

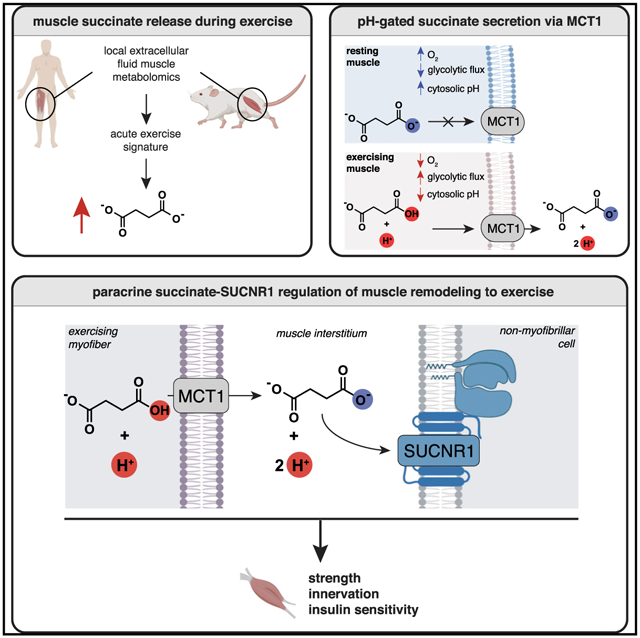
SUMMARY
In response to skeletal muscle contraction during exercise, paracrine factors coordinate tissue remodeling, which underlies this healthy adaptation. Here we describe a pH-sensing metabolite signal that initiates muscle remodeling through exercise. In mice and humans, exercising skeletal muscle releases the mitochondrial metabolite succinate into the local interstitium and circulation. Selective secretion of succinate is facilitated by its transient protonation, which occurs upon muscle cell acidification. In the protonated monocarboxylic form, succinate is rendered a transport substrate for monocarboxylate transporter 1, which facilitates pH-gated release. Upon secretion, succinate signals via its cognate receptor SUCNR1 in non-myofibrillar cells in muscle tissue to control muscle-remodeling transcriptional programs. This succinate-SUCNR1 signaling is required for paracrine regulation of muscle innervation, muscle matrix remodeling, and muscle strength in response to exercise training. In sum, we define a bioenergetic sensor in muscle that utilizes intracellular pH and succinate to coordinate tissue adaptation to exercise.
Graphical Abstract
In Brief
Reddy et al. identify a bioenergetic sensor that uses pH and succinate to regulate muscle tissue adaptation to exercise.
INTRODUCTION
Skeletal muscle remodeling in response to exercise has multiple systemic health benefits in mice and humans. This muscle adaptation requires paracrine processes, including local neurotrophic signaling, angiogenesis, and extracellular matrix remodeling. Initiation of this paracrine activity depends on upstream signals originating from contracting muscle that are poorly defined. Metabolite abundance and transport are regulated on rapid timescales that make metabolites an appropriate signaling modality for initiating rapid physiological adaptation (Martínez-Reyes and Chandel, 2020). In mice and humans, the muscle-remodeling response begins rapidly upon acute exercise (Ghosh et al., 2010; Hawley et al., 2014; Kemi et al., 2002; Sylow et al., 2017). Therefore, we hypothesized that metabolite signals originating from exercising muscle could be essential for activating paracrine processes that control muscle remodeling.
Here we apply metabolomics approaches to mice and humans to define a metabolite pH sensor in muscle that communicates the local energetic status to the paracrine tissue environment and controls tissue remodeling. We show that muscle selectively secretes the mitochondrial metabolite succinate into the muscle interstitium and circulation upon exercise. Because of its unique physiochemical properties, succinate is exported from muscle cells through acute regulation of its protonation status by local pH. When protonated, succinate is transformed from a dicarboxylate that is trapped in the cell to a monocarboxylate. In this protonated monocarboxylate form, succinate is rendered a transport substrate for the plasma membrane transporter monocarboxylate transporter 1 (MCT1), which facilitates pH-gated release. Upon pH-gated secretion during exercise, succinate signals via its cognate receptor succinate receptor 1 (SUCNR1) in non-myofibrillar cells in muscle tissue to coordinate muscle remodeling. Finally, we demonstrate that muscle innervation, extracellular matrix (ECM) remodeling, and increased strength initiated upon exercise depend critically on this newfound succinate-SUCNR1 signaling cascade.
RESULTS AND DISCUSSION
Mouse and Human Muscle Secretes Succinate during Exercise
We used a comparative metabolomics approach to determine metabolites that are selectively and locally secreted from exercising muscle in mice in vivo (Figure 1A; Figures S1A and S1B; STAR Methods). We applied acute exercise intervention, which is known to initiate muscle remodeling, improve insulin sensitivity, and provide systemic health benefits (Ghosh et al., 2010; Hawley et al., 2014; Kemi et al., 2002; Sylow et al., 2017). Mice walked on a treadmill or were subjected to a ramped running protocol until exhaustion. Following intervention, samples were rapidly isolated from the tibialis anterior (TA) muscle, a major muscle engaged by treadmill running (Henríquez-Olguin et al., 2019). Samples were gated on three criteria: (1) metabolite accumulation immediately post-exercise in local muscle extracellular fluid (Figure 1B; Table S1A; see Figures S1A and S1B and STAR Methods for a description of interstitial fluid isolation and QC); (2) metabolite enrichment in muscle interstitial fluid compared with total muscle tissue (Figure 1C; Table S1B); (3) local enrichment in local muscle extracellular fluid compared with bulk circulation (Figure 1D; Table S1C). A single metabolite fulfilled all three criteria for a local extracellular metabolite signature of acute exercise: the mitochondrial metabolite succinate. We additionally determined the same parameters in the gastrocnemius (GA) muscle, another muscle group engaged by treadmill running, and found that succinate had a comparable signature (Figures 1E-1G).
Figure 1. Succinate Is Released Selectively by Exercising Muscle in Mice and Humans.

(A) Comparative metabolomics approach to identify exercise-responsive metabolites released locally by muscle. Extracellular fluids are a combination of interstitial fluids and local circulation.
(B–D) Summary results from the comparative approach, illustrating all annotated metabolites (gray), metabolites fulfilling each individual criterion (black), and metabolites fulfilling all criteria (red).
(B) Metabolites accumulated in muscle extracellular fluid after exercise were defined as fold change > 2 and −log10p>3 versus sedentary extracellular fluid (n = 8).
(C) Metabolites enriched post-exercise in extracellular fluid compared with whole muscle (n = 8).
(D) Metabolites selectively enriched in local muscle extracellular fluid post-exercise were defined as fold change > 2 and −log10p > 3 versus post-exercise bulk plasma (n = 8).
(E) Local release of succinate post-exercise occurs in tibialis anterior (TA) and gastrocnemius (GA) muscle. A selective increase in muscle extracellular fluid is observed in both muscle groups but not in whole muscle (n = 8).
(F) Selective accumulation of succinate in muscle extracellular fluid post-exercise. The relative change in abundance in muscle and interstitial fluid when sedentary and post-exercise is determined separately. Then, to query relative accumulation in extracellular fluid versus muscle, the ratio of relative differences is plotted (n = 8).
(G) Local accumulation of succinate in muscle extracellular fluid post-exercise. The relative change in abundance in plasma and interstitial fluid when sedentary and post-exercise is determined separately. Then, to query relative accumulation in extracellular fluid versus bulk plasma, the ratio of relative differences is plotted (n = 8).
(H) Experimental design to quantify succinate release by human exercising muscle.
(I) Femoral artery and vein succinate concentration during human exercise (n = 10).
(J) Femoral artery-vein difference in succinate concentration during human exercise (n = 10).
(K) Comparison of post-exercise enrichment of mitochondrial TCA cycle metabolites in extracellular fluid compared with whole muscle (n = 8). The relative change in abundance in muscle and extracellular fluid when sedentary and post-exercise is determined separately. Then, to query relative accumulation in interstitial fluid versus muscle, the ratio of relative differences is plotted.
Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.005 (two-tailed Student’s t test for pairwise comparisons, one-way ANOVA for multiple comparisons involving one independent variable).
We next examined whether succinate was also released by exercising muscle in humans. We recruited 25- to 35-year-old, healthy, non-obese, non-smoking men. These individuals were catheterized in the femoral artery and vein to allow local sampling of blood entering and draining from exercising muscles, as described in Kleinert et al. (2018). Following this, individuals exercised on a bicycle ergometer for 60 min at 67% maximal oxygen uptake (VO2 max), with local blood collected during exercise and recovery (Figure 1H; Table S2; STAR Methods). Muscle venous succinate concentrations rose to ~200 μM upon exercise and then decreased rapidly upon recovery (Figure 1I). Notably, changes in succinate concentration were concomitant with elevations in arterial and venous lactate (Figure S1C). The arterial-venous difference in metabolite abundance determines whether a tissue is a net producer or consumer of a given metabolite (Kiens et al., 1993). We found a highly significant arterial-venous difference during exercise, with substantially more succinate found in exercising venous blood, indicating that exercising muscle is a source of elevated succinate (Figure 1J; Figure S1D). Taken together, local and selective release of succinate by muscle is a rapid response to exercise in mice and humans.
Muscle Cells Secrete Succinate through a pH-Gated Mechanism via MCT1
Identification of succinate as a metabolite that is released from exercising muscle was unexpected because the plasma membrane of most mammalian cells is thought to be impermeable to mitochondrial dicarboxylates, including succinate (Ehinger et al., 2016; Hems et al., 1968; MacDonald et al., 1989; Mills et al., 2018). However, a long-standing and curious observation is that blood succinate levels are substantial and dynamic, responding to interventions like hypoxia (Hochachka et al., 1975), exercise (Hochachka and Dressendorfer, 1976), and exposure to cold temperatures (Mills et al., 2018). In this context, because of its apparent lack of permeation through the plasma membrane, extracellular succinate is widely considered to be a proxy for cellular rupture and non-specific leakage (Murphy and O’Neill, 2018).
However, this supposition is inconsistent with the selectivity of succinate release observed here, which precludes non-specific cell rupture (Figure S1E) and is indicative of a defined export mechanism for this metabolite. Succinate release into the muscle interstitium and blood was atypical compared with most mitochondrial dicarboxylates (Figure 1K). Based on this observation, we considered the physiochemical properties of succinate compared with other mitochondrial carboxylates that are not released from muscle in the same context (Figures 2A and 2B). We noted that succinate is distinct from other mitochondrial carboxylates based on its high monocarboxylic pKa (Figure 2A). This property would uniquely render a substantial proportion of succinate in the monocarboxylate form at the physiological pH of exercising muscle. Indeed, during exercise, muscle pH has been reported in the 6.4–6.8 range, depending on intensity and duration, and rapidly re-alkalinizes post-exercise (Hagberg, 1985; Robergs et al., 2004; Sahlin et al., 1981). We hypothesized that the protonated, monocarboxylic form of succinate could be the transported form during exercise (Figure 2C).
Figure 2. pH-Gated Secretion of Succinate.

(A) Monocarboxylic pKa characteristics of TCA cycle metabolites. Distinctly, succinate exists in the monocarboxylic form at physiological acidic pH.
(B) Relationship between the TCA cycle metabolite monocarboxylic pKa and post-exercise enrichment in interstitial fluid (n = 8).
(C) A model for pH-gated release of the monocarboxylic form of succinate during exercise.
(D) C2C12 myotube hypoxia drives selective succinate secretion (n = 6).
(E) Promotion of glycolytic flux through acute inhibition of mitochondrial oxidative phosphorylation drives intracellular acidification in C2C12 myotubes. (n = 4).
(F) Promotion of glycolytic flux through acute inhibition of mitochondrial oxidative phosphorylation with antimycin A drives selective succinate secretion in C2C12 myotubes (n = 6).
(G) Promotion of glycolytic flux through acute inhibition of mitochondrial oxidative phosphorylation with atpenin A5 drives selective succinate secretion in C2C12 myotubes (n = 6).
(H) Monensin equilibrates intracellular pH with extracellular pH through H+/Na+ antiport.
(I) Monensin prevents intracellular acidification by acute inhibition of mitochondrial oxidative phosphorylation in C2C12 myotubes (n = 4).
(J) Monensin-driven cytosolic alkalinization prevents succinate secretion initiated by acute inhibition of mitochondrial oxidative phosphorylation in C2C12 myotubes (n = 6).
