

Abstract
In view of the recent global pandemic caused by COVID-19 intense efforts have been devoted worldwide towards the development of an effective treatment for this disease. Recently, PDE4 inhibitors have been suggested to attenuate the cytokine storm in COVID-19 especially tumour necrosis factor alpha (TNF-α). In our effort we have explored the 2-substituted pyrrolo[2,3-b]quinoxalines for this purpose because of their potential inhibitory properties of PDE-4 / TNF-α. Moreover, several of these compounds appeared to be promising in silico when assessed for their binding affinities via docking into the N-terminal RNA-binding domain (NTD) of N-protein of SARS-CoV-2. A rapid and one-pot synthesis of this class of molecules was achieved via the Cu-catalyzed coupling-cyclization-desulfinylation of 3-alkynyl-2-chloroquinoxalines with t-butyl sulfinamide as the ammonia surrogate under ultrasound irradiation. Most of these compounds showed good to significant inhibition of TNF-α in vitro establishing a SAR (Structure Activity Relationship) within the series. One compound e.g. 3i was identified as a promising hit for which the desirable ADME and acceptable toxicity profile was predicted in silico.
Keywords: Pyrrolo[2,3-b]quinoxaline; Catalysis; Ultrasound; In silico study; COVID-19
Graphical Abstract

1. Introduction
Being pathogens with very long single-strained RNA the Human Corona Viruses (HCoVs) are capable of causing a number of diseases via various pathogenic mechanisms. Indeed, due to the higher mutation rate of RNA viruses over DNA viruses they can undergo easy mutation thereby infecting various animal and human species. Among the six HCoVs such as HCoV-NL63 and HCoV-229E as Alpha-CoVs and HCoV-OC43, HCoV-HKU1, SARS-CoV and MERS-CoV as Beta-CoVs reported till 2019 end the SARS-CoV (Severe acute respiratory syndrome Corona Virus) and MERS-CoV (Middle East respiratory syndrome Corona Virus) were identified as the most aggressive HCoVs that may cause severe and fatal respiratory infections and / or multi-organ failure [1,2]. The outbreak of SARS-CoV-1 was initially witnessed in southern China during 2002-2003 and re-emerged in Guangdong province of China in December 2003. While the outbreak of MERS-CoV occurred in 2012 in Jeddah of Saudi Arabia the SARS-CoV-2 caused a global outbreak during 2019-2020 called “COVID-19” (coronavirus disease 2019) [3] resulting in a severe blow to the world health and economy. A large number of people have died in various countries till date and the number of new infections are increasing exponentially every day [4]. Due to the lack of definitive vaccines and therapeutic drugs there was an urgent need in devoting efforts to identify effective therapeutics via employing diverse approaches. Accordingly, an immunomodulant as well as anti-malaria drug chloroquine (A, Fig 1 ) was examined and the in vitro studies [5] indicated its effectiveness against SARS-CoV and MERS-CoV [6,7]. A close analogue of A i.e. the drug hydroxychloroquine was also explored as an experimental treatment for COVID-19 [8]. Favipiravir or T-705 (B, Fig. 1), the first approved drug in China [9] and explored in Japan, India etc is a pyrazine based anti-influenza drug that causes selective inhibition of RNA-dependent RNA polymerase of influenza virus [10]. The other agents that are currently in clinical trials for SARS-CoV-2 mostly include Remdesivir, Indinavir, Saquinavir, Darunavir, ASC09, Ritonavir, Lopinavir etc [11].
Fig. 1.

Example of drugs and agents that were explored against coronavirus.
