

Abstract
This work reports the amy1 gene cloning from Massilia timonae CTI-57, and its successful expression in Escherichia coli Rosetta™ (DE3) from the pTRCHis2B plasmid. The recombinant AMY1 protein had 47 kDa, and its modeled structure showed a monomer composed of three domains. An N-terminal domain with the characteristic (β/α)8-barrel structure of α-amylases, which contained the catalytic amino acid residues. The second domain was small, and the C-terminal domain was similar to those found in the barley α-amylase. A phylogenetic analysis demonstrated a high sequence identity of the studied protein with bacterial and plant α-amylases from the GH13_6 subfamily. This is the first characterized bacterial α-amylase from this glucoside hydrolase subfamily. Besides starch, the enzyme was also active against maltodextrin, amylopectin, and blocked p-nitrophenyl α-d-maltoheptaoside, but could not use β-cyclodextrin or 4-nitrophenyl α-d-glucopyranoside. The KM for highly pure grade soluble starch from potato and Vmax values were 0.79 mg/mL and 0.04 mg/min, respectively. The calcium ion showed to be essential for the purified enzyme’s activity, while EDTA, molybdenum, cobalt, and mercury were strong inhibitors. The enzyme was almost fully active in SDS presence. The enzyme’s optimal pH and temperature were 6.0 and 60 °C, respectively, and its denaturation Tm was 79 °C. A TLC analysis revealed that glucose and maltose are products of the enzyme’s action on starch. In conclusion, this work described the M. timonae GH13_6 subfamily α-amylase, which showed to be thermostable and anionic detergent-resistant.
Electronic supplementary material
The online version of this article (10.1007/s13205-020-02505-w) contains supplementary material, which is available to authorized users.
Keywords: α-Amylase, Massilia timonae, Recombinant expression, Hydrolase
Introduction
The global market of industrial enzymes is expected to reach nearly US$6.2 billion by 2020 (Singh et al. 2016). Amylases represent 25% of this market (Gurung et al. 2013; Singh et al. 2016). The industrial starch enzymatic hydrolysis is eco-friendly, because it produces little residues (Tomasik and Horton 2012; Singh et al. 2016; Läufer 2017).
Amylases are used in detergent industry, which require enzymes with activity in extreme conditions of pH and temperature (Prakash and Jaiswal 2010; Gurung et al. 2013; Niyonzima and More 2014). Amylases are also widely used in food industry, in baking, brewing, and in starch processing for glucose production (Lévêque et al. 2000; Raveendran et al. 2018). Besides, there are applications of amylases in pharmaceutical, textile, paper, and analytical industries (Gupta et al. 2003; Souza and Magalhães 2010; Gurung et al. 2013; Raveendran et al. 2018). Another application of amylases is in biofuel production, to obtain fermentable sugars from starch (Läufer 2017). The increasing demand in various industries requires enzymes with suitable characteristics for each application, and the search of new amylases and improvement of the existing ones are of great value (Mehta and Satyanarayana 2016; Gopinath et al. 2018; Sindhu et al. 2017; Chapman et al. 2018).
Amylases are classified according to their action pattern (Sindhu et al. 2017). β-amylases and glucoamylases are exoamylases. Filamentous fungi constitute the primary source of glucoamylases, while β-amylases are generally of plant origin (Pandey et al. 2000). α-Amylases are endo-acting enzymes, which hydrolyze internal α-1,4-glycosidic linkages of starch (Gupta et al. 2003; van der Maarel et al. 2002). This enzyme’s action releases oligosaccharides of various lengths and dextrins (El-Fallal et al. 2012; Gupta et al. 2003; van der Maarel et al. 2002; Sindhu et al. 2017). α-Amylases may be derived from bacteria, archaea, and eukatyote (Gupta et al. 2003; Mehta and Satyanarayana 2016).
Industrial amylases are produced mainly by microorganisms (Sun et al. 2010). Bacteria of genus Bacillus and fungi of genera Aspergillus, Penicillium, and Rhizopus are the primary sources of amylases (Sun et al. 2010; Gurung et al. 2013). Microbial enzymes are preferred due to their stability and higher catalytic activity (Gurung et al. 2013). Besides, microorganisms grow abundantly on low-cost substrates, and their production is not affected by seasonal fluctuations (Gurung et al. 2013; Singh et al. 2016).
In a previous study in our laboratory (Santos et al. 2019), a strain of Massilia timonae (CTI-57), which was isolated from maize grains with rotten symptoms, presented amylolytic activity in selective solid medium. M. timonae is a Gram-negative, aerobic, rod-shaped, non-fermenting, mesophilic bacterium from the phylum Proteobacteria and order Burkholderiales (La Scola et al. 1998; Lindquist et al. 2003). The Massilia genus species were first isolated from clinical samples and subsequently isolated from many environmental samples (La Scola et al. 1998; Lindquist et al. 2003; Nagy et al. 2005; Gallego et al. 2006; Fahlgren et al. 2011). Species of the Massilia genus are reported to be able to grow using starch as a carbon source and to produce polyhydroxyalkanoates, suggesting their ability to produce amylases (Cerrone et al. 2011). A search in the M. timonae CCUG 45783 sequenced genome was conducted, and five amylase genes were found. A gene coding for a cyclodextrinase was already cloned, and the recombinant enzyme was characterized (Santos and Barbosa-Tessmann 2019). This present study targeted the α-amylase protein GenBank EKU83291.1 from this bacterium. This protein is almost identical to the protein GenBank AWL05875.1, which is a member of the plant α-amylase subfamily GH13_6.
α-Amylases are classified inside glycoside hydrolase family 13 (GH13) in the Carbohydrate-Active Enzymes database (CAZy) (Lombard et al. 2014). According to the amino acid sequence, this family has been divided in 42 subfamilies (Stam et al. 2006). The subfamily GH13_6 is closely related to the GH13_7 subfamily. These subfamilies are represented mainly by plants and Archaea α-amylases, respectively (Janeček et al. 1999, 2014; Jones et al. 1999; Janeček 2016). In phylogenetic analysis, the α-amylase subfamilies GH13_6 and GH13_7, together with the subfamily GH13_5, which includes bacterial liquefying enzymes as the ones from Bacillus species, belong altogether to one evolutionary cluster (Janeček et al. 2014; Janeček 2016).
The α-amylases from the GH13_6 and GH13_5 subfamilies have their structure typically organized in three domains (Janeček et al. 2014). The N-terminal domain, referred as A domain, is highly conserved and composed of a barrel of eight β-strands encircled by eight α-helices. This domain has the three highly conserved catalytic residues in its C-terminal β-strands. The B domain, of variable length, protrudes between the third β-strand and the third α-helix of the N-terminal domain and has the substrate and calcium-binding sites. The active site is formed between the A and B domains (van der Maarel et al. 2002; El-Fallal et al. 2012; Janeček et al. 2014). The C-terminal domain, stated as C domain, is organized in antiparallel β-strands (El-Fallal et al. 2012; Janeček et al. 2014).
This work described the cloning of a gene from the M. timonae CTI-57 genomic DNA, which codes for a homologous protein to the M. timonae CCUG 45783 EKU83291.1 GH13_6 α-amylase subfamily. This gene was expressed in Escherichia coli, and the recombinant enzyme was purified and characterized.
Materials and methods
Bacterial strains
E. coli TOP10™ was used for molecular cloning, while E. coli Rosetta™ (DE3) and E. coli Bl21 Star™ (DE3) were used as expression hosts. The M. timonae CTI-57 strain was previously obtained from maize grains presenting rotten symptoms, which were collected from the soil as harvest leftovers, during August 2015, at the Irrigation Technical Center (CTI), at the main campus of the Universidade Estadual de Maringá, Paraná, Brazil (Santos et al. 2019). This isolate produced a hydrolysis halo when grown in nutrient agar containing starch. This strain was being maintained in Nutrient slant agar culture at 4 °C and in Nutrient medium (6 g/L meat peptone; 0.5 g/L MgSO4; 0.5 g/L KCl; pH 7.0) with 50% glycerol at − 20 °C. However, this strain was of difficult maintenance, and it died during this work execution.
Genomic DNA extraction
A three-day old colony, grown at 37 °C on nutrient agar (6 g/L meat peptone; 0.5 g/L MgSO4; 0.5 g/L KCl; pH 7.0; 20 g/L agar) was used to inoculate 5 mL of liquid Luria–Bertani (LB) medium (10 g/L tryptone; 5 g/L yeast extract; 10 g/L NaCl), which were incubated for 24 h at 37 °C, under orbital agitation of 100 rpm. After that, the cells of 1.5 mL of the obtained culture were harvested by centrifugation (12,000 g, 1 min). The cells pellet was washed two times with 500 μL of TE buffer (10 mM Tris; 1 mM Na2EDTA; pH 8.0) and finally resuspended in 200 μL of TE buffer, boiled for 10 min, and centrifuged for 1 min (10,000×g). The supernatant was considered as the source of genomic DNA and was stored at − 20 °C.
Cloning of the α-amylase gene and construction of the expression plasmids
Previously, in a study conducted in our laboratory (Santos and Barbosa-Tessmann 2019), five amylolytic enzyme genes were identified in the sequenced and annotated genome of M. timonae CCUG 45783, a species from the Human Microbiome Project (GenBank AN: EKU82996.1, EKU83004.1, EKU82989.1, EKU82293.1, EKU83291.1). Primer pairs were designed targeted for each one of the five genes, and if a signal peptide is present, the part of the gene coding for the mature forms of the proteins, i.e., without the signal peptide, were considered. When these primers were used in PCR reactions, one pair did amplify a cyclodextrinase gene (GenBank EKU82989.1), which recombinant coded protein was characterized (Santos and Barbosa-Tessmann 2019).
