

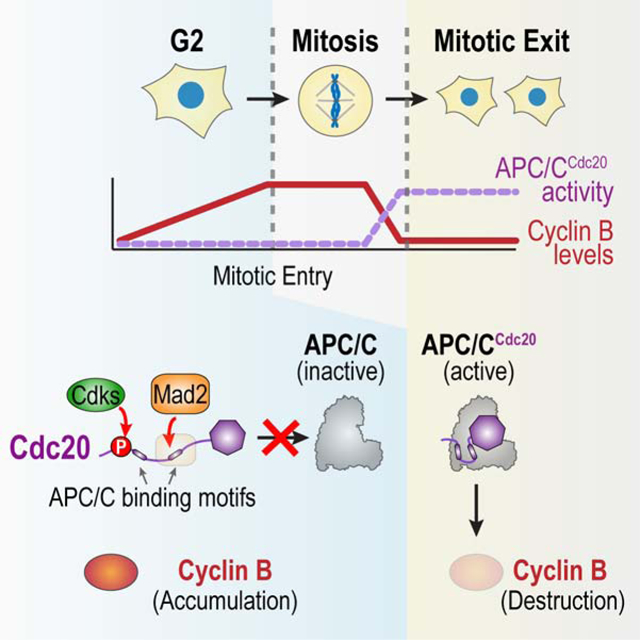
SUMMARY
In the eukaryotic cell cycle, a threshold level of cyclin B accumulation triggers the G2-to-M transition and subsequent cyclin B destruction triggers mitotic exit. The Anaphase Promoting Complex/Cyclosome (APC/C) is the E3 ubiquitin ligase that, together with its co-activator Cdc20, targets cyclin B for destruction during mitotic exit. Here we show that two pathways act in concert to protect cyclin B from Cdc20-activated APC/C in G2, in order to enable cyclin B accumulation and the G2-to-M transition. The first pathway involves the Mad1-Mad2 spindle checkpoint complex, acting in a distinct manner from checkpoint signaling after mitotic entry but employing a common molecular mechanism–the promotion of Mad2-Cdc20 complex formation. The second pathway involves cyclin-dependent kinase phosphorylation of Cdc20, which is known to reduce Cdc20’s affinity for the APC/C. Cooperation of these two mechanisms, which target distinct APC/C binding interfaces of Cdc20, enables cyclin B accumulation and the G2-to-M transition.
Keywords: Mitosis, cell division, germline, spindle checkpoint, Mad2, Mad1, APC/C, Cdc20, Cdk1, cyclin B3
Graphical Abstract
eTOC Blurb
Lara-Gonzalez et al. show that the spindle checkpoint protein Mad2 acts in concert with cyclin-dependent kinase phosphorylation to promote the G2-to-M transition in the cell cycle. The two mechanisms act in parallel to suppress the cyclin degradation machinery in G2, thereby allowing cyclin B accumulation and transition into M phase.
INTRODUCTION
The rise and decay in Cdk1-cyclin B activity drives eukaryotic cells into and out of M phase in the cell cycle (Morgan, 2007; Ferrell, 2013). While Cdk1 protein levels are relatively constant during the cell cycle, cyclin B periodically accumulates until it reaches a critical threshold that, through a series of feedback mechanisms, leads to high Cdk1-cyclin B kinase activity (Morgan, 2007; Ferrell, 2013). High Cdk1-cyclin B kinase activity drives striking cellular changes associated with mitosis—genome compaction, nuclear envelope breakdown, centrosome separation, bipolar spindle formation, and assembly of kinetochores that couple chromosomes to spindle microtubules (Morgan, 2007). Once all chromosomes have attached to spindle microtubules, cyclin B degradation is initiated to drive mitotic exit. The APC/C is the multi-subunit E3 ubiquitin ligase that, when bound to its co-activator Cdc20, targets cyclin B as well as securin, an inhibitor of sister chromatid separation, for proteolytic degradation (Peters, 2006; Pines, 2011; Alfieri et al., 2017).
To achieve the periodic accumulation and decline of cyclin B levels that demarcates M phase in the cell cycle, APC/CCdc20 must be restrained to enable sufficient cyclin B accumulation to trigger the G2-to-M transition (Lara-Gonzalez et al., 2017). Once mitosis is initiated, APC/CCdc20 must also be restrained until all chromosomes are connected to the spindle, in order to prevent errors in chromosome segregation. Recent work has greatly advanced our understanding of how unattached chromosomes inhibit APC/CCdc20 during mitosis, via the spindle checkpoint pathway that is activated at kinetochores (Musacchio, 2015; Corbett, 2017). The kinetochore-based spindle checkpoint produces a diffusible inhibitor of the APC/CCdc20, known as the mitotic checkpoint complex. A central component of the mitotic checkpoint complex is the protein Mad2, which exists in two conformations: open and closed (Luo et al., 2002; Sironi et al., 2002). The closed form of Mad2 is bound to related peptide motifs from its cellular partners, Mad1 and Cdc20. When the spindle checkpoint is active, Mad1-Mad2 complexes concentrate at unattached kinetochores where they recruit soluble open Mad2 and catalyzes its conversion to form a closed complex with Cdc20. Kinetochore-generated Mad2-Cdc20 complex then associates with Mad3 (known as BubR1 in vertebrates)–Bub3 to generate the mitotic checkpoint complex (Musacchio, 2015; Corbett, 2017). The mitotic checkpoint complex is a potent inhibitor of already-active APC/CCdc20 that acts by binding to the APC/CCdc20 holocomplex and interfering with its ability to recruit E2 ubiquitin-conjugating enzymes and to recognize substrates (Izawa and Pines, 2015; Alfieri et al., 2016; Yamaguchi et al., 2016). Early in mitosis, prior to assembly of mature kinetochores, Mad1-Mad2 complex localized at nuclear pores is proposed to generate the mitotic checkpoint complex via a similar reaction (Rodriguez-Bravo et al., 2014).
In contrast to APC/CCdc20 regulation by kinetochores in mitosis, how APC/CCdc20 is restrained during G2 to allow cyclin B accumulation and the G2-to-M transition is not understood. The APC/C inhibitor Emi1 was initially proposed to block APC/CCdc20 activity in G2 (Reimann et al., 2001). However, subsequent work showed that Emi1 targets APC/C bound by its other co-activator, Cdh1, and its primary function is to prevent re-replication of DNA (Di Fiore and Pines, 2007; Machida and Dutta, 2007). Emi2, related to Emi1, was shown to inhibit APC/CCdc20 during oocyte meiotic arrest (Schmidt et al., 2005; Tung et al., 2005); however, knockout analysis has shown that Emi2 is dispensable for somatic divisions in mammals (Gopinathan et al., 2017). In addition, Emi proteins are not as widely conserved as cyclin B and the APC/C. Other mechanisms suggested to restrain APC/CCdc20 include the N-terminal region of BubR1(Mad3) (Malureanu et al., 2009), phosphorylation of Cdc20 by cyclin-dependent kinases (Hein and Nilsson, 2016), and requirement for phosphorylation of core APC/C subunits by the mitotic kinases Cdk1 and Plk1 to promote Cdc20 binding (Fujimitsu et al., 2016; Qiao et al., 2016; Zhang et al., 2016). The last idea has strong support from biochemical, structural and Xenopus egg extract studies—egg extracts rapidly cycle between S and M phases without an intervening G2 phase—but its importance in wider cellular contexts has not been assessed. Thus, in contrast to the mechanistic understanding of how active APC/CCdc20 is regulated by the spindle checkpoint after mitotic entry, there is less clarity on how APC/CCdc20 is restrained to allow sufficient cyclin B accumulation for the G2-to-M transition.
Here, by analyzing proliferation in the C. elegans germline, and supporting key findings in human cultured cells, we define two parallel-acting mechanisms that target Cdc20 to suppress APC/CCdc20 activation and enable the G2-to-M transition. The first mechanism involves the Mad1-Mad2 complex acting in a manner distinct from its well-defined spindle checkpoint function that requires kinetochore localization. The second mechanism involves phosphorylation of Cdc20 that reduces its binding affinity for the APC/C. Inhibition of both mechanisms disrupts the G2-to-M transition to a similar extent as inhibition of core mitotic entry circuit factors, such as Cdk1 and cyclin B. In addition to identifying a dual inhibitory mechanism that restrains APC/CCdc20 to enable the G2-to-M transition, these results reveal a conserved checkpoint signaling-independent function of the Mad1-Mad2 complex.
RESULTS
Mad2 has a spindle checkpoint-independent function that is essential for viability and fertility in C. elegans
When the spindle checkpoint is activated, unattached kinetochores catalyze formation of the mitotic checkpoint complex, containing Mad2 and Mad3(BubR1) that binds to and inhibits Cdc20-activated APC/C (Musacchio, 2015; Corbett, 2017) (Fig. 1A). We confirmed equal importance of MAD-2 and MAD-3 for spindle checkpoint signaling in C. elegans by generating deletions of mad-2 and mad-3 using CRISPR/Cas9 (Figs. S1A,B), and characterizing null mutant embryos using a checkpoint assay that overcomes the drug impermeability of C. elegans embryos (Essex et al., 2009; Kim et al., 2017). In this assay, monopolar spindles with unattached kinetochores are generated during the second embryonic division by inhibition of a protein required for centriole duplication (Figs. 1B,C). Spindle checkpoint activation at the unattached kinetochores extends mitotic duration ~6–7 fold (Figs. 1B,C) (Kim et al., 2017); this extension was not observed in embryos lacking either MAD-2 or MAD-3 (Figs. 1B,C). (Movie S1).
Figure 1. Mad2 contributes to viability and fertility independently of the kinetochore-activated spindle checkpoint.
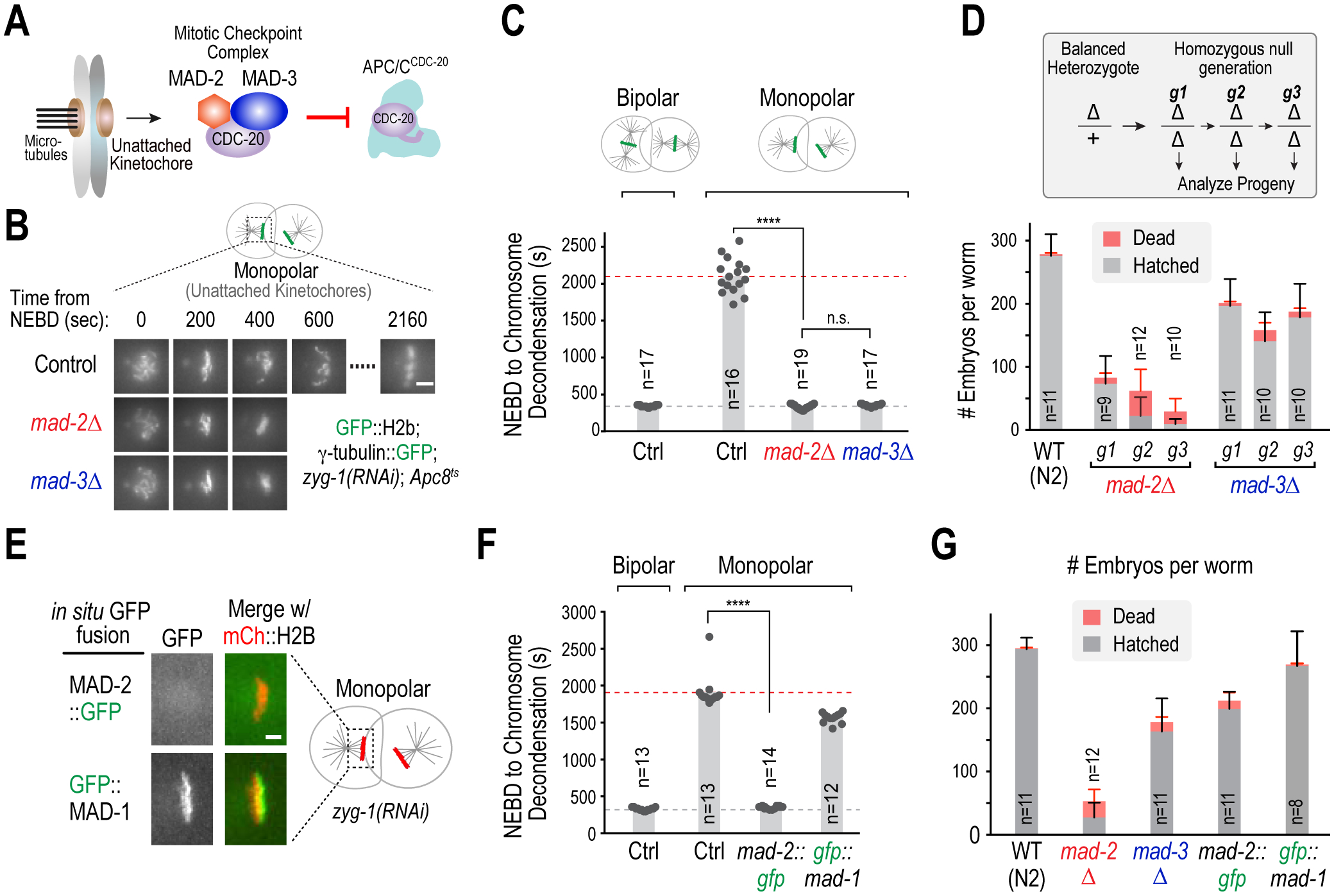
(A) Schematic of spindle checkpoint signaling: unattached kinetochores generate the mitotic checkpoint complex, containing Mad2 and Mad3, that inhibits APC/CCdc20. (B) Spindle checkpoint assay in C. elegans embryos. Null embryos for mad-2 or mad-3 were depleted of ZYG-1, an essential centriole duplication component, in order to generate monopolar spindles in the second embryonic division. Combining this treatment with a weak APC/C mutant (Apc8ts) results in a robust checkpoint-dependent mitotic arrest. Images show example stills of two-cell embryos undergoing mitosis under the indicated conditions. Scale bar, 5 μm. (C) Quantification of mitotic duration for the conditions in 1B. n is number of embryos scored. (D) Fertility and embryo viability analysis of first, second and third homozygous mutant generations (g1, g2 and g3). n is number of adults whose progeny were scored. Error bars are 95% confidence intervals. (E) Localization of indicated in situ tagged GFP fusions to unattached kinetochores on monopolar spindles. The GFP tag interferes with the ability of MAD-2 to localize to unattached kinetochores. Scale bar, 2 μm. (F) & (G) Spindle assembly checkpoint assay (F) and viability and fertility analysis (G) for the indicated conditions. Progeny from long-term propagated strains (>3 generations) were analyzed. n is the number of adults scored for progeny number. Error bars are 95% confidence intervals. **** represents p<0.0001 from Mann-Whitney tests; non-significant (n.s.) is p>0.05. See also Figure S1.
Despite their equivalent requirement for checkpoint signaling, the phenotypes observed in mad-2Δ and mad-3Δ worms were strikingly different. While mad-2Δ progeny of heterozygous mothers developed into adults, they exhibited significantly lower fertility and embryonic viability compared to mad-3Δ worms; after three generations, mad-2Δ worms produced a few, mostly dead, embryos (Fig. 1D). By contrast, mad-3Δ worms laid a constant number of progeny that was ~2/3 of that in control worms. Numerous developmental abnormalities were observed for mad-2Δ but not mad-3Δ animals (Fig. S1C), consistent with prior analysis of mad-2 and mad-3 mutants that partially deleted their coding regions (Kitagawa and Rose, 1999; Stein et al., 2007) and with RNAi-mediated depletion of MAD-2 and MAD-3 (Figs. S1D–F). These observations indicate that MAD-2 has a checkpoint signaling-independent role in fertility and viability.
Serendipitous support for the conclusion that MAD-2 has a checkpoint-independent function came from in situ tagging of MAD-2 and its binding partner MAD-1 with GFP (Figs. 1E; S1G). In situ GFP-tagged MAD-1 localized to kinetochores and supported checkpoint signaling, fertility, and embryonic viability. By contrast, the MAD-2::GFP fusion failed to localize to kinetochores and was inactive in checkpoint signaling (Figs. 1E,F), but nonetheless exhibited fertility and embryonic viability similar to mad-3Δ worms (Fig. 1G). These results suggest that mad-2::gfp is a separation-of-function allele that supports fertility and viability but not spindle checkpoint signaling. They also suggest that the role of MAD-2 in fertility and embryonic viability does not require its localization to kinetochores.
Mad2 is a conserved regulator of the G2-to-M transition
The most prominent phenotype of mad-2Δ worms was an abrupt reduction in fertility, suggesting that MAD-2 is important for germline development. To analyze the effect of MAD-2 and MAD-3 inhibition on germline development, we used an assay in which the introduction of food triggers germ cell proliferation in larvae (Fig. S2A). In C. elegans, the germline arises from a pair of progenitor cells that are quiescent in G2 phase of the cell cycle (Fukuyama et al., 2006). Feeding at the L1 larval stage triggers proliferation of these progenitors, producing nascent germlines with 20–30 cells within 20h (Fig. 2A).
Figure 2. Mad2 promotes the G2 to M transition.