(K) Monensin-driven cytosolic alkalinization prevents succinate secretion initiated by hypoxia in C2C12 myotubes. pH values denote clamped extracellular pH (n = 6).
(L) Promotion of glycolytic flux through acute inhibition of mitochondrial oxidative phosphorylation with atpenin A5 drives succinate secretion from intact EDL muscle, which is prevented by monensin (n = 5).
Data are represented as mean ± SEM. *p or #p < 0.05, **p < 0.01, ***p < 0.005 (two-tailed Student’s t test for pairwise comparisons, one-way ANOVA for multiple comparisons involving one independent variable).
To test this hypothesis, we first determined whether manipulations that drive intracellular acidification are sufficient to facilitate selective release of succinate from muscle cells. Muscle acidification during exercise occurs because of elevated glycolytic lactate production to maintain cellular ATP, in part as a consequence of local hypoxia (Hagberg, 1985; Robergs et al., 2004; Sahlin et al., 1981). Indeed, subjecting muscle cells to physiologic hypoxia increases lactate production and lowers the cytosolic pH (Bright and Ellis, 1992; Hagberg, 1985). Remarkably, hypoxia was sufficient to selectively drive succinate release from C2C12 myotubes (Figure 2D). Conversely, we acutely inhibited mitochondrial oxidative phosphorylation, which is known to elevate glycolytic flux and drive cellular acidification (Mookerjee et al., 2015). These interventions rapidly promoted cytosolic acidification (Figure 2E) and concomitant selective release of succinate from muscle cells (Figures 2F and 2G). In parallel, we used established methods to equilibrate the extracellular and cytosolic pH to clamp the cytosolic pH at 6.4 (see STAR Methods for details). This intervention alone resulted in a modest increase in succinate release (Figure S1F). However, in combination with inhibition of oxidative phosphorylation with atpenin A5, cytosolic acidification significantly potentiated succinate release (Figure S1F). These findings suggest that, in addition to cytosolic acidification, a pH and succinate gradient across the plasma membrane facilitates succinate export. This supposition is examined in a later section.
To determine whether cytosolic acidification is required for succinate release under these conditions, we applied monensin, a plasma membrane Na+/H+ antiporter that alkalinizes the cytosolic pH (Lin et al., 2003; Figure 2H). Co-incubation with monensin prevented cellular acidification by pharmacological inhibition of oxidative phosphorylation (Figure 2I) and inhibited succinate release (Figure 2J). Inhibition of succinate release by monensin resulted in simultaneous accumulation of intracellular succinate (Figure S1G) and a further decrease in mitochondrial respiration (Figure S1H), indicating specific inhibition of release at the plasma membrane by this intervention. Similarly, monensin treatment during hypoxia prevented succinate release (Figure 2K). To examine whether pH-dependent release of succinate occurs in intact muscle, we subjected isolated muscle groups to similar acute metabolic perturbations ex vivo. We first examined whether glycolytic conditions promoting lactate production were sufficient to drive succinate release in isolated muscle. Subjecting isolated TA, EDL, and soleus muscle to high-glucose conditions was sufficient to drive succinate release (Figure S1I). Moreover, treatment of muscles ex vivo with atpenin A5 to inhibit oxidative phosphorylation potentiated succinate release in EDL and TA muscle (Figure S1I). The EDL is particularly amenable to acute pharmacological perturbation ex vivo, so we examined whether inhibition of intracellular acidification affected succinate release. Remarkably, monensin treatment inhibited succinate release in EDL muscle (Figure 2L).
To examine the molecular basis for pH-dependent succinate release by muscle cells and muscle tissue, we determined which plasma membrane transporters are highly expressed in muscle and have the capacity to transport carboxylates. We applied proteomic mass spectrometry (MS) to TA and GA muscle, which are muscles that can export succinate (Figure 1E). From this comparative analysis, the annotated plasma membrane succinate transporter SLC13A3 (Markovich and Murer, 2004) was undetectable in muscle. Instead, the only plasma membrane transporters known to transport carboxylate-containing molecules identified were MCT1 and MCT4, neither of which are thought to transport succinate (Figure 3A; Table S3A).
Figure 3. pH-Gated Secretion of Succinate through MCT1.

(A) Muscle proteomics identified the MCT transporter family as abundant plasma membrane carboxylic acid transporters in TA and GA muscle.
(B) A model of pH-gated release of the monocarboxylic form of succinate through MCT1.
(C) Depletion of MCT1 inhibits secretion of succinate in C2C12 myotubes initiated by pharmacological acidification. All samples are normalized to the scr. vehicle condition, so siMCT1 does not affect basal succinate release (n = 6).
(D–F) Pharmacological inhibition of MCT1 inhibits secretion of succinate in C2C12 myotubes initiated by pharmacological acidification. All inhibitors were added in parallel with atpenin A5. AZD3965 and AR-C155858 are specific MCT1 inhibitors, whereas α-CHCA inhibits all MCTs (n = 6).
(G) Depletion of MCT1 prevents hypoxic secretion of succinate by C2C12 myotubes (n = 6).
(H) Recombinant human MCT1 facilitates pH-dependent succinate transport in Xenopus oocytes. All rates were obtained in the first 10 min following succinate addition, which corresponds to the linear phase of uptake (n = 6).
(I) Succinate transport by recombinant human MCT1 is inhibited by AZD3965. All rates were obtained in the first 10 min following succinate addition, which corresponds to the linear phase of uptake (n = 5–8).
Data are represented as mean ± SEM. *p or #p < 0.05, **p < 0.01, ***p < 0.005 (two-tailed Student’s t test for pairwise comparisons, one-way ANOVA for multiple comparisons involving one independent variable).
MCT1 and MCT4 transport molecules that are structurally similar to succinate; i.e., short-chain monocarboxylates. However, MCT1 and MCT4 are strictly selective for a single carboxylate group over dicarboxylates (Halestrap, 2012). Based on our observation of pH dependence for succinate release by muscle cells, we hypothesized that MCTs could facilitate selective release when succinate is in the protonated monocarboxylate form because of lowered intracellular pH (Figure 3B). Such a regulated release of succinate would utilize pH as a “gate” for its export into the extracellular space. Small interfering RNA (siRNA)-mediated reduction in MCT1, but not MCT2 or MCT4, significantly inhibited succinate release induced by pharmacological cellular acidification (Figure 3C; Figures S2A-S2D). Notably, siMCT1 did not affect extracellular succinate levels in cells not subjected to cellular acidification (Figure 3C). Based on these findings, we treated muscle cells with established MCT1 inhibitors (Halestrap, 2012), all of which substantially inhibited succinate release initiated by cellular acidification (Figures 3D-3F; Figure S2E). Similarly, pharmacological inhibition of MCT1 in mature isolated EDL muscle tissue prevented succinate release initiated by glycolytic operation (Figure S2F) and inhibition of oxidative phosphorylation (Figure S2G). Moreover, siMCT1 prevented succinate secretion initiated by hypoxia (Figure 3G).
We next directly examined pH-dependent transport of succinate by MCT1 by expressing the recombinant human protein in Xenopus oocytes. This system facilitates functional MCT1 expression and membrane localization and is widely used to establish its monocarboxylate transport properties (Bröer et al., 1999; Nancolas et al., 2015). We found that recombinant MCT1 facilitated succinate transport at pH 6.4 (Figure 3H; Figures S2H and S2I). Succinate transport by MCT1 was strongly pH dependent and inhibited by the pharmacological MCT1 inhibitor AZD3965 (Figures 2H and 2I). Notably, the kinetics of succinate transport by MCT1 in this system were lower than those observed for lactate (Figure S2J). These data are in line with MCT1 having a higher capacity for lactate transport over some 4-carbon monocarboxylates (Bröer et al., 1998) and therefore appear to distinguish the kinetics of MCT1-mediated succinate transport as lower in comparison with its canonical substrate. Taken together, our findings indicate that MCT1 facilitates selective release of succinate by muscle cells through a pH-gated transport mechanism (Figure 3B).
Non-myofibrillar Cells in Muscle Respond to Succinate-SUCNR1 Signaling to Drive Muscle-Remodeling Transcriptional Programs
Having shown that muscle cells possess a mechanism for selective release of succinate and that succinate release is exercise responsive, we next considered whether this export process could initiate muscle tissue remodeling driven by exercise. We hypothesized that succinate may facilitate this type of regulation because it is an endogenous ligand for the G-protein-coupled receptor SUCNR1 (He et al., 2004). Local extracellular succinate levels range from less than 20 μM at rest to 60–200 μM during exercise, followed by rapid renormalization post-exercise (Figure 1I). The half-maximal response (EC50) for succinate-induced activation of SUCNR1 is reported to be in the range of 28–56 μM, and ~99% responses are achieved at 200 μM or higher (He et al., 2004). Therefore, the local extracellular succinate concentrations achieved by exercise correlate with the dynamic regulation range of succinate-SUCNRI signaling.
SUCNR1 agonism is upstream of numerous potential phosphorylation cascades that elicit rapid cellular adaptation; however many of these remain incompletely defined (Mills and O’Neill, 2014). Therefore, we first explored whether regulation of SUCNR1-mediated signaling was relevant in the acute muscle response to exercise and the specific phosphorylation cascades modulated by SUCNR1 in this context. We performed unbiased phosphoproteomics of TA skeletal muscle from wild-type (WT) and SUCNR1KO mice immediately following exercise, when succinate release is maximal. Remarkably, we observed significant enrichment of several kinase pathways for which the phosphorylation status depended on SUCNR1 during exercise (Figure 4A; Tables S3B and S3C). Two of these pathways are established downstream of SUCNR1 agonism (PKA and mitogen-activated protein kinase [MAPK]; Ariza et al., 2012; Mills and O’Neill, 2014). We additionally observed enrichment for CDK2-dependent phosphorylation cascade targets, which have not been linked previously to SUCNR1 agonism. These data indicate that succinate-SUCNR1 phosphorylation signaling is rapidly responsive to acute exercise in whole muscle.
Figure 4. Succinate-SUCNR1 Signaling Controls Non-cell-autonomous Transcriptional Responses to Acute Exercise.

(A) Phosphoproteomics analysis of TA muscle immediately following exercise, comparing the relative abundance of phosphorylation of the WT and SUCNR1 KO. Sites exhibiting over 50% elevation in the WT versus SUCNR1 KO are highlighted and were subjected to kinase enrichment analysis. Top enriched pathways and those known to be downstream of SUCNR1 agonism are highlighted. A full list is provided in Table S3 (n = 4).
(B) SUCNR1 is expressed in muscle tissue but not mature myotubes or immortalized C2C12 myoblasts, whereas non-myofibrillar cells resident in muscle tissue express SUCNR1 (n = 3–4).
(C) TA muscle RNAScope ISH to establish cellular localization of SUCNR1. Red, SUCNR1; pdgfrα stromal cell marker; yellow, desmin myofiber marker. SUCNR1 exhibits strong colocalization with PDGFRα and anti-localization with desmin. Three representative fields of view are shown.
(D) Manders’ colocalization coefficient of TA muscle RNAScope ISH data from (C). Coefficients of less than 0.1 are considered completely anti-colocalized, which is validated by the anti-colocalization control of pdgfrα and desmin (distinct cell type markers). SUCNR1 is completely anti-colocalized with desmin (myofibers) and colocalizes with pdgfrα (stromal cells) (n = 3).
(E) A model of non-cell-autonomous regulation of muscle remodeling post-exercise via SUCNR1.
(F) The SUCNR1-dependent transcriptional landscape post exercise in muscle satellite cells (CD45−, CD31−, Sca1−, Vcam). Example significant pathways show decreased expression in SUCNR1 KO (highlighted). See Table S4 for a full list.
(G) The SUCNR1-dependent transcriptional landscape after exercise in stromal cells (CD45−, CD31−, Sca1+, PDGFRa+). Example significant pathways show decreased expression in SUCNR1 KO (highlighted). See Table S4 for a full list.
(H) The SUCNR1-dependent transcriptional landscape after exercise in muscle hematopoietic cells (CD45). Example significant pathways show decreased expression in SUCNR1 KO (highlighted). See Table S4 for a full list.
(I) The SUCNR1-dependent transcriptional landscape after exercise in endothelial cells (CD45−, CD31+). Example significant pathways show decreased expression in SUCNR1 KO (highlighted). See Table S4 for a full list.