In this present work, we have chosen to work with the M. timonae α-amylase GenBank EKU83291.1. First, several homologous genes from different M. timonae sequenced genomes were retrieved from GenBank, aligned, and primers were designed targeting the coding region, without the 5´ region coding for the signal peptide. To later subclone the gene into the pTrcHis2B expression plasmid, the following primer pair was designed. The forward primer (5´-TTGGATCCaGCSACCCCGCCGGCCCAGTCC) contained a restriction site for the enzyme BamHI (underlined) and had no initial codon ATG, because it would be given by the expression vector pTrcHis2B. An extra A (small letter) was added to maintain the correct reading frame. The reverse primer (5´-TTTCTAGAcgTTTGATCCAGACCGCGTAAT) contained a restriction site for the enzyme XbaI (underlined). The stop codon was deleted, while the bases CG (small letter) were added to keep the reading frame, and give to the expressed protein a poly-histidine tag to facilitate the purification. To later subclone the gene to the pET21a(+) expression plasmid, the following primer pair was designed. The forward primer (5´-TTCATATGGCSACCCCGCCGGCCCAGTCC) had a restriction site for the enzyme NdeI (underlined). The NdeI restriction sequence gave the initial ATG. The reverse primer (5´-TTAAGCTTTTTGATCCAGACCGCGTAAT) had a restriction site for the enzyme HindIII (underlined). The stop codon was deleted so that the recombinant protein could have a poly-histidine tag for further purification.
The PCR reaction was performed with 1 U of Accu Taq™ DNA Polymerase (Sigma Aldrich, USA), enzyme buffer [50 mM Tris–HCl, 15 mM ammonium sulfate (pH 9.3, adjusted with NH4OH), 2.5 mM MgCl2, 1% Tween 20], 0.2 mM of each dNTP, 25 pmol of each primer, and 2 μL of the obtained genomic DNA supernatant, in a final volume of 25 μL. The used conditions consisted of an initial incubation of 10 min at 94 °C, followed by 25 cycles of 1.5 min at 94 °C, 1.5 min at 60 °C, and 2 min at 68 °C. After that, the reactions were maintained for 10 min at 68 °C for a final extension of products.
The PCR obtained products were cloned into the pCR2.1® plasmid from the TOPO TA Cloning kit (Thermo Fisher Scientific, USA) or pGEM®-T plasmid from the pGEM®-T Vector Systems (Promega, USA), after the user guide instructions. The recombinant plasmids were transformed into E. coli TOP10™ (Chung et al. 1989) and recovered by alkaline lysis (Sambrook and Russel 2001). Then, the truncated genes were transferred from the pCR2.1® or pGEM®-T vectors to the pTrcHis2B and pET21a(+) expression vectors, using digestion and ligation technology (Sambrook and Russel 2001). The recombinant plasmids were transformed in E. coli TOP10™ strain (Chung et al. 1989), recovered by alkaline lysis, and analyzed by restriction digestion (Sambrook and Russel 2001).
The cloned gene, in the pCR2.1® vector, which contained the BamHI and XbaI restriction sites in the 5ʹ and 3ʹ ends, respectively, was sequenced at the Center for Human Genome Studies (CEGH), at the University of São Paulo. The sequencing reactions were performed with primers designed using the sequence of the GenBank EKU83291.1 α-amylase gene from the M. timonae CCUG 45783, and primers targeting the plasmid regions flanking the insert. The contig was generated using the BioEdit program (Hall 1999).
In silico analysis
A search in the CAZy database (https://www.cazy.org/) (Lombard et al. 2014) was conducted to find the protein’s family and subfamily. This search retrieved an GH13_6 plant subfamily α-amylase from M. timonae (AWL05875.1), which is almost identical with the EKU83291.1 and the one from the present study. Considering the phylogenetic proximity of α-amylases from the subfamilies GH13_5, GH13_6, and GH13_7 (Stam et al. 2006; Janeček et al. 2014; Janeček et al., 2016), proteins from these subfamilies were retrieved from GenBank, UniProt, and CAZy databases and aligned using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/) to check for the level of identity among our protein sequence with those proteins sequences (Supplementary Table 1). A phylogenetic tree was constructed using the neighbor-joining method (Saitou and Nei 1987) in the MEGAX program (Kumar et al. 2018). The confidence limits of the branching were assessed using Bootstrap analyses with 1000 heuristic replicates (Felsenstein 1985). Values higher than 70% in this test were considered reliable phylogenetic grouping among proteins.
A structural model for the protein coded by the cloned gene from M. timonae CTI-57 was obtained with the Modeller v9.16 program (Webb and Sali 2016), using as a template the Hordeum vulgare (barley) α-amylase (PDB 1AMY), a member of the GH13_6 subfamily (Kadziola et al. 1994). This template was selected by a search using the GenTHREADER prediction method inside the PSIPRED server (https://bioinf.cs.ucl.ac.uk/psipred/) and a search in the PDB database. The best models among 250 generated in each run were selected based on the Modeller DOPE Score. The stereo-chemical quality of the obtained models was evaluated using the Procheck software, and the percentage of secondary structure was obtained using VADAR version 1.8 (https://vadar.wishartlab.com/). The DiANNA 1.1 webserver (https://clavius.bc.edu/~clotelab/DiANNA/) was used for disulfide bond prediction. The PISA server (https://www.ebi.ac.uk/pdbe/pisa/) was used to verify macromolecular interfaces and probable multimeric structures. The modeled PDB structure was visualized and colored using the CCP4MG program (McNicholas et al. 2011). A search for structural domains was performed in the Conserved Protein Domain Database (CDD) from NCBI (https://www.ncbi.nlm.nih.gov) and Pfam from EMBL-EBI (https://pfam.xfam.org/).
The secretion signal peptide presence and the molecular weight and pI were obtained using the SignalP-5.0 Server (https://www.cbs.dtu.dk/services/SignalP/) and Compute pI/Mw tool from ExPASy (https://www.expasy.org/), respectively. The RNA folder Web Server program (https://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi) was used to predict the mRNA 5´secondary structure with its minimal free energy and the Rare Codon Analyzer program (https://www.biologicscorp.com/tools/RareCodonAnalyzer) was used to predict mRNA Codon Adaptation Index (CAI) value.
Expression analysis and purification of the recombinant α-amylase
For the expression analysis, the E. coli strain Rosetta™ (DE3) was transformed (Chung et al. 1989) with 50 ng of the pTrcHis2B-amy1 or pET21a(+)-amy1 vectors, and the E. coli strain Bl21 Star™ (DE3) was transformed with 50 ng of the pET21a(+)-amy1 vector. The obtained bacteria were transferred to 10 mL of LB medium containing 50 µg/mL ampicillin and incubated overnight, at 37 °C, with orbital agitation (100 rpm). An aliquot of 100 µL (1% inoculum) of each culture was transferred to tubes containing 10 mL of LB medium with 50 µg/mL ampicillin. An aliquot of 100 µL of the E. coli Bl21 Star™ (DE3) transformed with the pET21a(+)-amy1 vector was also transferred to a tube containing 10 mL of the auto-induction ZYM-5052 medium (Studier 2005) containing 50 µg/mL ampicillin and prepared slightly modified as described in Santos and Barbosa-Tessmann (2019). All tubes were incubated at 37 °C with agitation (100 rpm). After 4 h of incubation, when the optical density (OD) reached values of 0.6–0.8 at 600 nm, isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to a final concentration of 1 mM to the LB medium cultures. All cultures were then incubated at 20 °C, for 18 h, under agitation (100 rpm). The cells were harvested by centrifugation (2,000 g, for 10 min, at 4 °C). The obtained bacterial pellet was resuspended in 10 mL of lysis buffer (50 mM Tris pH 7.5, 50 mM NaCl, 2 mM CaCl2, 1 mM PMSF) and the cells were disrupted through sonication (30 cycles of –5 s on and 5 s off—with an amplitude of 30%). The samples were centrifuged at 10,000×g for 5 min. The enzyme expression was verified in the obtained supernatant using the starch-iodine complex distaining enzyme assay and SDS-PAGE analysis.
For the enzyme purification, E. coli Rosetta™ (DE3) was transformed (Chung et al. 1989) with 50 ng of the pTrcHis2B-amy1. The obtained bacteria were transferred to 10 mL of LB medium containing 50 µg/mL ampicillin and incubated overnight, at 37 °C, with orbital agitation (100 rpm). An aliquot of 500 µL of the obtained culture was transferred to a 250 mL Erlenmeyer containing 50 mL of LB medium with 50 µg/mL ampicillin. After 4 h of incubation, at 37 °C, with agitation (100 rpm), IPTG was added to a final concentration of 1 mM, and the culture was incubated at 20 °C, for 18 h, under orbital agitation (100 rpm). After cell collection and lysis as described above, the supernatant was applied into a HisTrap™ HP column (GE Healthcare Life Sciences, USA) loaded with Ni2+ and equilibrated with the equilibration buffer (50 mM Tris buffer, pH 7.5, 50 mM NaCl, 20 mM imidazole). After washing the column with 20 mL of equilibrium buffer, the recombinant protein was eluted with the same equilibration buffer, but containing a 50–150 mM imidazole gradient instead. Fractions of 1.5 mL were collected, and aliquots of 60 μL of each elution fraction were used for the enzyme assay, with the starch-iodine complex distaining method (Palanivelu 2001). Fractions containing the enzyme were pooled and dialyzed against 50 mM Tris buffer, pH 7.5, with or without 2 mM CaCl2. For storage of the sample, glycerol and CaCl2 were added up to 50% (v/v) and 2 mM, respectively, unless otherwise specified. The samples were kept at -20 °C until use.
Enzyme assays
The starch-iodine complex distaining method (Palanivelu 2001) was used to measure the amylase activity. For that, a sample aliquot of 500 μL was added to 500 μL of 0.1% (w/v) regular soluble starch, and 1.0 mL of 100 mM Tris buffer, pH 6.0. The reaction was incubated for 10 min at 60 °C, and then stopped with addition of 500 μL of 0.1 M HCl. For the remaining starch evaluation, an aliquot of 500 μL of iodine solution [KI 2.0% (w/v); I2 0.2% (w/v)] was added, followed by absorbance reading at 690 nm. One enzyme unit was defined as the amount of enzyme that caused a 20% reduction in the absorbance/min, under the assay conditions.
