


Abstract
Cancer immunotherapy targeting co-inhibitory pathways by checkpoint blockade shows remarkable efficacy in a variety of cancer types. However, only a minority of patients respond to treatment due to the stochastic heterogeneity of tumor microenvironment (TME). Recent advances in single-cell RNA-seq technologies enabled comprehensive characterization of the immune system heterogeneity in tumors but posed computational challenges on integrating and utilizing the massive published datasets to inform immunotherapy. Here, we present Tumor Immune Single Cell Hub (TISCH, http://tisch.comp-genomics.org), a large-scale curated database that integrates single-cell transcriptomic profiles of nearly 2 million cells from 76 high-quality tumor datasets across 27 cancer types. All the data were uniformly processed with a standardized workflow, including quality control, batch effect removal, clustering, cell-type annotation, malignant cell classification, differential expression analysis and functional enrichment analysis. TISCH provides interactive gene expression visualization across multiple datasets at the single-cell level or cluster level, allowing systematic comparison between different cell-types, patients, tissue origins, treatment and response groups, and even different cancer-types. In summary, TISCH provides a user-friendly interface for systematically visualizing, searching and downloading gene expression atlas in the TME from multiple cancer types, enabling fast, flexible and comprehensive exploration of the TME.
INTRODUCTION
Cancer is a leading cause of death worldwide (1). In recent years, cancer immunotherapy has emerged as one of the most promising therapeutic strategies and demonstrated remarkable efficacy in tumor elimination and control (2). One major obstacle for immunotherapy is that only a small fraction of patients can benefit from the treatment due to the highly complex and heterogeneous tumor microenvironment (TME; 3). Therefore, it is vital to investigate the detailed cell-type compositions and characterize gene expression dynamics in TME, which could potentially improve the utility of cancer immunotherapy.
Single-cell RNA sequencing (scRNA-seq) has been increasingly adopted to investigate cell phenotypes, states, functions and crosstalk in the TME (4). It provides an unprecedented resolution to decipher the heterogeneous populations in TME, allowing identification of novel cell-types and discovery of unknown associations (5). For example, Zheng et al. characterized the infiltrated T cells of liver cancer using scRNA-seq and identified LAYN as a marker for expanded tumor Treg and exhausted CD8 T-cells (6). Guo et al. discovered a ‘pre-exhausted’ stage of T cells and bimodal distribution of TNFRSF9 in Tregs from non-small-cell lung cancer (NSCLC), suggesting previously unknown heterogeneity of the tumor infiltrated T-cells (7). A recent study performed on melanoma patients treated with checkpoint therapy showed that patients with high TCF7+CD8+ T cells are associated with favorable clinical outcomes after treatment (8). These studies proved that single-cell transcriptomics enabled cancer biologists and oncologists to understand the TME heterogeneity better and provided novel clinical implications. However, the rapidly accumulated tumor scRNA-seq data have also posed significant computational challenges for data integration and reuse.
There have been efforts to systematically collect and curate single-cell datasets, such as CancerSEA, scRNASeqDB, SCPortalen, PanglaoDB and JingleBells (9–13). Only CancerSEA is cancer-related, although it solely focuses on cancer cells without considering immune or stromal cells in the TME. Moreover, most of these databases contain a limited number of cells. CancerSEA (9) explores the functional heterogeneity of only 41 900 cancer cells, and SCPortalen (11) only has 67 146 cells combining human and mouse datasets. Large scale repositories, such as Single Cell Portal from the Broad Institute (14) and Single Cell Expression Atlas from European Bioinformatics Institute (EMBL-EBL; 15), provide greater numbers of datasets. Still, they are not cancer-focused and have limited and often inconsistent cell-type annotations across datasets. So far, there are still no comprehensive, intuitive, and convenient web resources with user-friendly interactive features for researchers to explore public tumor scRNA-seq datasets.
Here, we present Tumor Immune Single Cell Hub (TISCH), a comprehensive and curated web resource aiming to decipher the complex components of the TME at single-cell resolution. TISCH builds a scRNA-seq atlas of 76 high-quality tumor datasets across 27 cancer types, which were mainly collected from Gene Expression Omnibus (GEO; 16) and ArrayExpress (17). Three additional PBMC datasets from healthy donors were included to provide baseline expression levels for immune cells. The TISCH atlas comprises nearly 2 million cells, of which 378K were malignant cells, and 1566K were non-malignant cells. These datasets were uniformly processed with a standardized workflow, including quality control, batch effect removal, clustering, differential expression analysis, curated multi-level cell-type annotation, malignant cell classification and functional enrichment analysis. TISCH provides a user-friendly interface to support interactive exploration and visualization of each dataset or across multiple datasets at both single-cell and annotated cluster levels. The continued maintenance and update of TISCH promise to be of great utility to the immuno-oncology community.
