image correspond to the height profiles directly below each image.
Upon co-incubation with htt-exon1(46Q) in the presence of POPC, curcumin’s ability to slow aggregation was dampened (Figure 5). While there was initially a significant reduction in the level of oligomers, the number of oligomers formed at 5 and 8 h co-incubation time points was not significantly different from that of incubations of htt-exon1(46Q) in the presence of POPC (Figure 5B). There was, however, a larger distribution of oligomer sizes, yet the mode size of oligomers was comparable to those formed in the presence of POPC but in the absence of curcumin at 1, 3, and 5 h (Figure 5C). At 8 h, the oligomer height distribution was shifted toward larger sizes. The promotion of larger oligomers by curcumin that was observed in the absence of lipid was much more subtle in the presence of POPC due to the larger distribution of oligomer sizes. Similar populations of fibrils were detected after 1 and 3 h from co-incubations with curcumin to control in the presence of POPC, and a small (but significant; p < 0.05) reduction in the fibril population was observed at 5 and 8 h (Figure 5D). Fibrils formed in the presence of curcumin were similar in morphology to the control (Figures 5E and 7A). The average height along the contour of the fibril was larger that that of the control fibrils, with a mode average height along the fibril contour of 8.5–9 nm; however, the distribution of these heights was also much broader, suggesting that lipids may be incorporated into the fibril structure.
When EGCG was co-incubated with htt-exon1(46Q) in the presence of POPC vesicles, EGCG was still an effective inhibitor of fibril formation (Figure 5). The number of oligomers per unit area was significantly (p < 0.01 at all time points) reduced compared to that of the control (Figure 5B), and no fibrils were observed at any time point (Figure 5D). While the number of oligomers was reduced, the morphologies of the observed oligomers formed in the presence of POPC vesicles and EGCG were very different compared with those in the control (Figure 5C). There were a number of small oligomers (~2–3 nm in height), but there was also a large population of larger aggregates that could be considerably more than 10 nm in height (Figure 7A) and occupy large amounts of surface area (Figure S6). The size of these large aggregates was heterogeneous, and the ability of EGCG to promote large, globular aggregates of htt was not weakened by the presence of POPC. However, unlike the large aggregates observed when EGCG was incubated with htt in the absence of lipid, these species were more rounded and smooth in appearance.
htt-exon1(46Q) incubated in the presence of TBLE exhibited oligomer formation after incubation for 1 h with the population of oligomers increasing with time (Figure 6A,B). Unlike incubations in the absence of lipids or with POPC vesicles, the oligomers that formed in the presence of TBLE increased in size with time [going from a mode height of 5–6 nm at 1 h to a mode height of 7.5–8.5 nm after incubation for 8 h (Figure 6C)]. This increase in height is consistent with the observed trend that slowing fibril formation can promote large oligomeric species of htt to form. The number of fibrils per unit area (with TBLE) was smaller compared with that of incubations performed with POPC vesicles (Figure 6C); however, fibril numbers were comparable with htt-exon1(46Q) incubations without lipids. Closer inspection of the AFM images suggests that the fibrils formed in the presence of TBLE were shorter than those grown in the absence of lipids, which could result in the reduced intensity of the ThT signal. That is, while the number of fibrils is comparable, the longer fibrils formed in the absence of TBLE would have more surface area (due to more monomers per fibril) to bind ThT, producing a stronger signal. Despite this apparent difference in fibril length, the average height along the fibril contours (mode of 6.5–7 nm) of fibrils formed in the presence of TBLE did not change with time and was comparable to that of fibrils formed in the absence of lipids and POPC (Figures 6E and 7B).
When co-incubated with htt-exon1(46Q) in the presence of TBLE, curcumin no longer reduced the level of aggregation compared to the control (Figure 6). After 1 h, a significant increase in the htt oligomer population was observed for co-incubations with curcumin (Figure 6B). The large number of oligomers per unit area was stable at later time points, and the numbers were comparable to that of the control. The height distributions of oligomers formed from co-incubations of htt with curcumin in the presence of TBLE were very comparable to that of the control at all time points (Figure 6C). As TBLE in general reduces the level of htt fibril formation, the size of htt oligomers did increase with time. Consistent with the ThT analysis, the number of fibrils formed from co-incubations of curcumin in the presence of TBLE was significantly larger compared with that of the control at 5 and 8 h (Figure 6D). The average height along the contour of the fibril was larger for control fibrils, with a mode average height along the fibril contour of 8.5–9 nm (Figure 6E); however, the distribution of these heights was also much broader. The mode average height along the contour was 5–6.5 nm for fibrils formed from co-incubations with curcumin in the presence of TBLE. The thicknesses along these fibrils formed in the presence of TBLE (in the absence and presence of curcumin) were quite varied (Figure 7B).