The SARS-CoV possess a membrane comprising of four viral proteins [e.g. spike glycoprotein (S), membrane glycoprotein (M), nucleocapsid proteins (N) and an envelope protein (E)] that play a key role during the host cell entry and viral morphogenesis and release of S-glycoprotein on the virus surface for attaching the virus to a host receptor i.e. ACE-II [12]. In 2016, the nucleocapsid (N)-RNA binding domain was explored as a target protein for the virtual screening of molecules towards the identification of potential hits against human coronavirus (CoV-OC43) [13]. Accordingly, the quinoline derivative (C, Fig. 1) was identified as the most promising hit and the crystallographic structure of C bound with HCoV-OC43 was deposited. Recently, a crystallographic structure of Apo-nucleocapsid (RNA-binding domain) of SARS-CoV-2/COVID-19 (PDB: 6M3M) has revealed the potential drug targeting sites [14]. In earlier study using HCoV-OC43 as a model for CoV (in 2014) Hou et al presented the 3D structure of HCoV-OC43 N-terminal domain (N-NTD) complexed with ribonucleoside 5′-monophosphates to identify a distinct ribonucleotide-binding pocket [15]. Indeed, a new inhibitor PJ34 (D, Fig. 1) was identified by targeting this pocket through virtual screening and the crystal structure of the N-NTD–PJ34 complex was determined. Nevertheless, efforts towards identification of small organic molecule based inhibitor against nucleocapsid (N) of SARS-CoV-2 is rather uncommon. In our earlier effort we have explored alkynyl substituted chloroquinoxaline derivatives E (Fig. 2 ) as potential ligands for N-protein of SARS-CoV-2 in silico [16]. In further continuation of this research we became interested in exploring the related 2-substituted pyrrolo[2,3-b]quinoxalines F (Fig. 2) as potential agents for COVID-19. Notably, several of these compounds showed high inhibition (> 90% at 30 µM) of PDE4B (phosphodiesterase 4B) in vitro previously though the assay results remained inconclusive due to the interference caused by the direct luciferase inhibition of these compounds [17]. Recently the PDE4 inhibitors, known treatment for asthma, chronic obstructive pulmonary disease (COPD), psoriasis etc have been predicted to be a valuable therapeutic option for COVID-19 (Fig. 2) due to their upstream inhibition of multiple cytokine signaling pathways along with the regulation of the pro-inflammatory/anti-inflammatory balance [18]. Indeed, because of targeting an early stage inflammatory response and ameliorating lung inflammation, PDE4 inhibitors might be effective for the initial phase of COVID-19 pneumonia prior to the onset of cytokine storm and severe multi-organ dysfunction [19].
Fig. 2.

Previously and currently explored quinoxaline derivatives E and F as potential ligands for N-protein of SARS-CoV-2 in silico.
2. Results and discussion
To validate our predictive rational in favor of selecting the template F the related docking studies were carried out in silico. The iGEMDOCK version 2.1 software [20] (a graphical automatic drug design system for docking, screening and analysis) was used for this purpose. It is a program for computing ligand conformation and orientation relative to the active site of the protein. The in silico docking simulation studies were performed to evaluate the molecular interactions of a series of compounds e.g. 3a-j derived from F along with the reference compound C (Fig 1) with the SARS-CoV-2 nucleocapsid protein N-terminal RNA binding domain (PDB: 6M3M). The corresponding binding energies and the interaction diagrams are presented in Table 1 and 2 , respectively. Notably, the results obtained via iGEMDOCK were further validated using the two other docking programs such as DOCKTHOR [21] and SWISSDOCK [22] (Table 1).
Table 1.
The binding energies of molecules 3a-j and C with the SARS-CoV-2 N-terminal RNA binding domain residues.
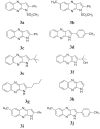 | |||
| Molecules | Estimated Total Energy (kcal/mol) | ||
| GEMDOCK | DOCKTHOR | SWISSDOCK | |
| 3a | -78.86 | -7.84 | -6.40 |
| 3b | -78.42 | -7.77 | -6.17 |
| 3c | -86.05 | -8.12 | -6.85 |
| 3d | -86.00 | -7.83 | -6.91 |
| 3e | -68.90 | -7.68 | -6.37 |
| 3f | -73.11 | -7.23 | -6.11 |
| 3g | -72.08 | -7.94 | -6.18 |
| 3h | -64.23 | -7.44 | -6.19 |
| 3i | -89.37 | -7.94 | -6.33 |
| 3j | -85.33 | -8.10 | -6.98 |
| C | -81.02 | -6.90 | -6.75 |
Table 2.
The interaction diagrams (ribbon representation) of molecules 3a-j and the reference compound C with the SARS-CoV-2 N-terminal RNA binding domain residues.
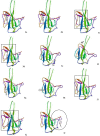 |
*The box indicates compounds with similar binding modes into the target protein. The circle indicates a different binding site for the reference compound C.
Among the compounds examined (Table 1) the molecule 3c, 3d and 3j showed superior binding energies across all three docking programs i.e. GEMDOCK, DOCKTHOR and SWISSDOCK used whereas 3i and 3g showed encouraging results in case of GEMDOCK and DOCKTHOR. Moreover, the interaction diagrams (Table 2) indicated 3c, 3d, 3i and 3j had similar docking pose in the same site of the target protein. These molecules also showed similar docking interactions with the residues TYR71, LYS75 and TRP80 (Table 3 ). Indeed, the nitrogen of the central fused ring was involved in the H-bonding interaction with TRP80 in all these cases whereas π-π interactions was observed with TYR71 and LYS75 in most of the cases. The 2D and 3D interaction diagram of compound 3c and 3i with SARS-CoV-2 nucleocapsid protein N-terminal RNA binding domain is shown in Fig 3 (see the suppl data for interaction diagram with other molecules). Overall, these molecules showed similar interactions with the residues in the active site and their binding energies fall in the similar range.