MATERIALS AND METHODS
Data collection and meta information curation
We developed a text-mining-based data parsing workflow and collected tumor scRNA-seq datasets of human from GEO (16) and ArrayExpress (17). We searched the single-cell-related keywords such as ‘single cell RNA sequencing’ or ‘scRNAseq’ or ‘single cell’ or ‘single-cell’, as well as the technology-related keywords like ‘microfluidics’, ‘10X Genomics’ and ‘SMARTseq’, and the tumor-related keywords such as ‘tumor’ or ‘cancer’ or ‘carcinoma’ in the description page of GEO or ArrayExpress. Each dataset was then manually confirmed and curated. A total of 118 cancer-related scRNA-seq datasets were obtained initially and were further filtered to keep the datasets with >1000 high-quality cells. To expand the utility of TISCH, we also included the scRNA-seq datasets of mice treated with immunotherapy and three scRNA-seq datasets of human peripheral blood mononuclear cells (PBMC) from 10X Genomics. Overall, the TISCH database contains 76 high-quality tumor datasets across 27 cancer types and three PBMC datasets (Supplementary Table S1). We downloaded the expression matrix of the raw count, TPM or FPKM (if available) for each dataset. We collected sample information from databases or the original studies, such as the patient ID, tissue origin, treatment condition, response groups and the original cell-type annotation. Notably, we processed each cancer type separately if a dataset contained multiple cancer types. The source code for processing all the collected scRNA-seq datasets are deposited at the Github repository (https://github.com/DongqingSun96/TISCH/tree/master/code)
Data quality control
We applied a standardized analysis workflow based on MAESTRO v1.1.0 (18) for processing all the collected datasets, including quality control, batch effect removal, cell clustering, differential expression analysis, cell-type annotation, malignant cell classification and gene set enrichment analysis (GSEA; Figure 2). The raw count, TPM or FPKM table was used as input for the standardized workflow. The quality of cells was determined by two metrics: the number of total counts (UMI) per cell (library size) and the number of detected genes per cell. Low-quality cells were filtered out if the library size was <1000, or the number of detected genes was <500 (Supplementary Figure S1A).
Figure 2.

Overview of the TISCH workflow and features. TISCH automatically parsed and curated tumor single-cell RNA-seq datasets from GEO or Array Express databases. All datasets were then uniformly processed with a standardized workflow, including quality control, batch effect removal, cell clustering, differential expression analysis, and cell type annotation at multiple levels. Each dataset in TISCH is displayed with relevant study information, including species, treatment, the number of patients and cells, technology platform, stage and related study. In the Dataset module, TISCH provides two functions: single-dataset exploration and multiple-dataset comparison. In the Gene module, TISCH allows single gene expression visualization across multiple datasets and cell types. TISCH also supports the downloading of expression matrices, DE gene tables and meta-information for each dataset.
Batch effect evaluation and correction
To systematically evaluate the batch effects for each dataset, we employed an entropy-based metric (19,20) to quantify the mixing of the data across batches. In most datasets, samples from different patients are usually affected by batch effects. We constructed a k-NN (k = 30) graph based on the Euclidean distance between cells in the UMAP coordinates for each dataset with more than one patient. For each cell j, we computed the distribution of patients in its nearest neighbors. The measure of the mixing between patients Hj is defined as:
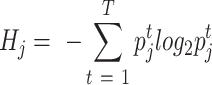 |
where  is the percentage of cells from patient t in the 30 nearest neighbors of cell j and T is the number of patients. High entropy means that the most similar cells in one cell’s neighborhood are from different patients. By contrast, low entropy means that the most similar cells are from the same patient, indicating the existence of a potential batch effect. However, it should be noticed that for the datasets, which mainly contain malignant cells, the low entropy could arise from the heterogeneity of malignant cell expression between different tumors (21). We thus separated the collected datasets into three groups. (i) For datasets mainly containing malignant cells (malignant % > 75%), there is no need to remove the batch between different patients as it reflects the difference between distinct tumors. (ii) For datasets with a median entropy lower than 0.7, we corrected the batch effect using Seurat v3.1.2 (22; Supplementary Figure S1B,C). The median entropies were shifted towards higher values after batch effect removal, indicating the potential batch effects were significantly corrected. (iii) Datasets with a median entropy higher than 0.7 were considered less affected by the batch effect (Supplementary Figure S1B). We evaluated the batch effects based on sample tissue origins for datasets without patient information or with only one patient. Only two datasets CRC_GSE120909_mouse_aPD1 and NET_GSE140312 have potential batch effects from tissue origins. The batch effects were also removed by Seurat v3.1.2, as described.
is the percentage of cells from patient t in the 30 nearest neighbors of cell j and T is the number of patients. High entropy means that the most similar cells in one cell’s neighborhood are from different patients. By contrast, low entropy means that the most similar cells are from the same patient, indicating the existence of a potential batch effect. However, it should be noticed that for the datasets, which mainly contain malignant cells, the low entropy could arise from the heterogeneity of malignant cell expression between different tumors (21). We thus separated the collected datasets into three groups. (i) For datasets mainly containing malignant cells (malignant % > 75%), there is no need to remove the batch between different patients as it reflects the difference between distinct tumors. (ii) For datasets with a median entropy lower than 0.7, we corrected the batch effect using Seurat v3.1.2 (22; Supplementary Figure S1B,C). The median entropies were shifted towards higher values after batch effect removal, indicating the potential batch effects were significantly corrected. (iii) Datasets with a median entropy higher than 0.7 were considered less affected by the batch effect (Supplementary Figure S1B). We evaluated the batch effects based on sample tissue origins for datasets without patient information or with only one patient. Only two datasets CRC_GSE120909_mouse_aPD1 and NET_GSE140312 have potential batch effects from tissue origins. The batch effects were also removed by Seurat v3.1.2, as described.