Data are represented as mean ± SEM.
The SUCNR1-dependent signaling cascades described above are upstream of a diverse range of distinct transcriptional responses whose nature crucially depends on the identity of the target cell. Therefore, we next determined the relevant cell populations for which acute transcriptional regulation by SUCNR1 is relevant following exercise. A recent study suggested that SUCNR1 in muscle myotubes regulates fiber type remodeling (Wang et al., 2019). However, we found that, although SUCNR1 is detectable in whole muscle, it is not expressed in purified muscle myotubes or isolated C2C12 myoblasts (Figure 4B). Instead, we determined that SUCNR1 expression in whole muscle is attributable to major non-myofibrillar resident cell types, including stromal, endothelial, and satellite cell populations (Figure 4B). To corroborate this, we performed RNAScope in situ hybridization (ISH) localization of the SUCNR1 transcript in intact muscle. Because RNAScope ISH provides extremely sensitive detection of transcripts in intact tissue (Wang et al., 2012, 2013), we reasoned that this would unequivocally localize cell populations expressing SUCNR1 in intact muscle. Remarkably, the SUCNR1 transcript was undetectable in muscle fibers, exhibiting complete anti-colocalization with the myofibrillar marker desmin (Figures 4C and 4D). Instead, the SUCNR1 signal correlated with non-myofibrillar stromal populations based on strong colocalization with pdgfrα (Figures 4C and 4D). In line with these findings, we found succinate to have no direct stimulatory effect on isolated muscle myoblast differentiation (Figure S3A), myoblast proliferation rate (Figures S3B-S3D), or transcriptional control of myosin heavy chains in differentiated myotubes (Figure S3E). Together with the lack of detectable expression of SUCNR1 in mature muscle cells (Figures 4B-4D), our data indicate an absence of cell-autonomous succinate-SUCNR1 regulation of muscle cell remodeling.
We hypothesized that succinate secreted from muscle cells could control cell non-autonomous transcriptional regulation in muscle tissue via SUCNR1 (Figure 4E). To explore this hypothesis, we purified major resident cell types found in muscle tissue: endothelial, hematopoietic (resident immune cells), stromal, and satellite (Wang et al., 2020). WT and SUCNR1 knockout (KO) mice were subjected to acute treadmill exercise, which drives succinate release (Figures 1B-1D; Figure S3F), and cell populations were isolated immediately post-exercise, when succinate release is maximal. Pure cell populations were subjected to RNA sequencing (RNA-seq) to define the transcriptional response in each cell type that requires succinate-SUCNR1 signaling (Tables S4A-S4D). We then performed pathway analyses to implicate biological processes that depend on SUCNR1 based on reduced transcript levels in SUCNR1 KO cells compared with the WT (Table S4E). Several cell-type-specific transcriptomics responses that depend on SUCNR1 were uncovered by this analysis (Figures 4F-4I; Tables S4A-S4D). In satellite cells and stromal cells, we observed significant decreases in transcripts involved in neuronal projections and axon guidance because of SUCNR1 KO (Figures 4F and 4G; Table S4E). SUCNR1 KO satellite cells also exhibited decreased expression of cell-cell adhesion transcripts, whereas stromal cells lacking SUCNR1 had decreased expression of transforming growth factor β (TGF-β) signaling and EGFR signaling transcripts (Figures 4F and 4G; Table S4E). Among these transcripts were established factors involved in muscle remodeling, including GDF6, a growth factor that mediates muscle repair (Hisamatsu et al., 2016). Hematopoietic (immune) and endothelial cell populations exhibited fewer systematic changes as a consequence of SUCNR1 ablation, with select protein glycosylation and ECM-receptor transcripts exhibiting decreased expression (Figures 4H and 4I; Table S4E). Together, our data show that lack of succinate-SUCNR1 signaling results in a coordinated decrease in transcripts involved in innervation, muscle regeneration, and ECM formation in multiple muscle-resident, non-myofibrillar cell types.
The Succinate Secretion/SUCNR1 Pathway Mediates Muscle Adaptation to Exercise Training
Our data support a model where succinate released from exercising muscle acts as an upstream regulator of the paracrine response to acute exercise; in particular, innervation and remodeling of muscle ECM (Figure 4). Initiation of these processes is critical for the muscle response to exercise training. Therefore, we next examined the role of succinate-SUCNR1 signaling on exercise training-induced muscle remodeling. To do so, we applied a voluntary resistance wheel running protocol. Resistance wheel training elicits systemic health benefits within 3 weeks, including elevated insulin sensitivity and increased muscle strength (Figure 5A; Ghosh et al., 2010; Roemers et al., 2018). We applied this training protocol to WT and SUCNR1 KO mice. Sedentary mice displayed identical grip strength (Figure 5B). Interestingly, SUCNR1 KO mice ran more than WT mice throughout the training protocol (Figure 5C). Despite this, SUCNR1 KO mice exhibited a striking absence of strength gains in response to voluntary exercise, unlike the substantial gains observed in WT mice (Figures 5D and 5E).
Figure 5. Muscle Adaptation to Exercise Training Requires Succinate-SUCNR1 Signaling.

(A) Resistance wheel training protocol.
(B) Grip strength of untrained WT and SUCNR1 KO mice (n = 15 WT, n = 12 KO).
(C) Distance run by WT and SUCNR1 KO mice throughout the training regimen (n = 15 WT, n = 12 KO).
(D and E) Absolute (D) and percentage (E) change in grip strength during resistance wheel training (n = 15 WT, n = 12 KO).
(F) Protein abundance differences between WT and SUCNR1 KO trained TA muscle. Pathways enriched in proteins exhibiting more than 20% differences between genotypes are highlighted (n = 4).
(G) Actin and myosin chain protein abundance differences between WT and SUCNR1 KO trained TA muscle (n = 4). Fast-twitch myosin chains are depleted in SUCNR1 KO trained TA muscle.
(H) ECM protein pathway members exhibiting decreased abundance in trained SUCNR1 KO TA muscle (n = 4).
(I) Inflammatory complement pathway members exhibiting increased abundance in trained SUCNR1 KO TA muscle (n = 4).
(J) Quantification of innervation in whole TA muscle by 3D tubb3 staining (n = 5).
(K) Representative compressed 2D projections of muscle tubb3 volume. The top row and bottom row are representative top view and side view projections, respectively.
(L) Experimental design to correlate maximal succinate release by human exercising muscle and post-exercise insulin sensitivity.
(M) Correlation between maximal femoral venous succinate concentration in human exercising muscle and post-exercise insulin sensitivity (n = 10).
(N) Insulin tolerance test of WT and SUCNR1 KO mice 3 h following acute treadmill running (n = 6–7).
Data are represented as mean ± SEM. *p or #p < 0.05, **p < 0.01, ***p < 0.005 (two-tailed Student’s t test for pairwise comparisons, one-way ANOVA for multiple comparisons involving one independent variable).
To understand the molecular basis of lack of training-induced muscle strength adaptation, we examined the proteomes of TA muscle from WT and SUCNR1 KO mice. Trained SUCNR1 KO muscle exhibited decreased protein abundance of major pathways involved in muscle adaptation to exercise compared with the WT (Figure 5F; Table S5A). First, myosin complex protein abundance was decreased in SUCNR1 KO mice. In addition to overall lower levels of these proteins, substantial remodeling of myosin chain complexes was observed in genotypes. SUCNR1 KO trained muscle had significantly lower levels of fast twitch myosin heavy chains while displaying remodeling of non-skeletal muscle myosin chains and tropomyosins (Figure 5G). As a class, ECM organization proteins were also depleted in SUCNR1 KO trained muscle, including major collagens and laminins (Figures 5F and 5H; Table S5A). These protein families play key roles in myogenesis, force transmission, and innervation in muscle (Sanes, 2003). Conversely, SUCNR1 KO trained muscle exhibited significant elevations of complement pathway markers of tissue inflammation (Figures 5F and 5I; Table S5A), many of which are associated with insulin resistance (Phieler et al., 2013). Importantly, these differences were not observed between genotypes in muscle of untrained mice, indicating an exercise-dependent requirement for SUCNR1 in mediating these adaptations (Figures S3G-S3I; Table S5B).
Our proteomics analysis revealed that succinate-SUCNR1 signaling is required for maintaining ECM components involved in muscle innervation following exercise training. Moreover, our analyses of the SUCNR1-dependent transcriptome post-exercise demonstrated decreased expression of transcripts involved in axon guidance and neuronal projections (Figures 4F and 4G). Neuronal adaptation in muscle is a paracrine consequence of exercise training that plays a key role in increasing muscle strength (Folland and Williams, 2007; Gabriel et al., 2006). We therefore hypothesized that a basis of compromised training-induced muscle strength in SUCNR1 KO mice could be attributable to impaired muscle innervation. To examine this, we performed three-dimensional whole-tissue-mount immunolabeling of the neural-specific tubulin TUBB3 in TA muscle from WT and SUCNR1 KO mice. Remarkably, we found that resistance muscle training (Figure 5A) resulted in substantial increases in TA muscle neural staining that were abrogated in SUCNR1 KO mice (Figures 5J and 5K; Video S1). We additionally monitored whole-tissue vascularization (Figures S4A and S4B; Video S1) and muscle fiber volume (Figures S4C and S4D; Video S1) and found no differences between genotypes.
Taken together, these data demonstrate that succinate-SUCNR1 signaling plays a central role in exercise training-induced muscle innervation, muscle fiber type and ECM remodeling, and muscle strength. It is noteworthy that neural adaptations in muscle are an early paracrine response to resistance training that can improve muscle strength independent of hypertrophy (Folland and Williams, 2007). Moreover, motor neurons directly affect target myotube fiber content and function (Buller et al., 1960), whereas sympathetic innervation of muscle and afferent signaling by sympathetic nerves can regulate muscle strength, regeneration, and blood flow to muscle (Barker and Saito, 1981; Beitzel et al., 2004). Therefore, it is likely that the substantial effects of succinate release and SUCNR1 signaling on strength and muscle remodeling are at least partly attributable to modulation of muscle innervation.
Finally, we further elaborated our findings that SUCNR1 signaling antagonizes chronic muscle inflammation, which is associated with insulin resistance (Figure 5I). In mice and humans, a single bout of exercise improves insulin sensitivity for hours post-exercise (Richter et al., 1989), and our data indicate an anti-inflammatory role of succinate-SUCNR1 signaling. Therefore, we examined whether the concentration of local extracellular succinate during exercise is predictive of post-exercise insulin sensitivity (Figure 5L). Individuals were sampled as described in Figure 1E and Table S2A and, following recovery, were subjected to a hyperinsulinemic-euglycemic clamp (Table S2B). Notably, we found a significant correlation between peak exercise succinate concentration in muscle femoral vein and insulin sensitivity post-exercise (Figure 5M; Table S2C). This association between peak venous succinate and insulin sensitivity was distinct among all covariates tested (Table S2; Figures S4E and S4F) and occurred in the absence of correlations with peak VO2 (Figure S4G) and peak muscle load (Figure S4H). Based on these findings, we examined whether succinate-SUCNR1 signaling regulates post-exercise insulin sensitivity. We found that post-exercise insulin sensitivity was blunted in SUCNR1 KO mice (Figure 5N), which also exhibited modest basal glucose insensitivity (Figure S4I). Notably, previous examinations of SUCNR1 KO mice have established glucose insensitivity that depends on obesogenesis initiated by high-fat feeding, which occurs without changes in insulin tolerance (McCreath et al., 2015). Therefore, the role of extracellular succinate-SUCNR1 signaling in insulin sensitivity observed here may be specific to the exercise response.
