


Abstract
ProThermDB is an updated version of the thermodynamic database for proteins and mutants (ProTherm), which has ∼31 500 data on protein stability, an increase of 84% from the previous version. It contains several thermodynamic parameters such as melting temperature, free energy obtained with thermal and denaturant denaturation, enthalpy change and heat capacity change along with experimental methods and conditions, sequence, structure and literature information. Besides, the current version of the database includes about 120 000 thermodynamic data obtained for different organisms and cell lines, which are determined by recent high throughput proteomics techniques using whole-cell approaches. In addition, we provided a graphical interface for visualization of mutations at sequence and structure levels. ProThermDB is cross-linked with other relevant databases, PDB, UniProt, PubMed etc. It is freely available at https://web.iitm.ac.in/bioinfo2/prothermdb/index.html without any login requirements. It is implemented in Python, HTML and JavaScript, and supports the latest versions of major browsers, such as Firefox, Chrome and Safari.
INTRODUCTION
Thermodynamic data for proteins are important for understanding the mechanism of protein folding, delineating the factors influencing the stability of proteins and mutants, development of computational tools, relating mutational effects on structure, stability, function and diseases as well as to design new stable mutants for different applications (1–5). With the advancements in high throughput biophysical techniques, the availability of thermodynamic data of proteins is increasing rapidly. The efficient compilation of these thermodynamic data with sequence and structure information could serve as a valuable resource for protein researchers.
We have developed the thermodynamic database for proteins and mutants, ProTherm, and updated continuously till 2006 (6–10). It has been effectively used to relate physicochemical properties with protein stability (11–13), development of computational tools for predicting the change in melting temperature (14–17) and free energy change upon mutation (18–28) and understanding the role of protein stability in disease causing mutations (29–31). Recently, Kulandaisamy et al. (2020) developed a thermodynamic database, MPTherm, specifically for membrane proteins (32).
In this work, we have updated and reconstructed the database, ProThermDB, which contains the experimental thermodynamic parameters for proteins and mutants along with sequence and structure information, experimental methods and conditions, literature information and visualization of proteins and mutants. We have included >14 500 new data (84% increase) and among them 2200 are published in the last three years. In addition, we have included organism-based data and cell line data in the current version of the database. Furthermore, we have provided a user-friendly web interface to search the database and download the required data. It is cross-linked with UniProt (33) and Protein Data Bank (PDB) (34) as well as to the PubMed so that users can get the source of each entry directly. Besides, we have included a separate page for individual entry, where users can get protein information, experimental conditions, literature information, and visualization of three-dimensional structure of the protein with mutation information. We have also rectified the known errors reported in the literature (35). The ProThermDB is freely available at https://web.iitm.ac.in/bioinfo2/prothermdb/index.html.
CONTENTS OF THE DATABASE
ProThermDB provides two types of thermodynamic data for wild-type and mutants proteins based on experimental methods: (i) experimental thermodynamic data obtained with purified proteins using traditional methods such as circular dichroism (CD), differential scanning calorimetry (DSC) and fluorescence spectroscopy, and (ii) data produced by mass spectrometry (MS)-based high-throughput techniques such as thermal proteome profiling (TPP) (36) and limited proteolysis (LiP) (37) using a whole-cell approach without protein purification. Further, data from high-throughput techniques are grouped together based on the source organism and cell lines.
Each entry in ProThermDB is identified with a unique accession number and it includes the following information:
Protein information: protein name, source, UniProt ID, length of the protein sequence, enzyme commission number, molecular weight, mutation based on UniProt and/or PDB, and type of mutation such as single, double and multiple.
Structural information: PDB codes for both wild-type and mutant structures (if available), number of chains, chain name, mutation details (wild-type and mutant residues along with mutant position), solvent accessibility and secondary structure of wild-type residue and 3D visualization of the mutation using JSmol interface (38). We utilized the SIFTS database for mapping residue positions between UniProt and PDB (39). Further, we observed that 95% of data have the same protein constructs used in thermodynamic experiments and reported in PDB.
Experimental conditions: temperature (T), pH, buffer name, additives, measurement, method and remarks.
Literature information: PubMed identifier, name of the author(s), journal name, year of publication, location of data and keywords.