Cell clustering and differential gene analysis
For each dataset, the MAESTRO workflow identified the top 2000 variable features and employed PCA for dimension reduction, KNN, and Louvain algorithm for identifying clusters (23,24). To better capture the cellular difference and variabilities for datasets with different cell numbers, we adjusted the number of principal components and the resolution for graph-based clustering, which were both increased with the cell number (Supplementary Table S2). The uniform manifold approximation and projection (UMAP) were utilized to reduce the dimension further and visualize the clustering results (25). We applied the Wilcoxon test to identify differentially expressed (DE) genes of each cluster compared to all other cells based on the log-transformed fold change (|logFC| > = 0.25) and false discovery rate (FDR < 1e-05).
Cell-type annotation
The clusters of malignant cells were determined by combining three approaches. First, we took the cell-type annotations provided by the original studies. Second, we checked the malignant cell makers’ expression distribution from the initial research, such as epithelial markers, EMT genes, if available (26). Third, we ran InferCNV v1.2.1 (27) to predict cell malignancy based on the predicted copy number variation and separated the cells into malignant and non-malignant clusters (Supplementary Figure S1D). Among the collected datasets, 38 datasets include malignant cells, of which 10 datasets were annotated with the original cell type annotation, 25 datasets were annotated based on malignant gene signatures, and 3 datasets were annotated by inferCNV (AEL_GSE142213, ALL_GSE132509, MM_GSE141299). For the other normal clusters, we automatically annotated the cell clusters with a marker-based annotation method employed in MAESTRO using the DE genes between clusters. The marker genes of each cell type were collected from the published resources (28–30) and curated manually (Supplementary Tables S3 and S4). We calculated the average logFC of the marker genes for each cell type in each cluster and took it as a cell-type score Sc. Each cluster will be assigned a specific cell type Cj, which has the highest score among all input cell-type signatures.
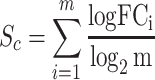 |
(1) |
 |
(2) |
Where M is the set of all collected cell types, m is the number of marker genes for a certain cell type c in M.  is the logFC of marker gene i in cell type c, which is derived based on the differential gene analysis for each cluster. We used a parameter cutoff for
is the logFC of marker gene i in cell type c, which is derived based on the differential gene analysis for each cluster. We used a parameter cutoff for 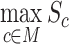 to optimize the capacity of the marker-based cell-type annotation and set the default value to 0.6 based on nine datasets with original cell-type annotation. The automatic cell-type annotation
to optimize the capacity of the marker-based cell-type annotation and set the default value to 0.6 based on nine datasets with original cell-type annotation. The automatic cell-type annotation 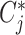 is predicted as:
is predicted as:
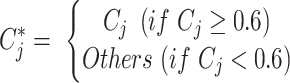 |
(3) |
We retained 18 common cell types at the major-lineage level, such as B cells (B), CD8+ T cells (CD8T), conventional CD4+ T cells (CD4Tconv; Supplementary Figure S2A and Supplementary Table S3). To gain more detailed insights into immune cell heterogeneity, we further collected and curated the sub-lineage signatures (Supplementary Table S4) and generated minor-lineage level annotation. For example, typical CD8+ T cells at the major-lineage level could be further separated into naïve CD8+ T cells (CD8Tn), central memory CD8+ T cells (CD8Tcm), effector memory CD8+ T cells (CD8Tem) and effector CD8+ T cells (CD8Teff). After automatic cell-type annotation, we performed manual corrections to all the annotated cell types by combining them with original annotation and malignant cell identification in the previous step. Based on the major-lineage level annotations and malignant cell identity, all the cells were classified into three types, malignant cells, immune cells and stromal cells, which was defined as malignancy level annotation. For each dataset, we also provided a dot plot for marker gene expression across all the cell-types to confirm the accuracy of the cell-type annotation (Supplementary Figure S2B).
Functional enrichment analysis
To characterize the functions of distinct cell-type populations, we performed gene set enrichment analysis (31,32) according to the rank of genes based on the fold-change from the differential analysis. We totally collected 16 626 gene-sets for GSEA, including 186 Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (33), 50 hallmark pathways, 10 192 Gene Ontology terms (BP: 7530; CC: 999; MF: 1663), 4872 immunologic signatures, 189 oncogenic signatures and 1137 transcriptional factor targets from the Molecular Signatures Database (MSigDB v7.1; 34). Significant up-regulated, and down-regulated pathways (FDR ≤ 0.05) in each cluster were identified and visualized to enable the functional enrichment analysis between different clusters. In addition, we also performed functional enrichment analysis of each cell-type between pre- and post-treatment, or responder and non-responder for the datasets with treatment information. Notably, we performed hierarchical clustering on the enriched GO terms due to the high similarity across ontology terms. The term with the longest paths to the root within a GO subtree will be considered as a representative term and labeled in the heatmap (35). This analysis was fulfilled by GSEA v4.0.3 for Linux, and figures were generated by the ComplexHeatmap R package v1.99.5 (36). In addition to performing GSEA at the cluster level, we also employed Single-Cell Signature Explorer (37) to calculate gene-set enrichment scores at the single-cell level. Only the visualizations on hallmark pathways are available in TISCH due to the limited computational resource.