Conclusions
Together, our findings define a bioenergetic mechanism in muscle that senses intracellular pH, using succinate to communicate the local energetic status to the peripheral tissue environment. Because of its distinct physiochemical properties with respect to other mitochondrial dicarboxylates, succinate can be exported from muscle cells through acute regulation of its protonation status by local pH. When protonated, succinate is transformed from a dicarboxylate, which is trapped in the cell, to a monocarboxylate. In the protonated monocarboxylate form, succinate is rendered a transport substrate for the plasma membrane transporter MCT1, which facilitates pH-gated release. Upon pH-gated secretion during exercise, succinate signals via its cognate receptor SUCNR1 in non-muscle cells present in muscle tissue to coordinate muscle remodeling. Neurotrophic and muscle ECM remodeling initiated upon exercise depend essentially on paracrine succinate-SUCNR1 signaling, which is also required for major physiologic and molecular adaptations to exercise training. Although our data demonstrate a role of succinate-SUCNR1 paracrine regulation in muscle, it is possible that autocrine regulation could also play a role in some contexts. Although we could not detect SUCNR1 in isolated muscle cells, the process of isolation and culturing could initiate cellular adaptation, leading to loss of SUCNR1 expression. Moreover, although we did not detect SUCNR1 in intact muscle by ISH, it is possible that, following certain types of exercise training or fiber type remodeling, that SUCNR1 is expressed in these cell types as well.
It is noteworthy that a recent study investigated the effects of orally administered sodium succinate on muscle metabolism. In that study, dietary succinate altered skeletal muscle outputs such as strength and time to muscle fatigue in a SUCNR1-dependent fashion (Wang et al., 2019). These adaptations were attributed to signaling through myofibrillar SUCNR1. Alternatively, it is tempting to attribute the effects of dietary succinate to the same paracrine cascade initiated by exercise-induced, muscle-derived succinate determined in this work. When considering this possibility, it should be noted that orally administered succinate has major and rapid regulatory effects on systemic glucose homeostasis (De Vadder et al., 2016), whole-body energy expenditure (Mills et al., 2018), and intestinal nutrient assimilation (de Vadder and Mithieux, 2018). These systemic regulatory circuits are linked to muscle metabolism. Therefore, any acquired effects on muscle tissue following several weeks of dietary succinate supplementation must take these upstream pathways into account when considering mechanisms of action. In any case, it will be interesting to examine the extent to which dietary succinate initiates regulation of muscle innervation and ECM remodeling similar to the local and transient release of succinate by exercising muscle.
Our findings also propose a general model of how the intracellular energetic status can be communicated systemically through pH-gated succinate release. This mode of systemic integration reconciles the nature of succinate as an intracellular metabolite with the expanding list of physiological processes that can be regulated by extracellular succinate and/or SUCNR1, including energy expenditure (Mills et al., 2018), inflammation (Littlewood-Evans et al., 2016; Peruzzotti-Jametti et al., 2018), and the renin-angiotensin system (Deen and Robben, 2011). It is also consistent with the hypothesis that extreme pathologies involving tissue ischemia or local pH gradients between astrocytes and neurons could drive succinate release via MCT1 (Andrienko et al., 2017; Molnár et al., 2008). Our model predicts that instances of physiological hypoxia or intracellular acidification will engage succinate release as a general mechanism to initiate non-cell-autonomous modes of regulation.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Edward Chouchani (EdwardT_Chouchani@dfci.harvard.edu).
Materials Availability
Materials and data generated from this study is available upon request from Edward Chouchani (EdwardT_Chouchani@dfci.harvard.edu).
Data and Code Availability
The published article includes all datasets generated or analyzed during this study.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse Lines
Animal experiments were performed according to procedures approved by the Institutional Animal Care and Use Committee of the Beth Israel Deaconess Medical Center. Unless otherwise stated, mice used were male C57BL/6J (8 ± 12 weeks of age; Jackson Laboratories) and housed in a temperature-controlled (23°C) room on a 12-h light-dark cycle. Male WT and SUCNR1KO littermate matched controls were used.
Human exercise study recruitment
Ten men were recruited for the study, which was approved by the Copenhagen Ethics Committee (Reg. number H-16040740) and performed in accordance with the Declaration of Helsinki. Informed written consent was received from each participant prior to study inclusion. All subjects were healthy, moderately physically active and with no family history of diabetes. The subjects were 27 ± 0.8 (means ± SEM) years old, with a body weight of 85 ± 2.5 kg, body mass index (BMI) of 24 ± 0.5 kg·m-2 and maximal oxygen uptake (VO2 peak) of 49 ± 1 ml·kg-1·min-1. Maximal oxygen uptake was measured by an incremental exercise test on a Monark Ergomedic 839E bicycle ergometer (Monark). Before the trial subjects consumed a eucaloric controlled diet for 3 days to eliminate dietary differences between subjects. The daily energy requirements were individually determined from weighed dietary registrations and predicted equations from WHO/FAO/UNU. All food items were weighed and prepared in the metabolic kitchen and menus were delivered to the subjects and ingested at home. The diet consisted of 60 energy-percent (E%) carbohydrate, 25E% fat and 15E% protein.
Cell lines
C2C12 murine myoblasts (obtained from ATCC) were in growth media consisting of DMEM and 10% fetal bovine serum and 1% penicillin/streptomycin. Upon reaching confluency, growth media was exchanged for differentiation media consisting of DMEM containing 2% horse serum. Seven days post differentiation cells appeared mostly as phase-bright fused myotubes (Ramesh et al., 2019).
Isolation of muscle satellite cells
Mouse satellite cells were isolated as previously described (Sampath et al., 2018). Briefly, gastrocnemius and tibialis anterior muscles were dissected and digested with Collagenase type 2 and Dispase (Worthington Biochemical Corporation) for a total of 90-120 min. Cell suspension was strained with a 40 μm filter and stained cells the following antibodies: anti-CD45 (BD Biosciences), anti-CD11b (BD Biosciences), anti-CD31 (eBiosciences) and anti-Sca1 (BD Biosciences). Next, cells were incubated with streptavidin microbeads (Miltenyi Biotech), SA-Texas Red (Invitrogen), PE labeled anti-integrin α7 antibody labeled with phycoerythrin (AbLab) and anti-CD34 eFluor660 (eBioscience). Dead cells were identified using propidium iodide staining. Cells were sorted on a FACS Aria 2 and routinely yielded > 95% CD34+ α7 integrin+ cells from a single round of sorting. Scatterplots were analyzed using FlowJo (TreeStar). Live cells were plated in collagen coated plates (Corning) and maintained in growth media (50:50 DMEM /F10, 15% FBS, 1% penicillin/streptomycin, and 2.5 ng/ml bFGF). For in vitro differentiation, satellite cells were washed with PBS and subjected to differentiation media (DMEM supplemented with 5% horse serum) for three days. Depending on experimental condition, vehicle (PBS) or succinate (50, 100 and 200 μM) were added to growth and differentiation media.
METHOD DETAILS
Acute exercise training protocol
Eight-week old mice were acclimated to a motorized treadmill (Columbus Instruments) for three days prior to forced running protocol. Training sessions consisted of mice walking on the treadmill for 5 min at 12 m/min followed by a 1 min rest and then 5 min at 12 m/min and 5 min at 14 m/min. On the day of running, all mice were subjected to the training protocol. Mice in the sedentary group were taken off the treadmill after the training protocol was complete while mice in the exercise group remained on the treadmill. After the training period, exercised mice remained on the treadmill and the speed was ramped up 2 m/min every 4 min, until a final speed of 26 m/min was reached. Exercised mice were removed from the treadmill once exhausted, which was deemed to be the point where they refused to step on the treadmill.
Sample preparation for metabolite profiling
Following intervention, animals were sacrificed immediately by cervical dislocation and tissues were rapidly extracted and immediately snap frozen in liquid nitrogen for metabolite profiling. For metabolite extraction, tissues were weighed and then homogenized with extraction buffer which consisted of 80% methanol containing inosine15N4, thymine-d4 and glycocholate-d4 internal standards (Cambridge Isotope Laboratories) at a 4:1 volume to wet weight ratio. Samples were then centrifuged twice (5 min, 10,000 g, 4°C). Supernatant was then further diluted with 80% HPLC grade methanol at a ratio of 1:10 extracted metabolite to methanol. Interstitial fluid was extracted from muscles immediately upon extraction using a rapid isolation procedure described previously (Spinelli et al., 2017; Sullivan et al., 2019). Muscles were subjected to centrifugation (10 min, 800 g, 4°C) following placement in a 20 μm nylon mesh filter (EMD Millipore). Metabolites were extracted by adding extraction buffer at a ratio of 1:100 interstitial fluid to methanol. Samples were then centrifuged twice (5 min, 10,000 g, 4°C) and supernatant was collected. For plasma collection, blood was taken via cheek bleed from mice, collected into a heparin column (Becton Dickson), centrifuged (10 min, 1,000 g, 4°C), and subsequently snapfrozen. Metabolites were extracted from plasma by adding extraction buffer in a 1:4 ratio plasma to methanol. Samples were then centrifuged twice (5 min, 10,000 g, 4°C) and supernatant was collected. For media metabolite extraction, media samples were collected and centrifuged (1 min, 21,100 g, 4°C) to remove any debris or cell contaminants. Metabolites from media were extracted by adding extraction buffer in a ratio of 1:4 media to methanol. Samples were then centrifuged twice (5 min, 10,000 g, 4°C) and supernatant was collected.
Metabolite analyses by mass spectrometry
All extracted samples were then subjected to LC/MS analysis as previously described (Mills et al., 2018). Metabolite extracts were loaded onto a Luna-HILIC column (Phenomenex) using an UltiMate-3000 TPLRS LC with 10% mobile phase A (20 mM ammonium acetate and 20 mM ammonium hydroxide in water) and 90% mobile phase B (10 mM ammonium hydroxide in 75:25 v/v acetonitrile/methanol). A 10 min linear gradient to 99% mobile phase A was used to separate metabolites. Subsequent analysis was carried out using a QExactive HF-X mass spectrometer (Thermo Fisher Scientific). Negative and positive ion modes were used with full scan analysis over m/z 70-750 m/z at 60,000 resolution, 1e6 AGC, and 100 ms maximum ion accumulation time. Additional MS settings were: ion spray voltage, 3.8 kV; capillary temperature, 350°C; probe heater temperature, 320°C; sheath gas, 50; auxiliary gas, 15; and S-lens RF level 40. Targeted processing of a subset of known metabolites and isotopologues was conducted using TraceFinder software version 4.1 (Thermo Fisher Scientific). Compound identities were confirmed using reference standards. In all case metabolite abundance was normalized using internal standards and relative changes were assessed by comparison with metabolite extracted from the same sample type (i.e., tissue, IF, plasma).
Human exercise study protocol
After three days on the control diet, subjects ingested a light standardized breakfast (1.6 M J) at 6 A.M at home. Subjects arrived at the laboratory at 8 A.M, and after 10-15 min of supine rest, teflon catheters were inserted into the femoral artery in one leg and the femoral vein of the other leg under local anesthesia. After continued supine rest, basal blood samples were obtained from the femoral artery and vein at 10 A.M (0 min). Subjects then initiated a 60 min exercise bout on the Monark Ergomedic 839E bicycle ergometer at 67 ± 1% of maximal oxygen uptake (VO2max). VO2 peak was determined by an incremental test performed on a bike ergometer (Monark) at least 1 week prior to the experiment. Femoral arterial and femoral venous samples were collected simultaneously during exercise at 20, 40 and 60 min and again after 10 and 120 min of resting supine recovery without food intake. Femoral arterial blood flow at rest was measured by high frequency 12-3 MHz linear array transducer in Power Doppler mode interfaced to an Affiniti70 ultrasound machine (Phillips Ultrasound). During exercise, leg blood flow was estimated based on pulmonary oxygen uptake and the A-V difference for oxygen as described (Jorfeldt and Wahren, 1971). The pulmonary oxygen consumption was measured using a MasterScreen CPX system (Becton).
Euglycemic hyperinsulinemic clamp
Two hours after completion of the exercise trial, a 2 h euglycemic, hyperinsulinemic clamp was initiated. Subjects were clamped at their individual ambient plasma glucose level obtained prior to initiation of the insulin infusion. Insulin was infused at 1.4 mU.kg−1.min−1 resulting in a plasma insulin concentration 100 μU/ml. Plasma glucose concentration was measured every ~7 min and a variable glucose infusion (20% Fresenius Kabi, SE) into a forearm vein was adjusted to clamp euglycaemia as previously described (DeFronzo et al., 1979). Insulin sensitivity was calculated as the glucose infusion rate over the last 30 min of the 120 min clamp.
Human blood sample preparation
Blood was sampled in heparinized syringes and transferred to EDTA-containing tubes and centrifuged at 4°C at 3000 g for 5 min. Plasma was aspirated, frozen, and stored at −80°C until analysis.