Table 3.
The residues of the binding site involved in the interaction with the selected molecules.
| Compounds | Binding site interacting residuesa |
| 3c | TYR71, LYS75, TRP80 |
| 3d | TYR71, LYS75, TRP80 |
| 3g | LYS18, TYR71, TRP80 |
| 3i | TYR71, LYS75, TRP80 |
| 3j | ASP16, TYR71, LYS75, TRP80 |
| C | TRP5, THR96, ASP92, ASN101 |
The underlined residue was involved in H-bonding interaction.
Fig. 3.

2D and 3D interaction diagram of SARS-CoV-2 nucleocapsid protein N-terminal RNA binding domain with (A) compound 3c and (B) compound 3i.
In the light of usefulness of pyrrolo[2,3-b]quinoxaline derivatives as potential ligands for SARS-CoV-2 it was desirable to expedite their convenient access for further study. The known synthesis of pyrrolo[2,3-b]quinoxaline derivatives [23,24,25] include the Pd-mediated intramolecular ring closure of 2-alkynyl-3-trifluoroacetamidoquinoxalines [24] or action of amines on 2-chloro-3-alkynylquinoxalines [25]. In our earlier effort the 2-substituted pyrrolo[2,3-b]quinoxalines having free NH were prepared directly via the Cu-catalyzed coupling-cyclization of 3-alkynyl-2-chloroquinoxalines with methanesulfonamide in a single pot [17]. While the methodology afforded the desired products in good yields the duration of the reaction was 4-8h. It was therefore desirable to establish a faster access to this class of compounds. The use of ultrasound irradiation is a well-known option in accelerating the reaction rate considerably thereby reducing the reaction time significantly. It is therefore not surprising that the ultrasound irradiation being a useful alternative to the conventional energy sources such as heat, light, or ionizing radiation has found wide applications in organic synthesis [26], [27], [28], [29]. Indeed, the ultrasound-assisted reactions have become common approaches in the area of green and sustainable chemistry [26] because of (i) their environmentally friendly nature (via minimization of waste generation), [27] (ii) decreased energy requirements [28] and (iii) increased efficiency and effectiveness. Thus we became interested in adopting this approach in our current effort.
Initially, we examined the reaction of 2-chloro-3-(phenylethynyl)quinoxaline (1a) with methanesulfonamide (NH2SO2CH3) (2a) under ultrasound irradiation at 80°C without altering the other reaction conditions used earlier [17]. The reaction was carried out in the presence of Cu(OAc)2 and Et3N in DMF using a laboratory ultrasonic bath SONOREX SUPER RK 510H model producing irradiation of 35 kHz (entry 1, Table 4 ). While the reaction was completed within 2h the yield of desired product 3c was not particularly high. The yield was not improved when 2a was replaced by other sulfonamides such as benzene sulfonamide (2b) (entry 2, Table 4) or p-toluene sulfonamide (2c) (entry 3, Table 4). Notably, the product yield was increased considerably when t-butyl sulfinamide (racemic) (2d) was used (entry 4, Table 4) and the reaction was completed within 1.5 h. Encouraged by this observation we continued the optimization studies. However, change of base from Et3N to DBU decreased to product yield (entry 5, Table 4) whereas increase of reaction time did not improve the yield significantly (entry 6, Table 4). The reaction was also performed under silent conditions (entry 7, Table 4). While the coupling-cyclization proceeded under this condition the product 3c was isolated in inferior yield even after 4h. All these reactions were carried out using slight excess of stoichiometric amount of 2. In order to understand the role of sulfinamide the reaction was carried out using half equivalent of 2d when the reaction did not reach to the completion affording 3c in inferior yield (entry 8, Table 4). Further, these reactions were catalyzed by 1 mol% of Cu(OAc)2 whereas decrease in catalyst quantity was found to be counterproductive (entry 9, Table 4). Similarly, the effect of quantity of base i.e. 1.5 equivalent of Et3N used was also examined and was found to have negative impact on the product yield when used in decreased quantity i.e. 1 equivalent or less. Notably, while the current coupling-cyclization-desulfinylation was found to be insensitive to the presence of air however maintaining the anhydrous atmosphere was necessary in order to avoid the partial hydrolysis of the starting chloro derivative (1a) as a side reaction. Thus the reaction does not require the use of any closed reaction vessel thereby avoiding the pressure development inside the vessel. Nevertheless, the condition of entry 4 of Table 1 appeared to be optimum for the preparation of 3c under ultrasound irradiation.
Table 4.