Gene conversion
To generate the consistent gene symbol across different genome assemblies and species, we converted genes of each human and mouse dataset into GRCh38.p13 and GRCm38.p6, respectively. Besides, we converted the GRCm38 mouse homologous genes to GRCh38 human genes using ‘getLDS’ function of biomaRT package v2.42.0 (38), which enables the gene search across different species in TISCH. For those genes with one-to-many relations between species, only one homology mapping was retained randomly.
Gene visualization across cancer types and cell types
In the Gene module, we converted both raw count and FPKM to the TPM matrix to ensure the expression level is relatively comparable between different datasets. The expression level 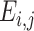 of a gene i in the cell j was quantified as
of a gene i in the cell j was quantified as 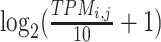 . TPM values were divided by 10 to lower the impact of varying dropout rates between genes (21,39).
. TPM values were divided by 10 to lower the impact of varying dropout rates between genes (21,39).
In addition, datasets with a large number of cells (>10 000) will usually consume high memory and take long response time to generate expression visualization figures across multiple datasets. To ensure the quick response for users when searching a gene across multiple cancer types and cell types, we applied a sub-sampling procedure for 49 datasets with >10 000 cells. For each gene, we sorted the cells according to the expression level of the gene in each cluster with >200 cells. Every ten cells were assigned into a bin and the median of the ten cells was calculated to represent the expression level of the bin. For clusters with <200 cells, all the cells were kept directly. Each point in the gene expression violin plot represents a bin, and the distribution of bins was shown between different cell-types and datasets. This method collapsed large datasets into almost one-tenth of the original ones, significantly improving the speed of read-in and generating the violin plots for gene expression visualization in the Gene module.
Web portal for the database
Based on the uniformly processed scRNA-seq datasets, we build the TISCH web portal to present the analysis results in a user-friendly way. All the processed and annotated datasets can be searched, visualized and downloaded from the web portal. The front-end display is achieved through HTML and CSS, and the back-end data are organized and queried by the MySQL database management system v8.0.20. The interaction between the front-end and back-end is enabled through JavaScript and Python. All the charts in TISCH are generated by Highcharts v8.1.2 and in-house Python and R scripts. TISCH database is deployed with the Apache2 HTTP server and is freely available at http://tisch.comp-genomics.org without any registration or login. All the functions of TISCH have been tested in Google Chrome and Apple Safari browsers.
RESULTS
Dataset summary in TISCH
The current TISCH database contains 2 045 746 cells from 79 datasets ranging 27 cancer types, with 378 392 malignant cells and 1 667 354 non-malignant cells. There are 76 tumor-related datasets in TISCH, including 17 tumor datasets with immunotherapy treatment (12 human datasets and five mouse datasets; Figure 1). Three additional PBMC datasets from healthy donors are also included to provide baseline expression levels for immune cells. On average, each dataset has 26 455 cells, with one largest dataset from NSCLC have over 200K cells (Supplementary Table S1). In total, TISCH covered 68 287 genes for human datasets and 18 789 genes for mouse datasets, with an average of 18 411 genes covered per dataset.
Figure 1.

Summary of TISCH datasets. TISCH includes 79 high-quality single-cell datasets, covering nearly 2 million cells across 27 cancer types. Datasets on human and mouse tumors are indicated as green and orange in the inner circle, respectively. Datasets with immunotherapy are labeled in red. The number of cells for each dataset is shown inside the parenthesis.
Utility of TISCH
TISCH presents all the analysis results, including clustering, differential gene identification, cell-type annotation and GSEA, in a user-friendly interface for public accessing. TISCH provides two modules for users to visualize the datasets (Figure 2). The Dataset module supports the detailed exploration of an individual dataset. In addition, it also supports multiple gene expression visualizations across multiple datasets at the single-cell level. The Gene module allows single gene visualization across multiple different scRNA-seq datasets at the cell-type level.
Single-dataset exploration
In the Dataset module, TISCH supports the advanced search for datasets of interest to explore the cell-type composition, gene expression distribution, functional status of each cell-type and comparison between different tissue origins or treatment groups. If users focus on one specific cancer type, they can click the corresponding tissue icon on the Home page to query related datasets. In the forwarding Dataset page, users can further narrow down the query results according to other criteria, such as species, treatment and included cell-types. TISCH will return the datasets satisfying the criteria with relevant study information, including the number of patients and cells, technology platform, treatment, stage and the related publication.
For each scRNA-seq dataset, the pre-analyzed results of the dataset will be shown in four different tabs, including the overview, gene, GSEA and download tabs. In the overview tab (Figure 3A), two UMAP plots with cells colored by the cell clusters and cell-type annotations will be displayed on the top. TISCH allows users to choose cell-type annotations from three levels, malignancy level, major-lineage level and minor-lineage level (Supplementary Figure S2A; see ′Materials and Methods’ section). Besides, other meta information, such as patient information, tissue origin, treatment condition and cell-type annotation from the original study can also be displayed if available. To confirm cell-type annotation accuracy, TISCH provides a dot plot to show the marker gene expression level for all the annotated cell types (Supplementary Figure S2B). Users could optionally click the right UMAP plot to visualize the expression of cell-type-specific markers for each dataset. In the middle of the overview page, TISCH displays a pie plot to show the total number of cells in each cell-type and a bar plot to show the cell-type proportion across patients (Figure 3A). The two plots can provide an intuitive overview of the cell-type and patient statistics for each dataset. On the bottom of the overview page, the top differentially expressed genes for each cluster are provided for users to discover each cell-type’s potential markers. TISCH also allows users to search for interested genes and see their relative logFC in different cell-types.