When EGCG was co-incubated with htt-exon1(46Q) in the presence of TBLE, the formation of aggregates was robustly inhibited, as the number of oligomers per unit area was significantly reduced compared to that of the control (Figure 6). Oligomer and fibril formation was significantly (p < 0.01) decreased at all time points (Figure 6B,D). While the number of aggregates was reduced, the morphologies of the observed aggregates formed in the presence of EGCG were very different compared with those observed in control experiments and experiments with curcumin (Figures 6C and 7B). Similar to the case for incubations in the presence of POPC, aggregates distinguished as oligomers from our automated scripts were often large, conglomerate-looking in morphology when incubated with TBLE vesicles. These aggregates were larger than the typical oligomers formed by htt, occupying a much larger area of the surface, and more heterogeneous (Figure S7). Again, the lipid surfaces did not impede the ability of EGCG to prevent htt fibril formation and promote large globular aggregates. These large, nonfibrillar aggregates tended to be smooth and rounded similar to those observed with EGCG and htt in the presence of POPC. At the 8 h time point, some of these larger aggregates had a flat, peripheral feature. However, this feature was distinct from the peripheral feature observed for EGCG and htt incubation in the absence of lipids as these flat regions were between 5 and 10 nm in height, suggesting that these might be lipid bilayer stacks.
As the presence of lipid vesicles altered the ability of curcumin and EGCG to inhibit fibril formation, we next performed a series of colorimetric membrane binding assays using lipid/polydiacetylene (PDA) vesicles. These vesicles were made with either POPC or TBLE as the lipid component. Lipid/PDA vesicles have varied colorimetric responses (CRs) when exposed to proteins or small molecules depending on the extent of interaction.(52,53) The CR in the lipid/PDA vesicles is related to transitions of the PDA polymer backbone structure, which is affected by changes in tension associated with exogenous molecules interacting with and/or inserting into the vesicle.(53) By measurement of the absorbance of both the blue (640 nm) and red (500 nm) wavelengths of lipid/PDA vesicles upon exposure to htt-exon1(46Q), the percent CR was obtained, which directly corresponds to the protein–lipid interaction. Importantly, the %CR response takes into account background experiments performed with the small molecules, so the response associated with curcumin or EGCG in the absence of htt is easily removed to obtain the %CR associated specifically with the htt. These background experiments with just curcumin or EGCG can be compared with neat buffer to determine the extent to which each small molecule interacts with the lipids, as well (all independent PDA assays are shown in Figure S8). With POPC/PDA vesicles, curcumin by itself invoked a minimal %CR, demonstrating that curcumin has little interaction with POPC (Figure 8A). However, the %CR of curcumin alone was extensive with TBLE/PDA vesicles, starting as high as 45% and reaching >60% within hours, suggesting that curcumin strongly binds to TBLE (Figure 8C). EGCG, which is known to bind lipids,(57) interacted with both POPC and TBLE, with a slightly stronger interaction with TBLE (Figure 8A,C). htt-exon1(46Q) by itself had a stronger (~3-fold) interaction with TBLE/PDA vesicles than with POPC/PDA vesicles. The addition of curcumin or EGCG weakened the interaction of htt-exon1(46Q) with both POPC/PDA and TBLE/PDA vesicles (Figure 8B,D).
Figure 8.
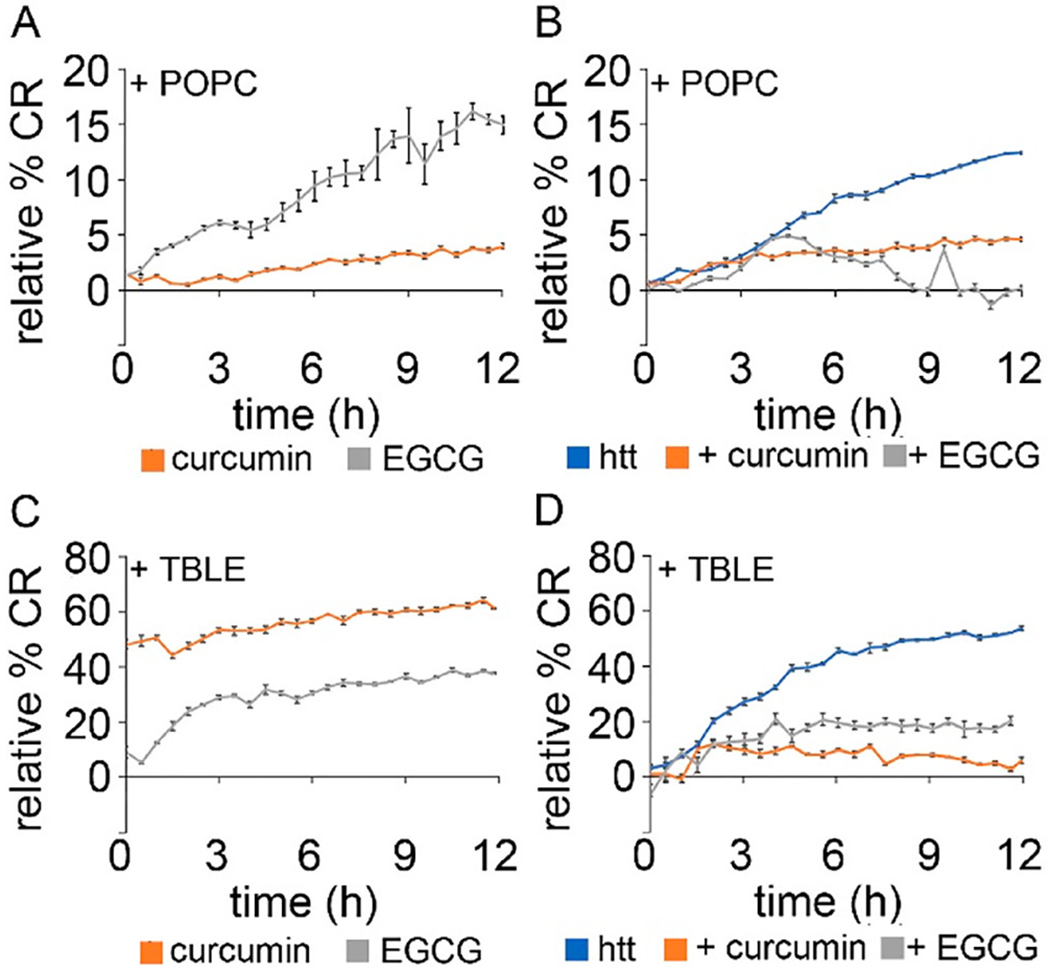
Lipid binding assays for (A) PDA/POPC vesicles exposed to either curcumin or EGCG, (B) PDA/POPC vesicles exposed to htt-exon1(46Q) with either curcumin or EGCG, (C) PDA/TBLE vesicles exposed to either curcumin or EGCG, and (D) PDA/TBLE vesicles exposed to htt-exon1(46Q) with either curcumin or EGCG. Error bars are provided for every sixth data point and represent the standard error of the mean.