Alternatively, the amylase activity was evaluated using the dinitrosalicylic acid (DNS) method for reducing sugars release (Miller 1959). For this, a sample aliquot of 500 μL was added to 1.5 mL of 0.5% (w/v) regular starch in 50 mM Tris buffer, pH 6.0. The reaction was incubated for 10 min at 60 °C, and then stopped with addition of 500 μL of the DNS solution [1% (w/v) DNS; 0.4 M NaOH; 1 M potassium sodium tartrate]. The reactions were boiled for 5 min, had 2.5 mL of deionized water added, and the absorbance was read at 540 nm. A reaction with substrate and without enzyme was run and the absorbance reading of the contaminant reducing sugars was decreased from the absorbance obtained in the enzyme’s presence. A calibration curve was drawn using glucose as a standard (0.25–2 μmol/mL of reaction or 0.25–2 mM). One enzyme unit was defined as the amount of enzyme that releases 1 μmol of reducing sugar/min, under the assay conditions.
SDS-PAGE and zymogram
Protein concentration was evaluated with the method of Bradford (1976), using albumin as standard. An aliquot (7.2 μg) of the purified protein was precipitated with trichloroacetic acid (TCA), and the obtained pellet was resuspended in 20 μL of the sample dilution buffer [20% (v/v) glycerol, 1% (w/v) SDS, 0.03 mg/mL bromophenol blue, 125 mM Tris, pH 6.8, 0.72 M β-mercaptoethanol]. Aliquots (10 μg) of the cell homogenate and the supernatant obtained after centrifugation were treated in the same manner. The samples were boiled for 10 min and then loaded on a discontinuous denaturing 7.5% polyacrylamide gel (SDS-PAGE), pH 8.9, with a 4.5% stacking polyacrylamide gel, pH 6.8. An aliquot of 1 μL of the broad range molecular marker (BioRad, USA) was mixed with 19 μL of the sample dilution buffer, boiled for 10 min, and also loaded in the same gel. The gel was run in the Tris–glycine buffer system and stained with Coomassie brilliant blue R-250.
For the zymogram analysis, a 7.5% PAGE gel was run, as described above, except that the samples were not TCA precipitated or boiled, and SDS and β-mercaptoethanol were not added to the gel or samples. Aliquots of 7 μL of the homogenate (6.5 μg) and the purified enzyme (0.16 μg; 0.4 U) were applied into the gel, which was run in the Tris–glycine buffer system without SDS. After the run, a stationary incubation of the gel was performed in 50 mL of 50 mM Tris buffer, pH 6.0, containing 2 mM CaCl2, and 0.5% regular soluble starch (w/v), at 4 °C, for 1 h, with a subsequent incubation for 2 h, at 40 °C, with 60 rpm of orbital agitation in the same solution. Finally, the gel was stained with the same iodine solution used in the enzyme assay.
Substrate specificity and kinetic parameters
For evaluation of enzyme’s substrate specificity, aliquots of 2.4 μg of the purified enzyme, in the presence of 2 mM CaCl2, were evaluated with the DNS method, using as substrates regular soluble starch, maltodextrin (dextrose equivalent 4.0–7.0, Sigma–Aldrich 419672, USA), amylopectin from maize (Sigma–Aldrich 10120, Germany), and β-cyclodextrin (Sigma–Aldrich W402826, Germany), as described in the enzyme assays item.
The enzyme activity with the synthetic substrates 4-nitrophenyl α-D-glucopyranoside (p-NPG, Sigma-Aldrich N1377) and blocked p-nitrophenyl α-D-maltoheptaoside (BpNPG7, Sigma-Aldrich N5665) was also evaluated. For that, an aliquot of 50 μL (6.5 μg) of the purified enzyme was added to 450 μL of 50 mM Tris buffer, pH 6.0, containing 2 mM CaCl2 and 4 mM of the substrates. The reaction was incubated for 2 h at 60 °C in a rotatory mixer (10 rpm). After that, 500 μL of 100 mM Tris, pH 8.0, were added and the absorbance was read at 410 nm. A calibration curve was drawn in the same conditions, without the incubation, using p-nitrophenol (p-NP) (6–60 μm). One enzyme unit was defined as the amount of enzyme that releases 1 μmol of p-NP under the assay conditions.
The initial velocities of the enzyme activity were determined with the starch-iodine complex distaining method. For that, an aliquot of 0.48 μg of the purified enzyme (4.4 U) in 500 μL of 50 mM Tris, pH 7.5, with 2 mM CaCl2, was added to 500 μL of different concentrations, from 0.2 to 2.0 mg/mL, of the highly pure grade soluble starch from potato (Sigma–Aldrich cat. Nº 33615), and to 1.0 mL of 100 mM Tris, pH 6.0. Residual starch concentrations, after enzyme action for 10 min at 60 °C, were calculated with a calibration curve made with starch (0.2–2.0 mg/mL) as a standard. The initial velocities were expressed as mg of starch consumed/min. KM and Vmax values were obtained by non-linear regression using GraphPad Prism® Version 6.01.
Analysis of the enzyme reaction products
An aliquot of 250 μL (0.95 U) of the purified enzyme was added to 750 μL of 1.0% (w/v) highly pure grade soluble starch from potato prepared in 100 mM Tris buffer, pH 6.0, containing 2 mM CaCl2, and incubated for 16 h at 40 °C in a rotatory mixer (10 rpm). A control reaction was prepared with 750 μL of the substrate solution with 250 μL of 100 mM Tris buffer containing 2 mM CaCl2, and was incubated in the same manner as the sample. Ten μL of the sample and control reactions were applied on a thin-layer chromatography plate (TLC) (Silica gel, Sigma-Aldrich, Germany), along with 10 μL of the following standards at 100 mM: D-(+)-anhydrous glucose (G1) and D-(+)-maltose monohydrate (G2). The solvent system used in the ascending chromatography was butanol/ethanol/water (5:3:2). After air-drying the plate, the spots were developed by spraying them with H2SO4 and methanol (1:9) containing 0.2% orcinol, followed by heating at 100 °C (Benassi et al. 2014).
Effect of potential activators and inhibitors, pH, and temperature on the purified enzyme
All reactions were performed with the starch-iodine complex distaining method, incubated at 60 °C for 10 min, in 100 mM Tris buffer, pH 6.0, containing 2 mM CaCl2, except in the CaCl2 effect evaluation on the enzyme activity, where no calcium was added. The activity was calculated as the percentage of the reaction with no addition of ions or detergents, unless stated otherwise. Putative activators and inhibitors were investigated by adding metal ions or detergents in the enzyme assay up to a final concentration of 5 or 10 mM for solids and 0.5 or 1% (v/v) for liquids.
The enzyme’s optimum pH was determined by performing the starch-iodine complex distaining enzyme assay in 100 mM Tris buffer, at pH values—5.0, 5.5, 6.0, 6.5, 7.0, 7.5, and 8.0. A 24 h pH stability time course was conducted by incubating the purified enzyme at pHs 5.0, 6.0, 7.0, and 8.0, at room temperature. Aliquots were collected with time and used in the enzyme assay, under the standard conditions. The results were calculated as the percentage of the highest activity reaction (100%).
The purified enzyme’s optimum temperature was determined by varying the temperature from 30 to 80 °C with increments of 10 °C in the starch-iodine complex distaining method. The thermal stability was evaluated by incubating the enzyme from 30 to 85 °C with increments of 5 or 10 °C, in a thermal cycler, during 30 min, before the enzyme assay in the starch-iodine distaining method standard conditions. The temperature in which the enzyme loses 50% of its activity (Tm) after incubation was calculated with the second-order polynomial regression equation obtained curve. A thermal stability 24 h time course was carried out at 60 and 70 °C. All the results were calculated as the percentage of the highest activity reaction (100%).
Statistical analysis
When indicated, averages and standard deviations (SD) were submitted to ANOVA and compared using the Tukey test, in the SASM–Agri program (Canteri et al. 2001). Identical letters indicate no difference among averages (α = 0.01).
Results and discussion
Sequence analysis of the M. timonae AMY1 protein
A partial α-amylase gene from M. timonae CTI-57 genomic DNA, without the signal peptide and the stop codon, was amplified, cloned into the pCR2.1® plasmid, and sequenced. This gene, named amy1, was the first α-amylase gene cloned from M. timonae and is the first bacterial gene cloned from the GH13_6 plant subfamily of α-amylases. The other GH13_6 subfamily known gene from a bacterial sample is the gene that codes the AmyM, a maltohexaose-forming α-amylase from Corallococcus sp. strain EGB (Li et al. 2015). The M. timonae CTI-57 partial amplified gene had 1,203 bp, coding a 401 amino acids protein. The obtained gene sequence was deposited in GenBank under the accession number MN990470.
The predicted molecular mass and isoelectric point of the amy1 gene encoded protein, without the signal peptide, were 43.6 kDa and 6.25, respectively. The protein sequence alignment (Fig. 1) showed that the M. timonae CTI-57 AMY1 protein shared 90.52% of identity with the α-amylase EKU83291.1 from M. timonae CCUG 45783, and 43.12% with the barley α-amylase (Hordeum vulgare, GenBank P04063.3). The three invariably conserved catalytic residues in all α-amylases (Asp179, Glu204, and Asp281, given positions in our protein) were identified (Fig. 1). The α-amylases comprise a large variety of enzymes, known to have highly variable sequences among them. However, seven short sequences are conserved (CSR—Conserved Sequence Region) (Janeček et al. 2014; Janeček and Gabriško 2016), which are present in the M. timonae CTI-57 AMY1 protein (Fig. 1). One of the most notable features of the GH13_6 α-amylases subfamily is the presence of a glycine at the end of the CSR II, the carbonyl group of which serves as a specific ligand for the calcium ion (Janeček et al. 2014). This glycine is conserved in the M. timonae CTI-57 AMY1, and could play a similar role (Fig. 1).