Effect of catalysts/base on the coupling-cyclization of 1a to 3c.a
 | |||
| Entry | 2; Z = | Time | %Yieldb |
| 1 | 2a; MeSO2 | 2 | 51 |
| 2 | 2b; PhSO2 | 2 | 46 |
| 3 | 2c; p-MeC6H4SO2 | 2 | 40 |
| 4 | 2d; tBuSO | 1.5 | 67 |
| 5 | 2d | 1.5 | 32c |
| 6 | 2d | 2 | 68 |
| 7 | 2d | 4 | 49d |
| 8 | 2d | 1.5 | 30e |
| 9 | 2d | 1.5 | 36f |
All the reactions were carried out by using 1a (2.5 mmol), 2 (2.8 mmol), Cu(OAc)2 (0.025 mmol) and Et3N (3.77 mmol) in DMF (3 mL) under ultrasound.
Isolated yield.
DBU was used in place of Et3N.
The reaction was performed under silent conditions.
The reaction was performed using 1.4 mmol of 2d.
The reaction was performed using 0.014 mmol of Cu(OAc)2.
The optimized reaction conditions were then used to prepare all the target compounds 3c-j (Table 1) and results are presented in Table 5 . A range of 2-chloro-3-alkynyl quinoxaline derivatives (1) were employed for this purpose and the ultrasound assisted reaction proceeded well in all these cases affording the corresponding 2-substituted pyrrolo[2,3-b]quinoxalines (3c-j) in good to acceptable yield. Notably and expectedly, unlike the previous report [17] the use of 2d did not afford the corresponding 1-alkylsulfonyl substituted analogues such as 3a and 3b (Table 1) under the conditions employed. Thus access of these compounds were made via the earlier non-sonochemical method [17]. Nevertheless, all the synthesized pyrrolo[2,3-b]quinoxaline derivatives (3) were characterized by the spectral (1H and 13C NMR and Mass) data. The partial 1H and 13C NMR data of a representative compound 3e is presented in Fig 4 . It is evident that the methyl protons of t-butyl group appeared at 1.46 in the 1H NMR spectra whereas the C-3 proton and NH of the pyrrolo[2,3-b]quinoxaline ring appeared at 6.65 and 9.13 δ respectively. The methyl carbons of t-butyl group appeared at 31.4 ppm in the 13C NMR spectra whereas the carbon bearing all these Me groups appeared at 40.5 ppm. The 13C NMR signals at 99.4 and 150.9 ppm were due to the C-3 and one of the ring junction carbon as shown in Fig 4, respectively. The presence of NH moiety was further supported by the IR absorption near 3200 cm−1.
Table 5.
Ultrasound assisted synthesis of compound 3c-j.a
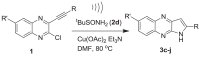 | |||
| 1; R’, R = | Time | Product (3) | %Yieldb |
| 1a; H, Ph | 1.5 | 3c | 67 |
| 1b; H, C6H4Me-p | 1.5 | 3d | 71 |
| 1c; H, t-Bu | 2 | 3e | 59 |
| 1d; H, CMe2OH | 1.5 | 3f | 63 |
| 1e; H, n-Bu | 1.5 | 3g | 73 |
| 1f; H, H | 2 | 3h | 60 |
| 1g; Me. Ph | 4 | 3i | 65 |
| 1h; Me, C6H4Me-p | 1.5 | 3j | 62 |
All the reactions were carried out by using 1 (2.5 mmol), 2d (2.8 mmol), Cu(OAc)2 (0.025 mmol) and Et3N (3.77 mmol) in DMF (3 mL) under ultrasound.
Isolated yield.
Fig. 4.

Partial representation of 1H and 13C NMR spectral data of compound 3e.
Based on results of Table 2 and previous reports [17, 30] a plausible Cu-catalytic cycle depicting the ultrasound assisted coupling-cyclization-desulfinylation of 1 with 2d is presented in Scheme 1. The catalyst Cu(OAc)2 participated in all three steps i.e. in the initial C-N coupling followed by subsequent cyclization (via formation of a second C-N bond) and then in the final N-S bond cleavage. In the first catalytic cycle the chloro group of 1 was activated due to the coordination of the proximate nitrogen of the pyrrolo[2,3-b]quinoxaline ring with the Cu(II) species under ultrasound irradiation. Subsequent ultrasound assisted displacement of the chloro group by the sulfinamide reactant 2d afforded the intermediate E-1 with the release of Cu catalyst to complete the first catalytic cycle. The next catalytic cycle was initiated with the activation of the triple bond of E-1 by coordination to the Cu(II) species to form the π-complex E-2. The increased nucleophilicity of the sulfinamide nitrogen in the form of corresponding anion generated in the presence of Et3N under ultrasound facilitated its intramolecular nucleophilic attack to the metal-coordinated triple bond in an endo-dig fashion. As a result the metal-vinyl species E-3 was generated that upon subsequent in situ protonation afforded E-4. Finally, the intermediate E-5 was generated from E-4 with the regeneration of Cu-catalyst thereby completing the second catalytic cycle. The conversion of E-5 to the product 3 involved the ultrasound assisted cleavage of N-S bond facilitated by the Cu(II) species via coordination with the oxygen atom of S=O group [31] (see Scheme S-1, Supplementary data). In order to gain further evidence on the involvement of intermediate E-5 attempt was made to detect the formation of the corresponding compound i.e. 1-(t-butylsulfinyl)-2-phenyl-1H-pyrrolo[2,3-b]quinoxaline. Indeed, this compound was detected and isolated in low yield when the reaction of entry 4 of table 4 was stopped after 45 min. The disappearance of this compound from the reaction mixture on further progress of the reaction clearly indicated intermediacy of E-5 in the current transformation. Notably, we failed to isolate the alkyne derivative corresponding to E-2 even after several attempts perhaps due to its high reactivity under the conditions employed.