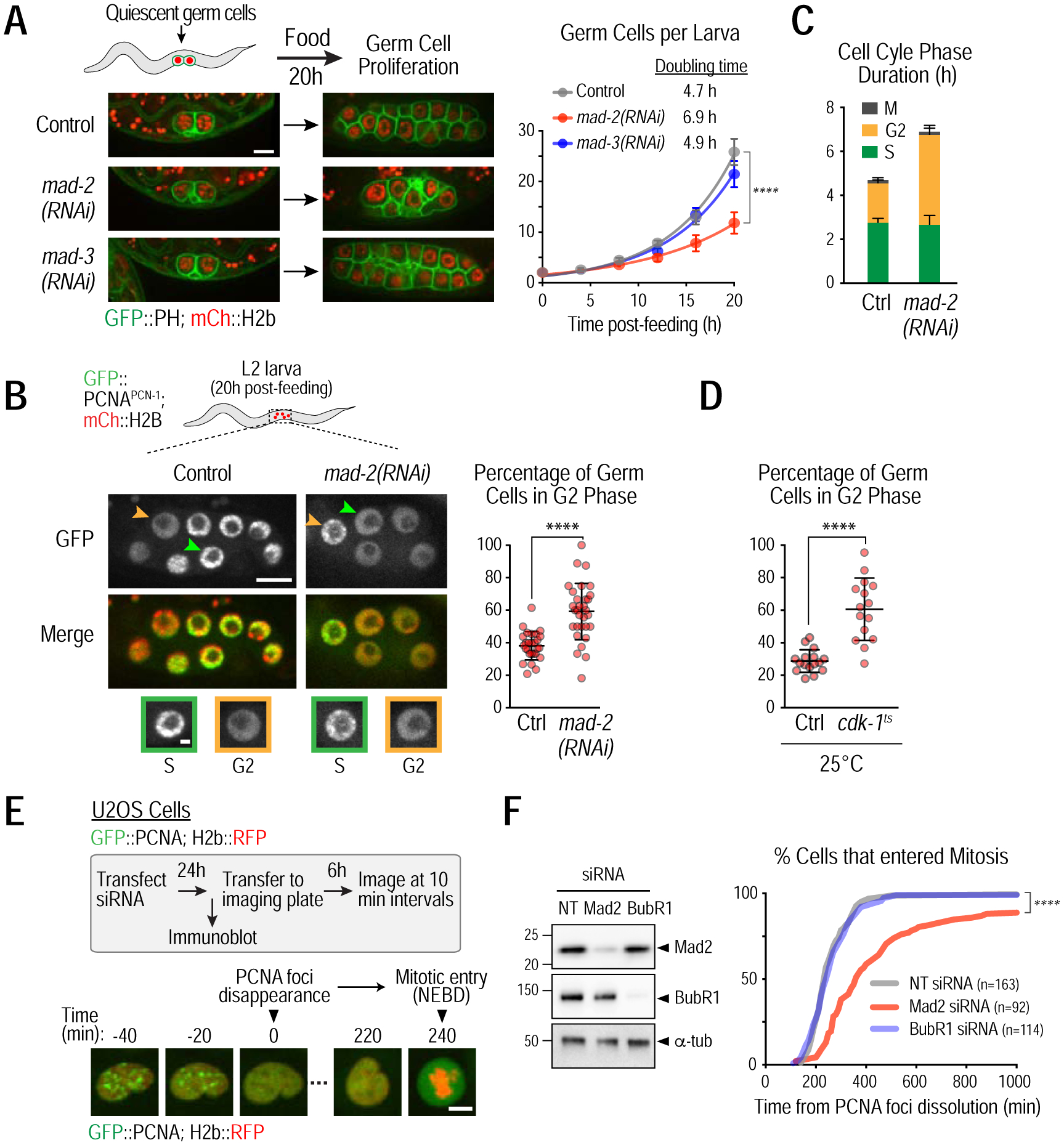
(A) Representative images (left) and quantification (right) of germ cell proliferation in the indicated conditions; germ cells are visualized using plasma membrane (GFP::PH) and chromatin (mCherry::H2b) markers expressed under a germ cell-specific promoter. >17 larvae were analyzed for each time point. Bar, 5 μm. (B) (left) Cell cycle phase analysis of germ cells, based on GFP::PCNAPCN−1 nuclear localization. Representative images of L2 larvae; arrowheads point to S (orange) and G2 (green) phase germ cells that are magnified in insets below. Bar, 5 μm; inset bar, 1 μm. (right) Quantification of proportion of G2 phase nuclei in individual larval germlines. (C) Duration of S and G2 phases calculated based on doubling times from Fig. 2B. (D) Quantification of proportion of G2 phase nuclei in individual larval germlines in the presence or absence of a weakening cdk-1 mutation (cdk-1ts). Experiments were done at the restrictive temperature of 25°C. (E) (top) Schematic of G2-to-M transition analysis in human U2OS cells and (bottom) stills from a time-lapse movie of the nuclear region in a single cell. The time between dissolution of PCNA foci and nuclear envelope breakdown (NEBD) provides a quantitative measure for the G2-to-M transition in single cells. Bar, 10 μm. (F) (left) Immunoblot assessing protein depletion efficiency in human U2OS cells. NT is a non-targeting siRNA used as a control. BubR1 is the human orthologue of MAD-3. α-tubulin serves as a loading control. (right) Percent cells in mitosis as a function of time after PCNA foci dissolution for the indicated conditions. Error bars are 95% confidence intervals (A,C) or standard deviation (B,D). **** represents p<0.0001 from Mann-Whitney tests (A,B,D) or Kolmogorov-Smirnov test (F); non-significant (n.s.) is p>0.05. See also Figure S2.
Depletion of MAD-2 or MAD-3 did not affect germ cell quiescence in larvae held for up to 4 days in the absence of food (not shown). By contrast, MAD-2 depletion led to a significant reduction in the rate of germ cell proliferation, whereas MAD-3 depletion had no effect (Fig. 2A); similar results were obtained using mutants instead of RNAi (Fig. S2B). Exponential fits revealed that MAD-2 depletion increased germ cell doubling time by ~50%, from 4.7h to 6.9h. Thus MAD-2 has a checkpoint signaling-independent function in germ cell proliferation.
Germ cells have a negligible G1 phase and initiate S-phase immediately after nuclei reform in telophase (Fig. S2C; Movie S2) (Fox et al., 2011; Seidel and Kimble, 2015). To determine which cell cycle phase (S, G2 or M) was affected by MAD-2 depletion, we imaged GFP-tagged PCNAPCN−1, which concentrates in replication foci during S-phase but is diffuse in G2 nuclei (Fig. 2B). In control larvae, ~60% of germ cells were in S-phase, and ~40% were in G2; only a minor percentage (3.5%) were in M-phase (Fig. S2D; (Fox et al., 2011)). In mad-2(RNAi) larvae, the proportions were reversed with ~40% of germ cells in S-phase and ~60% in G2 (Figs. 2B; S2D). Combining these numbers with the doubling time measurements indicated that in mad-2(RNAi) the duration of S phase was unchanged whereas G2 duration more than doubled (Fig. 2C). Thus, MAD-2 promotes the G2-to-M transition. In support of this conclusion, a similar phenotype was observed for a temperature-sensitive mutant of CDK-1 (cdk-1(ne2257) (Shirayama et al., 2006) (Figs. 2D; S2E).
To determine if the role of Mad2 in promoting the G2-to-M transition is conserved, we analyzed human U2OS cells expressing fluorescent PCNA and histone H2b; the interval between dissolution of PCNA foci and nuclear envelope breakdown (NEBD) provides a measure for the G2-to-M transition in individual cells (Leonhardt et al., 2000; de Groot et al., 2015) (Fig. 2E). Depletion of Mad2, but not BubR1, the human Mad3 homolog, significantly delayed the G2-to-M transition (Figs. 2F; S2F; Movie S3), while both depletions abolished spindle checkpoint signaling (Fig. S2G). The delay caused by Mad2 depletion was suppressed by expression of siRNA-resistant Myc-Mad2, indicating that it is not due to an off-target effect (Fig. S2H–I). Thus, Mad2 has a conserved role in promoting the G2-to-M transition and this function is distinct from its role in the spindle checkpoint after mitotic entry. Notably, MAD-2 depletion did not affect cell cycle progression during rapid embryonic divisions in C. elegans that cycle between S and M phases (not shown), suggesting that it acts in cell cycles with a defined G2 phase.
Mad2 acts in the cytoplasm in a complex with Mad1 to promote the G2-to-M transition
To determine how Mad2 promotes the G2-to-M transition, we focused on C. elegans germ cells, where loss of the checkpoint function of Mad2 has a relatively minor effect. Mad2 is present in a 2:2 hetero-tetrameric complex with Mad1 and in a free monomeric pool (Chung and Chen, 2002; Sironi et al., 2002; Shah et al., 2004; De Antoni et al., 2005) (Fig. 3A). In addition to localizing to unattached kinetochores in mitosis, the Mad1-Mad2 complex localizes in interphase to nuclear pores, through a direct interaction of Mad1 with the nuclear pore basket component Tpr (NPP-21 in C. elegans) (Chen et al., 1998; Campbell et al., 2001; Lee et al., 2008), and to the nucleoplasm, via a nuclear localization sequence (NLS) in Mad1 (Scott et al., 2005).
Figure 3. Mad1-Mad2 targets Cdc20 in the cytoplasm to promote the G2-to-M transition.
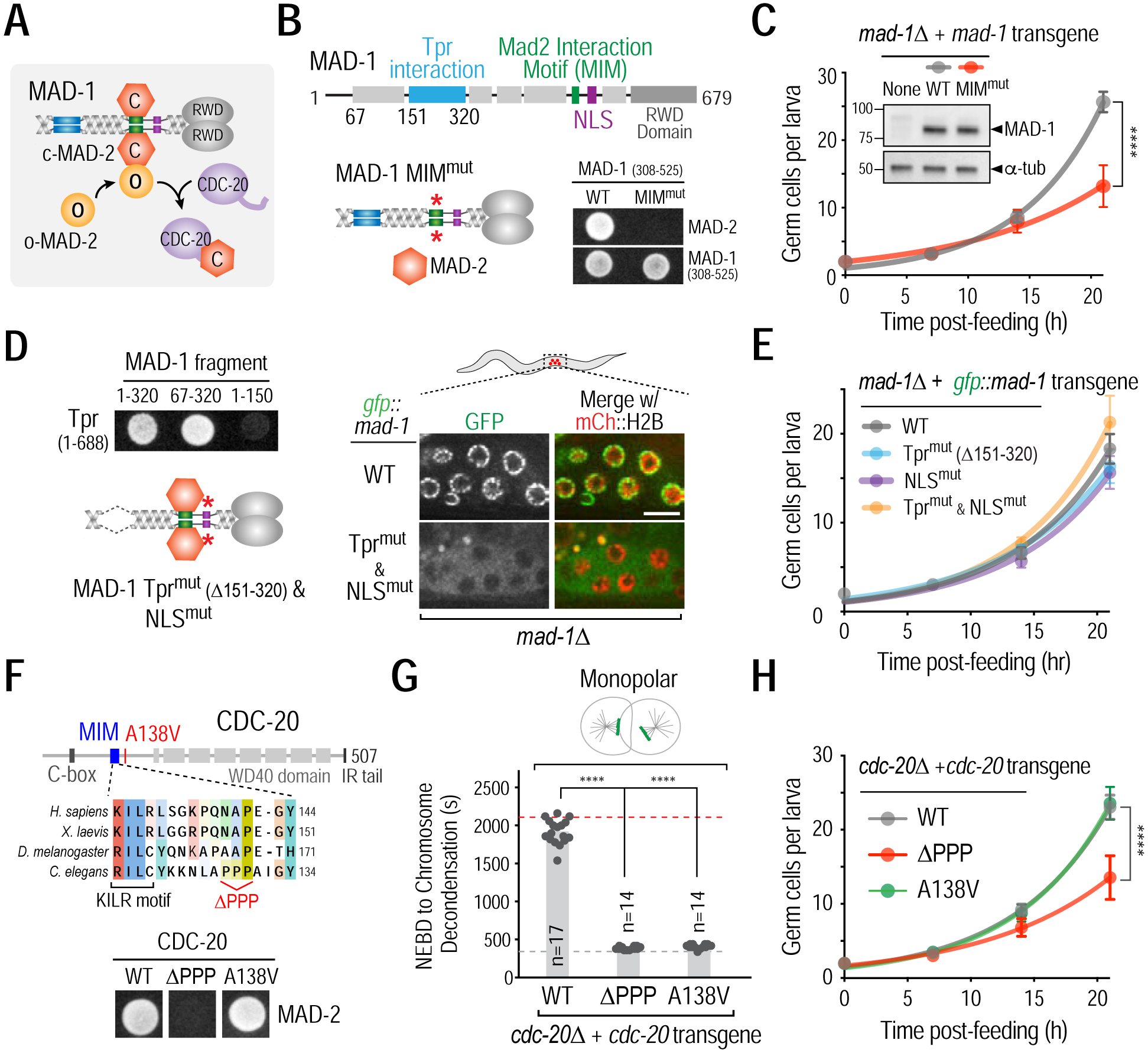
(A) Schematic of Mad1-Mad2-mediated catalysis of Mad2-Cdc20 complex formation. (B) (top) Schematic of MAD-1 primary structure highlighting MAD-2 interaction motif (MIM, green), Tpr interaction domain (sky blue), and nuclear localization signal (NLS, purple). (bottom left) Schematic of MIMmut MAD-1; see Fig. S3A for details on MIM mutant generation; (bottom right) Yeast two-hybrid data showing the effect of mutating the MIM in MAD-1. (C) Effect of mutating MAD-1’s MIM on germ cell proliferation. Inset shows immunoblot of mad-1Δ strains expressing WT or MIMmut MAD-1 from single copy transgene insertions. α-tubulin serves as a loading control. (D) (left top) Yeast two hybrid data mapping the interaction of MAD-1 with Tpr (NPP-21) to residues 151–320 and (left bottom) schematic of MAD-1 mutant engineered to disrupt Tpr interaction and nuclear transport; see Fig. S3A for details on NLS mutant generation; (right) Disruption of MAD-1 nuclear periphery and nuclear interior localization in germ cells by deletion of the Tpr interaction domain and mutation of the NLS. Bar, 5 μm. (E) Effect of disrupting MAD-1 nuclear periphery and/or nuclear interior localization on germ cell proliferation. (F) (top) Schematic of the CDC-20 region important for MAD-2 and APC/C-binding (KILR motif); the ΔPPP mutant selectively disrupts MAD-2 binding. (bottom) Yeast-two hybrid data showing the effect of the CDC-20 ΔPPP mutant on its interaction with MAD-2. (G) & (H) Checkpoint signaling (G) and germ cell proliferation (H) analysis in the indicated CDC-20 mutants. Dashed lines indicate average mitotic duration in bipolar (grey) and monopolar (red) controls. (C,E,H) Germ cell proliferation in 15 or more larvae was quantified at each time point; (G) n is number of embryos scored for mitotic duration. All error bars are 95% confidence intervals. **** represents p<0.0001 from Mann-Whitney tests. See also Figures S3 and S4.
To determine if complex formation between MAD-2 and MAD-1 is required for promoting the G2-to-M transition, we replaced endogenous MAD-1 with a mutant that disrupts the MAD-2 interaction motif (MIM; Fig. 3B; S3A) (Luo et al., 2002; Sironi et al., 2002)). MAD-1 MIMMut localized to unattached kinetochores and to the nuclear periphery similarly to wild-type (WT) MAD-1 (Figs. S3B,C) but was defective in spindle checkpoint signaling (Fig. S3D). Notably, MAD-1 MIMMut reduced the rate of germ cell proliferation (Fig. 3C) and compromised fertility (Fig. S3E) to a similar extent as mad-2Δ (Fig. 1D). By contrast, replacing MAD-1 with a mutant that cannot bind Tpr and lacked the NLS (MAD-1 Tprmut & NLSmut), and therefore failed to localize to the nuclear periphery or interior (Figs. 3D; S3F–I), did not affect the rate of germ cell proliferation (Figs. 3E; S3J). Thus, MAD-2 must interact with MAD-1 to promote the G2-to-M transition and this function can be executed in the cytoplasm.
Mad2 interacts with Cdc20 in order to promote the G2-to-M transition
The Mad1–Mad2 complex interacts with free Mad2 and converts it from an open to a closed conformation that forms a complex with Cdc20 (Fig. 3A) (De Antoni et al., 2005). To test if MAD-2 association with CDC-20, which is expressed in the cytoplasm of germ cells (Fig. S4A), is important for promoting the G2-to-M transition, we engineered a CDC-20 mutant (CDC-20 ΔPPP) that specifically compromised its ability to bind to MAD-2 (Hwang et al., 1998) (Fig. 3F). Replacing endogenous CDC-20 with CDC-20 ΔPPP mutant abrogated spindle checkpoint signaling without affecting normal mitotic duration, indicating that CDC-20 ΔPPP retained the ability to normally activate the APC/C (Figs. 3G; S4B,C). CDC-20 ΔPPP reduced the rate of germ cell proliferation and caused a fertility defect that mimicked mad-2 loss (Figs. 3H; S4D). By contrast, a different CDC-20 mutant (CDC-20 A138V; Stein et al., 2007; Kim et al., 2017) that disrupts spindle checkpoint signaling but maintains the ability to interact with MAD-2, did not affect the rate of germ cell proliferation (Figs. 3F–H; S4B–D). Thus, the ability of MAD-2 to promote the G2-to-M transition requires MAD-2 to bind to CDC-20.
The ability of the Mad1-Mad2 complex to catalyze the conformational change in Mad2 that promotes Mad2-Cdc20 complex formation involves dimerization of free Mad2 with Mad1-bound (Fig. 3A; (De Antoni et al., 2005)). When expressed in the absence of endogenous MAD-2, a mutant version of MAD-2 that cannot dimerize (the MAD-2 R133E; Q134A mutant, De Antoni et al., 2005) resulted in severe fertility and viability defects (Figs. S4E–H). Thus, a MAD-1–MAD-2 template-mediated formation of a MAD-2–CDC-20 complex is important for a normal G2-to-M transition.
The Mad1-Mad2 complex promotes the G2-to-M transition by inhibiting APC/CCdc20
MAD-2 binding to CDC-20 interferes with an APC/C binding region in CDC-20 called the KILR motif (Tang et al., 2001; Fang, 2002; Izawa and Pines, 2012) (Fig. 3F). This fact suggests that MAD-1–MAD-2 promotes the G2-to-M transition by preventing CDC-20 from associating with and activating the APC/C. A prediction of this model is that reducing APC/C activity should alleviate the effect of MAD-2 inhibition on the G2-to-M transition.