Fig. 1.

Sequence alignment. Clustal Omega sequence alignment of the M. timonae CTI-57 AMY1 (GenBank MN990470), M. timonae CCUG 45783 (GenBank EKU82989.1), and H. vulgare (GenBank P04063.3) α-amylases. The secretion sequences are in small letters and brown color. The catalytic amino acid residues in the active site are in bold green and have a black triangle (▲) on the top. The seven relatively conserved sequences among α-amylases (Janeček et al. 2014; Janeček and Gabriško 2016) are highlighted in bright yellow and boxed. The amino acid residues that bind to calcium are in bold magenta and have a black diamond (♦) on the top. Cys residues involved in the predicted disulfide bridge (Cys201-Cys232) are in blue and have a black square (■) on the top
A search for domains in the CDD and Pfam platforms indicated that the M. timonae CTI-57 AMY1 protein has an alpha-amylase catalytic domain (Family: Alpha-amylase, PF00128, and Clan CL0058 – Tim barrel glycoside hydrolase superfamily) with an e-value of 1.3 × e−09, and an alpha-amylase C-terminal beta-sheet domain (Family: Alpha-amyl_C2, PF07821, and Clan CL0369 – Glycoside hydrolase domain superfamily) with an e-value of 1.8 × e−19. The catalytic and C-terminal beta-sheet domains correspond to the domains A and C of the α-amylase family, respectively (El-Fallal et al. 2012). Those platforms could not recognize the B domain, which is smaller in α-amylases. Proteins containing the A domain are classified into the Glycoside hydrolase family 13 (GH13), according to CAZy classification. The alignment and domain analysis results together confirm that the studied protein is an α-amylase.
A phylogenetic tree was built (Fig. 2) based on the amino acids sequence similarities of proteins found in databases to verify the evolutionary relationship between the M. timonae and other α-amylases. Sequences of α-amylases from the subfamilies GH13_5, GH13_6, and GH13_7 were analyzed (Fig. 2; Supplementary Table 1). The protein purified and characterized in this study (M. timonae CTI-57 AMY1 protein), grouped together with proteins of the GH13_6 subfamily (Fig. 2). Two other prominent clades of amylases were also formed, one with Bacillus proteins (GH13_5 subfamily) and another with Archaea and Flavobacteria proteins (GH13_7) (Fig. 2). The first Archaeal related flavobacterium α-amylase was described by Li et al. (2014) and later better characterized by Yin et al. (2017). Furthermore, the close relationship of Flavobacteriaceae α-amylase with Archaeal α-amylases is well reviewed in the literature (Janeček et al. 1999; Jones et al. 1999). An Empedobacter (Flavobacteria) α-amylase clustered together with the M. timonae CTI-57 AMY1 protein and probably corresponds to a GH13_6 subfamily protein, different to the GH13_7 Flavobacteria and Archaea α-amylases. The bacterial analyzed proteins in our phylogenetic analysis belonged to the phyla Proteobacteria, Firmicutes, Spirochaetes, and Bacteroidetes (Flavobacteria).
Fig. 2.

The phylogenetic analysis. The optimal tree with the sum of branch length = 8,39,250,649 is shown. The bootstrap test values are shown below or above the branches. The tree is drawn to scale, with branch lengths equal to evolutionary distances. The analysis involved 46 amino acid sequences. The ambiguous positions were removed and there was a total of 729 positions in the final dataset. The M. timonae CTI-57 protein is indicated in bold red. All sequences from the GH13_6 subfamily share more than 40% of identity among them, but share lower identity with members of the other GH13 subfamilies. Proteobacteria phylum species are boxed in blue, Bacteroidetes phylum species in yellow, Firmicutes phylum species in purple, Spirochaetes phylum species in pink, plant species in green, and Archaea species in brown
The modeled structure of the M. timonae CTI-57 AMY1 α-amylase, using as a template the H. vulgare α-amylase from the GH13_6 subfamily (PDB 1AMY, Kadziola et al. 1994), is shown in Fig. 3. This model was the one with the best DOPE score and superposes well with the template (Fig. 3a). The obtained model shows that the M. timonae CTI-57 AMY1 protein has three domains: an N-terminal catalytic domain, the A domain, composed of (β/α)8-barrel, an irregular B domain, protruding between the third β-strand and the third α-helix of the N-terminal domain, and a small C-terminal domain, the C domain, constituted of five antiparallel β-strands (Fig. 3b). An analysis of the obtained M. timonae CTI-57 AMY1 model in the PISA and VADAR servers evidenced a secondary structure composed of α-helix (31%), β-conformation (22%), coil (46%), and turns (17%) (Fig. 3c) and that this protein is a monomer, as this form has the lowest, and therefore, more stable Gibbs free energy. The obtained structure showed the predicted disulfide bond between the residues Cys201-Cys232, which is shown in Fig. 3b, c. The active site is located between the A and B domains, and the critical catalytic residues are shown in Fig. 3d. Near the active site, there is a Ca2+ binding site, where three of the corresponding Ca2+ binding residues present in the template were also identified in the M. timonae CTI-57 AMY1 sequence (Figs. 1, 3d). The Ca2+ ion binding in this enzyme’s site stabilizes the loop in the B domain and is conserved in α-amylases (Morishita et al. 1997). The other two putative Ca2+ binding sites present in the template sequence (Kadziola et al. 1994) were not identified in the M. timonae CTI-57 AMY1 amino acid sequence. The active site cleft has a predominantly negative environment (Fig. 3e), probably due to the negatively charged catalytic residues.
Fig. 3.

The homology structural model of the M. timonae CTI-57 AMY1 α-amylase. a A superposition of the modeled protein, in blue, with the used template (PDB 1AMY), in gold. b Domains A, B, and C are shown in blue, green, and red, respectively. The predicted disulfide bond formed between Cys201-Cys232 is shown in yellow with the Cys residues in cyan. The Ca2+ ion is displayed in orange. c The α-helices, β-strands, turns, and coils are shown in red, blue, pink, and gray, respectively. The A domain, constituted by the (β/α)8-barrel, can be seen in the center. The domain B protrudes between β3-strand and α3-helix of domain A. The C-terminal domain is constituted of five antiparallel β-strands. d The catalytic residues are shown in green and red. The Ca2+ coordinating residues are shown in magenta and red. e Electrostatic potential (red—negative regions; blue—positive regions) in the protein surface positioned as in C)
Recombinant enzyme expression and purification
There was a M. timonae CTI-57 AMY1 α-amylase expression from the pTrcHis2B plasmid using the E. coli Rosetta™ (DE3), which was grown in LB medium. The vector pTrcHis2B has a mini cistron, which provides a sufficient translational restart and, therefore, is useful for eukaryotic protein expression in E. coli. The truncated M. timonae CTI-57 amy1 mRNA has a codon adaptation index (CAI) value of 0.81, which is lower than the considered ideal value of 1.0, indicating that it could have a high number of rare codons. The better translation initiation provided by the pTrcHis2B plasmid, combined with the rare codons tRNAs presence in Rosetta™ (DE3) E. coli, may have been responsible for the improved α-amylase expression. Indeed, an analysis of the predicted structure of the M. timonae CTI-57 amy1 mRNA 5ʹ region initial 255 nucleotides (without the signal sequence) identified a minimum free energy of -94.50 kcal/mol, which demands a ribosomal effort to translate it.
The recombinant protein yield from the pTrcHis2B, in 50 mL LB medium, after affinity chromatography and dialysis, in buffer containing or not CaCl2, was 76.2 μg (1.52 μg/mL of culture medium), with a specific activity of 558.5 U/mg of protein, using the starch-iodine complex distaining assay. This yield is lower than the one found for the B. subtilis WB800 α-amylase of 3.35 mg in 50 mL of culture medium, after a two-step purification in gel filtration and ion-exchange chromatography (Chen et al. 2015) and for the Halothermothrix orenii α-amylase of 0.9 mg in 50 mL of culture, after Ni-affinity chromatography (Mijts and Patel 2002).
The expressed recombinant protein from the pTrcHis2B had three additional amino acids at the N-terminal end (Met-Asp-Pro) and 23 additional amino acids at the C-terminal end, including the His-tag (RLEQYLISEEDLQSAVDHHHHHH). The predicted expressed protein molecular mass was 46.8 kDa, and the isoelectric point was 5.98. The SDS-PAGE analysis confirmed the predicted molecular mass of approximately 47.0 kDa (Fig. 4a). Although being variable, most amylases have a molecular mass around 40–60 kDa (Gupta et al. 2003; Sindhu et al. 2017). In the zymogram analysis (Fig. 4b), a clear hydrolysis halo can be seen in the same position in the cell total proteins (homogenized) lane as well in the purified enzyme lane, indicating that the produced α-amylase was purified. An identical PAGE gel was performed and revealed with the silver staining method (Blum et al. 1987), but the purified M. timonae CTI-57 AMY1 protein could not be detected because a very small amount of the protein was loaded in the gel (Fig. 4c).
Fig. 4.