Experimental thermodynamic data: free energy of unfolding in water (ΔGH2O), free energy obtained with thermal denaturation (ΔG), melting temperature (Tm), the slope (m) and midpoint of denaturation (Cm). In order to account the effect of mutations on protein stability, we computed the change in free energy of unfolding in water (ΔΔGH2O), change in free energy upon thermal denaturation (ΔΔG) and change in melting temperature (ΔTm). The positive and negative values of ΔΔGH2O, ΔΔG and ΔTm indicate stabilizing and destabilizing mutations, respectively.
RECENT DEVELOPMENTS
In the latest version of the database, we have developed a new user-friendly web interface with several search and display options. It contains 31 580 entries, including 84% of new data. For each entry, a separate page is added to provide the information on protein sequence and structure details, experimental conditions, thermodynamic parameters, literature information, and visualization of three-dimensional structures. We have unified protein details based on UniProt and PDB databases and corrected all the known errors (35) in 10% of the data such as (i) mutation position in a sequence based on UniProt and PDB databases, (ii) unified the units of free energy values (in kcal/mol), temperatures (°C) and other thermodynamic parameters, (iii) rectified errors in stability data, (iv) protein names, (v) UniProt ID and (vi) references. We have also provided an option for ‘data upload’ to receive the PubMed ID or DOI (Digital Object Identifier) from the users and include the data in the database. In addition, we included the thermodynamic data obtained from MS-based high-throughput techniques based on source organisms and cell lines.
In the 'statistics page’, we furnished a brief report on the contents of the ProThermDB database (https://web.iitm.ac.in/bioinfo2/prothermdb/Statistics.html) using pie charts. The figures illustrate the percentage for each type of mutation (wild-type, single, double and multiple mutations), location of single mutations with respect to solvent accessibility and secondary structure and average stability of stabilizing and destabilizing point mutations. Further, we provide a 20 × 20 amino acid substitution matrix for the frequency of single mutations, and 20 × 20 substitution matrices for average stabilizing and destabilizing values for each type of single mutation with respect to ΔΔGH2O, ΔΔG and ΔTm.
DATABASE STATISTICS
Figure 1 illustrates the overall growth of ProThermDB, and the recent version includes 84% of new data. We also showed the contents of the database based on mutation type, solvent accessibility, and secondary structure in Table 1. The latest version contains 38%, 51% and 11% of wild type, single and multiple mutations, respectively, which is similar (<4%) to the previous version (Table 1). On the other hand, ProThermDB has an increase of 70%, 101% and 69% data in wild type, single and multiple mutations, respectively. According to solvent accessibility, 43%, 29% and 28% of single mutations are located in buried, partially buried and exposed regions of protein structures, respectively (Table 1). We observed an increase of 113%, 96% and 97% on the number of data in these locations compared to the previous version. Based on secondary structure 38% of single mutations adopt 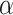 -helical conformation followed by 27% in coils and 21% in
-helical conformation followed by 27% in coils and 21% in 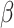 -strands (Table 1). Although the percentage of mutations is similar in previous and latest versions, the recent version has an increase of 124%, 73% and 119% of data in
-strands (Table 1). Although the percentage of mutations is similar in previous and latest versions, the recent version has an increase of 124%, 73% and 119% of data in 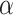 -helices, coils and
-helices, coils and 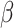 -strands, respectively.
-strands, respectively.
Figure 1.

Growth of the ProThermDB.
Table 1.
Comparison of data in ProThermDB with ProTherm 2006
| Data type | ProTherm 2006 | ProThermDB | % increase in ProThermDB |
|---|---|---|---|
| Total | 17113 | 31580 | 84.54 |
| Data type or Type of mutation | |||
| Wild-type | 7077 (41.35) | 12050 (38.16) | 70.27 |
| Single | 7969 (46.57) | 16028 (50.75) | 101.13 |
| Double | 1139 (6.66) | 2046 (6.48) | 79.63 |
| Multiple (≥3) | 928 (5.42) | 1456 (4.61) | 56.90 |
| Mutations in secondary structures a | |||
| Helix | 2470 (34.16) | 5540 (37.72) | 124.29 |
| Sheet | 1410 (19.50) | 3095 (21.07) | 119.50 |
| Turn | 1100 (15.21) | 2154 (14.67) | 95.82 |
| Coil | 2251 (31.13) | 3898 (26.54) | 73.17 |
| Mutations based on solvent accessibility a | |||
| Buried | 2969 (41.06) | 6313 (42.98) | 112.63 |
| Partially buried | 2172 (30.04) | 4254 (28.96) | 95.86 |
| Exposed | 2090 (28.90) | 4120 (28.05) | 97.13 |
Percentage of data for each category is shown in parenthesis.
aSecondary structure and solvent accessibility are computed using DSSP for proteins with known three-dimensional structures (40).