To test this prediction, we analyzed temperature-sensitive mutants in two different APC/C subunits, Apc8ts (mat-3(or344)) (Rappleye et al., 2002) and Apc6ts (emb-37(g48)) (Golden et al., 2000) (Fig. 4A), as well as a mutant of CDC-20 in its WD40 domain, D433N, which reduces APC/CCDC−20 activity (Kitagawa et al., 2002). Compromised APC/C function in all 3 mutants was evident from prolonged mitotic duration in one-cell embryos (Fig. 4A). All 3 mutants suppressed the germ cell proliferation defect resulting from MAD-2 depletion (Fig. 4B); the Apc8ts and Apc6ts mutants also partially suppressed the mad-2Δ fertility defect (Fig. S5A). Thus, the MAD-1–MAD-2 complex promotes the G2-to-M transition by catalyzing formation of a MAD-2–CDC-20 complex and suppressing APC/C activity, likely by sequestering CDC-20 to limit formation of active APC/CCDC−20 complexes.
Figure 4. Mad2 inhibits APC/CCdc20 to promote cyclin B accumulation.
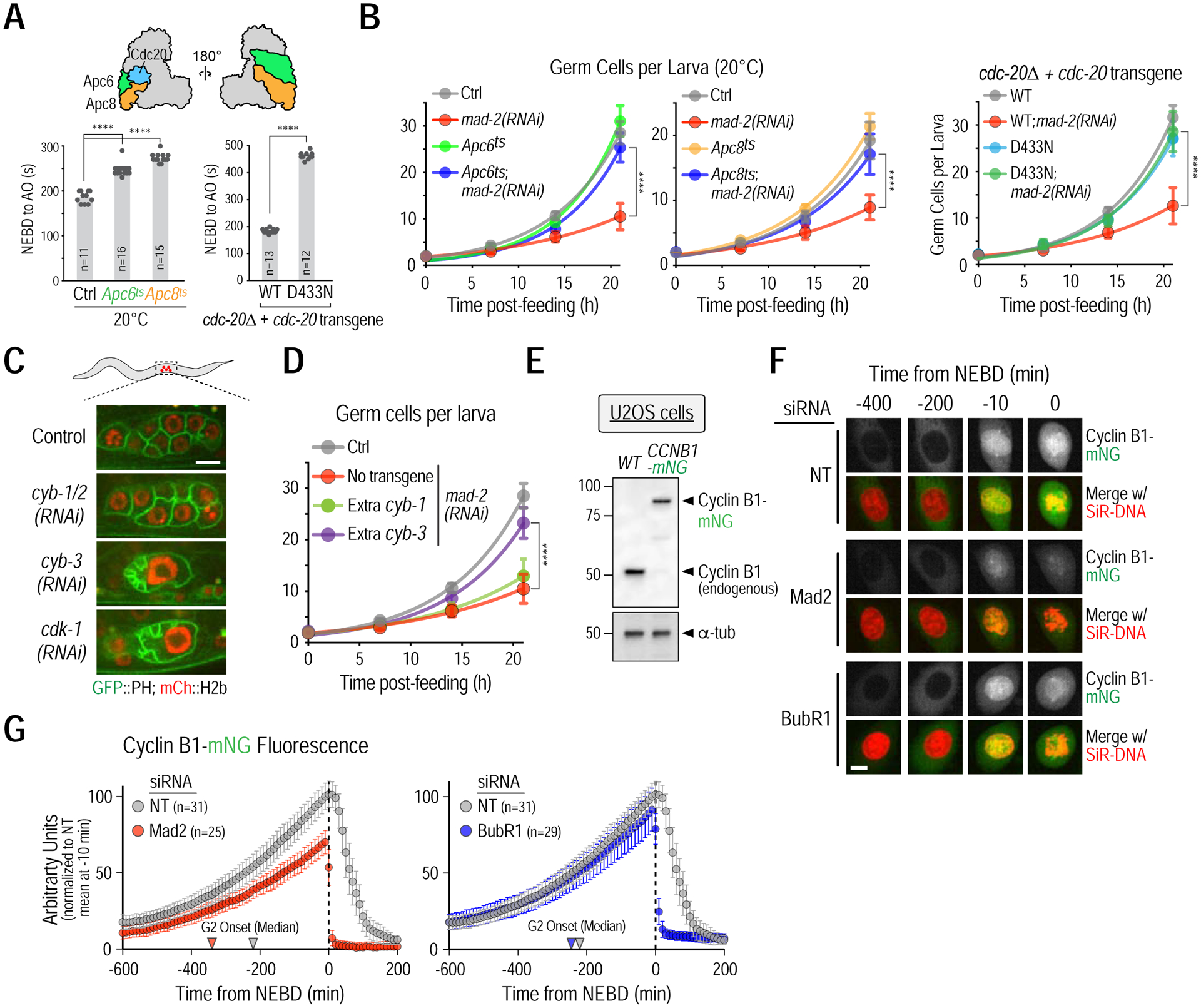
(A) (top) Schematic highlighting the two structural APC/C subunits, Apc6 and Apc8, and the co-activator Cdc20, mutations in which were used to weaken APC/CCDC−20 activity. (bottom) NEBD-anaphase onset (AO) timing in one-cell embryos for the indicated conditions. (B) Germ cell proliferation analysis for the indicated conditions. (C) Effect of cyclin B isoform depletions, conducted by soaking larvae in dsRNA, on germ cell proliferation. Due to high sequence homology, the dsRNA used to deplete cyb-1 is also expected to target cyb-2, hence it is refered to as cyb-1/2(RNAi). Bar, 5 μm. (D) Effect of doubling dosage of cyb-1 or cyb-3, using functional single copy transgene insertions. Data for control and mad-2(RNAi) are the same as in panel B, left. (E) Cyclin B1 immunoblot of the indicated U2OS cell line extracts; α-tubulin serves as a loading control. (F) Representative images of Cyclin B1-mNeonGreen accumulation for the indicated conditions. Bar, 10 μm. (G) Quantification of total cyclin B1-mNeonGreen fluorescence over time. Arrowheads point to the median G2 onset time from Fig. 1G. All error bars are 95% confidence intervals. Asterisks represent p<0.0001 from Mann-Whitney tests; non-significant (n.s.) is p>0.05. n is the number of embryos or cells analyzed. >14 larvae were analyzed in germ cell proliferation assays. See also Figure S5.
Mad1-Mad2 protects Cyclin B during its accumulation in G2
To determine if MAD-1–MAD-2 promoted the G2-to-M transition by protecting cyclin B from APC/CCdc20-mediated degradation during G2, we first identified the cyclin B isoform that promotes mitotic entry in C. elegans germ cells. C. elegans has 3 cyclin Bs: cyclin B1 (CYB-1), cyclin B2 (CYB-2, encoded by two closely related genes cyb-2.1 and cyb-2.2) and cyclin B3 (CYB-3) (Kreutzer et al., 1995; van der Voet et al., 2009). Inhibition of CYB-3, but not of CYB-1 and CYB-2, mimicked loss of CDK-1 (Fig. 4C), indicating that CYB-3 is the dominant cyclin B required for mitotic entry in the germline (Green et al., 2011; Yoon et al., 2012).
The sensitivity of germ cell divisions to larval immobilization methods necessary for live imaging precluded quantitative comparison of CYB-3 accumulation in control and MAD-2-depletions. Therefore, as an alternative approach, we tested if providing extra CYB-3 bypassed the need for MAD-1–MAD-2. A single copy transgene expressing CYB-3, under control of endogenous regulatory elements (Fig. S5C), rescued the lethality of a cyb-3 null mutant (Figs. S5D,E). When introduced into a wildtype background, the extra CYB-3 suppressed the germ cell proliferation defect of mad-2(RNAi) (Fig. 4D); extra CYB-3 also partially suppressed the fertility defect of mad-2Δ (Fig. S5F). By contrast, extra CYB-1 had no effect (Figs. 4D; S5C–F). These data are consistent with duplication of cyb-3 acting as a weak suppressor of mad-1Δ (Tarailo-Graovac et al., 2010).
In contrast to germ cells in motile C. elegans larvae, live imaging of cyclin B is feasible in human cells (Clute and Pines, 1999). In U2OS cells, we in situ-tagged cyclin B1 (encoded by CCNB1), the cyclin B isoform essential for mitotic entry in mammals, with mNeongreen (mNG) (Figs. 4E; S5G,H). In control cells, Cyclin B1-mNG steadily accumulated until NEBD and then declined as cells exited mitosis (Figs. 4F,G; Movie S4). The rate of cyclin B1 accumulation was significantly reduced in Mad2-depleted cells, which entered mitosis with ~70% of cyclin B1 levels at NEBD in control cells (Figs. 4F,G; Movie S4). These observations are reminiscent of a previous report that used Mad1 knockout RPE-1 cells (Rodriguez-Bravo et al., 2014). By contrast, depletion of BubR1 had no significant effect on cyclin B1 accumulation prior to NEBD, even though both BubR1 and Mad2 depletions resulted in equivalently rapid and premature degradation of cyclin B1 after mitotic entry (Figs. 4F,G; Movie S4). Collectively, the analysis in C. elegans germ cells and in human cells indicates that Mad1–Mad2 suppress APC/CCdc20 activity in order to protect cyclin B during its accumulation prior to the G2-to-M transition.
Cyclin-dependent kinase phosphorylation of Cdc20 acts in parallel to Mad1-Mad2 to enable the G2-to-M transition
The above data show that, while the Mad1-Mad2 complex plays a conserved role in promoting the G2-to-M transition, its inhibition delays but does not block mitotic entry. This observation suggested that there is an additional mechanism that helps restrain APC/CCdc20 in G2. Cdc20 phosphorylation by Cdk1/2 is one such potential mechanism, as phosphorylation reduces the binding affinity of Cdc20 for the APC/C (Fig. 5A) (Yudkovsky et al., 2000; Labit et al., 2012; Hein and Nilsson, 2016; Kim et al., 2017). The Cdk phosphorylation sites in Cdc20 are conserved and are located near the C-box, a key APC/C binding motif (Labit et al., 2012). In prior work we showed that CDC-20 was phosphorylated at three conserved sites in C. elegans, and that replacement of CDC-20 with a mutant (3A) that prevents phosphorylation significantly accelerated APC/C activation after NEBD in the early embryo (Kim et al., 2017).
Figure 5. Cdc20 phosphorylation and Mad2 act in parallel to suppress APC/CCdc20 activation in G2.
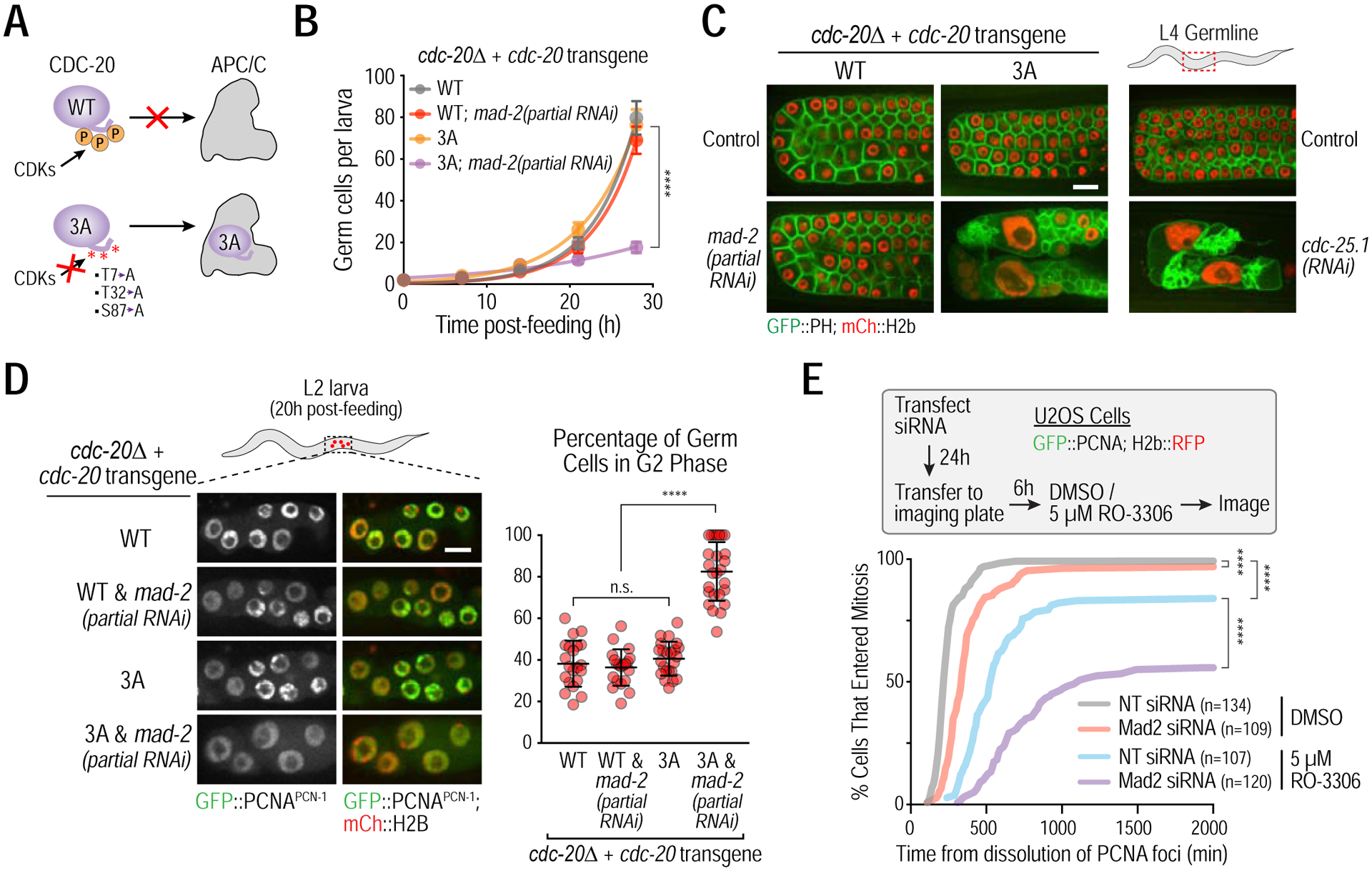
(A) Summary of the effect of Cdc20 phosphorylation by CDKs on its recruitment to the APC/C. 3A corresponds to a non-phosphorylatable CDC-20 mutant (Kim et al, 2017). (B) Germ cell proliferation analysis. As the CDC-20 phosphorylation mutant is synthetic lethal with spindle checkpoint inhibition in embryos, L1 larvae were subjected to post-hatching RNAi that results in partial MAD-2 inhibition. >15 larvae were quantified for each time point in germ cell proliferation analysis. Error bars are 95% confidence intervals. (C) Representative images of L4 larval stage germlines for the indicated conditions. Bar, 10 μm. (D) (left) Representative images and (right) plot of percentage of germ cells in G2, measured by GFP::PCNAPCN−1 imaging, 20h post-feeding of L1 larvae. Error bars are standard deviation. (E) (top) Experiment schematic and (bottom) plots of cumulative G2-to-M transition in U2OS cells as a function of time after PCNA foci dissolution for the indicated conditions. **** represents p<0.0001 from Mann-Whitney tests (B,D) or Kolmogorov-Smirnov test (E); non-significant (n.s.) is p>0.05. See also Figure S6.
To determine if Cdc20 phosphorylation acts in concert with Mad1–Mad2 to restrain APC/C activity in G2, we used C. elegans strains in which the sole source of CDC-20 was the non-phosphorylatable 3A mutant (Kim et al., 2017). To avoid the synthetic lethality that is observed when CDC-20 3A is combined with spindle checkpoint inhibition in embryos (Kim et al., 2017), we subjected L1 larvae to a post-hatching RNAi protocol that results in partial MAD-2 inhibition. Loss of CDC-20 phosphorylation or partial MAD-2 inhibition did not significantly affect germline development; however, combining the two perturbations led to a near-complete blockade of germ cell proliferation and was associated with a significant increase in nuclear size (Figs. 5B; S6A). Analysis of later larval stage germlines showed that combining CDC-20 3A with partial MAD-2 depletion resulted in a defect that was comparable to inhibition of essential mitotic entry factors such as CDC-25, CDK-1 and CYB-3 (Figs. 5C; S6B). A characteristic feature of this phenotype is giant nuclei, which likely arise from endoreduplication after failed mitotic entry (Figs. 5C; S6B) (Edgar and Orr-Weaver, 2001; Green et al., 2011). Analysis using the GFP::PCNAPCN−1 reporter indicated that the combined inhibition resulted in a high proportion of cells arrested in G2 (Fig. 5D). As expected based on MAD-3 not being required to promote the G2-to-M transition, MAD-3 inhibition did not synergize with loss of CDC-20 phosphorylation (Fig. S6C).
The next question we addressed was which cyclin-Cdk complex targets CDC-20 to promote the G2-to-M transition in germ cells. CYB-3–CDK-1 could promote its own accumulation by phosphorylating CDC-20 as part of a feedback loop or a different cyclin-Cdk complex could be responsible for CDC-20 phosphorylation. An attractive candidate for the cyclin in the kinase complex targeting CDC-20 in germ cells is CYB-1, which is expressed in germ cells (Fig. S6D) but whose inhibition has little effect on germ cell proliferation (Figs. 4C; S6E). In support of this idea, CYB-1 inhibition phenocopied the CDC-20 3A mutant when combined with partial MAD-2 inhibition (Fig. S6E).