Electrophoretic analyses. a SDS-PAGE gel. M Broad range molecular marker (BioRad, USA), H homogenized, S supernatant, AMY1 purified M. timonae CTI-57 α-amylase. The arrow indicates the purified enzyme. The gel was stained with Coomassie Blue. b The PAGE gel zymogram showing the activity of the native enzyme from the cell homogenized (H) and purified enzyme (AMY1) on soluble starch. The gel was stained with the iodine reagent and photographed. The bright halos indicate amylase activity. c The PAGE gel containing the cell homogenized (H) and purified enzyme (AMY1). The silver staining was used
Substrate specificity and kinetic parameters
The M. timonae CTI-57 AMY1 showed the highest activity against starch, followed by amylopectin and maltodextrin in the DNS test (Fig. 5a). There was no activity against β-cyclodextrin in the same test (Fig. 5a). In agreement, the Tepidimonas fonticaldi strain HB23 α-amylase was also more active against potato starch and less active against amylopectin and maltodextrin (Allala et al. 2019). The substrate preference of α-amylases for starch has been reviewed before (Gupta et al. 2003). The purified recombinant M. timonae CTI-57 AMY1 has this same preference as it was more active against larger size molecules with a greater number of α-1,4 linkages, such as starch and amylopectin. Regarding synthetic substrates, the purified enzyme showed activity against BpNPG7, but it had no activity against p-NPG, when the release of p-nitrophenol was monitored (Fig. 5b). The activity against the synthetic substrate BpNPG7 and the lack of activity against p-NPG, which has a single glucose unit, also corroborates the fact that this enzyme prefers larger substrates and could be considered an endoamylase.
Fig. 5.
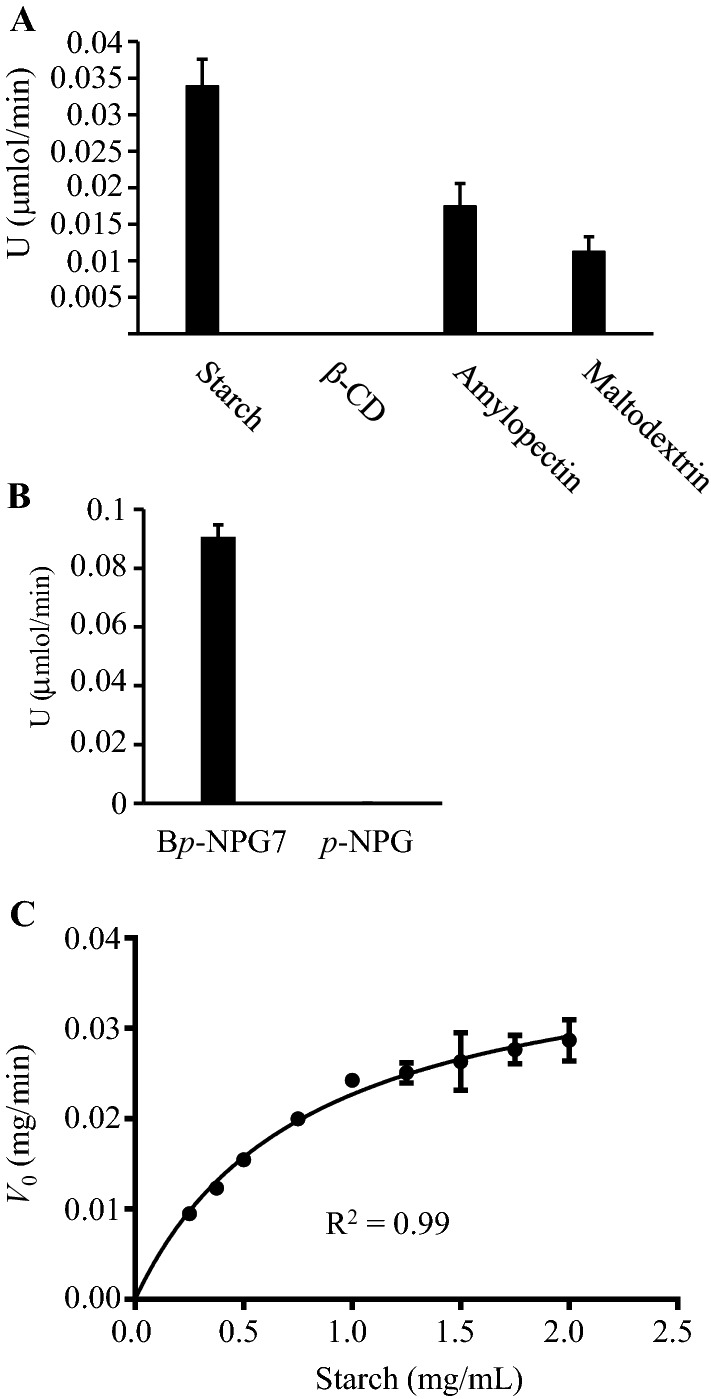
Substrate specificity and saturation curve of the purified recombinant M. timonae CTI-57 α-amylase AMY1. a Starch, β-cyclodextrin (β-CD), amylopectin, and maltodextrin at 0.5% were used in the DNS reaction method. b The substrates Bp-NPG7 and p-NPG were evaluated regarding p-NP release in a reaction. c Saturation curve varying starch concentration from 0.2 to 2.0 mg/mL. The initial velocity was measured as mg of starch that is degraded per min. All data represent the average and SD of three experimental sets
By measuring the initial velocities of starch consumption, it was possible to see that the enzyme followed the Michaelis–Menten kinetics (Fig. 5c). The purified recombinant M. timonae CTI-57 AMY1 α-amylase presented the following kinetics parameters: KM for starch of 0.79 mg/mL and Vmax of 0.04 mg/min of degraded starch. The KM values for bacterial α-amylases depends on the microorganism and are reported to be in the range of 0.3 to 14 mg/mL (Gupta et al. 2003). The M. timonae CTI-57 AMY1 α-amylase KM for starch is among the lower ones, and this indicates that this enzyme needs less substrate to reach half of the Vmax. The zymogram analysis results, indicates that this enzyme may have a high catalytic efficiency, where a small amount of the enzyme was able to degrade the starch present in the gel (Fig. 4b). Vmax estimates for bacterial α-amylases are rare in the literature and use different units, depending on the method used to measure the enzyme activity, which makes difficult the comparison among data.
Enzyme products
In the starch hydrolysis product analysis by the M. timonae CTI-57 AMY1 α-amylase, it is possible to see the production of glucose (G1) and maltose (G2) after 16 h of hydrolysis at 40 °C (Fig. 6). The obtained products reinforce the endo-acting mechanism of the purified α-amylase. According to our results, glucose and maltose were also detected on paper chromatography analysis of Micrococcus luteus α-amylase hydrolysis products (Ilori et al. 1997). Besides, several other bacterial α-amylases were also reported to produce glucose and maltose from starch hydrolysis (Gupta et al. 2003).
Fig. 6.
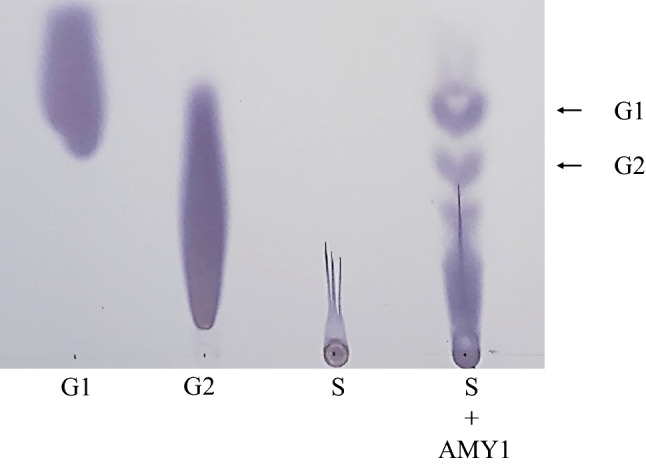
Starch Hydrolysis products by the M. timonae CTI-57 recombinant α-amylase AMY1. A TLC chromatogram with the hydrolysis products from highly pure grade soluble starch (S) incubated with the M. timonae CTI-57 recombinant enzyme (AMY1) at 40 °C for 16 h in a rotatory mixer at 10 rpm (S + AMY1). A control reaction with starch but without enzyme was also used (S). Standards: G1 glucose, G2 maltose
Effect of potential activators and inhibitors, pH, and temperature
The purified M. timonae CTI-57 AMY1 α-amylase was inactive in absence of Ca2+ (Table 1), which indicates that this cation is required for the enzyme’s activity. The enzyme was practically fully active with 2 mM Ca2+, and higher concentrations of Ca2+ did not cause a significant increase in the enzyme’s activity (Table 1). This calcium activation model suggests that there is a limit value in which all the enzyme molecules are already supplied with the required amount of Ca2+, and further increases do not improve the enzyme activity. Most α-amylases require Ca2+ for structural and catalytic activity (Gupta et al. 2003; Sivaramakrishnan et al. 2006). Calcium-independent α-amylases have also been reported, such as the Laceyella sp. DS3 α-amylase, which was activated by Ca2+ but was also active in this cation absence (El-Sayed et al. 2019). Therefore, there is a calcium variation dependency among α-amylases from different microorganisms.
Table 1.
Effect of activators and inhibitors on the purified α-amylase activity
| Substance | Relative activity (%)* | |||
|---|---|---|---|---|
| 0 mM | 2 mM | 5 mM | 10 mM | |
| CaCl2 | 0 | 100 | 110 ± 7.9 | 110 ± 4.3 |
| Control (2 mM CaCl2) | 100 ± 3.8a | 100 ± 4.9a | ||
| NaCl | 103 ± 6.1a | 80.9 ± 8.0b | ||
| KCl | 94.3 ± 8.0a | 82.5 ± 3.2a,b | ||
| NH4Cl | 89.8 ± 3.4a | 82.6 ± 4.8a,b | ||
| MgSO4∙7H2O | 57.6 ± 6.1b | 75.8 ± 6.7b | ||
| CoCl2∙6H2O | 58.7 ± 7.2b | 49.7 ± 5.1c | ||
| (NH4)6Mo7O24∙4H2O | 19.7 ± 3.8d,e | 25.8 ± 4.8d | ||
| HgCl2 | 44.3 ± 5.7b,c | 9.9 ± 4.0d,e | ||
| MnSO4∙H2O | 39.7 ± 4.9c,d | 53.8 ± 2.9c | ||
| EDTA | 11.3 ± 2.5e | 4.1 ± 4.1e | ||
| SDS | 104.3 ± 3.5a | 82.5 ± 2.4a,b | ||
*The data correspond to the average and standard deviation of four repetitions
Equal letters indicates no statistical differences at 1% (α = 0.01) of significance
The chelating agent EDTA caused a potent inhibition of the purified M. timonae CTI-57 AMY1 enzyme (Table 1), and this corroborates the result obtained by the Ca2+ ion on the enzyme’s activity. Although there are reports of amylases that are not affected by the EDTA presence, such as the T. fonticaldi strain HB23 α-amylase (Allala et al. 2019), most α-amylases are metalloenzymes, and EDTA causes inhibition of its activity, as reported for the B. subtilis DR8806 recombinant α-amylase (Emtenani et al. 2015). The calcium ion removal seems to cause reversible denaturation of α-amylases, due to loss of tridimensional structure (Prakash and Jaiswal 2010).