Figure 3.

Dataset module of TISCH. (A) The overview tab of the BLCA_GSE145281_aPDL1 dataset. Two UMAP plots with cells colored by cluster ID (left) and cell type (right) are displayed on the top of the tab. The pie plot and the bar plot show the cell number distribution of each cell type and the cell type proportion of each patient, respectively. The table below shows DE genes in each cluster. (B) The gene tab of the single-dataset module where expression of genes of interest can be visualized at single-cell and cell-type resolution. Two UMAP plots are to show the cell distribution of treatment response groups (left) and the expression of CXCL8 (right). The violin plot visualizes a comparison of CXCL8 expression between ‘Responder’ (orange) and ‘Non-responder’ (green) across cell types. The significance of the difference between the two groups in each cell type is evaluated through the Mann-Whitney U test and adjusted through Benjamini–Hochberg correction. ‘N.S.’ represents q (adjusted P-value) > 0.05, ‘*’ represents 0.01 < q ≤ 0.05, ‘**’ represents 0.001 < q ≤ 0.01, and ‘***’ represents q ≤ 0.001. (C) GSEA results of a single dataset. The enriched up- or down-regulated hallmark pathways in each cluster are visualized in heatmaps. (D) Multiple-dataset module, in which users can compare the gene expression across datasets at single-cell resolution. An example is presented to display the expression of LAYN and CCR8 at single-cell resolution in LIHC_GSE98638 and NSCLC_GSE99254.
In the gene tab (Figure 3B), TISCH provides a gene visualization function to search and compare multiple genes of interest simultaneously in the current dataset. UMAP plots that reflect the expression level of input genes at the single-cell resolution will be returned, enabling the exploration of the co-expression or mutually exclusive relationship between different genes. Besides, a violin plot will be displayed to show the distribution of the interested gene expression in different cell types. TISCH allows users to compare the expression of genes between different groups, such as tissue origins, treatment conditions or response groups if the meta-information is available (Figure 3B and Supplementary Figure S3D). The statistical significance between different groups was evaluated using the Mann–Whitney test for two groups or the Kruskal–Wallis test for three or more groups (Figure 3B). In addition to individual gene input, TISCH supports gene list upload so that users can explore the expression pattern of their interested gene signatures at both single-cell and cell-type level. Genes in the uploaded signature list will be collapsed by the mean or median of expression, which depends on users’ choices.
In the GSEA tab (Figure 3C), the pre-calculated GSEA results are available for users to characterize the functional differences between different cell types. We collected 16 626 gene sets from MSigDB (34), covering KEGG, hallmark, GO, immunological signatures, oncogenic signatures and transcriptional factor targets. Heatmaps will be shown to display the enriched up- or down-regulated pathways identified based on differential genes in each cluster. For the datasets with treatment information, TISCH also provides GSEA results for comparing functional pathways between different treatment conditions or treatment responses for each cell type. In addition, we integrated Single-Cell Signature Explorer (37) for computing GSEA pathway enrichment score at single-cell resolution. Users can optionally select a hallmark pathway of interest to visualize the single-cell-specific enrichment.
Besides the online search and visualization for each dataset, TISCH provides an easy way to download the data, including expression profiles, DE genes and related meta-information. The single-cell-level expression matrices are stored in compressed HDF5 format for a fast and flexible download. The top differential genes of each cluster displayed in the overview tab can also be downloaded. Moreover, TISCH provides three levels of cell-type annotations and curated meta-information at the single-cell resolution for downloading. All the figures shown on the web page can also be downloaded in high resolution. Users can utilize the downloaded data for further customized exploration.
To demonstrate an example of exploring the single-dataset module, we queried by cancer type ‘BLCA (Bladder Urothelial Carcinoma)’ and focused on the BLCA_GSE145281_aPDL1 dataset with anti-PDL1 treatment for further analysis. Studies have shown that the difference in patient's TME may lead to a distinct immunotherapeutic outcome (8,40), we thus compared the different abundance of the cell-type population between responder and non-responder groups. We observed that a higher proportion of monocytes or macrophages are present in the TME, with apparently more monocytes or macrophages in non-responders (Figure 3A, B). A previous study indicates that CXCL8, a major mediator of the inflammatory response, is highly expressed in myeloid cells than lymphoid cells, as well as in non-responders than responders (40). We confirmed this conclusion on BLCA_GSE145281_aPDL1 dataset (Figure 3B). Interestingly, a similar trend of highly expressed CXCL8 in myeloid cells of non-responders was also observed in an independent melanoma cohort SKCM_GSE120575_aPD1aCTLA4 (8; Supplementary Figure S3A–D). Hence, this single-dataset module enables quick and interactive gene expression visualization between different cell-types and treatment conditions.
Multiple-dataset comparison
In addition to single-dataset visualization, TISCH can also facilitate a comparative analysis of multiple datasets at single-cell resolution to explore the potential expression heterogeneity or homogeneity across multiple cohorts. Users can select multiple genes from multiple datasets and simultaneously compare the cell-type distribution and gene expression patterns (Figure 3D). Similar to single-dataset exploration, TISCH also allows the uploading of gene lists to visualize the averaged expression distribution of candidate gene signatures.