DISCUSSION
Amyloid formation is highly dependent on environmental factors, complicating efforts to design effective small molecules that inhibit disease-related protein aggregation. A major environmental factor that influences the aggregation of numerous amyloid-forming proteins is the presence of cellular and subcellular membranes comprised predominately of lipids. Here, we investigated how the presence of lipid vesicles altered the ability of two known amyloid inhibitors, curcumin and EGCG, to prevent htt fibrillization. First, we verified that both curcumin and EGCG inhibited fibril formation of htt-exon1(46Q) in the absence of lipid membranes. In particular, EGCG promoted unique htt aggregate species compared to controls, i.e., large, globular aggregates and thin protofilaments. Next, we determined that POPC enhances htt fibril formation whereas TBLE reduced the level of htt fibril formation. In the presence of lipid vesicles of either POPC or TBLE, curcumin became ineffective in inhibiting htt fibrillization and even promoted fibril formation in the presence of TBLE. In contrast, EGCG remained effective in preventing htt fibrillization in the presence of lipid vesicles; however, some small oligomers and large conglomerate aggregates still formed. Collectively, these results underscore the complexity of htt aggregation and targeting fibril formation with small molecules.
Surfaces, especially lipid membranes, are known to strongly influence amyloid formation, even promoting unique aggregation pathways.(50) Results presented here showing that the ability of curcumin and EGCG to alter htt aggregation is modified by the presence of lipid vesicles further support this notion. For htt, taking into account the influence of lipid membranes in the aggregation process of htt may play a key role in targeting the aggregation process as a therapeutic strategy, as htt and its aggregate forms are often intimately linked to a variety of membranes.(52) For example, htt localizes to specific subcellular compartments as a result of its normal function via lipid interactions,(66) participates in the transport of lipid vesicles (endocytic, synaptic, or lysosomal),(53,67–69) is associated with acidic phospholipids,(51) and localizes to brain membrane fractions.(70) htt also localizes to the ER,(66,71–74) mitochondria,(75–80) and plasma membrane.(51) Literature reports have suggested both enhanced(43) and inhibited(40,63,64,81) htt fibrillization in the presence of membranes. This discrepancy appears to be due to properties of the particular lipid systems used in these studies and can be modified by changes in membrane composition.(41,42)
Curcumin’s inability to effectively inhibit fibril formation in the presence of lipids may be due to a variety of reasons. In the assays performed here, it is important to note that htt aggregation is likely occurring both on the membrane surface and in solution, creating a complex system. Curcumin initially appeared to delay the onset of htt aggregation in the presence of POPC vesicles, but this ability dissipated with time. htt aggregation on POPC/POPS bilayers proceeds via a unique mechanism compared to aggregation in solution.(43) A plausible scenario is that curcumin is more effective against the pathway associated with aggregation in solution than that occurring on the POPC membrane. However, curcumin did reduce the overall level of interaction of htt with the POPC bilayer, and curcumin itself did not significantly interact with the lipid surface. Perhaps more interestingly, curcumin increased the level of fibril formation in the presence of TBLE vesicles. Unlike POPC, TBLE appears to reduce the level of htt fibril formation.(40,63,82) On the basis of the PDA assay, curcumin by itself strongly interacted with the vesicle surface, suggesting that the curcumin was sequestered within the vesicle. In addition, the interaction of htt with the TBLE vesicles was greatly inhibited by the presence of curcumin. Collectively, this suggests a scenario in which curcumin is quickly taken up by the TBLE vesicles, altering the membrane. This alteration reduces the amount of htt bound and aggregating on the TBLE vesicle surface, which is a slower fibril-forming pathway. As a result, there is a larger amount of htt aggregating in solution in the presence of curcumin than in controls of htt with just the TBLE vesicles. As the curcumin that would inhibit fibrillization in solution has been sequestered by the TBLE vesicles, the portion of htt in solution is free to aggregate. As a result, the level of fibrillization increased in the presence of curcumin and TBLE vesicles. While it has been investigated for its interaction with Aβ, it has also been suggested that curcumin binds to later stage aggregate species like protofibrils.(83,84) If curcumin also targets later stages of the aggregation process of htt, this too could contribute to its differing abilities to modify htt fibrillization in the presence and absence of lipids, especially if the aggregation process in the presence of lipids is altered.