Scheme 1.

The proposed Cu-catalytic cycle for the ultrasound assisted coupling-cyclization of 1 with 2d.
In view of the fact that inhibition of PDE4 may attenuate the cytokine storm in COVID-19 (through the upstream inhibition of pro-inflammatory molecules) particularly tumour necrosis factor alpha (TNF-α) [19] we examined the TNF-α inhibitory potential of all the synthesized compounds (3a-j) in vitro. All the compounds were tested initially at 10 μM concentration and the known TNF-α inhibitors rolipram [32] and thalidomide [33] was used as reference compounds in this assay (Table 6 ). While most of the compounds showed good to significant inhibition of TNF-α the compound 3c, 3d, 3i and 3j appeared to be promising (inhibition > 70%) among them. Besides, 3a, 3b, 3f and 3g were also found to be effective (inhibition > 60%). The nature and size of the substituent attached to the pyrrolo[2,3-b]quinoxaline ring at C-2 position appeared to have played a key role in the inhibition of TNF-α (Fig 5 ). The effectiveness of substituents was found to be in the order aryl moiety > long chain alkyl / hydroxyl alkyl > H / t-butyl. While the “Me” group was found be marginally better than “H” at C-5 position however activity was decreased when “H” was replaced by “MeSO2” group at N-1 position. Nevertheless, except 3e and 3h (inhibition < 60%) the IC50 values of rest of the compounds were determined (Table 6). Accordingly, the compound 3i with IC50 ~ 5.14 ± 0.34 µM (Fig. 6 ) was identified as the most potent inhibitor among them. While 3i was not better than the reference compound rolipram however it was found to be superior to another known inhibitor thalidomide (Table 6).
Table 6.
Inhibition of TNF-α by compounds 3a-j.
| Molecules | TNF-α inhibition | |
|---|---|---|
| % inhibition @ 10 μM | IC50 (μM) | |
| 3a | 66.5 | 7.12±0.48 |
| 3b | 67.9 | 7.02±0.72 |
| 3c | 76.1 | 5.47±0.69 |
| 3d | 75.6 | 5.92±0.89 |
| 3e | 56.8 | n.d. |
| 3f | 62.4 | 7.58±0.55 |
| 3g | 61.7 | 7.45±0.37 |
| 3h | 53.6 | n.d. |
| 3i | 77.3 | 5.14±0.34 |
| 3j | 75.4 | 5.65±0.29 |
| Rolipram | 97.7 | 0.91 ±0.02 |
| Thalidomide | n.d. | 198.91±6.32 |
n.d. = not determined.
Fig 5.

SAR summary of TNF-α inhibition of compound 3.
Fig. 6.

Concentration dependent inhibition of TNF-α by the compound 3i.
Next, an initial assessment about ADME (absorption, distribution, metabolism, and excretion) or pharmacokinetic properties of compound 3i along with the two other compounds 3c and 3j was carried out using Swiss ADME web-tool [34] and results are summarized in Table 7 (among the various descriptors only notable one are listed in the table). The desirable ADME was predicted for all the compounds including the high GI absorption. Indeed, a good to acceptable bioavailability (LogP > 3) (i.e. reasonable permeability through cell membranes and achieving required concentration at the site of interaction) is also predicted for these compounds. Notably, none of these compounds showed violation of Lipinski or Veber rule. Finally, to gain some initial idea about potential toxicity of the identified hit 3i the toxicity prediction of this compound was carried out using the pkCSM web-tool [35] (Table 8 ). Though the predicted AMES toxicity (mutagenicity or genetic toxicity) could be a possible concern the molecule was predicted to be safe in terms of hepatotoxicity, hERG I (human ether-a-go-go) inhibition, skin sensitisation etc. The molecule also appeared to be safe from the view point of predicted value of oral rat acute and chronic toxicity. Thus the molecule 3i may have medicinal value and deserves further evaluation towards the identification of prospective agent against COVID-19.