Acute muscle cell hypoxia experiments
C2C12 myoblasts were seeded at a density of 80,000 cells per well, 24 h prior to use. A minimal media was made consisting of 8.3 g/L DMEM, 4 mM L-glutamine, 1 mM pyruvate, 10 mM glucose, and 10 mM HEPES. Media was place in a sealed hypoxic chamber set to 0.1% oxygen tension, 24 h prior to use. On the day of the experiment, cells were placed in the hypoxic chamber for 2 h and previously degassed media was added to each well. After the 2 h, media from each well was collected inside the chamber, and snap frozen for metabolomic analysis.
Intracellular pH modulation
Pharmacological acidification of intracellular pH in C2C12 cells was achieved by acute inhibition of mitochondrial oxidative phosphorylation with 100 nM atpenin A5 (Cayman Chemical) and 0.5 mM antimycin A (Sigma Aldrich) for 1 h and 2 h, respectively. 100 μM monensin (Sigma Aldrich) were added 1h before administration of atpenin A5 or hypoxia to prevent cellular acidification. Intracellular pH acidification by extracellular equilibration was achieved as previously described (Nadtochiy et al., 2016). Briefly, cells were incubated with pH 6.4 medium supplemented with 30 mM NH4Cl and 10 μM 5-(N-ethyl-N-isopropyl) amiloride (EIPA) for 15 min, followed by reincubation in pH 6.4 medium with 10 μM EIPA alone for 4 hours.
Intracellular pH measurement
Intracellular pH was determined using the ratiometric fluorescent intracellular pH probe BCECF-AM (Invitrogen) (Khacho et al., 2014). Briefly, cells were loaded with BCECF (1 mM) for 30 min at 37°C. We used dual-excitation ratio of 480 nm and 440 nm and fixed emission at 535 nm to measure changes in BCECF-AM fluorescence. A calibration curve of the intracellular pH response in terms of the ratio of fluorescence intensities was performed using the high K+ nigericin technique.
MCT1 inhibition
Pharmacological inhibition of MCT1 in C2C12 cells was achieved by preincubation with 5 mM α-cyano-4-hydroxycinnamic acid, 100 nM AZD 3965, and 100 nM ARC155858 for 30 min, followed by hypoxia or acidification of intracellular pH experiments
Muscle cell siRNA
Opti-MEM media (Thermo Fisher Scientific) was incubated with lipofectamine (Life Technologies) for 10 min at room temperature. In a separate 1.5 mL tube, Opti-MEM media was incubated with 2 uL of siRNA (10 μM stock; Sigma Aldrich) for 5 min. The lipofectamine mixture was added dropwise to the siRNA mixture and left to incubate (20 min, room temperature). This final solution was added dropwise to a 12-well plate. C2C12 myoblasts were then added at a density of 40,000 cells/well. The media was changed 24 h later and the cells were used 48 h after plating. Knock-down efficiency was confirmed via qPCR.
Succinate transport assay in Xenopus laevis oocytes
Xenopus laevis oocytes were purchased from EcoCyte Bioscience and the transport assay was conducted as previously described (Broer et al., 1997; Markovich, 2008). Oocytes were injected with MCT1 cRNA (24 ng) prepared by in vitro transcription using the mMessage mMachine kit (Thermo Fisher Scientific) from the human MCT1 cloned in the plasmid pcDNA3.1 (GeneScript). Controls were injected with equivalent volume of water. Oocytes were cultured in modified Barth’s solution (88 mM NaCl, 1 mM KCl, 0.82 mM MgSO4, 0.4 mM CaCl2, 0.33 mM Ca(NO3)2, 2.4 mM NaHCO3, 10 mM HEPES, 10 U/ml penicillin, 10 μg/ml streptomycin, pH 7.4) for 3 days. For succinate uptake measurements, groups of 5 oocytes were incubated in OR2 buffer (82.5 mM NaCl, 2.5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 1 mM Na2HPO4, 5 mM HEPES, pH 6.4 or pH 7.4) containing [U13C] succinate or lactate at different concentrations for 10 min, which we established as the time point for linear kinetics of transport. Inhibition of MCT1 was achieved by incubating oocyte with 100 nM AZD3965 5 min prior to the addition of [U13C] succinate. Oocytes were washed three times in ice cold OR2 buffer, lysed with metabolite extraction buffer, and subjected to LC/MS analysis, as described above.
Ex vivo skeletal muscle experiments
Extensor digitorum longus and soleus muscles were rapidly isolated, transferred to 12 well plates containing DMEM supplemented with 25 mM glucose and maintained at 37°C - adapted from (Campos et al., 2018). For particular experiments, muscles were incubated with 10 μM AZD, 100 μM monensin for 30 min with or without 1 μM atpenin A5 for additional 60 min. Following interventions, media from each well was collected and snap frozen for metabolomic analysis.
Muscle tissue proteomics
Muscle tissues were homogenized in 100 mM HEPES pH 8.5, 8 M urea, 2% SDS, 1 p/15 mL protease and phosphatase inhibitors (Thermo Fisher Scientific). The mixture was clarified by centrifugation and the supernatant was subjected to BCA protein concentration measurement and reduced with 5 mM tris(2-carboxyethyl) phosphine (TCEP) and alkylated with 15 mM iodoacetamide. Proteins were then purified using the methanol-chloroform precipitation method and reconstituted in 200 mM EPPS at pH 8.5 and digested by Lys-C and trypsin overnight 1:100 enzyme-to-substrate ratio and an additional 6 h by trypsin the next day. Resulting peptides were labeled by TMT-pro 16plex (Thermo Fisher Scientific) following the SL-TMT protocol (Navarrete-Perea et al., 2018), and quench by adding 5% hydroxylamine. A ratio-check is then performed by mixing 1% sample from each TMT channel, and the TMT-labeled peptides were evenly mixed based on the ratio-check. The mixture was desalted using Sep-pak cartridges (Waters), dried, and fractionated using an Agilent 1100 quaternary HPLC system. Peptides were separated using a 50 min linear gradient from 18% to 40% acetonitrile in 10 mM ammonium bicarbonate, pH 8, into a total of 96 fractions that were consolidated into 8. Samples were dried and desalted via StageTip and reconstituted in 5% formic acid and 5% ACN for liquid chromatography tandem mass spectrometry (LC-MS/MS) analysis. The proteomics samples were either analyzed using an Orbitrap Fusion Lumos or an Orbitrap Eclipse mass spectrometer (Thermo Fisher Scientific) coupled with an Easy-nLC 1200 (Thermo Fisher Scientific) ultra-high pressure liquid chromatography (LC) pump were used to analyze the samples. The columns are packed in house with 100 mm inner diameter microcapillary column packed with 40 cm of GP-18 resin (2.6 mm, 200 A, Sepax). A FAIMSPro (Thermo Fisher Scientific) device for FAIMS separation of precursors (Schweppe et al., 2019). The FAIMS device was operated with default settings and multiple compensation voltages (40V/−60V/−80V).
For protein abundance measurements on the Orbitrap Fusion Lumos instrument, samples were analyzed using a single 135 min run with 9%–24% ACN at 500 nL min−1 flow rate. Fractionated peptides were injected. Under each FAIMS voltage, the full MS was operated with a scan range 375-1500 m/z at 120,000 resolution in the Orbitrap. Tandem mass spectra (MS2) were matched in real time to targeted proteins (Schweppe et al., 2020a). When a peptide match was observed to a targeted protein, a quantitative SPS-MS3 scan was acquired (McAlister et al., 2014). For protein abundance measurements on the Orbitrap Eclipse instrument with TMT, all samples were analyzed with a 180 min gradient consisting of 2% - 23% ACN, 0.125% FA at 500 nl/min flow rate. Under each FAIMS voltage, peptide ions were collected in data-dependent mode using a mass range of m/z 400 - 1600 using 2 s cycles. Resolution for MS1 was set at 120,000, with standard automatic gain control (AGC) target. Multiply-charged ions were selected and subjected to fragmentation at 35% normalized collisional energy (NCE) for MS2 with a dynamic exclusion of 120 s. Quantification was performed using multinotch SPS-MS3 as described previously (McAlister et al., 2014). For phosphoproteomics, unfractionated samples were analyzed with a 150 min gradient consisting of 2% - 27% ACN, 0.125% FA at 525 nl/min flow rate. The samples were first analyzed with multistage activation (MSA) based on 97.9763 Da neutral loss and quantified using SPSMS3, then analyzed again using high-resolution MS2 as described previously (Schweppe et al., 2020b). For label-free quantification, unfractionated samples were analyzed with a 180 min gradient consisting of 2% - 23% ACN, 0.125% FA at 500 nl/min flow rate. Peptide ions were collected in data-dependent mode using a mass range of m/z 400-1600 using 2 s cycles. Each ion is selected for further fragmentation in MS2. Maximum injection time was set to 50 ms and dynamic exclusion was 120 s.
The Comet algorithm (Eng et al., 2013) was used to search all MS/MS spectra against a database containing sequences of human (Mus musculus) proteins downloaded from UniProt (https://www.uniprot.org/). Reversed sequences were appended as decoys for FDR filtering, and common contaminant proteins (e.g., human keratins, trypsin) were included. Peptides were searched using following parameters: 25 ppm precursor mass tolerance; 1.0 Da product ion mass tolerance; fully tryptic digestion; up to three missed cleavages; variable modification: oxidation of methionine (+15.9949); static modifications: TMT (+304.2071) on lysine and peptide N terminus, carboxyamidomethylation (+57.0214637236) on cysteines. The target-decoy method was employed to control the false discovery rate (FDR) (Elias and Gygi, 2007; Huttlin et al., 2010; Peng et al., 2003). To distinguish correct and incorrect peptide identifications, linear discriminant analysis (LDA) was used to control peptide-level FDR to less than 1%. Peptides shorter than six amino acids were discarded. Protein-level FDR was also controlled to 1% and as a result the number of peptide reverse hits were further decreased. Peptides were matched to the least number of proteins. TMT reporter ion signal-to-noise ratios for all quantified peptides matched to the same protein were summed up to report protein abundance. Spectral counts were used to quantify proteins in label-free samples. KEGG pathway GO enrichment analyses were performed using Enrichr (https://amp.pharm.mssm.edu/Enrichr/; Chen et al., 2013; Kuleshov et al., 2016).
Phosphoproteomics
Samples were homogenized using TissueLyser II (QIAGEN) in a lysis buffer containing 50 mM HEPES pH 8.5, 8 M urea, 2% SDS, 150 mM NaCl plus 1X Roche complete protease and PhosStop phosphatase inhibitors. Following a BCA assay to estimate protein concentration, all lysates were reduced with 5 mM TCEP for 30 min at room temperature, then alkylated with 15 mM iodoacetamide for 30 min at room temperature in the dark. Proteins were precipitated by chloroform-methanol precipitation, digested, and 100 μg peptides from each sample were labeled by TMT-pro (Thermo Fisher Scientific) following the SL-TMT protocol (Navarrete-Perea et al., 2018). Samples were then combined according to a ratio-check, then desalted using a sep-pak cartridge (Waters). Ten percent of the resulting peptides were used for protein abundance analysis and fractionated using a High-pH fractionation kit (Thermo Fisher Scientiic) into 8 fractions and purified by stagetipping. The rest were subjected to phosphopeptide enrichment using the High-Select Fe-NTA kit (Paulo et al., 2018). The resulting phosphopeptides were desalted using a stage-tip and analyzed by mass spectrometry. Kinase enrichment analysis was performed using Enrichr (https://amp.pharm.mssm.edu/Enrichr/; Chen et al., 2013; Kuleshov et al., 2016).
Oocyte mass spectrometry
Proteins from control and MCT1 expressing oocytes were extracted and digested as described above. After digestion, peptides from each sample were then purified using stage-tips and 2 μg peptides were analyzed by mass spectrometry.