Further, the current version of ProThermDB contains 12 150 and 1 05 444 thermodynamic data from six organisms and five human cell lines, respectively. Figure 2 shows the distribution of thermodynamic data depends on source organisms and different cell lines that are derived from high throughput biophysical techniques. The Toxoplasma gondii has 5878 (49%) thermodynamic data followed by 2591 (21%) in Escherichia coli. K562 cell line of human has data for the maximum of 52% (Figure 2).
Figure 2.

Distribution of thermodynamic data obtained with whole-cell approaches based on (A) organisms and (B) cell lines.
We have compared the number and percentage of data in ProThermDB with MPTherm database based on the types of mutations and the results are presented in Supplementary Table S1. ProThermDB is dominated with single mutations followed by wild-type data whereas an opposite trend was observed in MPTherm. The percentage of double mutations is similar in both the databases. On the other hand, number of multiple mutations is less than double mutations in ProThermDB whereas MPTherm contains more number of multiple mutations than double mutations. Considering all the data together, number of data in ProThermDB is 4.4 times more than MPThermDB. Consequently, data for wild type, single, double and multiple mutations in ProThermDB are 3.2, 6.9, 5.7 and 2.1 times more than MPTherm (32).
Data retrieval from ProThermDB
The detailed information about the search and display options and an example for data retrieval from ProThermDB is illustrated in Figure 3. In this example, we build a query using a combination of multiple search options such as the protein name with ‘carbonic anhydrase 2’, secondary structure ‘helix’ (Figure 3A). Also, we selected the desired columns in the display options (Figure 3B). After submitting the query, the results are displayed in a table format (Figure 3C). We also provided an option to download the search results. In the result page, each entry accession number has a hyperlink for their respective external page, which contains the complete information (Supplementary Table S2). The structural visualization is available in the external page of each entry.
Figure 3.

An example of data retrieval from the ProThermDB database using different search and display options.
DATA AVAILABILITY
The ProThermDB database is developed using Python, HTML and JavaScript programming languages, and it supports the latest version of major browsers such as Firefox, Chrome, and Safari. The web interface is available at https://web.iitm.ac.in/bioinfo2/prothermdb/index.html. The database will be maintained and updated regularly. The updated information will be reflected on the homepage of the database. Any constructive comments and suggestions are welcome and should be sent to gromiha@iitm.ac.in or pbl.prothermdb2020@gmail.com.
Supplementary Material
ACKNOWLEDGEMENTS
We acknowledge the reviewers for their constructive comments. We thank the Indian Institute of Technology Madras and the High-Performance Computing Environment (HPCE) for computational facilities. Also, we thank all the members from Protein Bioinformatics Lab for discussions and valuable suggestions.
Contributor Information
Rahul Nikam, Department of Biotechnology, Bhupat and Jyoti Mehta School of BioSciences, Indian Institute of Technology Madras, Chennai 600 036, Tamilnadu, India.
A Kulandaisamy, Department of Biotechnology, Bhupat and Jyoti Mehta School of BioSciences, Indian Institute of Technology Madras, Chennai 600 036, Tamilnadu, India.
K Harini, Department of Biotechnology, Bhupat and Jyoti Mehta School of BioSciences, Indian Institute of Technology Madras, Chennai 600 036, Tamilnadu, India.
Divya Sharma, Department of Biotechnology, Bhupat and Jyoti Mehta School of BioSciences, Indian Institute of Technology Madras, Chennai 600 036, Tamilnadu, India.
M Michael Gromiha, Department of Biotechnology, Bhupat and Jyoti Mehta School of BioSciences, Indian Institute of Technology Madras, Chennai 600 036, Tamilnadu, India.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Indian Institute of Technology Madras.
Conflict of interest statement. None declared.