Here, we use an example to demonstrate the usage of the multiple-dataset module. It has been reported that LAYN and CCR8 are highly expressed in tumor-infiltrating Treg cells from colon cancer, non-small cell lung cancer and liver cancer (6,41). We observed the consistently high expression of LAYN and CCR8 in Treg cells from four independent datasets (LIHC_GSE98638, NSCLC_GSE99254, CRC_GSE108989 and CRC_GSE146771_Smartseq2; 6,7,20,28), suggesting the tumor homogeneity in terms of cell phenotype signatures (Figure 3D and Supplementary Figure S4). Besides the Treg cells, LAYN is also expressed in a subset of exhausted CD8T cells (Figure 3D and Supplementary Figure S4). As LAYN has been linked to immune suppressive function of tumor-infiltrating Treg and exhausted CD8T cells, this indicates the exhausted CD8T cells in the TME are highly heterogeneous and maybe in different exhaustion stage (6). Collectively, the comparative analysis of user-defined features across multiple datasets at single-cell resolution will provide a more detailed and comprehensive insight into the cell-type compositions and gene expression relationships in the TME.
Gene search across datasets
Although the Dataset module provides a detailed expression distribution for single or multiple datasets, it is often required to quickly locate which cell-type expresses the gene of interest across multiple tumor cohorts and different cancer types. In the Gene module, TISCH provides two ways of visualizing the gene expression from multiple cohorts (Figure 4A). The heatmap displays the input gene expression at the cell-type averaged level (Figure 4B). Simultaneously, the grid violin plot reflects the expression distribution of the input gene at single-cell or 10-cell-binned resolution (Figure 4C).
Figure 4.

Gene module of TISCH. (A) CCR8 gene searches across all cancer types and species. (B) The heatmap shows the expression of CCR8 in different cell types across all datasets with Treg cells. The color indicates the expression level of the gene. (C) The grid violin plot reflects the distribution of gene expression in different cell types across all datasets with Treg cells.
In the previous multiple-dataset module, we have already shown that CCR8 exhibits cell-type-specific expression in Treg cells from the colon, non-small cell lung and liver cancer TMEs. It is not clear whether CCR8 is expressed in other cell types or different cancer types. From the Gene module analysis, it is explicitly observed that CCR8 also shows highly specific expression in Treg cells for multiple other cancer types, such as melanoma, kidney and squamous cell carcinoma (Figure 4B). In addition, we observed a bimodal distribution of CCR8 expression in tumor-infiltrating Tregs cells from multiple cohorts, which is either due to the high drop-out rate of the scRNA-seq dataset, or caused by the heterogeneity within the Treg cells (Figure 4C). Therefore, the Gene module not only empowers the quick location of a specific gene expression pattern across different cell-types, but also helps researchers build a holistic picture of gene expression atlas among different cohorts and cancer-types.
DISCUSSION
Cancer immunotherapy has brought a paradigm shift to cancer treatment in recent years. Numerous scRNA-seq datasets have been generated to decipher the complex cell-type compositions and expression heterogeneity in the TME. However, a well-curated, uniformly processed and annotated data portal for TME scRNA-seq data reuse is still not available. In this context, we present TISCH as a comprehensive single-cell web portal for cancer biologists to investigate and visualize single-cell gene expression in the TME. TISCH shows several advantages compared to the existing single-cell tumor resources. First, TISCH is the most comprehensive TME single-cell data portal to our knowledge, including single-cell transcriptome atlas of around 2 million cells from 27 cancer types. The diverse cell types and cancer types present in TISCH enable users to systematically and holistically investigate the TME heterogeneity. Second, all the TISCH datasets were uniformly processed, annotated, and manually curated, which removes the barriers for cross-study comparisons and benefits the data-reuse. Finally, with the meta-information provided, TISCH allows comparisons between different patients, immunotherapy treatment groups and response groups, showing potential clinical indications for cancer therapy.
In summary, TISCH is a useful repository for TME single-cell transcriptomic data. It provides a user-friendly web resource for interactive gene expression visualization of cellular differences across multiple datasets at the single-cell resolution. TISCH will be a valuable resource for cancer biologists and immuno-oncologists to study gene regulation and immune signaling in the TME, identify novel drug targets and provide insights on therapy response. In the future, we will continue to pay efforts to improve TISCH. We will maintain the web resources regularly to integrate new datasets. We will also provide novel functions in TISCH, such as inferring gene–gene co-expression and cell–cell interactions based on expression correlations at the single-cell level. As the increasing numbers of public TME scRNA-seq data are available, we anticipate continued development and maintenance of the TISCH web resource will benefit the broader cancer research community.
Supplementary Material
ACKNOWLEDGEMENTS
The authors acknowledge X. Shirley Liu and Zexian Zeng from Dana Farber Cancer Institute for the helpful discussion and suggestions on the TISCH website. The authors acknowledge the authors from published studies to share their data on tumor profiling cohorts.
Contributor Information
Dongqing Sun, Shanghai Putuo District People's Hospital, School of Life Science and Technology, Tongji University, Shanghai 200060, China.
Jin Wang, Shanghai Putuo District People's Hospital, School of Life Science and Technology, Tongji University, Shanghai 200060, China.