Cdc20 removal and replacement is challenging in human cells, with depletion sufficient to block APC/C activation being difficult to achieve (Wolthuis et al., 2008). We therefore tested if partial Cdk1 inhibition synergized with Mad2 depletion, using the small molecule inhibitor RO-3306 (Vassilev et al., 2006) (Fig. 5E). Monitoring the G2-to-M transition in single cells (Fig. 2E), we found that 5 μM RO-3306 delayed entry into mitosis and caused a small fraction (~15%) of cells that dissolved PCNA foci not entering mitosis during the filming session. The effect of RO-3306 was significantly enhanced by Mad2 depletion, with ~40% of cells not entering mitosis and the remainder exhibiting a much greater delay than RO-3306 treatment alone (Fig. 5E). While this result does not directly implicate Cdc20 regulation by Cdk1 in human cells, it supports the notion that Mad2 acts in parallel with a Cdk1 activity-dependent mechanism to promote the G2-to-M transition.
DISCUSSION
The results shown here highlight two distinct mechanisms that target Cdc20 and act in concert to enable cyclin B accumulation and promote the G2-to-M transition. The first mechanism involves the Mad1-Mad2 complex and the second phosphorylation of Cdc20 by cyclin-dependent kinases (Fig. 6A). The findings lead to a model in which Cdc20 phosphorylation and Mad2 binding cooperatively reduce affinity of Cdc20 for the APC/C in G2 by targeting two conserved regions, the C-box and the KILR motif, that are employed by Cdc20 to bind to distinct parts of the Apc8B subunit of the APC/C (Fig. 6B) (Zhang et al., 2016). Mad2 binds adjacent to and sterically blocks the KILR motif whereas Cdc20 phosphorylation electrostatically inhibits interaction of the C-box with Apc8B (Izawa and Pines, 2012; Labit et al., 2012). This coordinated action prevents formation of active APC/CCdc20 complexes, thereby protecting cyclin B during its synthesis and enabling the G2-to-M transition. These findings illuminate a poorly understood feature of the eukaryotic cell cycle and identify a new conserved function for a central component of the spindle checkpoint pathway.
Figure 6. A dual mechanism targets APC/C binding motifs of Cdc20 to promote cyclin B accumulation and mitotic entry.
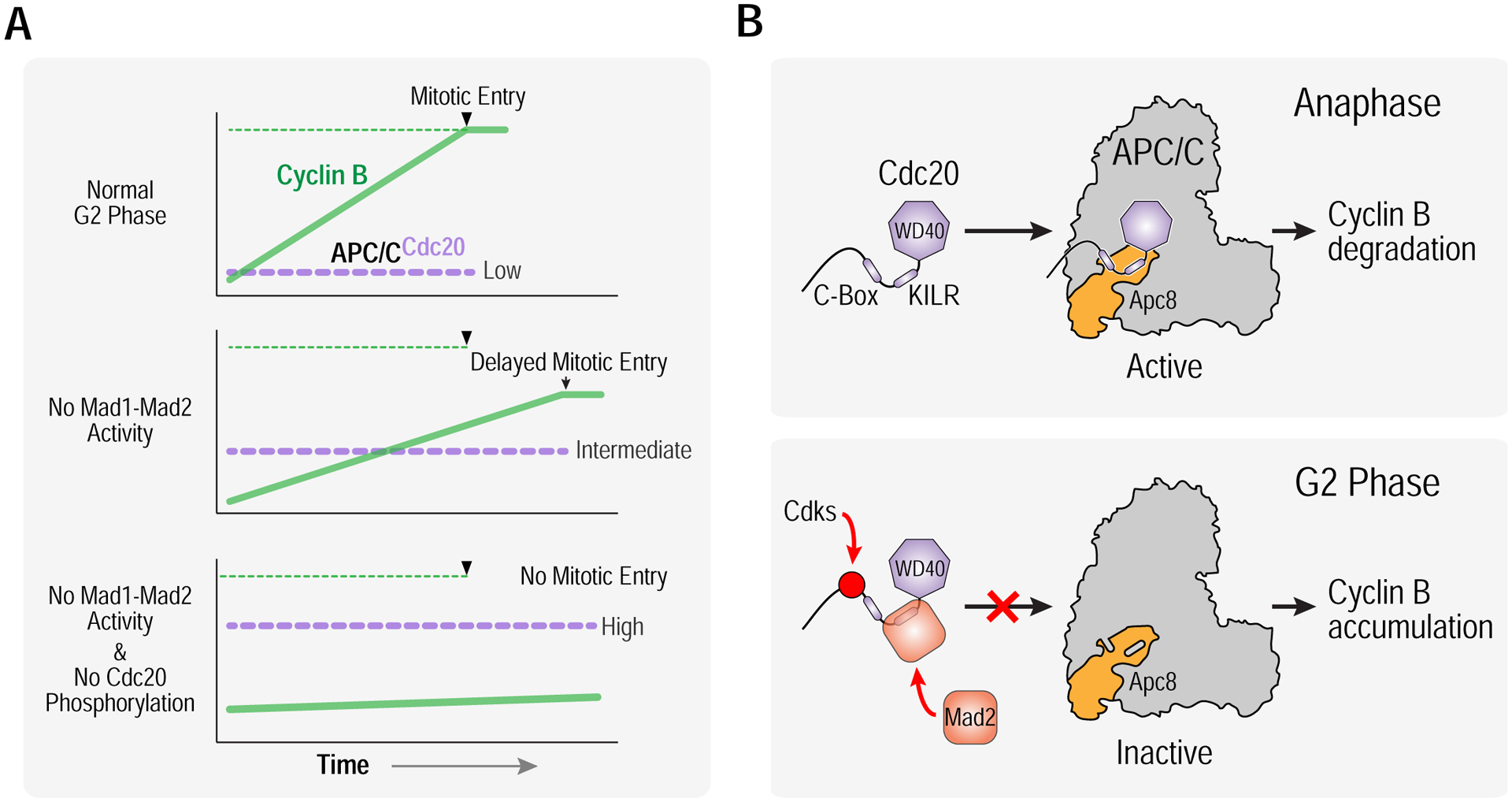
(A) Schematics highlighting normal G2 phase with cyclin B accumulation and transition into mitosis (top), delayed cyclin B accumulation with an extended G2 phase and entry into mitosis at reduced cyclin B levels in the absence of Mad1/Mad2 activity (middle), and a block to mitotic entry in the absence of both Mad1-Mad2 activity and Cdc20 phosphorylation by Cdks (bottom). (B) (top) Cdc20 binds and activates APC/C to degrade cyclin B at anaphase onset, employing its C-box and KILR motif to bind to distinct sites on the Apc8 subunit. (bottom) To keep APC/C in check during G2 phase, Mad1-Mad2 catalyzes Mad2 binding to the KILR motif and Cdks phosphorylate resides adjacent to the C-box, thereby coordinately suppressing binding affinity of Cdc20 for the APC/C, preventing the formation of active APC/CCdc20 complexes and allowing cyclin B to accumulate and trigger mitotic entry.
Distinct roles for Mad1-Mad2 in mitotic entry and in the spindle checkpoint
Using C. elegans and human cultured cells, we show that Mad1-Mad2 promotes mitotic entry by restricting the ability of APC/CCdc20 to target cyclin B for degradation. The Mad1-Mad2 reaction that promotes mitotic entry has different requirements from the Mad1-Mad2-based spindle checkpoint pathway. Mad1-Mad2 function in the spindle checkpoint requires docking onto kinetochores and/or nuclear pores, where kinase activities catalyze the formation of a Mad2-Cdc20 complex that associates with Mad3(BubR1)-Bub3 to produce the mitotic checkpoint complex, a potent inhibitor of already-active APC/CCdc20 (Musacchio, 2015; Corbett, 2017). In contrast, we show that the of Mad1-Mad2 in promoting the G2-to-M transition does not require kinetochore/nuclear pore docking or formation of the mitotic checkpoint complex and most likely acts by sequestering Cdc20 from forming active holocomplexes with the core APC/C (Fig. 6B).
Our data do not support the idea that an interphase form of the mitotic checkpoint complex is needed to inhibit APC/CCdc20 prior to the G2-to-M transition (Rodriguez-Bravo et al., 2014). This is evident from imaging of in situ-tagged cyclin B1 in human cells, where cyclin B1 levels prior to NEBD were similar between control and BubR1 depletion, even though cyclin B1 was immediately degraded following mitotic entry in the latter (Fig. 4G). Our findings also challenge a prior study where the N-terminus of BubR1, but not Mad2, was suggested to control cyclin B accumulation during mitotic entry in mouse embryonic fibroblasts (Malureanu et al., 2009); while the precise reasons for the difference will require future independent efforts to clarify, our results are supported by analysis in the highly divergent contexts of C. elegans germ cells and human cultured cells; in addition, we employed in situ-tagged cyclin B, in contrast to transfected cyclin B fusions in the prior study, which enabled rigorous comparison between different perturbations. While MCC containing BubR1 does exist at low levels prior to mitosis (e.g. Kim et al., 2018), we speculate that in interphase, where the affinity of Cdc20 for the APC/C is low, Mad2 is sufficient to sequester Cdc20, whereas the high affinity of Cdc20 for the APC/C in mitosis due to APC/C phosphorylation (see below) requires the full MCC to efficiently inhibit active APC/CCdc20.
In the spindle checkpoint, concentration of Mad1-Mad2 in the vicinity of kinase activities at the kinetochore accelerates the conformational transition of free Mad2 promoted by the Mad1-Mad2 complex (Kulukian et al., 2009; Faesen et al., 2017). Our results suggest that, prior to mitotic entry, Mad1-Mad2 complexes are acting in the cytoplasm to promote a similar association of free Mad2 with Cdc20. Inhibition of Mps1, the kinase activating Mad1-Mad2 in human cells at the kinetochore, does not affect the G2-to-M transition (R. Kabeche & A. Desai, unpublished data). Thus, either a different kinase activity is involved or the basal uncatalyzed rate of Mad2’s conformational transition by Mad1-Mad2 is sufficient to promote the G2-to-M transition. It will be important to elucidate in future work if and how the Mad1-Mad2 complex is regulated to promote the G2-to-M transition.
Direct targeting of Cdc20 is likely essential for the G2-to-M transition in cells with a defined G2 phase
In addition to Mad1-Mad2 catalyzed Mad2/Cdc20 complex formation, Cdc20 phosphorylation by Cdks, which reduces the affinity of its C-box-mediated interaction with the Apc8B subunit of the APC/C (Labit et al., 2012), is important to prevent APC/C activation prior to mitotic entry. Our data in C. elegans germ cells highlight the existence of a regulatory relay mechanism, in which a Cdk1-cyclin B1 complex phosphorylates Cdc20 to promote accumulation of cyclin B3, which is the major Cdk1 activator for mitotic entry in this tissue. In mammalian cells, where mitotic entry is driven by cyclin B1, cyclin A2-Cdk2, active in late S and G2 phases, has been proposed to be the kinase complex that phosphorylates Cdc20 (Hein and Nilsson, 2016), suggesting the existence of a conceptually similar relay mechanism. The severity of the defect in the G2-to-M transition when both Mad1–Mad2 and Cdc20 phosphorylation are simultaneously inhibited highlights the importance of keeping Cdc20 from activating the APC/C in order to enable entry into mitosis.
Recent work has identified a different mechanism that controls APC/CCdc20 activity that is intrinsic to the APC/C (Morgan, 2016). In this mechanism, a loop region of Apc1 limits binding of the C-box of Cdc20 until Cdk activity-dependent phosphorylation causes loop displacement (Fujimitsu et al., 2016; Qiao et al., 2016; Zhang et al., 2016). Our work suggests that, while this mechanism is likely sufficient for coupling APC/CCdc20 activation to Cdk activity in rapid embryonic divisions that alternate between S and M phases, it does not restrain APC/CCdc20 activation to a sufficient extent to enable the G2-to-M transition in C. elegans germ cells and human somatic cells that have a defined G2 phase. We speculate that an additional level of control is required in cells with a G2 phase because of Cdk-cyclin activity that is present, e.g. cyclin A2-Cdk2 in human cells (Yam et al., 2002) and cyclin B1-Cdk1 in C. elegans germ cells (Figs. S6D,E), that relieves the APC/C-intrinsic autoinhibition mechanism. Thus, to enable cyclin B accumulation in G2, it becomes essential to prevent Cdc20 from binding to the APC/C. In rapid embryonic cycles, Cdk activity (comprised of Cdk1-cyclin B) is only prominent in M-phase, thereby making the APC/C-intrinsic mechanism sufficient. Integration of the two mechanisms we describe here with genetic perturbation of the Cdk-mediated relief of APC/C autoinhibition will be an important future goal.
Our work does not address how APC/C activated by its other co-activator, Cdh1/Hct1/Fizzy-related (Schwab et al., 1997; Visintin et al., 1997), is suppressed during mitotic entry. As Mad2 does not bind to Cdh1, it is unlikely to directly control APC/CCdh1 activity. Emi1 and Cdk phosphorylation have both been suggested to inhibit APC/CCdh1 (Zachariae et al., 1998; Jaspersen et al., 1999; Grosskortenhaus and Sprenger, 2002). Notably, Emi1 inhibition of APC/CCdh1 is important to prevent re-replication (Di Fiore and Pines, 2007; Machida and Dutta, 2007), highlighting that APC/CCdh1 is suppressed during S and G2 phases. However, Emi1 is not conserved in species like C. elegans, suggesting that Cdk phosphorylation is also a potentially conserved mechanism suppressing APC/CCdh1, a possibility that will need to be addressed in future work.
In summary, we establish here the existence of two distinct mechanisms acting on Cdc20 to protect cyclin B during its accumulation and thereby enable the G2-to-M transition in the eukaryotic cell cycle. The fact that cells can still enter mitosis following loss of either mechanism and that these mechanisms likely do not operate in rapid embryonic divisions lacking a G2 phase, explains why they were not appreciated in prior efforts. Notably, one of the mechanisms we uncover involves a core spindle checkpoint complex acting outside of the context of chromosome segregation during mitosis. The mechanisms we describe are likely to broadly control cell division cycles with a defined G2 phase, during which genome integrity is ensured prior to commitment to mitosis.
STAR METHODS
Contact For Reagent And Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Arshad Desai (abdesai@ucsd.edu).