REFERENCES
- 1. Pfeil W. Protein Stability and Folding, Supplement 1: A Collection of Thermodynamic Data. 2001; NY: Springer. [Google Scholar]
- 2. Lorch M., Mason J.M., Clarke A.R., Parker M.J. Effects of core mutations on the folding of a β-sheet protein: implications for backbone organization in the I-state. Biochemistry. 1999; 38:1377–1385. [DOI] [PubMed] [Google Scholar]
- 3. Lorch M., Mason J.M., Sessions R.B., Clarke A.R. Effects of mutations on the thermodynamics of a protein folding reaction: implications for the mechanism of formation of the intermediate and transition states. Biochemistry. 2000; 39:3480–3485. [DOI] [PubMed] [Google Scholar]
- 4. Yamada Y., Banno Y., Yoshida H., Kikuchi R., Akao Y., Murate T., Nozawa Y.. Catalytic inactivation of human phospholipase D2 by a naturally occurring Gly901Asp mutation. Arch. Med. Res. 2006; 37:696–699. [DOI] [PubMed] [Google Scholar]
- 5. Singh S.M., Bandi S., Shah D.D., Armstrong G., Mallela K.M. Missense mutation Lys18Asn in dystrophin that triggers X-linked dilated cardiomyopathy decreases protein stability, increases protein unfolding, and perturbs protein structure, but does not affect protein function. PLoS One. 2014; 9:e110439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gromiha M.M., An J., Kono H., Oobatake M., Uedaira H., Sarai A.. ProTherm: thermo-dynamic database for proteins and mutants. Nucleic Acids Res. 1999; 27:286–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gromiha M.M., Uedaira H., An J., Selvaraj S., Prabakaran P., Sarai A.. ProTherm, thermodynamic database for proteins and mutants: developments in version 3.0. Nucleic Acids Res. 2002; 30:301–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sarai A., Gromiha M.M, An J., Prabakaran P., Selvaraj S., Kono H., Oobatake M., Uedaira H.. Thermodynamic databases for proteins and protein–nucleic acid interactions. Biopolymers. 2002; 61:121–126. [DOI] [PubMed] [Google Scholar]
- 9. Bava K.A., Gromiha M.M, Uedaira H., Kitajima K., Sarai A.. ProTherm, version 4.0: thermodynamic database for proteins and mutants. Nucleic Acids Res. 2004; 32:120–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kumar M.S., Bava K.A., Gromiha M.M., Prabakaran P., Kitajima K., Uedaira H., Sarai A.. ProTherm and ProNIT: thermodynamic databases for proteins and protein–nucleic acid interactions. Nucleic Acids Res. 2006; 34:D204–D206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ponnuswamy P.K., Gromiha M.M. On the conformational stability of folded proteins. J. Theor. Biol. 1994; 166:63–74. [DOI] [PubMed] [Google Scholar]
- 12. Gromiha M.M., Oobatake M., Kono H., Uedaira H., Sarai A.. Relationship between amino acid properties and protein stability: buried mutations. J. Protein Chem. 1999; 18:565–578. [DOI] [PubMed] [Google Scholar]
- 13. Shen B., Bai J., Vihinen M.. Physicochemical feature-based classification of amino acid mutations. Protein Eng. Des. Sel. 2008; 21:37–44. [DOI] [PubMed] [Google Scholar]
- 14. Saraboji K., Gromiha M.M., Ponnuswamy M.N. Average assignment method for predicting the stability of protein mutants. Biopolymers. 2006; 82:80–92. [DOI] [PubMed] [Google Scholar]
- 15. Masso M., Vaisman I.I. AUTO-MUTE 2.0: a portable framework with enhanced capabilities for predicting protein functional consequences upon mutation. Adv Bioinformatics. 2014; 2014:278385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pucci F., Bourgeas R., Rooman M.. Predicting protein thermal stability changes upon point mutations using statistical potentials: introducing HoTMuSiC. Sci. Rep. 2016; 6:23257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kulandaisamy A., Zaucha J., Frishman D., Michael Gromiha M.. MPTherm-pred: analysis and prediction of thermal stability changes upon mutations in transmembrane proteins. J. Mol. Biol. 2020; doi:10.1016/j.jmb.2020.09.005. [DOI] [PubMed] [Google Scholar]
- 18. Capriotti E., Fariselli P., Casadio R.. I-Mutant2. 0: predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005; 33:W306–W310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Parthiban V., Gromiha M.M., Schomburg D.. CUPSAT: prediction of protein stability upon point mutations. Nucleic Acids Res. 2006; 34:W239–W242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cheng J., Randall A., Baldi P.. Prediction of protein stability changes for single‐site mutations using support vector machines. Proteins. 2006; 62:1125–1132. [DOI] [PubMed] [Google Scholar]
- 21. Yin S., Ding F., Dokholyan N.V. Modeling backbone flexibility improves protein stability estimation. Structure. 2007; 15:1567–1576. [DOI] [PubMed] [Google Scholar]