EGCG remained effective in preventing htt fibril formation in the presence of lipid vesicles. The effectiveness of EGCG as an amyloid inhibitor in the presence of lipids appears to be protein specific, as EGCG was unable to prevent IAPP fibrillization on a lipid membrane(85) but was more effective in preventing Aβ aggregation in the presence of vesicles.(57) EGCG is known to be incorporated into lipid vesicles,(57) and we did observe that EGCG by itself invoked a colorimetric response in our PDA assays, verifying that it directly interacted with both POPC and TBLE. EGCG appears to target early events in htt aggregation,(34) and this also appears to be the case for EGCG’s interaction Aβ and α-synuclein aggregation.(27) This ability to intervene in the htt aggregation process at an early stage could be why EGCG remained an effective inhibitor in the presence of lipids. Even though EGCG remained effective in the presence of both lipid systems and promoted larger, nonfibrillar aggregates, the morphology of these large aggregates was distinct, suggesting that lipid vesicles did influence how EGCG interacts with htt.
Supplementary Material
Acknowledgments
Funding Sources
This work was supported by the National Institutes of Health (R15NS090380). M.B. was supported by a Ruby Distinguished Doctoral Fellowship from WVU. A.R.S, K.L.K., and B.J.L. were supported by the NSF-funded Chemistry Site Research Experience for Undergraduates (REU) program at WVU (CHE-1559654). I.H. was supported by the Research Apprenticeship Program (RAP) at WVU. G.A. was supported by the WVU Summer Undergraduate Research Experience (SURE) program.
ABBREVIATIONS
- Aβ
β-amyloid
- α-syn
α-synulceion
- AFM
atomic force microscopy
- Htt
huntingtin
- HD
Huntington’s disease
- AD
Alzheimer’s disease
- PD
Parkinson’s disease
Footnotes
Supporting Information. The following files are available free of charge.
Figures presenting ThT assay controls, additional trials of ThT assays, AFM images demonstrating the backgrounds associated with neat buffer, curcumin, EGCG, POPC, and TBLE, area analysis of htt aggregates, DLS analysis of lipid vesicle size, and additional trials of the PDA lipid binding assays
REFERENCES
- 1.Cline EN, Bicca MA, Viola KL, and Klein WL (2018) The Amyloid-beta Oligomer Hypothesis: Beginning of the Third Decade, J. Alzheimers Dis 64, S567–S610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martial B, Lefèvre T, and Auger M (2018) Understanding amyloid fibril formation using protein fragments: structural investigations via vibrational spectroscopy and solid-state NMR, Biophys. Rev 10, 1133–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adegbuyiro A, Sedighi F, Pilkington AW, Groover S, and Legleiter J (2017) Proteins Containing Expanded Polyglutamine Tracts and Neurodegenerative Disease, Biochemistry 56, 1199–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lotz GP, and Legleiter J (2013) The role of amyloidogenic protein oligomerization in neurodegenerative disease, J. Mol. Med 91, 653–664. [DOI] [PubMed] [Google Scholar]
- 5.Sengupta U, Nilson AN, and Kayed R (2016) The Role of Amyloid-β Oligomers in Toxicity, Propagation, and Immunotherapy, EBioMedicine 6, 42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campioni S, Mannini B, Zampagni M, Pensalfini A, Parrini C, Evangelisti E, Relini A, Stefani M, Dobson CM, and Cecchi C (2010) A causative link between the structure of aberrant protein oligomers and their toxicity, Nat. Chem. Biol 6, 140. [DOI] [PubMed] [Google Scholar]
- 7.Fraser PE, Nguyen JT, Surewicz WK, and Kirschner DA (1991) pH-dependent structural transitions of Alzheimer amyloid peptides, Biophys. J 60, 1190–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petkova AT, Buntkowsky G, Dyda F, Leapman RD, Yau WM, and Tycko R (2004) Solid State NMR Reveals a pH-dependent Antiparallel β-Sheet Registry in Fibrils Formed by a β-Amyloid Peptide, J. Mol. Biol 335, 247–260. [DOI] [PubMed] [Google Scholar]
- 9.Faller P, Hureau C, and Berthoumieu O (2013) Role of Metal Ions in the Self-assembly of the Alzheimer’s Amyloid-β Peptide, Inorg. Chem 52, 12193–12206. [DOI] [PubMed] [Google Scholar]
- 10.Kodali R, Williams AD, Chemuru S, and Wetzel R (2010) Aβ(1-40) Forms Five Distinct Amyloid Structures whose beta-Sheet Contents and Fibril Stabilities Are Correlated, J. Mol. Biol 401, 503–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Penney JB, Vonsattel JP, MacDonald ME, Gusella JF, and Myers RH (1997) CAG repeat number governs the development rate of pathology in Huntington’s disease, Ann. Neurol 41, 689–692. [DOI] [PubMed] [Google Scholar]