Ya Han, Shanghai Putuo District People's Hospital, School of Life Science and Technology, Tongji University, Shanghai 200060, China.
Xin Dong, Shanghai Putuo District People's Hospital, School of Life Science and Technology, Tongji University, Shanghai 200060, China.
Jun Ge, Shanghai Putuo District People's Hospital, School of Life Science and Technology, Tongji University, Shanghai 200060, China.
Rongbin Zheng, Shanghai Putuo District People's Hospital, School of Life Science and Technology, Tongji University, Shanghai 200060, China.
Xiaoying Shi, Shanghai Putuo District People's Hospital, School of Life Science and Technology, Tongji University, Shanghai 200060, China.
Binbin Wang, Shanghai Putuo District People's Hospital, School of Life Science and Technology, Tongji University, Shanghai 200060, China.
Ziyi Li, Shanghai Putuo District People's Hospital, School of Life Science and Technology, Tongji University, Shanghai 200060, China.
Pengfei Ren, Shanghai Putuo District People's Hospital, School of Life Science and Technology, Tongji University, Shanghai 200060, China.
Liangdong Sun, Department of Thoracic Surgery, Shanghai Pulmonary Hospital, School of Medicine, Tongji University, Shanghai 200433, China.
Yilv Yan, Department of Thoracic Surgery, Shanghai Pulmonary Hospital, School of Medicine, Tongji University, Shanghai 200433, China.
Peng Zhang, Department of Thoracic Surgery, Shanghai Pulmonary Hospital, School of Medicine, Tongji University, Shanghai 200433, China.
Fan Zhang, Clinical Translational Research Center, Shanghai Pulmonary Hospital, School of Life Science, Tongji University, Shanghai 200433, China.
Taiwen Li, State Key Laboratory of Oral Diseases, National Clinical Research Center for Oral Diseases, Chinese Academy of Medical Sciences Research Unit of Oral Carcinogenesis and Management, West China Hospital of Stomatology, Sichuan University, Chengdu, Sichuan 610041, China.
Chenfei Wang, Shanghai Putuo District People's Hospital, School of Life Science and Technology, Tongji University, Shanghai 200060, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [31801059, 81972551, 81702701]. Funding for open access charge: National Natural Science Foundation of China [31801059, 81972551, 81702701].
Conflict of interest statement. None declared.
REFERENCES
- 1. Collaborators G.B.D.R.F. Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016; 388:1659–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Finn O.J. Cancer immunology. N. Engl. J. Med. 2008; 358:2704–2715. [DOI] [PubMed] [Google Scholar]
- 3. Pardoll D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer. 2012; 12:252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Giladi A., Amit I.. Single-Cell Genomics: A stepping stone for future immunology discoveries. Cell. 2018; 172:14–21. [DOI] [PubMed] [Google Scholar]
- 5. Neu K.E., Tang Q., Wilson P.C., Khan A.A.. Single-Cell Genomics: Approaches and utility in immunology. Trends Immunol. 2017; 38:140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zheng C., Zheng L., Yoo J.K., Guo H., Zhang Y., Guo X., Kang B., Hu R., Huang J.Y., Zhang Q. et al.. Landscape of infiltrating T cells in liver cancer revealed by Single-Cell sequencing. Cell. 2017; 169:1342–1356. [DOI] [PubMed] [Google Scholar]
- 7. Guo X., Zhang Y., Zheng L., Zheng C., Song J., Zhang Q., Kang B., Liu Z., Jin L., Xing R. et al.. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 2018; 24:978–985. [DOI] [PubMed] [Google Scholar]
- 8. Sade-Feldman M., Yizhak K., Bjorgaard S.L., Ray J.P., de Boer C.G., Jenkins R.W., Lieb D.J., Chen J.H., Frederick D.T., Barzily-Rokni M. et al.. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell. 2018; 175:998–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yuan H., Yan M., Zhang G., Liu W., Deng C., Liao G., Xu L., Luo T., Yan H., Long Z. et al.. CancerSEA: a cancer single-cell state atlas. Nucleic Acids Res. 2019; 47:D900–D908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cao Y., Zhu J., Jia P., Zhao Z.. scRNASeqDB: A database for RNA-Seq based gene expression profiles in human single cells. Genes (Basel). 2017; 8:368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Abugessaisa I., Noguchi S., Bottcher M., Hasegawa A., Kouno T., Kato S., Tada Y., Ura H., Abe K., Shin J.W. et al.. SCPortalen: human and mouse single-cell centric database. Nucleic Acids Res. 2018; 46:D781–D787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Franzen O., Gan L.M., Bjorkegren J.L.M.. PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database (Oxford). 2019; 2019:baz046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ner-Gaon H., Melchior A., Golan N., Ben-Haim Y., Shay T.. JingleBells: A repository of Immune-Related Single-Cell RNA-Sequencing datasets. J. Immunol. 2017; 198:3375–3379. [DOI] [PubMed] [Google Scholar]