The anionic detergent SDS did not inhibit the enzyme (Table 1). Other amylases have been reported to be resistant to SDS inhibition, such as the Bacillus halodurans α-amylase (Murakami et al. 2007). The enzyme’s activity with neutral detergents addition, such as Tween 80 and Triton X-100, could not be determined because those reagents were not compatible with the starch-iodine distaining method. The purified enzyme activity in the presence of an anionic detergent indicates its possible use in the detergent industry, considering that anionic surfactants are the oldest and most widely used synthetic detergents and can be found in many personal- and household-care products (Kosswig 2005).
The monovalent cations Na+ and K+ present in the reaction assay at 10 mM caused a slight inhibition of the enzyme (Table 1). The divalent cations Mg2+, Mn2+, Co2+, Hg2+, and Mo2+, were inhibitors of the purified enzyme. Many metal cations, especially heavy metal ions, are reported to inhibit α-amylases (Gupta et al. 2003). In agreement, the Laceyella sp. DS3 α-amylase was reported to be inactivated by HgCl2 and MgSO4, and the T. fonticaldi strain HB23 α-amylase was utterly inhibited by Hg2+ and Mn2+ (El-Sayed et al. 2019; Allala et al. 2019). However, in opposition to our results, this latter enzyme was activated by Mg2+ (Allala et al. 2019). Therefore, the effect of metal ions on α-amylases activity varies between the source’s organisms.
The optimum pH for the M. timonae CTI-57 AMY1 was 6.0 (Fig. 7a). However, the enzyme was more than 80% active in pH 5.0 to 5.5 and was almost fully active in the range of pH 6.0 to 8.0. Most bacterial α-amylase has optimum pH on a slightly acidic to a neutral range of pH, mainly at 5.0 to 6.5 (Pandey et al. 2000; Gupta et al. 2003). This profile seems to repeat in recombinant enzymes, such as the Laceyella sp. DS3 and the B. subtilis WB800 recombinant α-amylases, which have optimum activity at pH 6.0 (El-Sayed et al. 2019; Chen et al.2015). This optimum acidic pH is essential for the starch industry, considering that the natural pH of the starch slurry is generally around 4.5 (Sivaramakrishnan et al. 2006). The optimum acidic pH is attributed to the ionization state of critical catalytic residues in the active site of α-amylases, such as the nucleophile aspartate, which should stay negatively charged, and the catalyst glutamic acid residue, which should have a proton to donate (Nielsen et al. 2001).
Fig. 7.
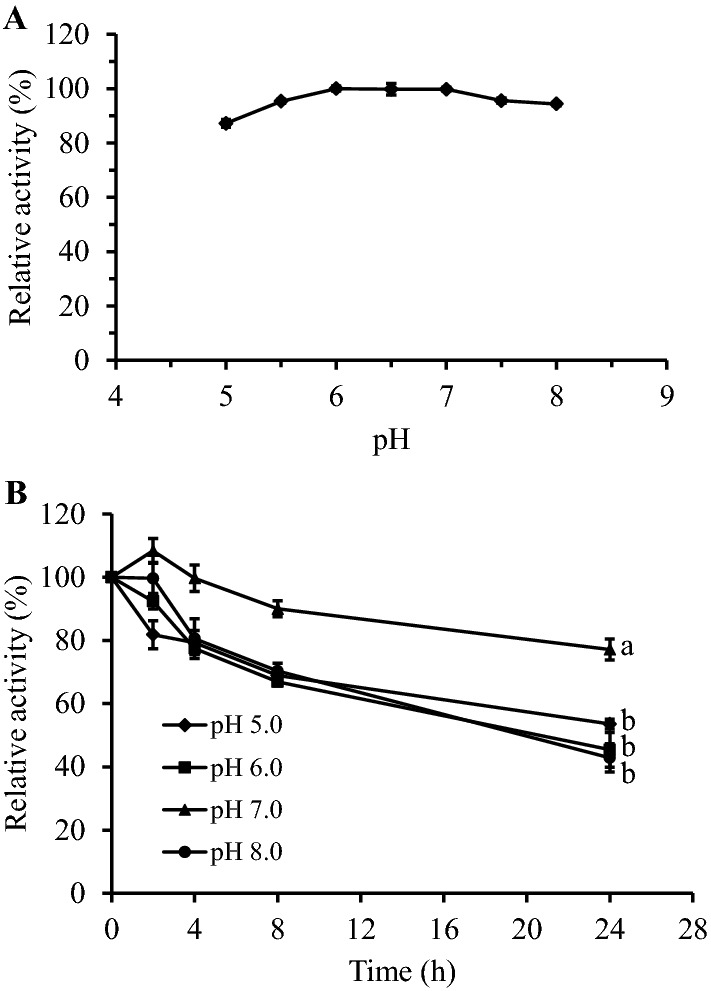
Effect of pH on the purified recombinant M. timonae CTI-57 α-amylase AMY1. a Optimum pH. b pH stability over time. Averages of the 24 h time point were submitted to ANOVA and compared using the Tukey test. Identical letters show no difference among averages (α = 0.01). The analysis was conducted with the starch-iodine distaining method. All data represent the average and SD of three experimental sets
The pH stability assay showed that the M. timonae CTI-57 AMY1 remained 53, 45, 77, and 42% active when incubated for 24 h in pH 5.0, 6.0, 7.0, and 8.0, respectively (Fig. 7b). This agrees with the α-amylases broad range of pH stability, from 4 to 11 (Gupta et al. 2003). However, in pH 7.0, the stability was statistically significant (α = 0.01) higher than in other pHs, including the optimum pH of 6.0. This stability pattern could be explained by the better preservation of the enzyme’s tertiary structure in pH 7.0, which could be altered in more acidic or basic pHs, due to changes in the ionic state of key residues. As all the enzymatic assays were conducted in the optimum pH (6.0), even after incubating in another pH, the catalytic residues’ charges could be restored during the reaction.
The recombinant M. timonae CTI-57 AMY1 enzyme showed to be very active in a wide range of temperatures, from 40 to 70 °C (Fig. 8a), but its optimum temperature in pH 6.0 was 60 °C, and in pH 7.5 was 70 °C. Regarding other recombinant α-amylases, the recombinant Laceyella sp. DS3 α-amylase was reported to have an optimum temperature of 55 °C (El-Sayed et al. 2019). Moreover, bacterial α-amylases are reported to be active in an extensive range of temperature, but several have high optima temperatures, what classifies them as thermophilic enzymes (Gupta et al. 2003; Sivaramakrishnan et al. 2006). The broader range of temperature in which the M. timonae CT57 AMY1 α-amylase could work and its resistance to the anionic detergent SDS could be useful in the detergent industry, considering the modern trend among consumers of using colder temperatures for doing laundry or dishwashing (El-Fallal et al. 2012).
Fig. 8.
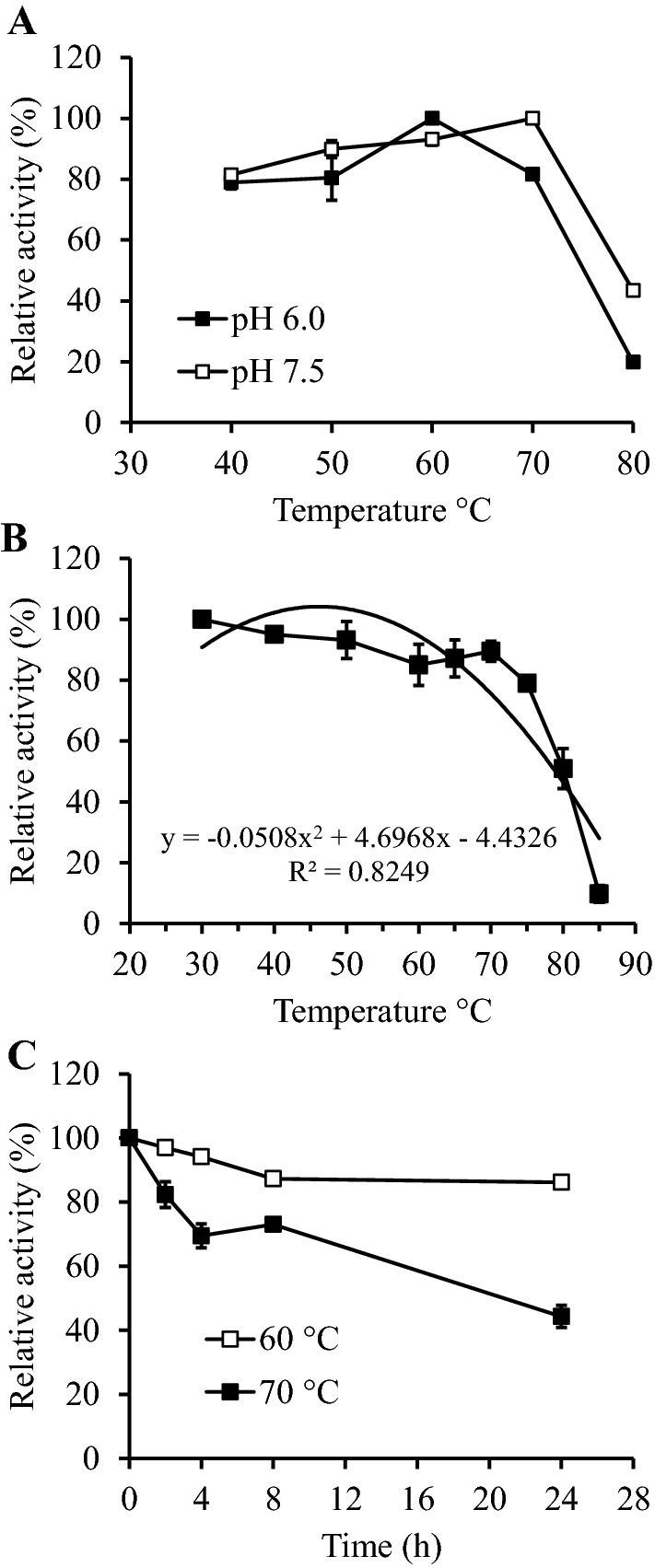
Effect of temperature on the purified recombinant M. timonae CTI-57 α-amylase AMY1. The analysis was conducted with the starch-iodine distaining method. a Optimum temperature. b Thermal stability. c Thermal stability over time. All data represent the average and SD of three experimental sets
In the thermal stability assay, the enzyme remained very active after 30 min of incubation, in temperatures ranging from 30 to 70 °C, but lost its activity when incubated at 85 °C, having a Tm of 79 °C, which was calculated from the second-order polynomial curve (Fig. 8b). In an activity time course of incubation at different temperatures, the recombinant M. timonae CTI-57 AMY1 enzyme retained 86.1 and 44.3% of its activity after incubation at 60 and 70 °C, respectively, for 24 h (Fig. 8c). Prakash and Jaiswal (2010) consider enzymes acting in a temperature equal or above 60 °C as thermostable, as most enzymes lose activity at temperatures of 50–60 °C. Considering the M. timonae CTI-57 AMY1 α-amylase Tm of 79 °C and stability when incubated at 60 °C for 24 h, it could be considered a thermostable enzyme, which is comparable to α-amylases of some bacteria from the Bacillus and Pyrococcus genera (Pandey et al. 2000; Sivaramakrishnan et al. 2006; Prakash and Jaiswal 2010; Mehta and Satyanarayana 2016).