- 22. Dehouck Y., Kwasigroch J.M., Gilis D., Rooman M.. PoPMuSiC 2.1: a web server for the estimation of protein stability changes upon mutation and sequence optimality. BMC Bioinformatics. 2011; 12:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pires D.E., Ascher D.B., Blundell T.L. mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics. 2014; 30:335–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Laimer J., Hofer H., Fritz M., Wegenkittl S., Lackner P.. MAESTRO-multi agent stability prediction upon point mutations. BMC Bioinformatics. 2015; 16:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Folkman L., Stantic B., Sattar A., Zhou Y.. EASE-MM: sequence-based prediction of mutation-induced stability changes with feature-based multiple models. J. Mol. Biol. 2016; 428:1394–1405. [DOI] [PubMed] [Google Scholar]
- 26. Steinbrecher T., Zhu C., Wang L., Abel R., Negron C., Pearlman D., Feyfant E., Duan J., Sherman W.. Predicting the effect of amino acid single-point mutations on protein stability—large-scale validation of MD-based relative free energy calculations. J. Mol. Biol. 2017; 429:948–963. [DOI] [PubMed] [Google Scholar]
- 27. Rodrigues C.H., Pires D.E., Ascher D.B. DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 2018; 46:W350–W355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Montanucci L., Capriotti E., Frank Y., Ben-Tal N., Fariselli P.. DDGun: an untrained method for the prediction of protein stability changes upon single and multiple point variations. BMC Bioinformatics. 2019; 20:335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Casadio R., Vassura M., Tiwari S., Fariselli P., Luigi Martelli P.. Correlating disease‐related mutations to their effect on protein stability: A large‐scale analysis of the human proteome. Hum. Mutat. 2011; 32:1161–1170. [DOI] [PubMed] [Google Scholar]
- 30. Stefl S., Nishi H., Petukh M., Panchenko A.R., Alexov E.. Molecular mechanisms of disease-causing missense mutations. J. Mol. Biol. 2013; 425:3919–3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Martelli P.L., Fariselli P., Savojardo C., Babbi G., Aggazio F., Casadio R.. Large scale analysis of protein stability in OMIM disease related human protein variants. BMC Genomics. 2016; 17:239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kulandaisamy A., Sakthivel R., Gromiha M.M. MPTherm: database for membrane protein thermodynamics for understanding folding and stability. Brief. Bioinform. 2020; bbaa064. [DOI] [PubMed] [Google Scholar]
- 33. UniProt Consortium UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019; 47:D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Burley S.K., Berman H.M., Kleywegt G.J., Markley J.L., Nakamura H., Velankar S.. Protein Data Bank (PDB): the single global macromolecular structure archive. Methods Mol. Biol. 2017; 1607:627–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang Y., Urolagin S., Niroula A., Ding X., Shen B., Vihinen M.. Pon-tstab: protein variant stability predictor. Importance of training data quality. Int. J. Mol. Sci. 2018; 19:1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Savitski M.M., Reinhard F.B., Franken H., Werner T., Savitski M.F., Eberhard D., Molina D.M., Jafari R., Dovega R.B., Klaeger S. et al.. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science. 2014; 346:1255784. [DOI] [PubMed] [Google Scholar]
- 37. Liu F., Fitzgerald M.C. Large-scale analysis of breast cancer-related conformational changes in proteins using limited proteolysis. J. Proteome Res. 2016; 15:4666–4674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hanson R.M., Prilusky J., Renjian Z., Nakane T., Sussman J.L. JSmol and the next-generation web-based representation of 3D molecular structure as applied to Proteopedia. Israel J. Chem. 2013; 53:207–216. [Google Scholar]
- 39. Dana J.M., Gutmanas A., Tyagi N., Qi G., O’Donovan C., Martin M., Velankar S.. SIFTS: updated structure integration with function, taxonomy and sequences resource allows 40-fold increase in coverage of structure-based annotations for proteins. Nucleic Acids Res. 2019; 47:D482–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kabsch W., Sander C.. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983; 22:2577–637. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The ProThermDB database is developed using Python, HTML and JavaScript programming languages, and it supports the latest version of major browsers such as Firefox, Chrome, and Safari. The web interface is available at https://web.iitm.ac.in/bioinfo2/prothermdb/index.html. The database will be maintained and updated regularly. The updated information will be reflected on the homepage of the database. Any constructive comments and suggestions are welcome and should be sent to gromiha@iitm.ac.in or pbl.prothermdb2020@gmail.com.