- 14. Ding J., Adiconis X., Simmons S.K., Kowalczyk M.S., Hession C.C., Marjanovic N.D., Hughes T.K., Wadsworth M.H., Burks T., Nguyen L.T. et al.. Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nat. Biotechnol. 2020; 38:737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Papatheodorou I., Moreno P., Manning J., Fuentes A.M., George N., Fexova S., Fonseca N.A., Fullgrabe A., Green M., Huang N. et al.. Expression Atlas update: from tissues to single cells. Nucleic Acids Res. 2020; 48:D77–D83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barrett T., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Holko M. et al.. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013; 41:D991–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Athar A., Fullgrabe A., George N., Iqbal H., Huerta L., Ali A., Snow C., Fonseca N.A., Petryszak R., Papatheodorou I. et al.. ArrayExpress update - from bulk to single-cell expression data. Nucleic Acids Res. 2019; 47:D711–D715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang C., Sun D., Huang X., Wan C., Li Z., Han Y., Qin Q., Fan J., Qiu X., Xie Y. et al.. Integrative analyses of single-cell transcriptome and regulome using MAESTRO. Genome Biol. 2020; 21:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Azizi E., Carr A.J., Plitas G., Cornish A.E., Konopacki C., Prabhakaran S., Nainys J., Wu K., Kiseliovas V., Setty M. et al.. Single-Cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell. 2018; 174:1293–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang L., Li Z., Skrzypczynska K.M., Fang Q., Zhang W., O’Brien S.A., He Y., Wang L., Zhang Q., Kim A. et al.. Single-Cell analyses inform mechanisms of Myeloid-Targeted therapies in colon cancer. Cell. 2020; 181:442–459. [DOI] [PubMed] [Google Scholar]
- 21. Puram S.V., Tirosh I., Parikh A.S., Patel A.P., Yizhak K., Gillespie S., Rodman C., Luo C.L., Mroz E.A., Emerick K.S et al.. Single-Cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell. 2017; 171:1611–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Butler A., Hoffman P., Smibert P., Papalexi E., Satija R.. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018; 36:411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M. 3rd, Hao Y., Stoeckius M., Smibert P., Satija R.. Comprehensive Integration of Single-Cell Data. Cell. 2019; 177:1888–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xu C., Su Z.. Identification of cell types from single-cell transcriptomes using a novel clustering method. Bioinformatics. 2015; 31:1974–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Becht E., McInnes L., Healy J., Dutertre C.A., Kwok I.W.H., Ng L.G., Ginhoux F., Newell E.W.. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2018; 37:38–44. [DOI] [PubMed] [Google Scholar]
- 26. Lambrechts D., Wauters E., Boeckx B., Aibar S., Nittner D., Burton O., Bassez A., Decaluwe H., Pircher A., Van den Eynde K. et al.. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 2018; 24:1277–1289. [DOI] [PubMed] [Google Scholar]
- 27. Patel A.P., Tirosh I., Trombetta J.J., Shalek A.K., Gillespie S.M., Wakimoto H., Cahill D.P., Nahed B.V., Curry W.T., Martuza R.L. et al.. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014; 344:1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang L., Yu X., Zheng L., Zhang Y., Li Y., Fang Q., Gao R., Kang B., Zhang Q., Huang J.Y. et al.. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature. 2018; 564:268–272. [DOI] [PubMed] [Google Scholar]
- 29. Yost K.E., Satpathy A.T., Wells D.K., Qi Y., Wang C., Kageyama R., McNamara K.L., Granja J.M., Sarin K.Y., Brown R.A. et al.. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 2019; 25:1251–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Newman A.M., Liu C.L., Green M.R., Gentles A.J., Feng W., Xu Y., Hoang C.D., Diehn M., Alizadeh A.A.. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 2015; 12:453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. et al.. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mootha V.K., Lindgren C.M., Eriksson K.F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstrale M., Laurila E. et al.. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003; 34:267–273. [DOI] [PubMed] [Google Scholar]
- 33. Kanehisa M., Goto S.. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000; 28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liberzon A., Birger C., Thorvaldsdottir H., Ghandi M., Mesirov J.P., Tamayo P.. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015; 1:417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bennett B.D., Bushel P.R.. goSTAG: gene ontology subtrees to tag and annotate genes within a set. Source Code Biol. Med. 2017; 12:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gu Z., Eils R., Schlesner M.. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016; 32:2847–2849. [DOI] [PubMed] [Google Scholar]
- 37. Pont F., Tosolini M., Fournie J.J.. Single-Cell Signature Explorer for comprehensive visualization of single cell signatures across scRNA-seq datasets. Nucleic Acids Res. 2019; 47:e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Durinck S., Spellman P.T., Birney E., Huber W.. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009; 4:1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tirosh I., Izar B., Prakadan S.M., Wadsworth M.H. 2nd, Treacy D., Trombetta J.J., Rotem A., Rodman C., Lian C., Murphy G. et al.. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016; 352:189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yuen K.C., Liu L.F., Gupta V., Madireddi S., Keerthivasan S., Li C., Rishipathak D., Williams P., Kadel E.E. 3rd, Koeppen H. et al.. High systemic and tumor-associated IL-8 correlates with reduced clinical benefit of PD-L1 blockade. Nat. Med. 2020; 26:693–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. De Simone M., Arrigoni A., Rossetti G., Gruarin P., Ranzani V., Politano C., Bonnal R.J.P., Provasi E., Sarnicola M.L., Panzeri I. et al.. Transcriptional landscape of human tissue lymphocytes unveils uniqueness of Tumor-Infiltrating T regulatory cells. Immunity. 2016; 45:1135–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.