Experimental Model And Subject Details
C. elegans strains
C. elegans strains were maintained in nematode growth media (NGM) plates seeded with the Escherichia coli OP50 strain at 20°C, with the exception of tempera ture-sensitive mutant for cdk-1 (cdk-1(ne2257)) that was maintained at 16°C. The list of strains used in this study is in the Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti MAD-2 | (Essex et al., 2009) | OD72 |
| Rabbit polyclonal anti MAD-3 | This study | OD219-b |
| Rabbit polyclonal anti MAD-1 (aa 430–679) | (Moyle et al., 2014) | OD195-b |
| Rabbit polyclonal anti Cyclin B1 | Cell Signaling | Cat #12231S; RRID AB_2783553 |
| Rabbit polyclonal anti CDC-20 (aa 1–160) | (Kim et al., 2017) | OD220-b |
| Mouse monoclonal anti alpha-tubulin (clone DM1A) | Sigma-Aldrich | Cat #T9026; RRID AB_477593 |
| Rabbit polyclonal anti-Mad2 | Bethyl Laboratories | Cat #A300–301A; RRID AB_2281536 |
| Sheep polyclonal anti-Mad2 | (Tighe et al., 2008) | SM2.2 |
| Rabbit polyclonal anti BubR1 | (Kim et al., 2018) | N/A |
| Goat anti rabbit IgG, HRP-conjugated | Jackson Immunoresearch | Cat #111-035-003; RRID AB_2313567 |
| Donkey anti mouse IgG, HRP-conjugated | Jackson Immunoresearch | Cat #715-035-150; RRID AB_2340770 |
| Goat anti-mouse IgG, AP-conjugated | Promega | Cat #S372B; RRID AB_430871 |
| Donkey anti-sheep IgG, HRP-conjugated | Thermo Fischer | Cat #A16041; RRID AB_2534715 |
| Bacterial and Virus Strains | ||
| Lentivirus: PGK promoter-Myc-Mad2-P2A-BSD | This study | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| RO-3306 | (Vassilev et al., 2006) | Sigma-Aldrich Cat #217699–5MG |
| Experimental Models: Cell Lines | ||
| U2OS | ATCC | HTB-96 |
| U2OS GFP-PCNA and H2b-RFP | (de Groot et al., 2015) | ODCL0033 |
| U2OS GFP-PCNA and H2b-RFP; Myc-Mad2 | This study | ODCL0094 |
| U2OS CCNB1-mNeoNGreen | This study | ODCL0034 |
| Experimental Models: Organisms/Strains | ||
| C. elegans N2 Bristol | Caenorhabditis Genetics Center | N2 |
| emb-27(g48ts)II | Caenorhabditis Genetics Center | GG48 |
| mat-3(or344)III | Caenorhabditis Genetics Center | HY601 |
| unc-119(ed3) ltIs38 [pAA1; pie-1/GFP::PH(PLC1delta1); cb-unc-119 (+)]III; ltIs37 [pAA64; pie-1/mCherry::his-58; cb-unc- 119 (+)]IV | (Green et al., 2011) | OD95 |
| mdf-1(gk2)V/nT1(qIs51)(IV;V) | (Moyle et al., 2014) | OD738 |
| ltSi219[pOD1248/pSW076; Pmex-5::GFP-PH(PLC1delta1)-operon-linker-mCherry-his-11; cb-unc-119(+)]I; unc-119(ed3)III | (Wang et al., 2015) | OD866 |
| ltSi310[pOD1577/pMM7C; Pmdf-1::mdf-1::mdf-1 3’UTR; cb-unc-119(+)]II; unc-119(ed3)?III; mdf-1(gk2)V | (Moyle et al., 2014) | OD1051 |
| ltSi608[pOD1583/pMM30; pmdf-1::GFP::mdf1::mdf-1 3’UTR; cb-unc-119(+)]II; unc-119(ed3)?III; mdf-1(gk2)V | (Moyle et al., 2014) | OD1208 |
| ltSi608[pOD1583/pMM30; pmdf-1::GFP::mdf1::mdf-1 3’UTR; cb-unc-119(+)]II; unc-119(ed3)?III; ltIs37 [pAA64; pie-1/mCherry::his-58; unc-119 (+)] IV; mdf-1(gk2) V | (Moyle et al., 2014) | OD1209 |
| unc-119(ed3)III; ltSi560[oxTi365; pPLG014; Pmex-5::GFP::his-11::tbb-2_3’UTR, tbg-1::gfp::tbb-2_3’UTR; cb-unc-119(+)]V | (Kim et al., 2015) | OD1702 |
| emb-27(g48)II; unc-119(ed3)?III; ltSi560 [oxTi365; pPLG014; Pmex-5::GFP::his-11::tbb-2_3’UTR, tbg-1::gfp::tbb-2_3’UTR; cb-unc-119(+)]V | This study | OD1900 |
| unc-119(ed3?) mat-3(or344)III; ltSi560 [oxTi365; pPLG014; Pmex-5::GFP::his-11::tbb-2_3’UTR, tbg-1::gfp::tbb-2_3’UTR; cb-unc-119(+)]V | (Kim et al., 2017) | OD2003 |
| ltSi661[pPLG029; Pmdf-1::mdf-1(Q496A, I497A, F498A, H499A, M500A)::mdf-1 3’UTR; cb-unc-119(+)]II; unc-119(ed3)?III mdf-1(gk2) V/nT1(qIs51) (IV;V) | This study | OD2061 |
| ltSi677 [pPLG034; Pmdf-1::GFP::mdf-1(delta 151–320)::mdf-1 3’UTR; cb-unc-119(+)]II; unc-119(ed3)?III; mdf-1(gk2)V/nT1(qIs51)(IV;V) | This study | OD2156 |
| ltSi677 [pPLG034; Pmdf-1::GFP::mdf-1(delta 151–320)::mdf-1 3’UTR; cb-unc-119(+)]II; unc-119(ed3)?III; ltIs37 [pAA64; pie-1/mCherry::his-58; unc-119 (+)]IV; mdf-1(gk2)V/nT1(qIs51)(IV;V) | This study | OD2157 |
| unc-119(ed3)?III; mdf-2(lt4::loxP::cb-unc-119(+)::loxP)IV | This study | OD2171 |
| san-1(lt6::loxP::cb-unc-119(+)::loxP)I; unc-119(ed3)?III | This study | OD2172 |
| unc-119(ed3)?III; mdf-2(lt4::loxP::cb-unc-119(+)::loxP)IV/nT1(qIs51)IV;V | This study | OD2174 |
| san-1(lt6::loxP::cb-unc-119(+)::loxP)I; unc-119(ed3)?III/hT2[bli-4(e937) let-?(q782) qIs48](I;III) | This study | OD2175 |
| unc-119(ed3?) mat-3(or344)III; mdf-1(gk2) ltSi560[oxTi365; pPLG014; Pmex-5::GFP::his-11::tbb-2_3’UTR, tbg-1::gfp::tbb-2_3’UTR; cb-unc-119(+)]V/nT1[qIs51](IV;V) | This study | OD2236 |
| ltSi587[pPLG024; Pmdf-2::mdf-2 delta intron 4::mdf-2 3’UTR; cb-unc-119(+)]II; unc-119(ed3)?III; mdf-2(lt4::loxP::cb-unc-119(+)::loxP)IV/nT1(qIs51)IV;V | This study | OD2300 |
| ltSi808[pPLG044; Pmdf-2::mdf-2 delta intron 4 R133E, Q134A::mdf-2 3’UTR; cb-unc-119(+)]II; unc-119(ed3)?III; mdf-2(lt4::loxP::cb-unc-119(+)::loxP)IV/nT1(qIs51)IV;V | This study | OD2453 |
| ltSi310[pOD1577/pMM7C; Pmdf-1::mdf-1::mdf-1 3’UTR; cb-unc-119(+)]II; unc-119(ed3?) mat-3(or344)III; mdf-1(gk2) ltSi560[oxTi365; pPLG014; Pmex-5::GFP::his-11::tbb-2_3’UTR, tbg-1::gfp::tbb-2_3’UTR; cb-unc-119(+)]V/nT1[qIs51](IV;V) | This study | OD2454 |
| ltSi661[pPLG029; Pmdf-1::mdf-1(Q496A, I497A, F498A, H499A, M500A)::mdf-1 3’UTR; cb-unc-119(+)]II; unc-119(ed3?) mat-3(or344)III; mdf-1(gk2) ltSi560[oxTi365; pPLG014; Pmex-5::GFP::his-11::tbb-2_3’UTR, tbg-1::gfp::tbb-2_3’UTR; cb-unc-119(+)]V/nT1[qIs51](IV;V) | This study | OD2458 |
| ltSi813[pPLG043; Pfzy-1::fzy-1 del 128–130::fzy-1 3’UTR; cb-unc-119(+)]I; fzy-1(lt20::loxP)/mIn1[mIs14 dpy-10(e128)]II; unc-119(ed3)?III | This study | OD2461 |
| san-1(lt6::loxP::cb unc-119(+)::loxP)I; unc-119(ed3?) mat-3(or344)III; ltSi560[oxTi365; pPLG014; Pmex-5::GFP::his-11::tbb-2_3’UTR, tbg-1::gfp::tbb-2_3’UTR; cb-unc-119(+)]V | This study | OD2577 |
| unc-119(ed3?) mat-3(or344)III; mdf-2(lt4::loxP::cb-unc-119(+)::loxP)IV; ltSi560 [oxTi365; pPLG014; Pmex-5::GFP::his-11::tbb-2_3’UTR, tbg-1::gfp::tbb-2_3’UTR; cb-unc-119(+)]V | This study | OD2579 |
| ltSi822[pPLG051; Pfzy-1::fzy-1 A138V::fzy-1 3’UTR; cb-unc-119(+)]I; fzy-1(lt20::loxP)/mIn1[mIs14 dpy-10(e128)]II; unc-119(ed3)?III | This study | OD2580 |
| ltSi813[pPLG043; Pfzy-1::fzy-1 del 128–130::fzy-1 3’UTR; cb-unc-119(+)]I;fzy-1(lt20::loxP)II; unc-119(ed3)?III; ltSi560 [oxTi365; pPLG014; Pmex-5::GFP::his-11::tbb-2_3’UTR, tbg-1::gfp::tbb-2_3’UTR; cb-unc-119(+)]V | This study | OD2583 |
| ltSi813[pPLG043; Pfzy-1::fzy-1 del 128–130::fzy-1 3’UTR; cb-unc-119(+)]I;fzy-1(lt20::loxP)II; unc-119(ed3?) mat-3(or344)III; ltSi560 [oxTi365; pPLG014; Pmex-5::GFP::his-11::tbb-2_3’UTR, tbg-1::gfp::tbb-2_3’UTR; cb-unc-119(+)]V | This study | OD2584 |
| ltSi822[pPLG051; Pfzy-1::fzy-1 A138V::fzy-1 3’UTR; cb-unc-119(+)]I; fzy-1(lt20::loxP)II; unc-119(ed3)?III; ltSi560 [oxTi365; pPLG014; Pmex-5::GFP::his-11::tbb-2_3’UTR, tbg-1::gfp::tbb-2_3’UTR; cb-unc-119(+)]V | This study | OD2585 |
| ltSi822[pPLG051; Pfzy-1::fzy-1 A138V::fzy-1 3’UTR; cb-unc-119(+)]I; fzy-1(lt20::loxP)II; unc-119(ed3?) mat-3(or344)III; ltSi560 [oxTi365; pPLG014; Pmex-5::GFP::his-11::tbb-2_3’UTR, tbg-1::gfp::tbb-2_3’UTR; cb-unc-119(+)]V | (Kim et al., 2017) | OD2586 |
| ltSi814[pPLG047; Pfzy-1::gfp::fzy-1::fzy-1 3’UTR; cb-unc-119(+)]I; unc-119(ed3)?III;; ltIs37 [pAA64; pie-1/mCherry::his-58; unc-119 (+)] IV | (Kim et al., 2017) | OD2591 |
| ltSi825[pPLG052; Pmdf-2::mdf-2 delta intron 4::gfp::mdf-2 3’UTR; cb-unc-119(+)]II; unc-119(ed3)III?; mdf-2(lt4::loxP::cb-unc-119(+)::loxP)IV/nT1[qIs51](IV;V) | This study | OD2663 |
| ltSi805[pPLG042; Pfzy-1::fzy-1::fzy-1 3’UTR; cb-unc-119(+)]I; fzy-1(lt20::loxP)/mIn1[mIs14 dpy-10(e128)]II; unc-119(ed3)?III | (Kim et al., 2017) | OD2664 |
| ltSi958[pPLG053; Pmdf-1::GFP::mdf-1 R518A, K519A, R520A, K521A::mdf-1 3’UTR; cb-unc-119(+)]II; unc-119(ed3)?III; mdf-1(gk2)V/nT1(qIs51)(IV;V) | This study | OD2685 |
| ltSi958[pPLG053; Pmdf-1::GFP::mdf-1 R518A, K519A, R520A, K521A::mdf-1 3’UTR; cb-unc-119(+)]II; unc-119(ed3)?III; ltIs37[pAA64; pie-1/mCherry::his-58; unc-119 (+)]IV; mdf-1(gk2)V/nT1(qIs51)(IV;V) | This study | OD2686 |
| ltSi959[pPLG054; Pmdf-1::GFP::mdf-1 d151–320 R518A, K519A, R520A, K521A::mdf-1 3’UTR; cb-unc-119(+)]II; unc-119(ed3)?III; mdf-1(gk2)V/nT1(qIs51)(IV;V) | This study | OD2687 |
| ltSi959[pPLG054; Pmdf-1::GFP::mdf-1 d151–320 R518A, K519A, R520A, K521A::mdf-1 3’UTR; cb-unc-119(+)]II; unc-119(ed3)?III; ltIs37[pAA64; pie-1/mCherry::his-58; unc-119 (+)]IV; mdf-1(gk2)V/nT1(qIs51)(IV;V) | This study | OD2688 |
| ltSi805[pPLG042; Pfzy-1::fzy-1::fzy-1 3’UTR; cb-unc-119(+)]I; fzy-1(lt20::loxP)II; unc-119(ed3)?III; ltSi560 [oxTi365; pPLG014; Pmex-5::GFP::his-11::tbb-2_3’UTR, tbg-1::gfp::tbb-2_3’UTR; cb-unc-119(+)]V | (Kim et al., 2017) | OD2692 |
| ltSi805[pPLG042; Pfzy-1::fzy-1::fzy-1 3’UTR; cb-unc-119(+)]I; fzy-1(lt20::loxP)II; unc-119(ed3?) mat-3(or344)III; ltSi560 [oxTi365; pPLG014; Pmex-5::GFP::his-11::tbb-2_3’UTR, tbg-1::gfp::tbb-2_3’UTR; cb-unc-119(+)]V | (Kim et al., 2017) | OD2693 |
| emb-27(g48)II; unc-119(ed3)?III; mdf-2(lt4::loxP::cb-unc-119(+)::loxP)IV/nT1[qIs51](IV;V) | This study | OD2832 |
| mat-3(or344) unc-119(ed3)?III; mdf-2(lt4::loxP::cb-unc-119(+)::loxP)IV/nT1[qIs51](IV;V) | This study | OD2835 |
| mdf-1(lt39[gfp::tev::loxP::3xFlag::mdf-1])V | (Wang et al., 2017) | OD2906 |
| unc-119(ed3?) mat-3(or344)III; ltIs37[pAA64; pie-1/mCherry::his-58; unc-119 (+)]IV | This study | OD2919 |
| unc-119(ed3)?III; ltIs37[pAA64; pie-1/mCherry::his-58; unc-119 (+)]IV; mdf-1(lt39[gfp::tev::loxP::3xFlag::mdf-1])V | This study | OD2920 |
| unc-119(ed3?) mat-3(or344)III; ltIs37[pAA64; pie-1/mCherry::his-58; unc-119 (+)]IV; mdf-1(lt39[gfp::tev::loxP::3xFlag::mdf-1])V | This study | OD3004 |
| ltSi1066[pPLG187; Pmex-5::gfp::ph::tbb-2 3’-UTR::operon linker::mCherry::his-11::tbb-2 3’-UTR; cb-unc-119(+)]II; unc-119(ed3)III | This study | OD3328 |
| ltSi1069[pPLG184; Pcyb-1::cyb-1::cyb-1 3’-UTR]II; unc-119(ed3)?III; mdf-2(lt4::loxP::cb-unc-119(+)::loxP)IV/nT1(qIs51)IV;V | This study | OD3336 |
| ltSi1066[pPLG187; Pmex-5::gfp::ph::tbb-2 3’-UTR::operon linker::mCherry::his-11::tbb-2 3’-UTR; cb-unc-119(+)]II; unc-119(ed3)?III; mdf-2(lt4::loxP::cb-unc-119(+)::loxP)IV/nT1(qIs51)IV;V | This study | OD3338 |
| san-1(lt6::loxP::cb-unc-119(+)::loxP)I; ltSi1066[pPLG187; Pmex-5::gfp::ph::tbb-2 3’-UTR::operon linker::mCherry::his-11::tbb-2 3’-UTR; cb-unc-119(+)]II; unc-119(ed3)?III | This study | OD3490 |
| ltSi1066[pPLG187; Pmex-5::gfp::ph::tbb-2 3’-UTR::operon linker::mCherry::his-11::tbb-2 3’-UTR; cb-unc-119(+)]II; mat-3(or344) unc-119(ed3)?III | This study | OD3493 |
| ltSi1093[pPLG207; Pmdf-1::mdf-1::mdf-1 3’UTR; cb-unc-119(+)]I; ltSi1066[pPLG187; Pmex-5::gfp::ph::tbb-2 3’-UTR::operon linker::mCherry::his-11::tbb-2 3’-UTR; cb-unc-119(+)]II; unc-119(ed3)?III; mdf-1(gk2) V/nT1(qIs51)(IV;V) | This study | OD3537 |
| ltSi1096[pPLG208; Pmdf-1::mdf-1(Q496A, I497A, F498A, H499A, M500A)::mdf-1 3’UTR; cb-unc-119(+)]I; ltSi1066[pPLG187; Pmex-5::gfp::ph::tbb-2 3’-UTR::operon linker::mCherry::his-11::tbb-2 3’-UTR; cb-unc-119(+)]II; unc-119(ed3)?III; mdf-1(gk2) V/nT1(qIs51)(IV;V) | This study | OD3538 |
| ltSi1112[pPLG217; Pmex-5::gfp::pcn-1::tbb-2 3’-UTR::operon linker::mCherry::his-11::tbb-2 3’-UTR; cb-unc-119(+)]II; unc-119(ed3)III | This study | OD3549 |
| ltSi805[pPLG042; Pfzy-1::fzy-1::fzy-1 3’UTR; cb-unc-119(+)]I; fzy-1(lt20::loxP) ltSi1066[pPLG187; Pmex-5::gfp::ph::tbb-2 3’-UTR::operon linker::mCherry::his-11::tbb-2 3’-UTR; cb-unc-119(+)]II/mIn1[mIs14 dpy-10(e128)]II; unc-119(ed3)?III | This study | OD3554 |
| mdf-2(lt104[mdf-2::gfp::tev::loxP::3xFlag])IV | This study | OD3637 |
| unc-119(ed3?)III; mdf-2(lt104[mdf-2::gfp::tev::loxP::3xFlag]) ltIs37[pAA64; pie-1/mCherry::his-58; unc-119 (+)]IV | This study | OD3655 |
| cyb-3(lt110)V/nT1[qIs51](IV;V). | This study | OD3737 |
| cyb-1(gk35) IV/nT1[qIs51](IV;V) | This study | OD3739 |
| unc-119(ed3?) mat-3(or344)III; ltIs37[pAA64; pie-1/mCherry::his-58; unc-119 (+)] mdf-2(lt105[mdf-2::gfp::tev::loxP::3xFlag])IV | This study | OD3740 |
| ltSi1112[pPLG217; Pmex-5::gfp::pcn-1::tbb-2 3’-UTR::operon linker::mCherry::his-11::tbb-2 3’-UTR; cb-unc-119(+)]II; cdk-1(ne2257) unc-119(ed3)?III | This study | OD3830 |
| ltSi1069[pPLG184; Pcyb-1::cyb-1::cyb-1 3’-UTR]II; unc-119(ed3)?III; cyb-1(gk35)IV/nT1[qIs51](IV;V) | This study | OD3837 |
| ltSi1138[pPLG240; Pcyb-3::cyb-3::cyb-3 3’UTR; cb-unc-119(+)]I; cyb-3(lt110)V/nT1[qIs51](IV;V) | This study | OD3838 |
| ltSi1138[pPLG240; Pcyb-3::cyb-3::cyb-3 3’UTR; cb-unc-119(+)]I; unc-119(ed3)?III; mdf-2(lt4::loxP::cb-unc-119(+)::loxP)IV/nT1(qIs51)IV;V | This study | OD3842 |
| cyb-2.2(tm1969)I; cyb-2.1(tm2027)IV | This study | OD3844 |
| ltSi1167[oxTi185; pPLG187; Pmex-5::gfp::ph::tbb-2 3’-UTR::operon linker::mCherry::his-11::tbb-2 3’-UTR; cb-unc-119(+)]I; unc-119(ed3)III | This study | OD3908 |
| ltSi1167[oxTi185; pPLG187; Pmex-5::gfp::ph::tbb-2 3’-UTR::operon linker::mCherry::his-11::tbb-2 3’-UTR; cb-unc-119(+)] ltSi1138[pPLG240; Pcyb-3::cyb-3::cyb-3 3’UTR; cb-unc-119(+)]I; unc-119(ed3)?III | This study | OD3910 |
| ltSi1167[oxTi185; pPLG187; Pmex-5::gfp::ph::tbb-2 3’-UTR::operon linker::mCherry::his-11::tbb-2 3’-UTR; cb-unc-119(+)]I; ltSi1069[pPLG184; Pcyb-1::cyb-1::cyb-1 3’-UTR]II; unc-119(ed3)?III | This study | OD3911 |
| cyb-1(lt125[cyb-1::LAP::mNeonGreen::loxP::3xFlag])IV | This study | OD3913 |
| ltSi1167[oxTi185; pPLG187; Pmex-5::gfp::ph::tbb-2 3’-UTR::operon linker::mCherry::his-11::tbb-2 3’-UTR; cb-unc-119(+)]I; emb-27(g48ts)II; unc-119(ed3)?III | This study | OD3942 |