- 12.Snell RG, Macmillan JC, Cheadle JP, Fenton I, Lazarou LP, Davies P, Macdonald ME, Gusella JF, Harper PS, and Shaw DJ (1993) Relationship between trinucleotide repeat expansion and phenotypic variation in Huntingtons disease, Nat. Genet 4, 393–397. [DOI] [PubMed] [Google Scholar]
- 13.Tobin AJ, and Signer ER (2000) Huntington’s disease: the challenge for cell biologists, Trends Cell Biol. 10, 531–536. [DOI] [PubMed] [Google Scholar]
- 14.Landrum E, and Wetzel R (2014) Biophysical underpinnings of the repeat length dependence of polyglutamine amyloid formation, J. Biol. Chem 289, 10254–10260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Legleiter J, Mitchell E, Lotz GP, Sapp E, Ng C, DiFiglia M, Thompson LM, and Muchowski PJ (2010) Mutant Huntingtin Fragments Form Oligomers in a Polyglutamine Length-dependent Manner in Vitro and in Vivo, J. Biol. Chem 285, 14777–14790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poirier MA, Li HL, Macosko J, Cai SW, Amzel M, and Ross CA (2002) Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrilization, J. Biol. Chem 277, 41032–41037. [DOI] [PubMed] [Google Scholar]
- 17.Ruggeri F, Vieweg S, Cendrowska U, Longo G, Chiki A, Lashuel H, and Dietler G (2016) Nanoscale studies link amyloid maturity with polyglutamine diseases onset, Sci. Rep 6, 31155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hands SL, and Wyttenbach A (2010) Neurotoxic protein oligomerisation associated with polyglutamine diseases, Acta Neuropathol. 120, 419–437. [DOI] [PubMed] [Google Scholar]
- 19.Morozova OA, Gupta S, and Colby DW (2015) Prefibrillar huntingtin oligomers isolated from HD brain potently seed amyloid formation, FEBS Lett. 589, 1897–1903. [DOI] [PubMed] [Google Scholar]
- 20.Olshina MA, Angley LM, Ramdzan YM, Tang J, Bailey MF, Hill AF, and Hatters DM (2010) Tracking Mutant Huntingtin Aggregation Kinetics in Cells Reveals Three Major Populations That Include an Invariant Oligomer Pool, J. Biol. Chem 285, 21807–21816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim SA, D’Acunto VF, Kokona B, Hofmann J, Cunningham NR, Bistline EM, Garcia FJ, Akhtar NM, Hoffman SH, Doshi SH, Ulrich KM, Jones NM, Bonini NM, Roberts CM, Link CD, Laue TM, and Fairman R (2017) Sedimentation Velocity Analysis with Fluorescence Detection of Mutant Huntingtin Exon 1 Aggregation in Drosophila melanogaster and Caenorhabditis elegans, Biochemistry 56, 4676–4688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim Yujin E., Hosp F, Frottin F, Ge H, Mann M, Hayer-Hartl M, and Hartl FU (2016) Soluble Oligomers of PolyQ-Expanded Huntingtin Target a Multiplicity of Key Cellular Factors, Mol. Cell 63, 951–964. [DOI] [PubMed] [Google Scholar]
- 23.Ramdzan YM, Nisbet RM, Miller J, Finkbeiner S, Hill AF, and Hatters DM (2010) Conformation Sensors that Distinguish Monomeric Proteins from Oligomers in Live Cells, Chem. Biol 17, 371–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sathasivam K, Lane A, Legleiter J, Warley A, Woodman B, Finkbeiner S, Paganetti P, Muchowski PJ, Wilson S, and Bates GP (2009) Identical oligomeric and fibrillar structures captured from the brains of R6/2 and knock-in mouse models of Huntington’s disease, Hum. Mol. Genet 19, 65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nie Q, Du XG, and Geng MY (2011) Small molecule inhibitors of amyloid beta peptide aggregation as a potential therapeutic strategy for Alzheimer’s disease, Acta Pharmacol Sin 32, 545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doig AJ, and Derreumaux P (2015) Inhibition of protein aggregation and amyloid formation by small molecules, Curr. Opin. Struc. Biol 30, 50–56. [DOI] [PubMed] [Google Scholar]
- 27.Bieschke J, Russ J, Friedrich RP, Ehrnhoefer DE, Wobst H, Neugebauer K, and Wanker EE (2010) EGCG remodels mature α-synuclein and amyloid-β fibrils and reduces cellular toxicity, P. Natl. Acad. Sci. USA 107, 7710–7715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rezai-Zadeh K, Arendash GW, Hou H, Fernandez F, Jensen M, Runfeldt M, Shytle RD, and Tan J (2008) Green tea epigallocatechin-3-gallate (EGCG) reduces β-amyloid mediated cognitive impairment and modulates tau pathology in Alzheimer transgenic mice, Brain. Res 1214, 177–187. [DOI] [PubMed] [Google Scholar]