Isolation of pure cell populations from muscle
The tibialis anterior, gastrocnemius, and quadricep muscles were isolated from each mouse and placed in a 1.5 mL tube containing 500 μL of phenol-free, serum-free DMEM (GIBCO). Tissues were then minced until disintegrated. Minced tissues were further digested (30 min, 37°C) in DMEM containing collagenase VIII (Sigma Aldrich) and dispase (Thermo Fisher Scientific). Tissue homogenate was filtered into a new 50 mL conical using a 70 μM filter and washed with phenol-free DMEM (GIBCO) containing 2% FBS and EDTA (staining buffer). Cells were then centrifuged (10 min, 1500 rpm, 4°C) and resuspended in staining buffer, twice. Buffer was aspirated and pellet was resuspended in 250 μL of staining buffer containing the following antibodies (Biolegend): CD45 BV605 (1:100), CD31 PE-Cy7 (1:100), CD140a APC (1:50), Sea1 PerCP-Cy5.5 (1:50), and Vcam1 PE (1:100). Cells were incubated for 20 min then washed twice (5 min, 1500 rpm, 4°C) and resuspended in 200 μL of staining buffer. Stained cells were then sorted using a FACS Aria II (Becton Dickinson). Cells were sorted twice to ensure purity.
Immunostaining
Cells were fixed with 100% methanol (chilled at −20°C) for 5 min and then permeabilized with 0.1% Triton in PBS for 15 min. Nonspecific binding was blocked with 2% BSA in PBS at room temperature for 60 min. Incubation with anti-MyHC (1:400; Sigma Aldrich) diluted in PBS with 0.1% BSA was performed overnight at 4°C. Cells were then incubated with Alexa Fluor 488 (Thermo Fisher Scientific) and DAPI (Invitrogen) diluted in PBS for 1 h.
Cell proliferation assay
C2C12 myoblasts were seeded at a density of 40,000 cells per well in a 12 wells plate, 24h prior to use. Cells were then brought into suspension using trypsin, followed by resuspension in fresh medium. Cell suspension was mixed with trypan blue (1:1) and transferred to a hemocytometer. Number of cells was counted 24, 48 and 72 h after the beginning of the experiment.
RNA-Seq analysis
Cell suspensions were stained with antibodies against CD45, CD31, Sca-1, CD140a (PDGFRα), and Vcam1 and sorted using FACS Aria II (Becton Dickson). DAPI was used for cellular viability. The following populations were isolated: hematopoietic (CD45+), endothelial (CD31+), stromal (CD45−CD31−PDGFRα+Sca-1+), and satellite (CD45−CD31−Sca-1−Vcaml+). One thousand cells from each population were double-sorted and collected into 5 μL Buffer TCL (QIAGEN) containing 1% beta-mercaptoethanol (Sigma Aldrich). Samples were placed on ice for 5 minutes, transferred to dry ice, and eventually stored at −80°C until submitted for sequencing. Smart-Seq2 libraries were prepared by the Broad Technology Labs and were sequenced using the Broad Genomics Platform (Picelli et al., 2014). Transcripts were quantified by the Broad Technology Labs computational pipeline using Cufflinks version 2.2.1 (Trapnell et al., 2012). For pathway analysis, differentially expressed genes were defined using the following cutoffs: expression (> 10), fold-change (> 2, WT versus KO), p value (< 0.05), and coefficient of variation (< 0.3). KEGG pathway and GO enrichment analyses were performed using Enrichr (https://amp.pharm.mssm.edu/Enrichr/; Eden et al., 2007, 2009; Chen et al., 2013; Kuleshov et al., 2016).
GTT and ITT
For insulin tolerance testing (ITT), 13-week old mice were fasted for 3.5h. At 2 h following initial fasting time, mice were subjected to a single bout of acute treadmill running for 45 min and then were allowed to rest of 45 min. Insulin (0.9 U/kg) was administered i.p. Blood glucose measurements were taken at 0, 15, 30, 60, 90, and 120 min using a glucometer. For glucose tolerance testing (GTT), mice were fasted for 6h and were administered glucose (1 g/kg) i.p. Blood glucose measurements were taken at 0, 15, 30, 60, 90, and 120 min using a glucometer.
RNA in situ hybridization
Tissues were fixed overnight in 4% paraformaldehyde at 4°C. Tissues were then washed three times in PBS and stored in 70% ethanol until paraffin embedded. Following embedding, tissues were sectioned into 5 μM thick slices and placed onto superfrost slides. In situ hybridization was performed using the RNAscope multiplex v2 assay and 4-plex ancillary kits from ACD. Slides were first deparaffinized by washing in xylene twice (room temperature, 5 min) followed by 2 methanol washes (room temperature, 2 min). Slides were then allowed to dry completely before H2O2 treatment (room temperature, 10 min). Slides were subsequently washed twice in dH2O (room temperature, 1 min). Samples were then incubated in RNAscope 1X Target Retrieval Reagent (100°C, 30 min). Following target retrieval, slides were rinsed in dH2O and then washed in methanol (room temperature, 3min). Slides were then allowed to dry before RNAscope Protease Plus reagent was applied to each section (40°C, 30 min). All 40°C incubations were performed in the HybEZ Oven. Slides were washed twice with dH2O. Following washes, RNAscope probes for desmin, pax7, pdgfrα, and SUCNR1 were administered to each section (40°C, 2 h). Slides were then washed twice with 1X Wash Buffer (room temperature, 2 min). Amp 1 was then applied to each section (40°C, 30 min). The slides were then washed twice with wash buffer (room temperature, 2min). These steps were repeated with Amp 2 (40°C, 30 min) and Amp 3 (40°C, 15 min). HRP-C1 reagent was then applied to each section (40°C, 15 min) followed by two washes (RT, 2 min). Opal 520 fluorophore was applied (1:750, 40°C, 30 min). Slides were washed twice and then HRP blocker was applied to each section (40°C, 15 min). These steps were repeated for HRP-C2 using opal 570, HRP-C3 using opal 620, and HRP-C4 using opal 690. All opal dyes were diluted 1:750 using TSA buffer. Samples were then mounted using ProLong Gold Antifade Mountant and allowed to dry overnight. Samples were then imaged using a fluorescent microscope. Colocalization analysis was performed in Fiji using the Coloc2 plugin for determination of the Manders’ colocalization coefficient.
Resistance training protocol
A resistance training wheel was created by attaching brass spacers to a standard running wheel as previously described (Call et al., 2010). Twelve-week old mice were singly housed and half of them were placed in cages containing a running wheel for 3 weeks. Wheels were weighted to 1 g of resistance at the start of the experiment and was increased by 1 g/week till a final weight of 3 g was reached. Distance run was continually recorded (Starr Life) and grip strength was assessed weekly in both trained and untrained mice using a force meter. Lean and fat mass were measured by MRI at the beginning and at the end of the 3 weeks. Mice were sacrificed after 3 weeks and the tibialis anterior and gastrocnemius were harvested for further processing.
iDISCO three-dimensional muscle imaging
Tibialis anterior and gastrocnemius muscles were fixed in 4% paraformaldehyde (Thermo Fisher Scientific). Muscles were then processed for whole-tissue clearing and imaging, using the iDISCO method (Renier et al., 2014). All washes were performed in a 2 mL tube with end-over-end shaking. Tissues were first dehydrated by washing in a series of methanol/H2O buffers: 20%, 40%, 60%, 80%, 100%, 100% (1 h each, room temperature). Samples were chilled (4°C) and then incubated in 66% dichloromethane (Sigma Aldrich)/33% methanol (o/n, room temperature). Tissues were then washed twice in 100% methanol (room temperature) and then chilled (4°C). Samples were bleached in fresh 5% H2O2 (Sigma Aldrich) diluted in methanol (o/n, 4°C). Tissues were then rehydrated by washing in a series of methanol/H2O buffers: 80%, 60%, 40%, 20%, PBS (1 h each, room temperature). Samples were then washed twice (1 h, room temperature) with PBS containing 2% Triton X-100 (Sigma Aldrich). In order to stain the whole tissue, samples were permeabilized (2 days, 37°C) using PBS containing 2% Triton X-100, glycine (Sigma Aldrich), and DMSO (Sigma Aldrich). Exogenous binding of antibodies was prevented by incubating samples in a blocking solution containing PBS, Triton X-100, DMSO, and FBS (2 days, 37°C). Following blocking, half of the samples were incubated with a TUBB3 antibody (1:2000, Biolegend) and the other half were incubated with a CD31 antibody (1:250, Biolegend) for 4 days at 37°C. Tissues were then washed 5 times with PBS containing 2% Tween-20 (Sigma Aldrich) and heparin (Sigma Aldrich). Samples were then incubated with secondary antibody (1:500, Invitrogen) for 4 days at 37°C. Tissues were then again washed 5 times with PBS containing 2% Tween-20. Samples were dehydrated by washing in a series of methanol/H2O buffers: 20%, 40%, 60%, 80%, 100%, 100% (1 h each, room temperature). Following dehydration, tissues were incubated in 66% dichloromethane/ 33% methanol (3 h, room temperature). Samples were then washed twice with 100% dichloromethane (15 min each, room temperature). Finally, tissues were cleared using dibenzyl ether (Sigma Aldrich). Whole tissues were then imaged at 0.66x magnification using a light sheet microscope. 3D reconstructions and renderings were made using the software Imaris (Bitplane). Further imaging analysis was performed using ImageJ.
Gene expression analysis
Stromal, endothelial, immune, and satellite cells were isolated from hindlimb muscles as previously described and collected into RLT buffer (QIAGEN) for RNA extraction. RNA was further purified using the RNAeasy Micro Kit (QIAGEN). RNA was then quantified using a Nanodrop 2000 UV-visible spectrophotometer. 100 ng of RNA was used to synthesize cDNA via reverse-transcription polymerase chain reaction (RT-PCR) using a high capacity cDNA reverse transcription kit (Thermo Fisher Scientific). cDNA was then diluted in a 1:4 ratio cDNA to water and used for real-time quantitative PCR (qPCR) analysis. qPCR reactions were performed in 384- well plates using GoTaq qPCR Master Mix (Promega). Samples were run on a Quant Studio 6 Flex Real-Time PCR machine (Applied Biosystems). Relative abundance of Sucnr1 was calculated by the ΔCt method using GAPDH as an endogenous control.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data analysis was performed in Excel, R, and Prism as described above. All data (unless otherwise noted) was presented as mean ± SEM. Unless otherwise noted, P values were calculated using a two-tailed Student’s t test for pairwise comparisons and one-way ANOVA for multiple comparisons involving a single variable. * p < 0.05; ** p < 0.01; *** p < 0.005. Details of statistical analyses and n values are found in the Figure Legends. For in vivo experiments, n indicates number of mice or humans. For all other experiments, n indicates biological replicates.