| ltSi310[pOD1577/pMM7C; Pmdf-1::mdf-1::mdf-1 3’UTR; cb-unc-119(+)]II; unc-119(ed3)?III; mdf-2(lt104[mdf-2::gfp::tev::loxP::3xFlag]) ltIs37[pAA64; pie-1/mCherry::his-58; unc-119(+)]IV; mdf-1(gk2)V | This study | OD4050 |
| ltSi976[pPLG063; Pmdf-1::mdf-1 delta 151–320::mdf-1 3’UTR; cb-unc-119(+)]II; unc-119(ed3)?III; mdf-2(lt104[mdf-2::gfp::tev::loxP::3xFlag]) ltIs37[pAA64; pie-1/mCherry::his-58; unc-119(+)]IV; mdf-1(gk2) V | This study | OD4051 |
| ltSi587[pPLG024; Pmdf-2::mdf-2 delta intron 4::mdf-2 3’UTR; cb-unc-119(+)]II; unc-119(ed3)? mat-3(or344)III; mdf-2(lt4::loxP::cb-unc-119(+)::loxP)IV; ltSi560[oxTi365; pPLG014; Pmex-5::GFP::his-11::tbb-2_3’UTR, tbg-1::gfp::tbb-2_3’UTR; cb-unc-119(+)]V | This study | OD4058 |
| ltSi808[pPLG044; Pmdf-2::mdf-2 delta intron 4 R133E, Q134A::mdf-2 3’UTR; cb-unc-119(+)]II; unc-119(ed3)? mat-3(or344)III; mdf-2(lt4::loxP::cb-unc-119(+)::loxP)IV; ltSi560[oxTi365; pPLG014; Pmex-5::GFP::his-11::tbb-2_3’UTR, tbg-1::gfp::tbb-2_3’UTR; cb-unc-119(+)]V | This study | OD4059 |
| ltSi822[pPLG051; Pfzy-1::fzy-1 A138V::fzy-1 3’UTR; cb-unc-119(+)]I; fzy-1(lt20::loxP) ltSi1066[pPLG187; Pmex-5::gfp::ph::tbb-2 3’-UTR::operon linker::mCherry::his-11::tbb-2 3’-UTR; cb-unc-119(+)]II/mIn1[mIs14 dpy-10(e128)]II; unc-119(ed3)?III | This study | OD4060 |
| ltSi1195[pPLG256; Pfzy-1::fzy-1 D433N::fzy-1 3’UTR; cb-unc-119(+)]I; fzy-1(lt20::loxP) ltSi1066[pPLG187; Pmex-5::gfp::ph::tbb-2 3’-UTR::operon linker::mCherry::his-11::tbb-2 3’-UTR; cb-unc-119(+)]II; unc-119(ed3)?III | This study | OD4062 |
| ltSi1195[pPLG256; Pfzy-1::fzy-1 D433N::fzy-1 3’UTR; cb-unc-119(+)]I; fzy-1(lt20::loxP)II; unc-119(ed3)?III; ltSi560 [oxTi365; pPLG014; Pmex-5::GFP::his-11::tbb-2_3’UTR, tbg-1::gfp::tbb-2_3’UTR; cb-unc-119(+)]V | This study | OD4063 |
| ltSi1200[pPLG262; Pmdf-1::GFP::mdf-1(Q496A, I497A, F498A, H499A, M500A)::mdf-1 3’UTR; cb-unc-119(+)]II; unc-119(ed3)?III; ltIs37[pAA64; pie-1/mCherry::his-58; unc-119 (+)]IV; mdf-1(gk2)V/nT1(qIs51)(IV;V) | This study | OD4071 |
| ltSi1088[oxTi185; pMO005; Pmex-5::mCherry::ph::tbb-2 3’-UTR::operon linker::mCherry::his-11::tbb-2 3’-UTR]I; ltSi608[pOD1583/pMM30; pmdf-1::GFP::mdf1::mdf-1 3’UTR; cb-unc-119(+)]II; unc-119(ed3)?III; mdf-1(gk2) V | This study | OD4083 |
| ltSi1088[oxTi185; pMO005; Pmex-5::mCherry::ph::tbb-2 3’-UTR::operon linker::mCherry::his-11::tbb-2 3’-UTR]I; ltSi677 [pPLG034; Pmdf-1::GFP::mdf-1(delta 151–320)::mdf-1 3’UTR; cb-unc-119(+)]II; unc-119(ed3)?III; unc-119 (+)]IV; mdf-1(gk2)V | This study | OD4084 |
| ltSi1088[oxTi185; pMO005; Pmex-5::mCherry::ph::tbb-2 3’-UTR::operon linker::mCherry::his-11::tbb-2 3’-UTR]I; ltSi958[pPLG053; Pmdf-1::GFP::mdf-1 R518A, K519A, R520A, K521A::mdf-1 3’UTR; cb-unc-119(+)]II; unc-119 (+)]IV; mdf-1(gk2)V | This study | OD4085 |
| ltSi1088[oxTi185; pMO005; Pmex-5::mCherry::ph::tbb-2 3’-UTR::operon linker::mCherry::his-11::tbb-2 3’-UTR]I; ltSi959[pPLG054; Pmdf-1::GFP::mdf-1 d151–320 R518A, K519A, R520A, K521A::mdf-1 3’UTR; cb-unc-119(+)]II; unc-119(ed3)?III; mdf-1(gk2)V | This study | OD4086 |
| unc-119(ed3)?III; cyb-1(lt125[cyb-1::LAP::mNeonGreen::loxP::3xFlag]) ltIs37[pAA64; pie-1/mCHERRY::his-58; unc-119 (+)]IV | This study | OD4100 |
| unc-119(ed3)?III; ltIs37[pAA64; pie-1/mCHERRY::his-58; unc-119 (+)]IV; cyb-3(lt135[mNeonGreen::tev::loxP::3xFlag::cyb-3])V | This study | OD4102 |
| ltSi813[pPLG043; Pfzy-1::fzy-1 del 128–130::fzy-1 3’UTR; cb-unc-119(+)]I; fzy-1(lt20::loxP) ltSi1066[pPLG187; Pmex-5::gfp::ph::tbb-2 3’-UTR::operon linker::mCherry::his-11::tbb-2 3’-UTR; cb-unc-119(+)]II/mIn1[mIs14 dpy-10(e128)]II; unc-119(ed3)?III | This study | OD4139 |
| ltSi965[pPLG058; Pfzy-1::fzy-1 T7A, T32A, S87A::fzy-1 3’UTR; cb-unc-119(+)]I; fzy-1(lt20::loxP) ltSi1066[pPLG187; Pmex-5::gfp::ph::tbb-2 3’-UTR::operon linker::mCherry::his-11::tbb-2 3’-UTR; cb-unc-119(+)]II; unc-119(ed3)?III | This study | OD4192 |
| ltSi805[pPLG042; Pfzy-1::fzy-1::fzy-1 3’UTR; cb-unc-119(+)]I; fzy-1(lt20::loxP); ltSi1112[pPLG217; Pmex-5::gfp::pcn-1::tbb-2 3’-UTR::operon linker::mCherry::his-11::tbb-2 3’-UTR; cb-unc-119(+)]II; unc-119(ed3?)III | This study | OD4225 |
| ltSi965[pPLG058; Pfzy-1::fzy-1 T7A, T32A, S87A::fzy-1 3’UTR; cb-unc-119(+)]I; fzy-1(lt20::loxP); ltSi1112[pPLG217; Pmex-5::gfp::pcn-1::tbb-2 3’-UTR::operon linker::mCherry::his-11::tbb-2 3’-UTR; cb-unc-119(+)]]II; unc-119(ed3?)III | This study | OD4226 |
| Oligonucleotides | ||
| CRISPR/Cas9 targeting sequences, see Table S1 | This paper | N/A |
| Primers for dsRNA production, see Table S2 | This paper | N/A |
| ON-TARGETplus Non-targeting Pool | Dharmacon | Cat # L-015475-00-0005 |
| Mad2 siRNA #1, CCUAUUGAAUCAGUUUCCAAUUU | (Ye et al., 2017) | N/A |
| Mad2 siRNA #2, CAGUAUAGGUAGGGAGAUAUU | (Ye et al., 2017) | N/A |
| ON-TARGETplus Human BUB1B siRNA - SMARTpool | Dharmacon | Cat # L-004101-00-0005 |
| Software and Algorithms | ||
| Fiji | (Schindelin et al., 2012) | RRID: SCR_002285 |
| Prism | Graphpad | RRID: SCR_002798 |
Cell lines
U2OS cells were maintained in McCoy’s 5A (Modified) Medium (Gibco), supplemented with 10% Fetal Bovine Serum in a humidified incubator at 37°C with 5% CO2.
Method Details
Generation of transgenic C. elegans strains
Transgenes were inserted at a single locus in the genome using a transposon-based strategy (Frokjaer-Jensen et al., 2008). Briefly, engineered transgenes were cloned into either pCFJ151 or pCF352 vectors and injected into strains EG6429 or EG6701 in order to generate single-copy insertions in chromosome II or I, respectively (Frokjaer-Jensen et al., 2008). Alternatively, transgenes cloned into pCFJ151 were injected onto strain EG8078 to obtain insertions in chromosome I (Frokjaer-Jensen et al., 2014). Transformants were selected on the basis of their ability to rescue the mobility defect of the parental strains and successful integrants were identified by PCR genotyping.
For the generation of mad-2, cyb-1 and cyb-3 transgene-based replacement systems, we engineered constructs containing the genomic locus for each gene, which included their promoter and 3’UTR (Fig. S4F; S5C). In the case of mad-2, intron 4 was removed because it contains repetitive sequences that made cloning and amplification difficult. All transgenes rescued the lethality of the respective null mutants (Fig. S4H; S5E). Replacement systems for MAD-1 and CDC-20 were described previously (Moyle et al., 2014; Kim et al., 2017).
CRISPR/Cas9-mediated genome editing
For the generation of mad-2 and mad-3 null alleles, we used a CRISPR/Cas9 strategy in which the coding sequence of each gene was replaced with the Caenorhabditis briggsae unc-119 sequence (Fig. S1A). We engineered plasmids encoding for single-guide RNAs (sgRNA) (Table S1) along with Cas9 (pDD162; Dickinson et al., 2013), as well as donor repair constructs containing C.b. unc-119 flanked by 2–3 kb homology arms flanking the sequence to be replaced. A mixture of these plasmids was microinjected into strain HT1593 [unc-119(ed3)III] and transformants selected 10 days later based on rescue of the unc-119(ed3) mobility defect. Genome editing was assessed by PCR, and successfully edited animals were backcrossed to N2 wild-type to remove any potential off-target mutations and balanced using either nT1[qIs51] for mad-2Δ (mdf-2(lt4)) or hT2[bli-4(e937) let-?(q782) qIs48] for mad-3Δ (san-1(lt6)). Both balancers express GFP in the pharynx in order to distinguish between homozygous mutant and heterozygous animals under a fluorescence stereomicroscope.
For the generation of a cyb-3 null allele, we used a co-CRISPR strategy in which dpy-10 was co-edited together with the cyb-3 locus (Arribere et al., 2014; Kim et al., 2014; Hattersley et al., 2018). N2 wild-type worms were injected with a mixture containing purified Cas9, tracrRNA, two crRNAs flanking the cyb-3 coding sequence and a crRNA targeting dpy-10. Four days later, dpy-10 mutants (which can be distinguished by their dumpy or roller phenotypes) were selected and screened by PCR to detect edits that removed the entire cyb-3 coding sequence, which were further confirmed by sequencing. The cyb-3 deletion (cyb-3(lt110)) was then backcrossed to N2 wild-type to remove the dpy-10 mutation and balanced using nT1[qIs51].
The null allele for cdc-20 (fzy-1(lt20)) was previously described (Kim et al., 2017). Null mutants for mad-1 (mdf-1(gk2)) and cyb-1 (cyb-1(gk35)), as well as temperature-sensitive mutants for Apc8 (Apc8ts; mat-3(or334)), Apc6 (Apc6ts; emb-27(g48)) and cdk-1 (cdk-1ts; cdk-1(ne2257)) were obtained from the Caenorhabditis Genetics Center (CGC). For the generation of in situ fluorescent tags for MAD-2, CYB-1 and CYB-3, we used the self-excising cassette method (SEC) (Dickinson et al., 2015). Generation of in situ fluorescently tagged MAD-1 was described previously (Kim et al., 2017; Wang et al., 2017).
RNA-mediated interference (RNAi)
Double-stranded RNAs (dsRNAs) used in this study are listed in Table S2. For embryo assays, dsRNAs were injected into L4 stage worms at a final concentration of 1 mg/ml. Worms were recovered at 20°C and embryos were analy zed 36–48h later. For embryonic lethality assays, L4s injected with dsRNAs were grown at 20°C for 24h, singled onto 35 mm plates and allowed to lay progeny for an additional 24h before the mothers were removed. The next day, after embryos had hatched in controls, embryonic lethality was scored.
For RNAi soaking (Tabara et al., 1998; Timmons et al., 2001), starved L1 larvae generated as described below, were transferred to a 5 μl drop of a mix containing 3.15 mM Spermidine, 0.055% gelatin and dsRNAs at a final concentration of 0.5 – 1.0 mg/ml in 1X soaking buffer (10.9 mM Na2HPO4, 5.5 mM KH2PO4, 1.26 mM NaCl and 4.7 mM NH4Cl), under RNAse-free conditions. Between 20–50 L1s were transferred to each drop. Soaked worms were incubated for 24h at 20°C in a humidifie d chamber, then recovered onto NGM plates with OP50 food at 20°C and imaged 7–72h late r.
Germ cell proliferation assays
For germ cell proliferation assays, L4 stage worms were injected with dsRNAs, recovered at 20°C and 24h later, adults were dissolved on NGM pl ates with no bacteria using a 50:50 mixture of 2N NaOH and concentrated household bleach. This treatment dissolves the worm bodies while leaving the embryos intact (Fig. S2A). The following day or two days later, after starved L1 larvae had hatched, worms were transferred onto NGM plates seeded with OP50 food and samples were taken every 4 or 7 hours in order to image germ cell proliferation. For experiments with the CDC-20 3A mutant, starved L1s were soaked in the indicated dsRNAs for 24h, recovered at 20°C and ima ged every 7 hours. For experiments with null mutants, balanced strains were maintained at 22°C and first homozygous generation worms were analyzed as described above.
Fertility/viability analysis
For scoring of progeny number over successive generations, mutants and, when applicable, their rescuing transgenes were maintained with balancer chromosomes (nT1[qIs51] (IV/V) for mad-1Δ and mad-2Δ, hT2[bli-4(e937) let-?(q782) qIs48] (I,III) for mad-3Δ and mIn1[mIs14 dpy-10(e128)]II for cdc-20Δ). All balancers used express GFP in the pharynx, which helps distinguish heterozygous balanced worms (GFP positive) from homozygous mutant worms (GFP negative; homozygotes for the balancer chromosome were either embryonic lethal or dumpy).
Balanced strains were propagated at 20°C. Homozygou s mutant L4s originating from balanced mothers (first homozygous generation, g1, Fig. 1D) were singled and scored for total number of progeny laid, which were phenotypically grouped into living larvae and dead embryos. Viable progeny (second homozygous generation, g2) were singled and scored similarly and the whole process was repeated once more (third homozygous generation, g3). When generation number is not indicated, the analysis was done on strains of the indicated genotype that were propagated for multiple generations.
Antibody generation
For MAD-3 antibody production, rabbits were immunized with Maltose-binding protein (MBP) fused to full-length MAD-3. Specific antibodies were affinity-purified from the sera, using MAD-3::His, in order to prevent the purification of antibodies against MBP (Desai et al., 2003). Antibody specificity was validated by western blot, against a lysate from mad-3Δ worms (Fig. S1B).
Immunoblotting
Gravid adult worms were transferred to tubes containing M9 buffer (22 mM KH2PO4, 42 mM Na2HPO4, 86 mM NaCl, and 1 mM MgSO4•7H2O), washed 4 times with M9 containing 0.1% Tween-20 and then resuspended on 1.5X sample buffer (87.5 mM Tris-HCl pH 6.8, 2.5% SDS, 150 mM DTT, 7.5% glycerol, bromophenol blue). After lysing by sonication and boiling, the equivalent of 8–12 worms was loaded onto 4–12% NuPAGE Bis-Tris Gels (Invitrogen). Proteins were then transferred to nitrocellulose membranes, probed with primary antibodies (see Key Resources Table) and detected using either horseradish (HRP)- conjugated secondary antibodies and WesternBright Sirius (Advansta) chemiluminescent substrate or, for the α-loading control, using alkaline phosphatase (AP)-conjugated secondary antibody and Western Blue® Stabilized Substrate for Alkaline Phosphatase (Promega). Membranes were imaged using a ChemiDoc MP imaging system (BioRad). For immunoblotting of U2OS samples, cells were collected by trypsinization, washed once with 1X PBS, resuspended on 1.5X sample buffer and boiled for 5 minutes before loading onto gels.