- 29.Palhano FL, Lee J, Grimster NP, and Kelly JW (2013) Toward the Molecular Mechanism(s) by Which EGCG Treatment Remodels Mature Amyloid Fibrils, J. Am. Chem. Soc 135, 7503–7510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lopez del Amo JM, Fink U, Dasari M, Grelle G, Wanker EE, Bieschke J, and Reif B (2012) Structural Properties of EGCG-Induced, Nontoxic Alzheimer’s Disease Aβ Oligomers, J. Mol. Biol 421, 517–524. [DOI] [PubMed] [Google Scholar]
- 31.Ehrnhoefer DE, Bieschke J, Boeddrich A, Herbst M, Masino L, Lurz R, Engemann S, Pastore A, and Wanker EE (2008) EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers, Nat. Strict. Mol. Biol 15, 558. [DOI] [PubMed] [Google Scholar]
- 32.Xu Y, Zhang Y, Quan Z, Wong W, Guo J, Zhang R, Yang Q, Dai R, McGeer PL, and Qing H (2016) Epigallocatechin Gallate (EGCG) Inhibits Alpha-Synuclein Aggregation: A Potential Agent for Parkinson’s Disease, Neurochem. Res 41, 2788–2796. [DOI] [PubMed] [Google Scholar]
- 33.Wobst HJ, Sharma A, Diamond MI, Wanker EE, and Bieschke J (2015) The green tea polyphenol (−)-epigallocatechin gallate prevents the aggregation of tau protein into toxic oligomers at substoichiometric ratios, FEBS Lett. 589, 77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ehrnhoefer DE, Duennwald M, Markovic P, Wacker JL, Engemann S, Roark M, Legleiter J, Marsh JL, Thompson LM, Lindquist S, Muchowski PJ, and Wanker EE (2006) Green tea (−)-epigallocatechin-gallate modulates early events in huntingtin misfolding and reduces toxicity in Huntington’s disease models, Hum. Mol. Genet 15, 2743–2751. [DOI] [PubMed] [Google Scholar]
- 35.Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, and Cole GM (2005) Curcumin Inhibits Formation of Amyloid β Oligomers and Fibrils, Binds Plaques, and Reduces Amyloid in Vivo, J. Biol. Chem 280, 5892–5901. [DOI] [PubMed] [Google Scholar]
- 36.Ono K, Hasegawa K, Naiki H, and Yamada M (2004) Curcumin has potent anti-amyloidogenic effects for Alzheimer’s β-amyloid fibrils in vitro, J. Neurosci. Res 75, 742–750. [DOI] [PubMed] [Google Scholar]
- 37.Pandey N, Strider J, Nolan WC, Yan SX, and Galvin JE (2008) Curcumin inhibits aggregation of α-synuclein, Acta Neuropathol. 115, 479–489. [DOI] [PubMed] [Google Scholar]
- 38.Dikshit P, Goswami A, Mishra A, Nukina N, and Jana NR (2006) Curcumin enhances the polyglutamine-expanded truncated N-terminal huntingtin-induced cell death by promoting proteasomal malfunction, Biochem. Bioph. Res. Co 342, 1323–1328. [DOI] [PubMed] [Google Scholar]
- 39.Verma M, Sharma A, Naidu S, Bhadra AK, Kukreti R, and Taneja V (2012) Curcumin prevents formation of polyglutamine aggregates by inhibiting Vps36, a component of the ESCRT-II complex, PLoS ONE 7, e42923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Burke KA, Hensal KM, Umbaugh CS, Chaibva M, and Legleiter J (2013) Huntingtin disrupts lipid bilayers in a polyQ-length dependent manner, BBA-Biomembranes 1828, 1953–1961. [DOI] [PubMed] [Google Scholar]
- 41.Chaibva M, Gao X, Jain P, Campbell WA, Frey SL, and Legleiter J (2018) Sphingomyelin and GM1 Influence Huntingtin Binding to, Disruption of, and Aggregation on Lipid Membranes, ACS Omega 3, 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gao X, Campbell WA, Chaibva M, Jain P, Leslie AE, Frey SL, and Legleiter J (2016) Cholesterol Modifies Huntingtin Binding to, Disruption of, and Aggregation on Lipid Membranes, Biochemistry 55, 92–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pandey NK, Isas JM, Rawat A, Lee RV, Langen J, Pandey P, and Langen R (2018) The 17-residue-long N terminus in huntingtin controls stepwise aggregation in solution and on membranes via different mechanisms, J. Biol. Chem 293, 2597–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Legleiter J, Fryer JD, Holtzman DM, and Kowalewski T (2011) The Modulating Effect of Mechanical Changes in Lipid Bilayers Caused by ApoE-Containing Lipoproteins on A beta Induced Membrane Disruption, ACS Chem. Neuro 2, 588–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yip CM, Darabie AA, and McLaurin J (2002) A beta 42-peptide assembly on lipid Bilayers, J. Mol. Biol 318, 97–107. [DOI] [PubMed] [Google Scholar]
- 46.Yip CM, and McLaurin J (2001) Amyloid-beta peptide assembly: A critical step in fibrillogenesis and membrane disruption, Biophys. J 80, 1359–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yates EA, Owens SL, Lynch MF, Cucco EM, Umbaugh CS, and Legleiter J (2013) Specific Domains of A beta Facilitate Aggregation on and Association with Lipid Bilayers, J. Mol. Biol 425, 1915–1933. [DOI] [PubMed] [Google Scholar]