The starch industry requires α-amylases to be active at high temperatures to reduce the process cost, and there has been a need and continual search for thermophilic and thermostable α-amylases (Sivaramakrishnan et al. 2006). In the industrial starch hydrolysis, two processes are conducted. The first one is the liquefaction, which is carried out by amylases that allows a rapid reduction in the solution viscosity and that is performed at pH 6.0 and high temperatures (80–110 °C) (van der Maarel et al. 2002; El-Fallal et al. 2012). The second process corresponds to the partially hydrolyzed starch saccharification, which uses enzymes that can release maltose and d-glucose and that is carried out at pH 4.5 and lower temperatures (60–70 ºC) (van der Maarel et al. 2002). α-Amylases are divided into two categories according to the degree of hydrolysis. Liquefying α-amylases, which do not produce free sugars and cause a more rapid reduction in the starch paste viscosity, and saccharifying α-amylases, which produce free sugars and reduce the starch viscosity less rapidly (El-Fallal et al. 2012; Tomasik and Horton 2012). Considering the M. timonae CTI-57 AMY1 α-amylase characteristics, such as optimum pH of 6.0, Tm of 79 °C, and starch specificity it could be considered as a liquefying enzyme.
Conclusion
In this study, a gene from a novel α-amylase from M. timonae CTI-57 was cloned. The gene was successfully expressed using the pTrcHis2B plasmid and the E. coli Rosetta™ (DE3) strain and the recombinant protein was characterized. This is the first experimentally characterized bacterial α-amylase from the plant α-amylases GH13_6 subfamily. The protein sequence had a high identity with proteins from other Massilia species, as well as with proteins from other bacteria and plants. The M. timonae CTI-57 AMY1 α-amylase was not inhibited by SDS and could have an application in the detergent industry. The biochemical characteristics of the purified protein, such as activity in pH 6.0, thermal stability, starch specificity, and endo-acting mechanism, classify it as a liquefying starch enzyme.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
The authors are thankful to the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Brazil, for the Project funding (grant 001) and the scholarship given to B. Y. Tagomori, respectively.
Funding
Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES)—Project funding (grant 001). Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq)—Scholarship to Bruna Yuki Tagomori.
Compliance with ethical standards
Conflict of interest
The authors declare no conflict of interest.
References
- Akinloye OA, Balogun EA, Kareem SO, Mosaku OS. Partial purification and some properties of α–glucosidase from Trichoderma longibrachiatum. Biokemistri. 2012;24(1):31–37. [Google Scholar]
- Allala F, Bouacem K, Boucherba N, Azzouz Z, Mechri S, Sahnoun M, Benallaoua S, Hacene H, Jaouadi B, Bouanane-Darenfed A. Purification, biochemical, and molecular characterization of a novel extracellular thermostable and alkaline α-amylase from Tepidimonas fonticaldi strain HB23. Int J Biol Macromol. 2019;132:558–574. doi: 10.1016/j.ijbiomac.2019.03.201. [DOI] [PubMed] [Google Scholar]
- Benassi VM, Pasin TM, Facchini FDA, Jorge JA, Polizeli MLTM. A novel glucoamylase activated by manganese and calcium produced in submerged fermentation by Aspergillus phoenicis. J Basic Microbiol. 2014;54:333–339. doi: 10.1002/jobm.201200515. [DOI] [PubMed] [Google Scholar]
- Blum H, Beier H, Gross HJ. Improved silver staining of plant proteins, RNA and DNA in polyacrylamide gels. Electrophoresis. 1987;8:93–99. [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of dye-binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Canteri MG, Althaus RA, Virgens Filho JS, Giglioti EA. SASM-AGRI - system for analysis and mean separation in agricultural assays using Scott-Knott, Tukey and Duncan methods. Braz J Agrocomp. 2001;1:18–24. [Google Scholar]
- Cerrone F, Sánchez-Peinado MDM, Rodríguez-Díaz M, González-López J, Pozo C. PHAs production by strains belonging to Massilia genus from starch. Starch Stäerke. 2011;63:236–240. [Google Scholar]
- Chapman J, Ismail AE, Dinu CZ. Industrial applications of enzymes: recent advances, techniques, and outlooks. Catalysts. 2018;8:238. [Google Scholar]
- Chen J, Chen X, Dai J, Xie G, Yan L, Lu L, Chen J. Cloning, enhanced expression and characterization of an α-amylase gene from a wild strain in B. subtilis WB800. Int J Biol Macromol. 2015;80:200–207. doi: 10.1016/j.ijbiomac.2015.06.018. [DOI] [PubMed] [Google Scholar]
- Chung CT, Niemela SL, Miller RH. One-step Preparation of competent Escherichia coli: transformation and storage of bacterial cells in the same solution. Proc Natl Acad Sci USA. 1989;86:2172–2175. doi: 10.1073/pnas.86.7.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Fallal A, Dobara MA, El-Sayed A, Omar N. Starch and microbial α-amylases: from concepts to biotechnological applications. In: Chang CF, editor. Carbohydrates—comprehensive studies on glycobiology and glycotechnology. London: IntechOpen; 2012. pp. 459–488. [Google Scholar]
- El-Sayed AKA, Abou-Dobara MI, El-Fallal AA, Omar NF. Heterologous expression, purification, immobilization and characterization of recombinant α-amylase AmyLa from Laceyella sp. DS3. Int J Biol Macromol. 2019;132:1274–1281. doi: 10.1016/j.ijbiomac.2019.04.010. [DOI] [PubMed] [Google Scholar]
- Emtenani S, Asoodeh A, Emtenani S. Gene cloning and characterization of a thermostable organic-tolerant α-amylase from Bacillus subtilis DR8806. Int J Biol Macromol. 2015;72:290–298. doi: 10.1016/j.ijbiomac.2014.08.023. [DOI] [PubMed] [Google Scholar]
- Fahlgren C, Bratbak G, Sandaa RA, Thyrhaug R, Zweifel UL. Diversity of airborne bacteria in samples collected using different devices for aerosol collection. Aerobiologia. 2011;27:107–120. [Google Scholar]
- Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- Gallego V, Sánchez-Porro C, García MT, Ventosa A. Massilia aurea sp. nov., isolated from drinking water. Int J Syst Evol Microbiol. 2006;56:2449–2453. doi: 10.1099/ijs.0.64389-0. [DOI] [PubMed] [Google Scholar]
- Gopinath SCB, Anbu P, Arshad MKM, Lakshmipriya T, Voon CH, Hashim U, Chinni SV. Biotechnological processes in microbial amylase production. BioMed Res Int. 2018;2017:1272193. doi: 10.1155/2017/1272193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, Gigras P, Mohapatra H, Chauhan GVK, B, Microbial α-amylases: a biotechnological perspective. Process Biochem. 2003;38:1599–1616. [Google Scholar]
- Gurung N, Ray S, Bose S, Rai V. A broader view: microbial enzymes and their relevance in industries, medicine, and beyond. BioMed Res Int. 2013;2013:329121. doi: 10.1155/2013/329121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- Ilori MO, Amund OO, Omidiji O. Purification and properties of an α-amylase produced by a cassava-fermenting strain of Micrococcus luteus. Folia Microbiol. 1997;42(5):445–449. doi: 10.1007/BF02826551. [DOI] [PubMed] [Google Scholar]
- Janeček Š. α-Amylases from Archaea: sequences, structures and evolution. In: Rampelotto P, editor. Biotechnology of extremophiles: grand challenges in biology and biotechnology. Cham: Springer; 2016. pp. 505–524. [Google Scholar]
- Janeček Š, Gabriško M. Remarkable evolutionary relatedness among the enzymes and proteins from the α-amylase family. Cell Mol Life Sci. 2016;73:2707–2725. doi: 10.1007/s00018-016-2246-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janeček Š, Lévêque E, Belarbi A, Haye B. Close evolutionary relatedness of α-amylases from Archaea and plants. Mol Evol. 1999;48:421–426. doi: 10.1007/pl00006486. [DOI] [PubMed] [Google Scholar]
- Janeček Š, Svensson B, MacGregor EA. α-Amylase: an enzyme specificity found in various families of glycoside hydrolases. Cell Mol Life Sci. 2014;71:1149–1170. doi: 10.1007/s00018-013-1388-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RA, Jermiin LS, Easteal S, Patel BKC, Beacham IR. Amylase and 16S rRNA genes from a hyperthermophilic archaebacterium. J Appl Microbiol. 1999;86:93–107. doi: 10.1046/j.1365-2672.1999.00642.x. [DOI] [PubMed] [Google Scholar]