Supplementary Material
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Brilliant Violet 605 anti-mouse CD45 | BioLegend | Cat#103140 |
| PE/Cyanine7 anti-mouse CD31 | BioLegend | Cat#102418 |
| APC anti-mouse CD140a | BioLegend | Cat#135908 |
| PerCP/Cyanine5.5 anti-mouse Ly-6A/E (Sca-1) | BioLegend | Cat#108124 |
| PE anti-mouse CD106 (Vcam1) | BioLegend | Cat#105713 |
| Anti-MyHC | Sigma | Cat# |
| RNAscope® 4-Plex Negative Control Probe | Advanced Cell Diagnostics | Cat#321831 |
| RNAscope® 4-Plex Positive Control Probe-Mm | Advanced Cell Diagnostics | Cat#321811 |
| RNAscope Probe - Mm-Sucnr1-C3 | Advanced Cell Diagnostics | Cat#437721-C3 |
| RNAscope Probe - Mm-Des | Advanced Cell Diagnostics | Cat#407921 |
| RNAscope Probe - Mm-Pax7-C2 | Advanced Cell Diagnostics | Cat#314181-C2 |
| RNAscope Probe - Mm-Pdgfra-C4 | Advanced Cell Diagnostics | Cat#480661-C4 |
| Opal 520 | Akoya Biosciences | Cat#FP1487001KT |
| Opal 570 | Akoya Biosciences | Cat#FP1488001KT |
| Opal 620 | Akoya Biosciences | Cat#FP1495001KT |
| Opal 690 | Akoya Biosciences | Cat#FP1497001KT |
| anti-Tubulin β-3 | BioLegend | Cat#802001 |
| LEAF Purified anti-mouse CD31 | BioLegend | Cat#102512 |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Invitrogen | Cat#A21245 |
| Goat anti-Rat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 | Invitrogen | Cat#A11077 |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Invitrogen | Cat#A11001 |
| Streptavidin MicroBeads | Miltenyi Biotec | Cat#130-048-101 |
| SA-Texas Red | Invitrogen | Cat#S872 |
| Biotin rat anti-mouse CD45 | BD Biosciences | Cat#553078 |
| Biotin rat anti-CD11b | BD Biosciences | Cat#553309 |
| CD31 monoclonal antibody | eBiosciences | Cat#13-0311-82 |
| Biotin rat anti-mouse Ly-6A/E | BD Biosciences | Cat#553334 |
| PE anti-integrin α7 antibody | AbLab | Cat#10ST215 |
| CD34 monoclonal antibody eFluor 660 | eBioscience | Cat#50-0341-82 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMEM | Corning | Cat#10-017-CV |
| Fetal Bovine Serum | Gemini Bio-Products | Cat#100-106 |
| Penicillin streptomycin | Fisher scientific | Cat#15-140-163 |
| Horse Serum, New Zealand origin | Fisher scientific | Cat#16050130 |
| Collagenase II | Worthington Biochemical Corporation | Cat#LS004176 |
| Natural Protease (Dispase) | Worthington Biochemical | Cat#LS02104 |
| Thymine-d4 | Cambridge Isotope | Cat#DLM-1089 |
| Inosine-15N4 | Cambridge Isotope | Cat#NLM-4264 |
| Glycocholate-d4 | Cambridge Isotope | Cat#DLM-2742 |
| Methanol | Fisher Scientific | Cat# A4124 |
| Ammonium acetate | Sigma | Cat#372331-100G |
| Ammonium hydroxide | Sigma | Cat# 338818-100ML |
| Acetonitrile | Fisher Scientific | Cat# A9964 |
| l-glutamine | Sigma | Cat#W368401 |
| HEPES | Sigma | Cat#H0887 |
| Pyruvate | Sigma | Cat#P2256 |
| Glucose | Sigma | Cat#G7021 |
| Atpenin A5 | Cayman Chemical | Cat#11898 |
| Antimycin | Sigma | Cat#A8674 |
| Monensin | Sigma | Cat#M5273 |
| Ammonium chloride | Sigma | Cat#254134 |
| EIPA | Sigma | Cat#337850 |
| BCECF-AM | Invitrogen | Cat#B1170 |
| Nigericin | VWR | Cat#89156-870 |
| α-cyano-4-hydroxycinnamic acid | Sigma | Cat#C2020 |
| AZD 3965 | Fisher Scientific | Cat#501364664 |
| ARC155858 | EMD Millipore | Cat#533436 |
| Opti-MEM | ThermoFisher Scientific | Cat#31985070 |
| Lipofectamine RNAiMAX | Invitrogen | Cat#13778-150 |
| Sodium Chloride | Fisher Scientific | Cat#S2711 |
| Potassium Chloride | Sigma | Cat#P9333 |
| Calcium Chloride | Sigma | Cat#499609 |
| Magnesium Chloride | Sigma | Cat#M8266 |
| Sodium Phosphate Dibasic | Sigma | Cat#S9763 |
| [U13C] succinate | Cambridge Isotopes | Cat#CLM-1571 |
| [U13C] lactate | Cambridge Isotopes | Cat#CLM-1578 |
| cOmplete Protease Inhibitor Cocktail | Sigma-Aldrich | Cat#CO-RO |
| PhosSTOP, Phosphatase Inhibitor | Sigma-Aldrich | Cat#PHOSS-RO |
| EPPS | Sigma-Aldrich | Cat#E9502 |
| Bond-Breaker TCEP Solution, Neutral pH | ThermoFisher Scientific | Cat#77720 |
| Iodoacetamide | Sigma-Aldrich | Cat#I1149 |
| Dithiothreitol (DTT) | Sigma-Aldrich | Cat#DTT-RO |
| Lambda phosphatase | Santa Cruz Biotechnology | Cat#sc-200312 |
| Lys-C | Wako Chemicals | Cat#125-05061 |
| Trypsin | Promega | Cat#V5113 |
| 16-plex TMT reagents | Thermo Fisher Scientific | Cat#A44520 |
| DMEM, high glucose, no glutamine, no phenol red | Life Technologies | Cat#31053028 |
| Collagenase VIII | Sigma | Cat#C2139 |
| Dispase II | Thermo Fisher Scientific | Cat#17105041 |
| Triton X-100 | Sigma | Cat#X100 |
| Bovine Serum Albumin | Sigma | Cat#A9647 |
| Trypan Blue | Sigma | Cat#T8154 |
| Buffer TCL | QIAGEN | Cat#1031576 |
| Beta-mercaptoethanol | Sigma | Cat#M6250 |
| Humulin R | Walgreens | Cat#Hi-210 |
| Paraformaldehyde | Fisher Scientific | Cat#50980488 |
| Dichloromethane | Fisher Scientific | Cat#NC0713665 |
| Xylenes | Fisher Scientific | Cat#AC396930010 |
| Hydrogen peroxide | Fisher Scientific | Cat#501657227 |
| Glycine | Sigma | Cat#G7126 |
| DMSO | Sigma | Cat#D8418 |
| Heparin | Sigma | Cat#H3393-50KU |
| Tween-20 | Sigma | Cat#P9416 |
| Dibenzyl Ether | Sigma | Cat#33630 |
| RLT Buffer | QIAGEN | Cat#79216 |
| GoTaq qPCR master mix | Promega | Cat#A6001 |
| ProLong Gold Antifade Moutnant | Life Technologies | Cat#P36930 |
| Critical Commercial Assays | ||
| mMessage mMachine kit | Life Technologies | Cat#AM1344 |
| High-Select Fe-NTA Phosphopeptide Enrichment Kit | ThermoFisher Scientific | Cat#A32992 |
| Pierce High pH Reversed-Phase Peptide Fractionation Kit | ThermoFisher Scientific | Cat#84868 |
| SmartSeq 2 | Illumina | |
| RNA scope multiplex v2 kit | Advanced Cell Diagnostics | Cat#323100 |
| RNAscope 4-Plex Ancillary Kit for Multiplex Fluorescent V2 | Advanced Cell Diagnostics | Cat#323120 |
| RNAeasy Micro Kit | QIAGEN | Cat#74004 |
| High capacity cDNA reverse transcription kit | Fisher Scientific | Cat# 4368813 |
| Experimental Models: Cell Lines | ||
| C2C12 cells | ATCC | Cat#CRL-1772 |
| Muscle satellite cells | This study | N/A |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J wild-type male mice | The Jackson Laboratories | Cat#000664 |
| SUCNR1 KO male mice | Mills et al., 2018 | N/A |
| Xenopus laevis oocytes | EcoCyte Biosciences | N/A |
| Oligonucleotides | ||
| MCT1 siRNA, SASI_Mm01_00112354 | Sigma | Cat#NM_009196 |
| MCT2 siRNA, SASI_Mm01_00096521 | Sigma | Cat#NM_011391 |
| MCT4 siRNA, SASI_Mm01_00119745 | Sigma | Cat#NM_001038653 |
| Sucnr1 FW 5′- GCAGAATGGCACAGAATTTATCT- 3′ | IDT | N/A |
| Sucnr1 Rev 5′- ACATTCCCAAGCAGTCCAA- 3′ | IDT | N/A |
| Gapdh FW 5′-AGGTCGGTGTGAACGGATTTG-3′ | IDT | N/A |
| Gapdh Rev 5′-TGTAGACCATGTAGTTGAGGT-3′ | IDT | N/A |
| Recombinant DNA | ||
| SLC16A1_OHu17492C_pcDNA3.1(+) | GenScript | Cat#OHu17492C |
| Software and Algorithms | ||
| Xcalibur | ThermoFisher Scientific | Cat#OPTON-30965 |
| TraceFinder | ThermoFisher Scientific | Cat#OPTON-30688 |
| R version 3.5.2 | R Project | https://www.r-project.org |
| RStuido Version 1.2.1335 | R Studio Team | https://rstudio.com |
| ImageJ | NIH | https://login.ezp-prod1.hul.harvard.edu/login?qurl=https://imagej.nih.gov%2fij%2f |
| GORILLA | Eden et al., 2007, 2009 | http://cbl-gorilla.cs.technion.ac.il |
| Prism 8 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Enrichr | Chen et al., 2013; Kuleshov et al., 2016 | https://amp.pharm.mssm.edu/Enrichr/ |
| Cufflinks version 2.2.1 | Trapnell et al., 2012 | https://github.com/cole-trapnell-lab/cufflinks |
| Imaris Version 9.3.1 | Bitplane | N/A |
| Other | ||
| Bicycle ergometer | Monark Exercise | Cat#839E |
| Animal Treadmill | Columbus Instruments | Cat# 1050-RM |
| 20 μm nylon mesh filter | EMD Millipore | Cat# NY2004700 |
| Heparin columns | Becton Dickinson | Cat# 365985 |
| Luna 5 μm NH2 column | Phenomenex | Cat#00F-4378-B0 |
| Q-Exactive HF-X mass spectrometer | ThermoFisher Scientific | Cat#0726042 |
| WPS-3000TBFC Biocompatible Well Plate Autosampler | ThermoFisher Scientific | Cat#5841.0020 |
| TCC-3000RS Thermostatted Column Compartment | ThermoFisher Scientific | Cat#5730.0000 |
| LPG-3400RS Quaternary Pump w/Degasser | ThermoFisher Scientific | Cat#5040.0036 |
| Ultrasound machine | Philips | Affiniti70 |
| Master Screen CPX System | Becton Dickinson | N/A |
| Hypoxia chamber | Coy Lab Products | N/A |
| Orbitrap Fusion Lumos Mass Spectrometer | ThermoFisher Scientific | Cat#IQLAAEGAAPFADBMBHQ |
| Orbitrap Eclipse mass spectrometer | ThermoFisher Scientific | Cat#FSN04-10000 |
| Multi-Therm heat-shake | Sigma-Aldrich | Cat#Z755753 |
| Sep-Pak C18 Cartridges | Waters | Cat#WAT054955 |
| TissueLyser II | QIAGEN | Cat#85300 |
| EASY-nLC 1200 System | ThermoFisher Scientific | Cat#LC140 |
| FAIMSPro | ThermoFisher Scientific | Cat#FMS02-10001 |
| FACS Aria II | Becton Dickinson | Cat# 643181 |
| Glucometer, ultra mini | One Touch | N/A |
| In-cage running wheels | Starr Life | N/A |
| Force Meter | Chatillon | Cat#DFE-002 |
| Nanodrop 2000 | Thermo Fisher | Cat#ND-2000 |
| Quant Studio 6 Flex Real-Time PCR machine | Applied Biosciences | Cat# 44-856-99 |
Highlights.
Mouse and human muscle selectively release succinate during exercise
Muscle cells release succinate by pH-gated secretion via MCT1
Extracellular succinate regulates paracrine responses to exercise through SUCNR1
SUCNR1 signaling mediates muscle remodeling responses to exercise training
ACKNOWLEDGMENTS
We thank Mike Murphy, Fred Goldberg, and Bruce Spiegelman for helpful discussions; Bill Kaelin for assistance with cellular hypoxia experiments; the Nikon Imaging Facility at Harvard Medical School for assistance with light and fluorescence microscopy; and the Harvard Center for Biological Imaging for assistance with in situ hybridization microscopy. We thank the Neurobiology Department and the Neurobiology Imaging Facility for consultations and instrument availability, which supported this work. This facility is supported in part by the Neural Imaging Center as part of NINDS P30 Core Center grant NS072030. This work was supported by the American Heart Association and NIH DK123095 (to E.T.C.), the JPB Foundation and NIH AR070334 (to D.M.), NIH NIGMS T32GM007753 (to O.K.Y.), Sao Paulo Research Foundation 2019/07221-9 (to L.H.M.B.), and the National Cancer Center (to H.X.). The human studies were supported by Novo Nordisk A/S.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2020.08.039.
DECLARATION OF INTERESTS
E.T.C. has filed for a patent based on data describing the role of SUCNR1 agonism and muscle remodeling in this work.