- 48.Jo EJ, McLaurin J, Yip CM, St George-Hyslop P, and Fraser PE (2000) alpha-synuclein membrane interactions and lipid specificity, J. Biol. Chem 275, 34328–34334. [DOI] [PubMed] [Google Scholar]
- 49.Eliezer D, Kutluay E, Bussell R, and Browne G (2001) Conformational properties of α-synuclein in its free and lipid-associated states11Edited by P. E. Wright, J. Mol. Biol 307, 1061–1073. [DOI] [PubMed] [Google Scholar]
- 50.Burke KA, Yates EA, and Legleiter J (2013) Biophysical insights into how surfaces, including lipid membranes, modulate protein aggregation related to neurodegeneration, Front. Neurol 4, 17–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kegel KB, Sapp E, Yoder J, Cuiffo B, Sobin L, Kim YJ, Qin ZH, Hayden MR, Aronin N, Scott DL, Isenberg G, Goldmann WH, and DiFiglia M (2005) Huntingtin Associates with Acidic Phospholipids at the Plasma Membrane, J. Biol. Chem 280, 36464–36473. [DOI] [PubMed] [Google Scholar]
- 52.Kegel-Gleason KB (2013) Huntingtin Interactions with Membrane Phospholipids: Strategic Targets for Therapeutic Intervention?, J. Huntingtons Dis 2, 239–250. [DOI] [PubMed] [Google Scholar]
- 53.Lee WC, Yoshihara M, and Littleton JT (2004) Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila model of Huntington’s disease, P. Natl. Acad. Sci. USA 101, 3224–3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gauci AJ, Caruana M, Giese A, Scerri C, and Vassallo N (2011) Identification of polyphenolic compounds and black tea extract as potent inhibitors of lipid membrane destabilization by Abeta(4)(2) aggregates, J. Alzheimers Dis 27, 767–779. [DOI] [PubMed] [Google Scholar]
- 55.Caruana M, Högen T, Levin J, Hillmer A, Giese A, and Vassallo N (2011) Inhibition and disaggregation of α-synuclein oligomers by natural polyphenolic compounds, FEBS Lett. 585, 1113–1120. [DOI] [PubMed] [Google Scholar]
- 56.Camilleri A, Zarb C, Caruana M, Ostermeier U, Ghio S, Högen T, Schmidt F, Giese A, and Vassallo N (2013) Mitochondrial membrane permeabilisation by amyloid aggregates and protection by polyphenols, BBA - Biomembranes 1828, 2532–2543. [DOI] [PubMed] [Google Scholar]
- 57.Malishev R, Nandi S, Kolusheva S, Levi-Kalisman Y, Klärner F-G, Schrader T, Bitan G, and Jelinek R (2015) Toxicity Inhibitors Protect Lipid Membranes from Disruption by Aβ42, ACS Chem. Neuro 6, 1860–1869. [DOI] [PubMed] [Google Scholar]
- 58.Muchowski PJ, Schaffar G, Sittler A, Wanker EE, Hayer-Hartl MK, and Hartl FU (2000) Hsp70 and Hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils, P. Natl. Acad. Sci. USA 97, 7841–7846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Burke KA, Godbey J, and Legleiter J (2011) Assessing mutant huntingtin fragment and polyglutamine aggregation by atomic force microscopy, Methods 53, 275–284. [DOI] [PubMed] [Google Scholar]
- 60.Burke KA, and Legleiter J (2013) Atomic force microscopy assays for evaluating polyglutamine aggregation in solution and on surfaces, Meth. Mol. Biol. (Clifton, N.J.) 1017, 21–40. [DOI] [PubMed] [Google Scholar]
- 61.Zheng F, Wu Z, and Chen Y (2012) A quantitative method for the measurement of membrane affinity by polydiacetylene-based colorimetric assay, Anal. Biochem 420, 171–176. [DOI] [PubMed] [Google Scholar]
- 62.Sokolovski M, Sheynis T, Kolusheva S, and Jelinek R (2008) Membrane interactions and lipid binding of casein oligomers and early aggregates, BBA - Biomembranes 1778, 2341–2349. [DOI] [PubMed] [Google Scholar]
- 63.Chaibva M, Jawahery S, Pilkington A. W. t., Arndt JR, Sarver O, Valentine S, Matysiak S, and Legleiter J (2016) Acetylation within the First 17 Residues of Huntingtin Exon 1 Alters Aggregation and Lipid Binding, Biophys. J 111, 349–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Burke KA, Kauffman KJ, Umbaugh CS, Frey SL, and Legleiter J (2013) The Interaction of Polyglutamine Peptides With Lipid Membranes is Regulated by Flanking Sequences Associated with Huntingtin, J. Biol. Chem 288, 14993–5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Burke KA, Yates EA, and Legleiter J (2013) Amyloid-Forming Proteins Alter the Local Mechanical Properties of Lipid Membranes, Biochemistry 52, 808–817. [DOI] [PubMed] [Google Scholar]
- 66.Atwal RS, Xia J, Pinchev D, Taylor J, Epand RM, and Truant R (2007) Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity, Hum. Mol. Genet 16, 2600–2615. [DOI] [PubMed] [Google Scholar]