- Kadziola A, Abe J, Svensson B, Haser R. Crystal and molecular structure of barley alpha-amylase. J Mol Biol. 1994;239:104–121. doi: 10.1006/jmbi.1994.1354. [DOI] [PubMed] [Google Scholar]
- Kosswig K. Surfactants. In Ullmann's encyclopedia of industrial chemistry. Weinheim: Wiley-VCH; 2005. [Google Scholar]
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35(6):1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Scola B, Birtles RJ, Mallet MN, Raoult D. Massilia timonae gen nov., sp. nov., isolated from blood of an immunocompromised patient with cerebellar lesions. J Clin Microbiol. 1998;36(10):2847–2852. doi: 10.1128/jcm.36.10.2847-2852.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Läufer A. Starch biorefinery enzymes. Adv Biochem Eng Biotechnol. 2017;166:137–152. doi: 10.1007/10_2016_60. [DOI] [PubMed] [Google Scholar]
- Lévêque E, Janeček Š, Haye B, Belarbi A. Thermophilic archaeal amylolytic enzymes. Enz Microb Technol. 2000;26:3–14. [Google Scholar]
- Li C, Du M, Cheng B, Wang L, Liu X, Ma C, Yang C, Xu P. Close relationship of a novel Flavobacteriaceae α-amylase with archaeal α-amylases and good potentials for industrial applications. Biotechnol Biofuels. 2014;7:18. doi: 10.1186/1754-6834-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Wu J, Zhang B, Wang F, Ye X, Huang Y, Huang Q, Cui Z. AmyM, a novel maltohexaose-forming α-amylase from Corallococcus sp strain EGB. Appl Environ Microbiol. 2015;81(6):1977–1987. doi: 10.1128/AEM.03934-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindquist D, Murrill D, Burran WP, Winans G, Janda MJ. Characteristics of Massilia timonae and Massilia timonae-like isolates from human patients, with an emended description of the species. J Clin Microbiol. 2003;41:192–196. doi: 10.1128/JCM.41.1.192-196.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard V, Ramulu HG, Drula E, Coutinho PM, Henrissat B. The Carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42:D490–D495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNicholas S, Potterton E, Wilson KS, Nobleb MEM. Presenting your structures: the CCP4MG molecular-graphics software. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):386–394. doi: 10.1107/S0907444911007281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta D, Satyanarayana T. Bacterial and archaeal α-amylases: diversity and amelioration of the desirable characteristics for industrial applications. Front Microbiol. 2016;7:1129. doi: 10.3389/fmicb.2016.01129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mijts BN, Patel BKC. Cloning, sequencing and expression of an αamylase gene, amyA, from the thermophilic halophile Halothermothrix orenii and purification and biochemical characterization of the recombinant enzyme. Microbiology. 2002;148:2343–2349. doi: 10.1099/00221287-148-8-2343. [DOI] [PubMed] [Google Scholar]
- Miller GL. Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Chem. 1959;31(3):426–428. [Google Scholar]
- Morishita Y, Hasegawa K, Matsuura Y, Katsube Y, Kubota M, Sakai S. Crystal structure of a maltotetraose-forming exo-amylase from Pseudomonas stutzeri. J Mol Biol. 1997;267:661–672. doi: 10.1006/jmbi.1996.0887. [DOI] [PubMed] [Google Scholar]
- Murakami S, Nishimoto H, Toyama Y, Shimamoto E, Takenaka S, Kaulpiboon J, Prousoontorn M, Limpaseni T, Pongsawasdi P, Aoki K. Purification and characterization of two alkaline, thermotolerant α-amylases from Bacillus halodurans 38C-2-1 and expression of the cloned gene in Escherichia coli. Biosci Biotechnol Biochem. 2007;71(10):2393–2401. doi: 10.1271/bbb.60666. [DOI] [PubMed] [Google Scholar]
- Nagy ML, Pérez A, Garcia-Pichel F. The prokaryotic diversity of biological soil crusts in the Sonoran desert (Organ Pipe Cactus National Monument, AZ) FEMS Microbiol Ecol. 2005;54:233–245. doi: 10.1016/j.femsec.2005.03.011. [DOI] [PubMed] [Google Scholar]
- Nielsen JE, Borchert TV, Vriend G. The determinants of a α-amylase pH-activity profiles. Protein Eng. 2001;14(7):505–512. doi: 10.1093/protein/14.7.505. [DOI] [PubMed] [Google Scholar]
- Niyonzima FN, More SS. Detergent-compatible bacterial amylases. Appl Biochem Biotechnol. 2014;174:1215–1232. doi: 10.1007/s12010-014-1144-3. [DOI] [PubMed] [Google Scholar]
- Palanivelu P. Analytical biochemistry and separation techniques. Madurai: Kalamani Printers; 2001. [Google Scholar]
- Pandey A, Nigam P, Soccol CR, Soccol VT, Singh D, Mohan R. Advances in microbial amylases. Biotechnol Appl Biochem. 2000;31:135–152. doi: 10.1042/ba19990073. [DOI] [PubMed] [Google Scholar]
- Prakash O, Jaiswal N. α-amylase: an ideal representative of thermostable enzymes. Appl Biochem Biotechnol. 2010;160:2401–2414. doi: 10.1007/s12010-009-8735-4. [DOI] [PubMed] [Google Scholar]
- Raveendran S, Parameswaran B, Ummalyma SB, Abraham A, Mathew AK, Madhavan A, Rebello S, Pandey A. Applications of microbial enzymes in food industry. Food Technol Biotechnol. 2018;56(1):16–30. doi: 10.17113/ftb.56.01.18.5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular cloning: a laboratory manual. 3. New York: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Santos FC, Barbosa-Tessmann IP. Recombinant expression, purification, and characterization of a cyclodextrinase from Massilia timonae. Protein Expres Purif. 2019;154:74–84. doi: 10.1016/j.pep.2018.08.013. [DOI] [PubMed] [Google Scholar]
- Santos FC, Castro FF, Apolonio TM, Yoshida L, Martim DB, Tessmann DJ, Barbosa-Tessmann IP. Isolation, diversity, and biotechnological potential of maize (Zea mays) grains bacteria. Genet Mol Res. 2019;18(3):gmr18320. [Google Scholar]
- Sindhu R, Binod P, Madhavan A, Beevi US, Mathew AK, Abraham A, Pandey A, Kumar V. Molecular improvements in microbial α-amylases for enhanced stability and catalytic efficiency. Bioresour Technol. 2017;245:1740–1748. doi: 10.1016/j.biortech.2017.04.098. [DOI] [PubMed] [Google Scholar]
- Singh R, Kumar M, Mittal A, Mehta PK. Microbial enzymes: industrial progress in 21st century. 3 Biotech. 2016;6(2):174. doi: 10.1007/s13205-016-0485-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivaramakrishnan S, Gangadharan D, Nampoothiri KM, Soccol CR, Pandey A. α-Amylases from microbial sources—an overview on recent developments. Food Technol Biotechnol. 2006;44(2):173–184. [Google Scholar]
- Souza PM, Magalhães PO. Applications of microbial α-amylase in industry—a review. Braz J Microbiol. 2010;41:850–861. doi: 10.1590/S1517-83822010000400004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stam MR, Danchin EGJ, Rancurel C, Coutinho PM, Henrissat B. Dividing the large glycoside hydrolase family 13 into subfamilies: towards improved functional annotations of a-amylase-related proteins. Protein Eng Des Sel. 2006;19(12):555–562. doi: 10.1093/protein/gzl044. [DOI] [PubMed] [Google Scholar]
- Studier FW. Protein production by auto-induction in high-density shaking cultures. Protein Expr Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Sun H, Zhao P, Ge X, Xia Y, Hao Z, Liu J, Peng M. Recent advances in microbial raw starch degrading enzymes. Appl Biochem Biotechnol. 2010;160:988–1003. doi: 10.1007/s12010-009-8579-y. [DOI] [PubMed] [Google Scholar]
- Tomasik P, Horton D. Enzymatic conversions of starch. Adv Carbohydr Chem Biochem. 2012;68:59–436. doi: 10.1016/B978-0-12-396523-3.00001-4. [DOI] [PubMed] [Google Scholar]
- Trevelyan WE, Procter DP, Harrison JS. Detection of sugars on paper chromatograms. Nature. 1950;166:444–445. doi: 10.1038/166444b0. [DOI] [PubMed] [Google Scholar]
- van der Maarel JEC, van der Veen B, Uitdehaag JC, Leemhuis H, Dijkhuizen L. Properties and applications of starch-converting enzymes of the α-amylase family. J Biotechnol. 2002;94(2):137–155. doi: 10.1016/s0168-1656(01)00407-2. [DOI] [PubMed] [Google Scholar]
- Webb B, Sali A. Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics. 2016;54:5.6.1–5.6.37. doi: 10.1002/cpbi.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H, Yang Z, Nie X, Li S, Sun X, Gao C, Wang Z, Zhou G, Xu P, Yang C. Functional and cooperative stabilization of a two-metal (Ca, Zn) center in α-amylase derived from Flavobacteriaceae species. Sci Rep. 2017;7:17933. doi: 10.1038/s41598-017-18085-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.