Table 7.
Computational ADME prediction of 3c, 3i and 3j.
| Properties | Molecules | ||
|---|---|---|---|
| (i) Physicochemical | 3c | 3i | 3j |
| Molecular Weight (g/mol) | 245.28 | 259.31 | 273.33 |
| Consensus Log Pa | 3.23 | 3.61 | 3.89 |
| Log S (ESOL)b | -4.01 (Moderately soluble) | -4.47 (Moderately soluble) | -4.58 (Moderately soluble) |
| (ii) Pharmacokinetics | |||
| GIc absorption | High | High | High |
| P-gpd substrate | yes | Yes | No |
| (iii) Druglikenss | |||
| Lipinski rule | No violation | No violation | No violation |
| Veber rule | No violation | No violation | No violation |
| Bioavailability score | 0.55 | 0.55 | 0.55 |
Log P: Lipophilicity.
Log S (ESOL): water solubility, calculated by ESOL method which is a Quantitative Structure-Property Relationship (QSPR) based model.
GI: Gastrointestinal.
P-gp: permeability glycoprotein.
Table 8.
Predicted toxicities of compound 3i.
| Property | Predicted Value | Unit |
|---|---|---|
| AMES toxicity | Yes | Categorical (Yes/No) |
| Max. tolerated dose (human) | 0.223 | Numeric (log mg/kg/day) |
| hERG I inhibitor | No | Categorical (Yes/No) |
| hERG II inhibitor | Yes | Categorical (Yes/No) |
| Oral Rat Acute Toxicity (LD50) | 2.525 | Numeric (mol/kg) |
| Oral Rat Chronic Toxicity (LOAEL) | 0.902 | Numeric (log mg/kg_bw/day) |
| Hepatotoxicity | No | Categorical (Yes/No) |
| Skin Sensitisation | No | Categorical (Yes/No) |
| T.Pyriformis toxicity | 0.314 | Numeric (log ug/L) |
| Minnow toxicity | -0.362 | Numeric (log mM) |
3. Conclusions
In conclusion, we have explored the 2-substituted pyrrolo[2,3-b]quinoxalines as potential agents against COVID-19. Several of these compounds appeared to be promising in silico when assessed for their binding affinities via docking into the N-terminal RNA-binding domain (NTD) of N-protein of SARS-CoV-2. A rapid synthesis of this class of molecules was achieved via the Cu-catalyzed coupling-cyclization-desulfinylation of 3-alkynyl-2-chloroquinoxalines with t-butyl sulfinamide as the ammonia surrogate under ultrasound irradiation. This one-pot method afforded the desired products in good yield, better than that of the previously reported method. Two N-sulfonyl analogues were also accessed via the reported method. Most of these compounds showed good to significant inhibition of TNF-α in vitro establishing a SAR (Structure Activity Relationship) within the series. In general the N-sulfonyl analogues were found to be less effective than the pyrrolo[2,3-b]quinoxalines having free NH group. One of the three best active compounds i.e. 3i was identified as a promising hit for which the desirable ADME and acceptable toxicity profile was predicted in silico. The compound 3i deserves further evaluation towards the identification of a prospective agent against COVID-19.
Credit Author Statement
Raviteja Chemboli, K. Deepti, K. R. S. Prasad and A. V. D. Nagendra Kumar were involved in the preparation, isolation, purification and characterization of all the target compounds presented in the current manuscript.
Ravikumar Kapavarapu and Alugubelli Gopi Reddy were involved in performing all the in silico studies as well as in vitro assays.
Mandava Venkata Basaveswara Rao and Manojit Pal were responsible conceptualization, coordination and overall supervision of the entire work presented in the submitted manuscript.
Declaration of Competing Interest
None.
Acknowledgement
The authors thank the management of Dr. Reddy's Institute of Life Sciences, Hyderabad, India, for continuous support and encouragement.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.molstruc.2020.129868.