- 67.Gauthier LR, Charrin BC, Borrell-Pages M, Dompierre JP, Rangone H, Cordelieres FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, and Saudou F (2004) Huntingtin Controls Neurotrophic Support and Survival of Neurons by Enhancing BDNF Vesicular Transport along Microtubules, Cell 118, 127–138. [DOI] [PubMed] [Google Scholar]
- 68.Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, and Goldstein LS (2003) Disruption of Axonal Transport by Loss of Huntingtin or Expression of Pathogenic PolyQ Proteins in Drosophila, Neuron 40, 25–40. [DOI] [PubMed] [Google Scholar]
- 69.Pal A, Severin F, Lommer B, Shevchenko A, and Zerial M (2006) Huntingtin-HAP40 complex is a novel Rab5 effector that regulates early endosome motility and is up-regulated in Huntington’s disease, J. Cell Biol 172, 605–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Suopanki J, Gotz C, Lutsch G, Schiller J, Harjes P, Herrmann A, and Wanker EE (2006) Interaction of huntingtin fragments with brain membranes - clues to early dysfunction in Huntington’s disease, J. Neurochem 96, 870–884. [DOI] [PubMed] [Google Scholar]
- 71.De Rooij K, Dorsman J, Smoor M, Den Dunnen J, and Van Ommen G (1996) Subcellular localization of the Huntington’s disease gene product in cell lines by immunofluorescence and biochemical subcellular fractionation, Hum. Mol. Genet 5, 1093–1099. [DOI] [PubMed] [Google Scholar]
- 72.Saudou F, Finkbeiner S, Devys D & Greenberg ME (1998) Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions, Cell 95, 55–66. [DOI] [PubMed] [Google Scholar]
- 73.Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, and MacDonald ME (2000) Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells, Hum. Mol. Genet 9, 2799–2809. [DOI] [PubMed] [Google Scholar]
- 74.Xia J, Lee DH, Taylor J, Vandelft M, and Truant R (2003) Huntingtin contains a highly conserved nuclear export signal, Hum. Mol. Genet 12, 1393–1403. [DOI] [PubMed] [Google Scholar]
- 75.Atwal RS, Desmond CR, Caron N, Maiuri T, Xia J, Sipione S, and Truant R (2011) Kinase inhibitors modulate huntingtin cell localization and toxicity, Nat. Chem. Biol 7, 453–460. [DOI] [PubMed] [Google Scholar]
- 76.Chang DT, Rintoul GL, Pandipati S, and Reynolds IJ (2006) Mutant huntingtin aggregates impair mitochondrial movement and trafficking in cortical neurons, Neurobiol. Dis 22, 388–400. [DOI] [PubMed] [Google Scholar]
- 77.Choo YS, Johnson GV, MacDonald M, Detloff PJ, and Lesort M (2004) Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release, Hum. Mol. Genet 13, 1407–1420. [DOI] [PubMed] [Google Scholar]
- 78.Gu M, Gash MT, Mann VM, Javoy-Agid F, Cooper JM, and Schapira AH (1996) Mitochondrial defect in Huntington’s disease caudate nucleus, Ann. Neurol 39, 385–389. [DOI] [PubMed] [Google Scholar]
- 79.Orr AL, Li S, Wang C-E, Li H, Wang J, Rong J, Xu X, Mastroberardino PG, Greenamyre JT, and Li X-J (2008) N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking, J. Neurosci 28, 2783–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Panov A, Gutekunst C-A, Leavitt B, Hayden M, Burke J, Strittmatter W, and Greenamyre J (2002) Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines, Nat. Neurosci 5, 731–736. [DOI] [PubMed] [Google Scholar]
- 81.Levy GR, Shen K, Gavrilov Y, Smith PES, Levy Y, Chan R, Frydman J, and Frydman L (2018) Huntingtin’s N-Terminus Rearrangements in the Presence of Membranes: A Joint Spectroscopic and Computational Perspective, ACS Chem Neurosci. [DOI] [PubMed] [Google Scholar]
- 82.Chaibva M, Burke KA, and Legleiter J (2014) Curvature Enhances Binding and Aggregation of Huntingtin at Lipid Membranes, Biochemistry 53, 2355–2365. [DOI] [PubMed] [Google Scholar]
- 83.Rao PP, Mohamed T, Teckwani K, and Tin G (2015) Curcumin Binding to Beta Amyloid: A Computational Study, Chem. Biol. Drug Des 86, 813–820. [DOI] [PubMed] [Google Scholar]
- 84.Martin TD, Malagodi AJ, Chi EY, and Evans DG (2019) Computational Study of the Driving Forces and Dynamics of Curcumin Binding to Amyloid-β Protofibrils, J. Phys. Chem. B 123, 551–560. [DOI] [PubMed] [Google Scholar]
- 85.Engel MFM, vandenAkker CC, Schleeger M, Velikov KP, Koenderink GH, and Bonn M (2012) The Polyphenol EGCG Inhibits Amyloid Formation Less Efficiently at Phospholipid Interfaces than in Bulk Solution, J. Am. Chem. Soc 134, 14781–14788. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.