Immunoprecipitation
C. elegans strains expressing either GFP::MAD-1 or MAD-2::GFP in the background of their respective null alleles were grown in liquid culture as described (Zanin et al., 2011). Worms were harvested from cultures synchronized at the gravid adult stage, flash-frozen and stored at −80°C. For each immunoprecipitation exper iment, 1g of worm pellet was used. Worms were thawed in 1 mL of lysis buffer (75 mM HEPES pH 7.4, 1.5 mM EGTA, 1.5 mM MgCl2, 150 mM KCl, 15% glycerol, 0.07% NP-40, cOmplete protease inhibitor cocktail (Roche)) and lysed by sonication. Extracts were cleared by centrifugation at 20.000 g for 20 min at 4°C and the supernatants were incubated with 40 μL of pre-equilibrated GFP-Trap beads (Chromotek) for 2 hr. at 4°C. Beads were then washed 6 times with wash buffer (50 mM HEPES pH 7.4, 1.0 mM EGTA, 1.0 mM MgCl2, 100 mM KCl, 10% glycerol, 0.05% NP-40, cOmplete protease inhibitor cocktail (Roche)) and eluted with 40 μL of 1.5X sample buffer (87.5 mM Tris-HCl pH 6.8, 2.5% SDS, 150 mM DTT, 7.5% glycerol, bromophenol blue). Samples were analyzed by immunoblotting, as described above.
Yeast two-hybrid analysis
Yeast-two hybrid assays were performed using the Matchmaker GAL4 Two-Hybrid System 3 (Clonetech). cDNAs encoding MAD-2, Tpr (NPP-21 in worms), CDC-20 or the various MAD-1 fragments were cloned into either pGBKT7 or pGADT7 vectors, transformed onto Saccharomyces cerevisiae AH109 or Y187 strains and selected on –Trp or –Leu dropout plates respectively. Transformed strains were mated, selected onto –Trp –Leu double dropout plates and interactions were tested on either –Trp –Leu –His (low stringency) or –Trp –Leu –His –Ade dropout plates (high stringency).
Brightfield imaging of larvae and adults
Strains growing on NGM plates with food were imaged using a stereo microscope (Nikon) coupled to a Dino-Eye Digital Eyepiece Camera (Dino-lite). Images were processed using a sharpening filter in Adobe Photoshop.
Fluorescence imaging of C. elegans embryos and germ cells
For time-lapse imaging, C. elegans embryos were dissected from adults in M9 media and mounted onto 2% agarose pads, covered with a coverslip and imaged at 20°C. Time-lapse imaging of one-cell embryos expressing GFP::H2b was performed on a deconvolution microscope (DeltaVision Elite; Applied Precision) equipped with a charge-coupled device camera (pco.edge 5.5 sCMOS; PCO) and a 60× 1.42NA PlanApo N objective (Olympus). 5×2μm z-stacks without binning were acquired at 10sec intervals with 2% illumination intensity (on an InsightSSI illuminator) and 100 ms exposure. Similar settings were used for imaging of monopolar mitoses in the two-cell embryo, except that a 20 sec interval imaging protocol was used.
Imaging of GFP::MAD-1 or MAD-2::GFP unattached kinetochore localization was performed on an Andor Revolution XD Confocal System (Andor Technology) with a spinning disk confocal scanner unit (CSU-10; Yokogawa) mounted on an inverted microscope (TE2000-E; Nikon), 100x or 60× 1.4 NA Plan Apochromat lenses, and outfitted with an electron multiplication back-thinned charged-coupled device camera (iXon, Andor Technology).
For imaging of germ cells, larvae were anesthetized with a mixture containing 0.2 mg/ml Tetramisole hydrochloride; 2 mg/ml Tricane (Sigma). 15 min later, larvae were transferred to 2% agarose pads using a mouth pipette, covered with a coverslip and imaged using an Andor microscope system (see above). For L1-L2 larvae, 26 × 1 μm series were taken, whereas for L4 germlines we used 40 × 1 μm series. Images were then processed using Fiji (Schindelin et al., 2012), and cells were counted through all z-planes.
Imaging analysis
For time-lapse experiments, NEBD was scored as the frame where free histone signal in the nucleus equilibrated with the cytoplasm, which was just before abrupt chromosome movements were evident. Anaphase onset was scored as the first frame with visible separation of sister chromatids. DNA decondensation was scored as the first frame where the histone intensity diminished and its area expanded. For cell cycle staging in germ cells, nuclei were scored as being in S-phase when the GFP::PCNAPCN−1 fluorescence was bright and punctate, or being in G2 when GFP::PCNAPCN−1 was dim and diffused. For quantification of GFP::MAD-1 and MAD-2::GFP nuclear periphery signals, images were imported to Fiji and a line-scan method was used (see Fig. S3G for details).
Cell cycle analysis of human U2OS cells
U2OS cells expressing GFP-PCNA and H2B-mRFP were previously described (de Groot et al., 2015). Cells growing at a 40–60% confluency were transfected with a mixture of siRNA oligos targeting the Mad2 3’-UTR (Ye et al., 2017), an ON-TARGETplus siRNA mix for BubR1 or a non-targeting siRNA mixture as a control (Dharmacon) at a final concentration of 100 nM using Lipofectamine RNAiMAX (Thermo Fisher). 24h after transfection, cells were seeded onto a 96-well microplate (Greiner, Cat. #655866); if used, RO-3306 (Vassilev et al., 2006) was added to a final concentration of 5 μM at this time. Movies were acquired on a CV1000 spinning disk confocal system (Yokogawa) with a 20x U-PlanApo 0.75 NA objective and 512×512 EM-CCD camera (Hamamatsu). Image acquisition and data analysis were performed using CellVoyager software. The humidity-controlled imaging chamber was maintained at 37°C and 5% CO 2. 4 fields per well were imaged and a 2×2 binning protocol was used. 4 × 3 μm z-sections in the GFP (20% power, 250 ms, 25% gain) and RFP (20% power, 150 ms, 25% gain) channels were captured in each field at 10-minute intervals for 60 hours. Cells were manually tracked from the disappearance of GFP-PCNA foci to the beginning of the next mitosis (nuclear envelope breakdown). GFP-PCNA foci appear in the nucleus during mid to late S-phase, and the first frame in which these foci are no longer visible was defined as the beginning of G2 phase. Results represent combined measurements of a minimum of 100 cells per condition from two independent experiments.
For spindle checkpoint assays, cells were treated with nocodazole (Sigma) at a final concentration of 0.2 μg/ml and imaged under similar conditions as described above, except that 5 minute intervals were used. Mitotic duration was determined as the interval between nuclear envelope breakdown and DNA decondensation.
Generation of lentivirus-expressed Myc-Mad2 cell line
For Mad2 siRNA rescue experiments, a Myc-Mad2 cDNA was cloned into a lentiviral vector, under the PGK promoter. The vector also contains a P2A sequence followed by a Blasticidin S deaminase gene for the selection of positive integrants using Blasticidin. Lentiviruses were generated using the Lenti-X Packaging Single Shots (VSV-G; Takara), following the manufacturer’s instructions. Viruses were used to infect U2OS cells expressing GFP-PCNA and H2b-mRFP and 48 h later, cells were treated with 10 μg/mL Blasticidin-S HCl (Gibco) until the appearance of resistant colonies. The polyclonal population was tested for expression of Myc-Mad2 and used for analysis.
Generation of in situ mNeongreen-tagged cyclin B1 cell line
For the generation of cyclin B1-mNeonGreen, a CRISPR-Cas9 strategy was used. Briefly, we engineered a construct encoding ~1000 bp of left homology arm corresponding to the cyclin B1 C-terminus, fused to the linker ASKLGAGAGAGAGAGAG and followed by the coding sequence for mNeonGreen. The construct also contained a P2A ribosome skipping peptide, a Neomycin resistance cassette downstream of the mNeonGreen sequence and 600 bp of right homology arm corresponding to the cyclin B1 3’-UTR (Fig. S5G).
U2OS cells were nucleofected with a mixture containing this construct and a plasmid encoding Cas9 and the gRNA targeting sequence ((pX459; Ran et al., 2013); Table S1) using Cell Line Nucleofector Solution V (Lonza). Two days after nucleofection, cells were seeded onto 96-well plates and selected using 400 μg/mL of G418 (Gibco). After two weeks, when colonies became visible, clones were transferred to 24-well plates, screened by immunoblotting with an anti-Cyclin B1 antibody (see Key Resources Table) to identify clones where endogenous cyclin B1 was replaced by cyclin B1-mNeonGreen. Selected clones were further subjected to sequencing and PCR genotyping (see Fig. S5H).
Cyclin B1 accumulation assays
U2OS cells expressing in situ mNeongreen-tagged cyclin B1 were transfected with siRNAs as described above and transferred to 96-well plates (Greiner, Cat. #655866). Two hours before imaging, SiR-DNA (Cytoskeleton, Inc) was added to a final concentration of 0.5 μM. Imaging was performed on a CQ-1 spinning disk confocal system (Yokogawa) with a 40x UPLSAPO 40×2 0.95 NA objective and a 2560 × 2160 pixel sCMOS camera. Image acquisition and data analysis were performed using CellVoyager software. The humidity-controlled imaging chamber was maintained at 37°C a nd 5% CO2. 6 fields per well were imaged and a 2×2 binning protocol was used. 6 × 3 μm z-sections in the 488 nm (25% power, 300 ms) and 640 nm (25% power, 100 ms) channels were captured in each field at 10-minute intervals for 48 hours.
For quantification of cyclin B1 accumulation, maximum intensity projections were generated using Fiji (Schindelin et al., 2012). An area was manually drawn around the entire cell and measured over time. The same area was copied to a region of the field with no cells in order to measure background. Background-corrected intensities were plotted relative to nuclear envelope breakdown using Prism (Graphpad).
Quantification And Statistical Analysis
Statistical parameters are reported in the Figures and Figure Legends. In figures, asterisks denote statistical significance as calculated by Mann-Whitney or Kolmogorov-Smirnov tests (•, p < 0.05; ••, p < 0.01; •••, p < 0.001; ••••, p < 0.0001). Statistical analysis was performed using Prism (Graphpad).
Data Code And Availability
This study did not generate datasets or code.
Supplementary Material
Movie S1. (Related to Figure 1). Spindle region of the AB cell of two-cell C. elegans embryos, induced to form monopolar spindles by preventing centriole duplication in the zygote using RNAi-mediated depletion of ZYG-1/Plk4. Monopolar spindles were imaged in the presence of the Apc8ts allele, in embryos deleted of mad-2 or mad-3. Embryos expressed GFP::H2b and γ-tubulin::GFP, which mark the chromosomes and spindle poles, respectively, and were filmed through mitosis at the permissive temperature of 20°C. Time stamp indicates time elapsed after NEBD in seconds. 5 × 2 μm z-stacks were acquired every 20 seconds and maximal intensity projections generated for each time frame. Playback rate is 10 frames per second, or 200X relative to real time.
Movie S2. (Related to Figure 2). Germ cells of L2 larvae expressing GFP::PCNAPCN−1 and mCherry::H2b; which mark replication foci (green) and chromatin (red) respectively. Larvae were mounted on agarose pads and filmed at 20°C. Note that the cell on top exiting mitosis starts accumulating PCNA very shortly after decondensing its chromosomes, consistent with the lack of G1 in germ cells. Time stamp indicates time elapsed after anaphase onset in minutes. 4 × 1 μm z-stacks were acquired every minute and maximal intensity projections generated for each time frame. Playback rate is 10 frames per second, or 600X relative to real time.
Movie S3. (Related to Figure 2). U2OS cells expressing GFP-PCNA and H2b-RFP; which mark replication foci (green) and chromatin (red) respectively. Cells were treated with either a non-targeting (NT), Mad2 or BubR1 siRNAs and filmed at 37°C. Time stamp indicates time elapsed after PCNA foci dissolution (G2 onset) in minutes. 4 × 3 μm z-stacks were acquired every 10 minutes and maximal intensity projections generated for each time frame. Playback rate is 8 frames per second, or 4800X relative to real time.
Movie S4. (Related to Fig. 4). U2OS cells expressing cyclin B1-mNeoNGreen and labeled with SiR-DNA, which marks the chromatin (red). Cells were treated with either a non-targeting (NT), Mad2 or BubR1 siRNAs and filmed at 37°C. Time stamp indicates time elapsed after NEBD in minutes. 6 × 3 μm z-stacks were acquired every 10 minutes and maximal intensity projections generated for each time frame. Playback rate is 10 frames per second, or 6000X relative to real time.
Highlights.
The spindle checkpoint protein Mad2 has a conserved role in the G2-to-M transition
Mad2’s role in G2-to-M requires Mad1 but is independent of kinetochores
Mad2 enables cyclin B accumulation by restraining its degradation by APC/C-Cdc20
Mad2 and Cdk phosphorylation act in parallel to inhibit APC/C-Cdc20 in G2
ACKNOWLEDGEMENTS:
We thank the Caenorhabditis Genetics Center for providing mutant strains; Rebecca Green for help with germline analyses; Dong Hyun Kim, Ruth Kabeche, Franz Meitinger and David Jenkins for advice on human cell experiments; Stephen Taylor and Don Cleveland for antibodies, Kevin Corbett for APC/C schematics and comments on the manuscript and Jonathon Pines for helpful discussions. This work was supported by an NIH grant (GM074215) to A.D. P.L-G. was supported by a Pew Latin American fellowship. A.D. and K.O. receive salary and other support from the Ludwig Institute for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS:
Authors declare no competing interests.