Appendix. Supplementary materials
Supplementary data
Supplementary data to this article can be found online at xxxxxxxxx
References
- 1.Liu C., Zhou Q., Li Y., Garner L.V., Watkins S.P., Carter L.J., Smoot J., Gregg A.C., Daniels A.D., Jervey S., Albaiu D. Research and Development on Therapeutic Agents and Vaccines for COVID-19 and Related Human Coronavirus Diseases. ACS Cent. Sci. 2020;6:315–331. doi: 10.1021/acscentsci.0c00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Wit E., Van Doremalen N., Falzarano D., Munster V.J. SARS and MERS: recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016;14:523–534. doi: 10.1038/nrmicro.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.–R., Zhu Y., Li B., Huang C.-L., Chen H.–D., Chen J., Luo Y., Guo H., Jiang R.–D., Liu M.–Q., Chen Y., Shen X.–R., Wang X., Zheng X.–S., Zhao K., Chen Q.–J., Deng F., Liu L.–L., Yan B., Zhan F.X., Wang Y.–Y., Xiao G.–F., Shi Z.–L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.World Health Organization; 2020. Coronavirus (COVID-19)https://who.sprinklr.com/ accessed on May 7. [Google Scholar]
- 5.Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M., Shi Z., Hu Z., Zhong W., Xiao G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Savarino A., Boelaert J.R., Cassone A., Majori G., Cauda R. Effects of chloroquine on viral infections: an old drug against today's diseases? Lancet Infect. Dis. 2003;3:722–727. doi: 10.1016/S1473-3099(03)00806-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Colson P., Rolain J.–M., Raoult D. Chloroquine for the 2019 novel coronavirus SARS-CoV-2. Int. J. Antimicrob. Agents. 2020;55 doi: 10.1016/j.ijantimicag.2020.105923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cortegiani A., Ingoglia G., Ippolito M., Giarratano A., Einav S. A systematic review on the efficacy and safety of chloroquine for the treatment of COVID-19. J. Critical Care. 2020 doi: 10.1016/j.jcrc.2020.03.005. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elfiky A.A. Anti-HCV, nucleotide inhibitors, repurposing against COVID-19. Life Sci. 2020;248 doi: 10.1016/j.lfs.2020.117477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Furuta Y., Gowen B.B., Takahashi K., Shiraki K., Smee D.F., Barnard D.L. Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antivir. Res. 2013;1002:446–454. doi: 10.1016/j.antiviral.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harrison C. Coronavirus puts drug repurposing on the fast track. Nature Biotechnol. 2020;38:379–381. doi: 10.1038/d41587-020-00003-1. [DOI] [PubMed] [Google Scholar]
- 12.Kumar V., Jung Y.S., Liang P.H. Anti-SARS coronavirus agents: a patent review (2008 - present) Expert Opin. Ther. Pat. 2013;23:1337–1348. doi: 10.1517/13543776.2013.823159. [DOI] [PubMed] [Google Scholar]
- 13.Chang C.K., Jeyachandran S., Hu N.J., Liu C.L., Lin S.Y., Wang Y.S., Chang Y.–M., Hou M.H. Structure-based virtual screening and experimental validation of the discovery of inhibitors targeted towards the human coronavirus nucleocapsid protein. Molecular BioSystems. 2016;12:59–66. doi: 10.1039/c5mb00582e. [DOI] [PubMed] [Google Scholar]
- 14.S. Chen, S. Kang, 6M3M: Structural insights of SARS-CoV-2 nucleocapsid protein RNA binding domain reveal potential unique drug targeting sites. RSCB PDB; doi: 10.2210/pdb6m3m/pdb. [DOI] [PMC free article] [PubMed]
- 15.Lin S.-Y., Liu C.-L., Chang Y.-M., Zhao J., Perlman S., Hou M.-H. Structural basis for the identification of the N-terminal domain of coronavirus nucleocapsid protein as an antiviral target. J. Med. Chem. 2014;57:2247–2257. doi: 10.1021/jm500089r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shahinshavali S., Hossain K.A., Kumar A.V.D.N., Reddy A.G., Kolli D., Nakhi A., Rao M.V.B., Pal M. Ultrasound assisted synthesis of 3-alkynyl substituted 2-chloroquinoxaline derivatives: Their in silico assessment as potential ligands for N-protein of SARS-CoV-2. Tetrahedron Lett. 2020;61 doi: 10.1016/j.tetlet.2020.152336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakhi A., Rahman Md.S., Kishore R., Meda C.L.T., Deora G.S., Parsa K.V.L., Pal M. Pyrrole[2,3-b]quinoxalines as inhibitors of firefly luciferase: Their Cu-mediated synthesis and evaluation as false positives in a reporter Gene Assay. Bioorg. Med. Chem. Lett. 2012;22:6433–6441. doi: 10.1016/j.bmcl.2012.08.056. [DOI] [PubMed] [Google Scholar]