REFERENCES
- Andrienko TN, Pasdois P, Pereira GC, Ovens MJ, and Halestrap AP (2017). The role of succinate and ROS in reperfusion injury - A critical appraisal. J. Mol. Cell. Cardiol 110, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariza AC, Deen PM, and Robben JH (2012). The succinate receptor as a novel therapeutic target for oxidative and metabolic stress-related conditions. Front. Endocrinol. (Lausanne) 3, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker D, and Saito M (1981). Autonomic innervation of receptors and muscle fibres in cat skeletal muscle. Proc. R. Soc. Lond. B Biol. Sci 212,317–332. [DOI] [PubMed] [Google Scholar]
- Beitzel F, Gregorevic P, Ryall JG, Plant DR, Sillence MN, and Lynch GS (2004). Beta2-adrenoceptor agonist fenoterol enhances functional repair of regenerating rat skeletal muscle after injury. J. Appl. Physiol 96, 1385–1392. [DOI] [PubMed] [Google Scholar]
- Bright CM, and Ellis D (1992). Intracellular pH changes induced by hypoxia and anoxia in isolated sheep heart Purkinje fibres. Exp. Physiol 77, 165–175. [DOI] [PubMed] [Google Scholar]
- Bröer S, Schneider HP, Bröer A, Rahman B, Hamprecht B, and Deitmer JW (1998). Characterization of the monocarboxylate transporter 1 expressed in Xenopus laevis oocytes by changes in cytosolic pH. Biochem. J 333, 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bröer S, Bröer A, Schneider HP, Stegen C, Halestrap AP, and Deitmer JW (1999). Characterization of the high-affinity monocarboxylate transporter MCT2 in Xenopus laevis oocytes. Biochem. J 341, 529–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buller AJ, Eccles JC, and Eccles RM (1960). Interactions between moto-neurones and muscles in respect of the characteristic speeds of their responses. J. Physiol 150, 417–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY,Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vadder F, and Mithieux G (2018). Gut-brain signaling in energy homeostasis: the unexpected role of microbiota-derived succinate. J. Endocrinol 236, R105–R108. [DOI] [PubMed] [Google Scholar]
- De Vadder F, Kovatcheva-Datchary P, Zitoun C, Duchampt A, Bäckhed E, and Mithieux G (2016). Microbiota-Produced Succinate Improves Glucose Homeostasis via Intestinal Gluconeogenesis. Cell Metab. 24, 151–157. [DOI] [PubMed] [Google Scholar]
- Deen PM, and Robben JH (2011). Succinate receptors in the kidney. J. Am. Soc. Nephrol 22, 1416–1422. [DOI] [PubMed] [Google Scholar]
- Eden E, Lipson D, Yogev S, and Yakhini Z (2007). Discovering motifs in ranked lists of DNA sequences. PLoS Comput. Biol 3, e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, and Yakhini Z (2009). GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehinger JK, Piel S, Ford R, Karlsson M, Sjövall F, Frostner EA, Morota S, Taylor RW, Turnbull DM, Cornell C, et al. (2016). Cell-permeable succinate prodrugs bypass mitochondrial complex I deficiency. Nat. Commun 7, 12317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folland JP, and Williams AG (2007). The adaptations to strength training : morphological and neurological contributions to increased strength. Sports Med. 37, 145–168. [DOI] [PubMed] [Google Scholar]
- Gabriel DA, Kamen G, and Frost G (2006). Neural adaptations to resistive exercise: mechanisms and recommendations for training practices. Sports Med. 36, 133–149. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Golbidi S,Werner I,Verchere BC, and Laher I (2010). Selecting exercise regimens and strains to modify obesity and diabetes in rodents: an overview. Clin. Sci. (Lond.) 119, 57–74. [DOI] [PubMed] [Google Scholar]
- Hagberg H (1985). Intracellular pH during ischemia in skeletal muscle: relationship to membrane potential, extracellular pH, tissue lactic acid and ATP. Pflugers Arch. 404, 342–347. [DOI] [PubMed] [Google Scholar]
- Halestrap AP (2012). The monocarboxylate transporter family–Structure and functional characterization. IUBMB Life 64, 1–9. [DOI] [PubMed] [Google Scholar]
- Hawley JA, Hargreaves M, Joyner MJ, and Zierath JR (2014). Integrative biology of exercise. Cell 159, 738–749. [DOI] [PubMed] [Google Scholar]
- He W, Miao FJ, Lin DC, Schwandner RT, Wang Z, Gao J, Chen JL, Tian H, and Ling L (2004). Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature 429, 188–193. [DOI] [PubMed] [Google Scholar]
- Hems R, Stubbs M, and Krebs HA (1968). Restricted permeability of rat liver for glutamate and succinate. Biochem. J 107, 807–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henríquez-Olguin C, Knudsen JR, Raun SH, Li Z, Dalbram E, Treebak JT, Sylow L, Holmdahl R, Richter EA, Jaimovich E, and Jensen TE (2019). Cytosolic ROS production by NADPH oxidase 2 regulates muscle glucose uptake during exercise. Nat. Commun 10, 4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisamatsu D, Ohno-Oishi M, Nakamura S, Mabuchi Y, and Naka-Kaneda H (2016). Growth differentiation factor 6 derived from mesenchymal stem/stromal cells reduces age-related functional deterioration in multiple tissues. Aging (Albany NY) 8, 1259–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochachka PW, and Dressendorfer RH (1976). Succinate accumulation in man during exercise. Eur. J. Appl. Physiol. Occup. Physiol 35, 235–242. [DOI] [PubMed] [Google Scholar]
- Hochachka PW, Owen TG, Allen JF, and Whittow GC (1975). Multiple end products of anaerobiosis in diving vertebrates. Comp. Biochem. Physiol. B 50, 17–22. [DOI] [PubMed] [Google Scholar]
- Kemi OJ, Loennechen JP, Wisloff U, and Ellingsen O (2002). Intensity-controlled treadmill running in mice: cardiac and skeletal muscle hypertrophy. J. Appl. Physiol 93, 1301–1309. [DOI] [PubMed] [Google Scholar]
- Kiens B, Essen-Gustavsson B, Christensen NJ, and Saltin B (1993). Skeletal muscle substrate utilization during submaximal exercise in man: effect of endurance training. J. Physiol 469, 459–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinert M, Clemmensen C, SjØberg KA, Carl CS, Jeppesen JF, Wojtaszewski JFP, Kiens B, and Richter EA (2018). Exercise increases circulating GDF15 in humans. Mol. Metab 9, 187–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44 (W1), W90–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HJ, Herman P, and Lakowicz JR (2003). Fluorescence lifetime-resolved pH imaging of living cells. Cytometry A 52, 77–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littlewood-Evans A, Sarret S, Apfel V, Loesle P, Dawson J, Zhang J, Muller A, Tigani B, Kneuer R, Patel S, et al. (2016). GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J. Exp. Med 213, 1655–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald MJ, Fahien LA, Mertz RJ, and Rana RS (1989). Effect of esters of succinic acid and other citric acid cycle intermediates on insulin release and inositol phosphate formation by pancreatic islets. Arch. Biochem. Biophys 269, 400–406. [DOI] [PubMed] [Google Scholar]
- Markovich D, and Murer H (2004). The SLC13 gene family of sodium sulphate/carboxylate cotransporters. Pflugers Arch. 447, 594–602. [DOI] [PubMed] [Google Scholar]
- Martínez-Reyes I, and Chandel NS (2020). Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun 11, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCreath KJ, Espada S, Gálvez BG, Benito M, de Molina A, Sepúlveda P, and Cervera AM (2015). Targeted disruption of the SUCNR1 metabolic receptor leads to dichotomous effects on obesity. Diabetes 64, 1154–1167. [DOI] [PubMed] [Google Scholar]
- Mills E, and O’Neill LA (2014). Succinate: a metabolic signal in inflammation. Trends Cell Biol. 24, 313–320. [DOI] [PubMed] [Google Scholar]
- Mills EL, Pierce KA, Jedrychowski MP, Garrity R, Winther S, Vidoni S, Yoneshiro T, Spinelli JB, Lu GZ, Kazak L, et al. (2018). Accumulation of succinate controls activation of adipose tissue thermogenesis. Nature 560, 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnár T, Barabás P, Héja L, Fekete EK, Lasztóczi B, Szabó P, Nyitrai G, Simon-Trompler E, Hajós F, Palkovits M, and Kardos J (2008). gamma-Hydroxybutyrate binds to the synaptic site recognizing succinate monocarboxylate: a new hypothesis on astrocyte-neuron interaction via the protonation of succinate. J. Neurosci. Res 86, 1566–1576. [DOI] [PubMed] [Google Scholar]
- Mookerjee SA, Goncalves RLS, Gerencser AA, Nicholls DG, and Brand MD (2015). The contributions of respiration and glycolysis to extracellular acid production. Biochim. Biophys. Acta 1847, 171–181. [DOI] [PubMed] [Google Scholar]
- Murphy MP, and O’Neill LAJ (2018). Krebs Cycle Reimagined: The Emerging Roles of Succinate and Itaconate as Signal Transducers. Cell 174, 780–784. [DOI] [PubMed] [Google Scholar]
- Nancolas B, Sessions RB, and Halestrap AP (2015). Identification of key binding site residues of MCT1 for AR-C155858 reveals the molecular basis of its isoform selectivity. Biochem. J 466, 177–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peruzzotti-Jametti L, Bernstock JD, Vicario N, Costa ASH, Kwok CK, Leonardi T, Booty LM, Bicci I, Balzarotti B,Volpe G, et al. (2018). Macrophage-Derived Extracellular Succinate Licenses Neural Stem Cells to Suppress Chronic Neuroinflammation. Cell Stem Cell 22, 355–368.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phieler J, Garcia-Martin R, Lambris JD, and Chavakis T (2013). The role of the complement system in metabolic organs and metabolic diseases. Semin. Immunol 25, 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter EA, Mikines KJ, Galbo H, and Kiens B (1989). Effect of exercise on insulin action in human skeletal muscle. J. Appl. Physiol 66, 876–885. [DOI] [PubMed] [Google Scholar]
- Robergs RA, Ghiasvand F, and Parker D (2004). Biochemistry of exercise-induced metabolic acidosis. Am. J. Physiol. Regul. Integr. Comp. Physiol 287, R502–R516. [DOI] [PubMed] [Google Scholar]
- Roemers P, Mazzola PN, De Deyn PP, Bossers WJ, van Heuvelen MJG, and van der Zee EA (2018). Burrowing as a novel voluntary strength training method for mice: A comparison of various voluntary strength or resistance exercise methods. J. Neurosci. Methods 300, 112–126. [DOI] [PubMed] [Google Scholar]
- Sahlin K, Edström L, Sjöholm H, and Hultman E (1981). Effects of lactic acid accumulation and ATP decrease on muscle tension and relaxation. Am. J. Physiol 240, C121–C126. [DOI] [PubMed] [Google Scholar]
- Sanes JR (2003). The basement membrane/basal lamina of skeletal muscle. J. Biol. Chem 278, 12601–12604. [DOI] [PubMed] [Google Scholar]
- Spinelli JB, Yoon H, Ringel AE, Jeanfavre S, Clish CB, and Haigis MC (2017). Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass. Science 358, 941–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MR, Danai LV, Lewis CA, Chan SH, Gui DY, Kunchok T, Dennstedt EA, Vander Heiden MG, and Muir A (2019). Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. eLife 8, e44235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylow L, Kleinert M, Richter EA, and Jensen TE (2017). Exercise-stimulated glucose uptake - regulation and implications for glycaemic control. Nat. Rev. Endocrinol 13, 133–148. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Flanagan J, Su N, Wang LC, Bui S, Nielson A, Wu X, Vo HT, Ma XJ, and Luo Y (2012). RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J. Mol. Diagn 14, 22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Portier BP, Gruver AM, Bui S, Wang H, Su N, Vo HT, Ma XJ, Luo Y, Budd GT, and Tubbs RR (2013). Automated quantitative RNA in situ hybridization for resolution of equivocal and heterogeneous ERBB2 (HER2) status in invasive breast carcinoma. J. Mol. Diagn 15, 210–219. [DOI] [PubMed] [Google Scholar]
- Wang T, Xu YQ, Yuan YX, Xu PW, Zhang C, Li F, Wang LN, Yin C, Zhang L, Cai XC, et al. (2019). Succinate induces skeletal muscle fiber remodeling via SUNCR1 signaling. EMBO Rep. 20, e47892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Yaghi OK, Spallanzani RG, Chen X, Zemmour D, Lai N, Chiu IM, Benoist C, and Mathis D (2020). Neuronal, stromal, and T-regulatory cell crosstalk in murine skeletal muscle. Proc. Natl. Acad. Sci. USA 117, 5402–5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all datasets generated or analyzed during this study.