REFERENCES
- Alfieri C, Chang L, Zhang Z, Yang J, Maslen S, Skehel M, and Barford D (2016). Molecular basis of APC/C regulation by the spindle assembly checkpoint. Nature 536, 431–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfieri C, Zhang S, and Barford D (2017). Visualizing the complex functions and mechanisms of the anaphase promoting complex/cyclosome (APC/C). Open Biol 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arribere JA, Bell RT, Fu BX, Artiles KL, Hartman PS, and Fire AZ (2014). Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics 198, 837–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MS, Chan GK, and Yen TJ (2001). Mitotic checkpoint proteins HsMAD1 and HsMAD2 are associated with nuclear pore complexes in interphase. J Cell Sci 114, 953–963. [DOI] [PubMed] [Google Scholar]
- Chen RH, Shevchenko A, Mann M, and Murray AW (1998). Spindle checkpoint protein Xmad1 recruits Xmad2 to unattached kinetochores. J Cell Biol 143, 283–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung E, and Chen RH (2002). Spindle checkpoint requires Mad1-bound and Mad1-free Mad2. Mol Biol Cell 13, 1501–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clute P, and Pines J (1999). Temporal and spatial control of cyclin B1 destruction in metaphase. Nat Cell Biol 1, 82–87. [DOI] [PubMed] [Google Scholar]
- Corbett KD (2017). Molecular Mechanisms of Spindle Assembly Checkpoint Activation and Silencing. Prog Mol Subcell Biol 56, 429–455. [DOI] [PubMed] [Google Scholar]
- De Antoni A, Pearson CG, Cimini D, Canman JC, Sala V, Nezi L, Mapelli M, Sironi L, Faretta M, Salmon ED, et al. (2005). The Mad1/Mad2 complex as a template for Mad2 activation in the spindle assembly checkpoint. Curr Biol 15, 214–225. [DOI] [PubMed] [Google Scholar]
- de Groot CO, Hsia JE, Anzola JV, Motamedi A, Yoon M, Wong YL, Jenkins D, Lee HJ, Martinez MB, Davis RL, et al. (2015). A Cell Biologist’s Field Guide to Aurora Kinase Inhibitors. Front Oncol 5, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai A, Rybina S, Muller-Reichert T, Shevchenko A, Shevchenko A, Hyman A, and Oegema K (2003). KNL-1 directs assembly of the microtubule-binding interface of the kinetochore in C. elegans. Genes Dev 17, 2421–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fiore B, and Pines J (2007). Emi1 is needed to couple DNA replication with mitosis but does not regulate activation of the mitotic APC/C. J Cell Biol 177, 425–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson DJ, Pani AM, Heppert JK, Higgins CD, and Goldstein B (2015). Streamlined Genome Engineering with a Self-Excising Drug Selection Cassette. Genetics 200, 1035–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson DJ, Ward JD, Reiner DJ, and Goldstein B (2013). Engineering the Caenorhabditis elegans genome using Cas9-triggered homologous recombination. Nat Methods 10, 1028–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar BA, and Orr-Weaver TL (2001). Endoreplication cell cycles: more for less. Cell 105, 297–306. [DOI] [PubMed] [Google Scholar]
- Essex A, Dammermann A, Lewellyn L, Oegema K, and Desai A (2009). Systematic analysis in Caenorhabditis elegans reveals that the spindle checkpoint is composed of two largely independent branches. Mol Biol Cell 20, 1252–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faesen AC, Thanasoula M, Maffini S, Breit C, Muller F, van Gerwen S, Bange T, and Musacchio A (2017). Basis of catalytic assembly of the mitotic checkpoint complex. Nature 542, 498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang G (2002). Checkpoint protein BubR1 acts synergistically with Mad2 to inhibit anaphase-promoting complex. Mol Biol Cell 13, 755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrell JE Jr. (2013). Feedback loops and reciprocal regulation: recurring motifs in the systems biology of the cell cycle. Curr Opin Cell Biol 25, 676–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox PM, Vought VE, Hanazawa M, Lee MH, Maine EM, and Schedl T (2011). Cyclin E and CDK-2 regulate proliferative cell fate and cell cycle progression in the C. elegans germline. Development 138, 2223–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frokjaer-Jensen C, Davis MW, Hopkins CE, Newman BJ, Thummel JM, Olesen SP, Grunnet M, and Jorgensen EM (2008). Single-copy insertion of transgenes in Caenorhabditis elegans. Nature genetics 40, 1375–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frokjaer-Jensen C, Davis MW, Sarov M, Taylor J, Flibotte S, LaBella M, Pozniakovsky A, Moerman DG, and Jorgensen EM (2014). Random and targeted transgene insertion in Caenorhabditis elegans using a modified Mos1 transposon. Nat Methods 11, 529–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimitsu K, Grimaldi M, and Yamano H (2016). Cyclin-dependent kinase 1-dependent activation of APC/C ubiquitin ligase. Science 352, 1121–1124. [DOI] [PubMed] [Google Scholar]
- Fukuyama M, Rougvie AE, and Rothman JH (2006). C. elegans DAF-18/PTEN mediates nutrient-dependent arrest of cell cycle and growth in the germline. Curr Biol 16, 773–779. [DOI] [PubMed] [Google Scholar]
- Golden A, Sadler PL, Wallenfang MR, Schumacher JM, Hamill DR, Bates G, Bowerman B, Seydoux G, and Shakes DC (2000). Metaphase to anaphase (mat) transition-defective mutants in Caenorhabditis elegans. J Cell Biol 151, 1469–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopinathan L, Szmyd R, Low D, Diril MK, Chang HY, Coppola V, Liu K, Tessarollo L, Guccione E, van Pelt AMM, et al. (2017). Emi2 Is Essential for Mouse Spermatogenesis. Cell Rep 20, 697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RA, Kao HL, Audhya A, Arur S, Mayers JR, Fridolfsson HN, Schulman M, Schloissnig S, Niessen S, Laband K, et al. (2011). A high-resolution C. elegans essential gene network based on phenotypic profiling of a complex tissue. Cell 145, 470–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosskortenhaus R, and Sprenger F (2002). Rca1 inhibits APC-Cdh1(Fzr) and is required to prevent cyclin degradation in G2. Dev Cell 2, 29–40. [DOI] [PubMed] [Google Scholar]
- Hattersley N, Lara-Gonzalez P, Cheerambathur D, Gomez-Cavazos JS, Kim T, Prevo B, Khaliullin R, Lee KY, Ohta M, Green R, et al. (2018). Employing the one-cell C. elegans embryo to study cell division processes. Methods Cell Biol 144, 185–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein JB, and Nilsson J (2016). Interphase APC/C-Cdc20 inhibition by cyclin A2-Cdk2 ensures efficient mitotic entry. Nat Commun 7, 10975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang LH, Lau LF, Smith DL, Mistrot CA, Hardwick KG, Hwang ES, Amon A, and Murray AW (1998). Budding yeast Cdc20: a target of the spindle checkpoint. Science 279, 1041–1044. [DOI] [PubMed] [Google Scholar]
- Izawa D, and Pines J (2012). Mad2 and the APC/C compete for the same site on Cdc20 to ensure proper chromosome segregation. J Cell Biol 199, 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izawa D, and Pines J (2015). The mitotic checkpoint complex binds a second CDC20 to inhibit active APC/C. Nature 517, 631–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen A, and Medema RH (2013). Genetic instability: tipping the balance. Oncogene 32, 4459–4470. [DOI] [PubMed] [Google Scholar]
- Jaspersen SL, Charles JF, and Morgan DO (1999). Inhibitory phosphorylation of the APC regulator Hct1 is controlled by the kinase Cdc28 and the phosphatase Cdc14. Curr Biol 9, 227–236. [DOI] [PubMed] [Google Scholar]
- Kim DH, Han JS, Ly P, Ye Q, McMahon MA, Myung K, Corbett KD, and Cleveland DW (2018). TRIP13 and APC15 drive mitotic exit by turnover of interphase- and unattached kinetochore-produced MCC. Nat Commun 9, 4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Ishidate T, Ghanta KS, Seth M, Conte D Jr., Shirayama M, and Mello CC (2014). A co-CRISPR strategy for efficient genome editing in Caenorhabditis elegans. Genetics 197, 1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Lara-Gonzalez P, Prevo B, Meitinger F, Cheerambathur DK, Oegema K, and Desai A (2017). Kinetochores accelerate or delay APC/C activation by directing Cdc20 to opposing fates. Genes Dev 31, 1089–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Moyle MW, Lara-Gonzalez P, De Groot C, Oegema K, and Desai A (2015). Kinetochore-localized BUB-1/BUB-3 complex promotes anaphase onset in C. elegans. J Cell Biol 209, 507–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa R, Law E, Tang L, and Rose AM (2002). The Cdc20 homolog, FZY-1, and its interacting protein, IFY-1, are required for proper chromosome segregation in Caenorhabditis elegans. Curr Biol 12, 2118–2123. [DOI] [PubMed] [Google Scholar]
- Kitagawa R, and Rose AM (1999). Components of the spindle-assembly checkpoint are essential in Caenorhabditis elegans. Nat Cell Biol 1, 514–521. [DOI] [PubMed] [Google Scholar]
- Kreutzer MA, Richards JP, De Silva-Udawatta MN, Temenak JJ, Knoblich JA, Lehner CF, and Bennett KL (1995). Caenorhabditis elegans cyclin A- and B-type genes: a cyclin A multigene family, an ancestral cyclin B3 and differential germline expression. J Cell Sci 108 (Pt 6), 2415–2424. [DOI] [PubMed] [Google Scholar]
- Kulukian A, Han JS, and Cleveland DW (2009). Unattached kinetochores catalyze production of an anaphase inhibitor that requires a Mad2 template to prime Cdc20 for BubR1 binding. Dev Cell 16, 105–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labit H, Fujimitsu K, Bayin NS, Takaki T, Gannon J, and Yamano H (2012). Dephosphorylation of Cdc20 is required for its C-box-dependent activation of the APC/C. EMBO J 31, 3351–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara-Gonzalez P, Kim T, and Desai A (2017). Taming the Beast: Control of APC/C(Cdc20)-Dependent Destruction. Cold Spring Harb Symp Quant Biol 82, 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Sterling H, Burlingame A, and McCormick F (2008). Tpr directly binds to Mad1 and Mad2 and is important for the Mad1-Mad2-mediated mitotic spindle checkpoint. Genes Dev 22, 2926–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonhardt H, Rahn HP, Weinzierl P, Sporbert A, Cremer T, Zink D, and Cardoso MC (2000). Dynamics of DNA replication factories in living cells. J Cell Biol 149, 271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Tang Z, Rizo J, and Yu H (2002). The Mad2 spindle checkpoint protein undergoes similar major conformational changes upon binding to either Mad1 or Cdc20. Mol Cell 9, 59–71. [DOI] [PubMed] [Google Scholar]
- Machida YJ, and Dutta A (2007). The APC/C inhibitor, Emi1, is essential for prevention of rereplication. Genes Dev 21, 184–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malureanu LA, Jeganathan KB, Hamada M, Wasilewski L, Davenport J, and van Deursen JM (2009). BubR1 N terminus acts as a soluble inhibitor of cyclin B degradation by APC/C(Cdc20) in interphase. Dev Cell 16, 118–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan DO (2007). The cell cycle : principles of control (Sunderland, MA: Oxford University Press; ). [Google Scholar]
- Morgan DO (2016). Cell division: Mitotic regulation comes into focus. Nature 536, 407–408. [DOI] [PubMed] [Google Scholar]
- Moyle MW, Kim T, Hattersley N, Espeut J, Cheerambathur DK, Oegema K, and Desai A (2014). A Bub1-Mad1 interaction targets the Mad1-Mad2 complex to unattached kinetochores to initiate the spindle checkpoint. J Cell Biol 204, 647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musacchio A (2015). The Molecular Biology of Spindle Assembly Checkpoint Signaling Dynamics. Curr Biol 25, R1002–1018. [DOI] [PubMed] [Google Scholar]
- Peters JM (2006). The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol 7, 644–656. [DOI] [PubMed] [Google Scholar]
- Pines J (2011). Cubism and the cell cycle: the many faces of the APC/C. Nat Rev Mol Cell Biol 12, 427–438. [DOI] [PubMed] [Google Scholar]
- Qiao R, Weissmann F, Yamaguchi M, Brown NG, VanderLinden R, Imre R, Jarvis MA, Brunner MR, Davidson IF, Litos G, et al. (2016). Mechanism of APC/CCDC20 activation by mitotic phosphorylation. Proc Natl Acad Sci U S A 113, E2570–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappleye CA, Tagawa A, Lyczak R, Bowerman B, and Aroian RV (2002). The anaphase-promoting complex and separin are required for embryonic anterior-posterior axis formation. Dev Cell 2, 195–206. [DOI] [PubMed] [Google Scholar]
- Reimann JD, Freed E, Hsu JY, Kramer ER, Peters JM, and Jackson PK (2001). Emi1 is a mitotic regulator that interacts with Cdc20 and inhibits the anaphase promoting complex. Cell 105, 645–655. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Bravo V, Maciejowski J, Corona J, Buch HK, Collin P, Kanemaki MT, Shah JV, and Jallepalli PV (2014). Nuclear pores protect genome integrity by assembling a premitotic and Mad1-dependent anaphase inhibitor. Cell 156, 1017–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A, Duncan PI, Rauh NR, Sauer G, Fry AM, Nigg EA, and Mayer TU (2005). Xenopus polo-like kinase Plx1 regulates XErp1, a novel inhibitor of APC/C activity. Genes Dev 19, 502–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab M, Lutum AS, and Seufert W (1997). Yeast Hct1 is a regulator of Clb2 cyclin proteolysis. Cell 90, 683–693. [DOI] [PubMed] [Google Scholar]
- Scott RJ, Lusk CP, Dilworth DJ, Aitchison JD, and Wozniak RW (2005). Interactions between Mad1p and the nuclear transport machinery in the yeast Saccharomyces cerevisiae. Mol Biol Cell 16, 4362–4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel HS, and Kimble J (2015). Cell-cycle quiescence maintains Caenorhabditis elegans germline stem cells independent of GLP-1/Notch. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah JV, Botvinick E, Bonday Z, Furnari F, Berns M, and Cleveland DW (2004). Dynamics of centromere and kinetochore proteins; implications for checkpoint signaling and silencing. Curr Biol 14, 942–952. [DOI] [PubMed] [Google Scholar]
- Shirayama M, Soto MC, Ishidate T, Kim S, Nakamura K, Bei Y, van den Heuvel S, and Mello CC (2006). The Conserved Kinases CDK-1, GSK-3, KIN-19, and MBK-2 Promote OMA-1 Destruction to Regulate the Oocyte-to-Embryo Transition in C. elegans. Curr Biol 16, 47–55. [DOI] [PubMed] [Google Scholar]
- Sironi L, Mapelli M, Knapp S, De Antoni A, Jeang KT, and Musacchio A (2002). Crystal structure of the tetrameric Mad1-Mad2 core complex: implications of a ‘safety belt’ binding mechanism for the spindle checkpoint. EMBO J 21, 2496–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein KK, Davis ES, Hays T, and Golden A (2007). Components of the spindle assembly checkpoint regulate the anaphase-promoting complex during meiosis in Caenorhabditis elegans. Genetics 175, 107–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabara H, Grishok A, and Mello CC (1998). RNAi in C. elegans: soaking in the genome sequence. Science 282, 430–431. [DOI] [PubMed] [Google Scholar]
- Tang Z, Bharadwaj R, Li B, and Yu H (2001). Mad2-Independent inhibition of APCCdc20 by the mitotic checkpoint protein BubR1. Dev Cell 1, 227–237. [DOI] [PubMed] [Google Scholar]
- Tarailo-Graovac M, Wang J, Tu D, Baillie DL, Rose AM, and Chen N (2010). Duplication of cyb-3 (cyclin B3) suppresses sterility in the absence of mdf-1/MAD1 spindle assembly checkpoint component in Caenorhabditis elegans. Cell Cycle 9, 4858–4865. [DOI] [PubMed] [Google Scholar]
- Tighe A, Staples O, and Taylor S (2008). Mps1 kinase activity restrains anaphase during an unperturbed mitosis and targets Mad2 to kinetochores. J Cell Biol 181, 893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmons L, Court DL, and Fire A (2001). Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene 263, 103–112. [DOI] [PubMed] [Google Scholar]
- Tung JJ, Hansen DV, Ban KH, Loktev AV, Summers MK, Adler JR 3rd, and Jackson PK (2005). A role for the anaphase-promoting complex inhibitor Emi2/XErp1, a homolog of early mitotic inhibitor 1, in cytostatic factor arrest of Xenopus eggs. Proc Natl Acad Sci U S A 102, 4318–4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Voet M, Lorson MA, Srinivasan DG, Bennett KL, and van den Heuvel S (2009). C. elegans mitotic cyclins have distinct as well as overlapping functions in chromosome segregation. Cell Cycle 8, 4091–4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT, Tovar C, Chen S, Knezevic D, Zhao X, Sun H, Heimbrook DC, and Chen L (2006). Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc Natl Acad Sci U S A 103, 10660–10665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visintin R, Prinz S, and Amon A (1997). CDC20 and CDH1: a family of substrate-specific activators of APC-dependent proteolysis. Science 278, 460–463. [DOI] [PubMed] [Google Scholar]
- Wang S, Tang NH, Lara-Gonzalez P, Zhao Z, Cheerambathur DK, Prevo B, Chisholm AD, Desai A, and Oegema K (2017). A toolkit for GFP-mediated tissue-specific protein degradation in C. elegans. Development 144, 2694–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Wu D, Quintin S, Green RA, Cheerambathur DK, Ochoa SD, Desai A, and Oegema K (2015). NOCA-1 functions with gamma-tubulin and in parallel to Patronin to assemble non-centrosomal microtubule arrays in C. elegans. Elife 4, e08649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolthuis R, Clay-Farrace L, van Zon W, Yekezare M, Koop L, Ogink J, Medema R, and Pines J (2008). Cdc20 and Cks direct the spindle checkpoint-independent destruction of cyclin A. Mol Cell 30, 290–302. [DOI] [PubMed] [Google Scholar]
- Yam CH, Fung TK, and Poon RY (2002). Cyclin A in cell cycle control and cancer. Cell Mol Life Sci 59, 1317–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M, VanderLinden R, Weissmann F, Qiao R, Dube P, Brown NG, Haselbach D, Zhang W, Sidhu SS, Peters JM, et al. (2016). Cryo-EM of Mitotic Checkpoint Complex-Bound APC/C Reveals Reciprocal and Conformational Regulation of Ubiquitin Ligation. Mol Cell 63, 593–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Q, Kim DH, Dereli I, Rosenberg SC, Hagemann G, Herzog F, Toth A, Cleveland DW, and Corbett KD (2017). The AAA+ ATPase TRIP13 remodels HORMA domains through N-terminal engagement and unfolding. EMBO J 36, 2419–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S, Kawasaki I, and Shim YH (2012). CDC-25.1 controls the rate of germline mitotic cell cycle by counteracting WEE-1.3 and by positively regulating CDK-1 in Caenorhabditis elegans. Cell Cycle 11, 1354–1363. [DOI] [PubMed] [Google Scholar]
- Yudkovsky Y, Shteinberg M, Listovsky T, Brandeis M, and Hershko A (2000). Phosphorylation of Cdc20/fizzy negatively regulates the mammalian cyclosome/APC in the mitotic checkpoint. Biochem Biophys Res Commun 271, 299–304. [DOI] [PubMed] [Google Scholar]
- Zachariae W, Schwab M, Nasmyth K, and Seufert W (1998). Control of cyclin ubiquitination by CDK-regulated binding of Hct1 to the anaphase promoting complex. Science 282, 1721–1724. [DOI] [PubMed] [Google Scholar]
- Zanin E, Dumont J, Gassmann R, Cheeseman I, Maddox P, Bahmanyar S, Carvalho A, Niessen S, Yates JR 3rd, Oegema K, et al. (2011). Affinity purification of protein complexes in C. elegans. Methods Cell Biol 106, 289–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Chang L, Alfieri C, Zhang Z, Yang J, Maslen S, Skehel M, and Barford D (2016). Molecular mechanism of APC/C activation by mitotic phosphorylation. Nature 533, 260–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. (Related to Figure 1). Spindle region of the AB cell of two-cell C. elegans embryos, induced to form monopolar spindles by preventing centriole duplication in the zygote using RNAi-mediated depletion of ZYG-1/Plk4. Monopolar spindles were imaged in the presence of the Apc8ts allele, in embryos deleted of mad-2 or mad-3. Embryos expressed GFP::H2b and γ-tubulin::GFP, which mark the chromosomes and spindle poles, respectively, and were filmed through mitosis at the permissive temperature of 20°C. Time stamp indicates time elapsed after NEBD in seconds. 5 × 2 μm z-stacks were acquired every 20 seconds and maximal intensity projections generated for each time frame. Playback rate is 10 frames per second, or 200X relative to real time.
Movie S2. (Related to Figure 2). Germ cells of L2 larvae expressing GFP::PCNAPCN−1 and mCherry::H2b; which mark replication foci (green) and chromatin (red) respectively. Larvae were mounted on agarose pads and filmed at 20°C. Note that the cell on top exiting mitosis starts accumulating PCNA very shortly after decondensing its chromosomes, consistent with the lack of G1 in germ cells. Time stamp indicates time elapsed after anaphase onset in minutes. 4 × 1 μm z-stacks were acquired every minute and maximal intensity projections generated for each time frame. Playback rate is 10 frames per second, or 600X relative to real time.
Movie S3. (Related to Figure 2). U2OS cells expressing GFP-PCNA and H2b-RFP; which mark replication foci (green) and chromatin (red) respectively. Cells were treated with either a non-targeting (NT), Mad2 or BubR1 siRNAs and filmed at 37°C. Time stamp indicates time elapsed after PCNA foci dissolution (G2 onset) in minutes. 4 × 3 μm z-stacks were acquired every 10 minutes and maximal intensity projections generated for each time frame. Playback rate is 8 frames per second, or 4800X relative to real time.
Movie S4. (Related to Fig. 4). U2OS cells expressing cyclin B1-mNeoNGreen and labeled with SiR-DNA, which marks the chromatin (red). Cells were treated with either a non-targeting (NT), Mad2 or BubR1 siRNAs and filmed at 37°C. Time stamp indicates time elapsed after NEBD in minutes. 6 × 3 μm z-stacks were acquired every 10 minutes and maximal intensity projections generated for each time frame. Playback rate is 10 frames per second, or 6000X relative to real time.