- 18.Bridgewood C., Damiani G., Sharif K., Watad A., Bragazzi N.L., Quartuccio L., Savic S., McGonagle D. Rationale for Evaluating PDE4 Inhibition for Mitigating against Severe Inflammation in COVID-19 Pneumonia and Beyond. IMAJ. 2020;22:335–339. [PubMed] [Google Scholar]
- 19.Dalamaga M., Karampela I., Mantzoros C.S. Commentary: Phosphodiesterase 4 inhibitors as potential adjunct treatment targeting the cytokine storm in COVID-19. Metab. Clin. Exp. 2020;109 doi: 10.1016/j.metabol.2020.154282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang J.-M., Chen C.-C. GEMDOCK: A generic evolutionary method for molecular docking. Proteins: Structure, Function and Bioinformatics. 2004;55:288–304. doi: 10.1002/prot.20035. http://gemdock.life.nctu.edu.tw/dock/ GEMDOCK stands for Genetic Evolutionary Method for molecular Docking, see: [DOI] [PubMed] [Google Scholar]
- 21.Dock Thor: A receptor-ligand docking program; https://www.dockthor.lncc.br/v2/
- 22.SwissDock: a web service to predict the molecular interactions that may occur between a target protein and a small molecule; http://www.swissdock.ch/docking
- 23.Iijima C., Hayashi E. Quinoxalines. XXI: Synthesis of 2-Substituted Furo [2, 3-b] quinoxaline and 2-Substituted Pyrrolo [2, 3-b] quinoxaline. Yakugaku Zasshi. 1977;97:712–718. doi: 10.1248/yakushi1947.97.7_712. [DOI] [PubMed] [Google Scholar]
- 24.Arcadi A., Cacchi S., Fabrizi G., Paris L.M. 2,3-Disubstituted pyrrolo[2,3-b]quinoxalines via aminopalladation–reductive elimination. Tetrahedron Lett. 2004;45:2431–2434. [Google Scholar]
- 25.Ames D.E., Brohi M.I. Alkynyl- and dialkynyl-quinoxalines. Synthesis of condensed quinoxalines. J. C. S. Perkin Trans. 1980;1:1384–1389. [Google Scholar]
- 26.Mason T.J. Ultrasound in synthetic organic chemistry. Chem. Soc. Rev. 1997;26:443–451. [Google Scholar]
- 27.Cella R., Stefani H.A. In: Green Techniques for Organic Synthesis and Medicinal Chemistry. Zhang W., Cue B.W., editors. John Wiley & Sons, Ltd; Chichester, UK: 2012. Ultrasonic Reactions. [DOI] [Google Scholar]
- 28.Pizzuti L., Franco M.S.F., Flores A.F.C., Quina F.H., Pereira C.M.P. Recent Advances in the Ultrasound-Assisted Synthesis of Azoles. Kidwai M., Mishra N.K., editors. Recent Advances in the Ultrasound-Assisted Synthesis of AzolesGreen Chem. - Environ. Benign Approaches. 2012 doi: 10.5772/35171. [DOI] [Google Scholar]
- 29.Puri S., Kaur B., Parmar A., Kumar H. Applications of Ultrasound in Organic Synthesis - A Green Approach. Curr. Org. Chem. 2013;17:1790–1828. [Google Scholar]
- 30.Prakash A., Dibakar M., Selvakumar K., Ruckmani K., Sivakumar M. Efficient indoles and anilines syntheses employing tert-butyl sulfinamide as ammonia surrogate. Tetrahedron Lett. 2011;52:5625–5628. [Google Scholar]
- 31.Weaver E.F., Keim W. A preliminary study of differential thermal analysis, conductance and cryoscopic behavior of dimethyl sulfoxide complexes. Proc. Indian Acad. Sci. 1960;70:123–131. For complexation of S=O group with Cu(OAc)2 see: [Google Scholar]
- 32.Prabhakar U., Lipshutz D., Bartus J.O., Slivjak M.J., Smith E.F., 3rd, Lee J.C., Esser K.M. Characterization of cAMP-dependent inhibition of LPS induced TNF a production by rolipram, a specific phosphodiesterase IV (PDE IV) inhibitor. Int. J. Immunopharmacol. 1994;16:805–816. doi: 10.1016/0192-0561(94)90054-x. [DOI] [PubMed] [Google Scholar]
- 33.Moreira A.L., Sampaio E.P., Zmuidzinas A., Frint P., Smith K.A., Kaplan G. Thalidomide exerts its inhibitory action on tumor necrosis factor alpha by enhancing mRNA degradation. J. Exp. Med. 1993;177:1675–1680. doi: 10.1084/jem.177.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Daina A., Michielin O., Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7:1–13. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pires D.E.V., Blundell T.L., Ascher D.B. pkCSM: predicting small-molecule pharmacokinetic properties using graph-based signatures. J. Med. Chem. 2015;58:4066–4072. doi: 10.1021/acs.jmedchem.5b00104. http://biosig.unimelb.edu.au/pkcsm/prediction see also. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data
Supplementary data to this article can be found online at xxxxxxxxx