


Abstract
The COVID-19 outbreak has become a global emergency since December 2019. Analysis of SARS-CoV-2 sequences can uncover single nucleotide variants (SNVs) and corresponding evolution patterns. The Global Evaluation of SARS-CoV-2/hCoV-19 Sequences (GESS, https://wan-bioinfo.shinyapps.io/GESS/) is a resource to provide comprehensive analysis results based on tens of thousands of high-coverage and high-quality SARS-CoV-2 complete genomes. The database allows user to browse, search and download SNVs at any individual or multiple SARS-CoV-2 genomic positions, or within a chosen genomic region or protein, or in certain country/area of interest. GESS reveals geographical distributions of SNVs around the world and across the states of USA, while exhibiting time-dependent patterns for SNV occurrences which reflect development of SARS-CoV-2 genomes. For each month, the top 100 SNVs that were firstly identified world-widely can be retrieved. GESS also explores SNVs occurring simultaneously with specific SNVs of user's interests. Furthermore, the database can be of great help to calibrate mutation rates and identify conserved genome regions. Taken together, GESS is a powerful resource and tool to monitor SARS-CoV-2 migration and evolution according to featured genomic variations. It provides potential directive information for prevalence prediction, related public health policy making, and vaccine designs.
INTRODUCTION
As early as December 2019, several cases of human Coronavirus Disease 2019 (COVID-19) were reported in Wuhan, Hubei province in China. This is the seventh global outbreak of coronavirus utmost severe type in human and was caused by a novel coronavirus, the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) (1). Up to date of 22 July 2020, the pandemic of SARS-CoV-2 has caused over 13.3 million confirmed infection, including nearly 578 000 deaths all over the world. Since early 2020, abundant of efforts have been invested to SARS-CoV-2 related studies. Even though there were very limited SARS-CoV-2 complete genome data, many individual studies reported that the virus had gradually accumulated genomic sequence alterations, which were commonly seen in different widely spread viral strains. Particularly, single-nucleotide variants (SNVs) and other types of mutations were found in diverse countries/areas and across multiple time points (2,3) within the 29 903-nucleotide (nt) SARS-CoV-2 complete genome. The SNVs might have potential influence on the viral infectivity, or affect the biological functions, or shape protein structures, such as A23403G causing an AA change on Spike protein: D614G, and mutations on the viral Nsp6 and ORF10 (4–6). Now more than 66 000 viral genomic sequences of hCoV-19 have been submitted and shared via the online platform, the Global Initiative on Sharing Avian Influenza Data (GISAID) (https://www.gisaid.org/) as of July 2020 (7,8). The availability of most updated genome sequences brings us opportunities to discover newly emerging viral mutations that significantly contributing to SARS-CoV-2 transmission. Hence, an advanced systematic investigation of global SARS-CoV-2 mutations is needed to uncover the relationship between genomic features and geographical locations over the time of outbreak. Moreover, the study of co-occurrence correlations among the mutations will provide a better understanding of how mutations orchestrate the spreading and evolution of SARS-CoV-2 at genomic level.
So far several databases have been published with focus on mutations of SARS-CoV-2 and their relative functions, such as SARS-CoV-2 project of ViPR (9) and the GISAID (7,8). The ViPR is a virus pathogen platform which provides options of searching and downloading mutation information of SARS-CoV-2. As analytic tools for sequence alignment and visualization, it lacks enough analytical results connecting sequence mutations with countries/areas and associated timelines of occurrence, which are critical to explore the potential transmission paths of SARS-CoV-2. The GISAID also features data searching, downloading and related tools integrated with an online app Next hCoV-19 based on Nextstrain (10), which yields both the phylogenetic analysis and geographical information of the SARS-CoV-2 samples. However, as a Flu and SARS-CoV2 genomic database, the GISAID is dedicated on viral clades, which were determined based on several current genetic marker variants, instead of systematic analysis on all individual mutations. Given no clues whether new viral clades would create during most recent viral evolution, we must monitor all possible viral sequence variants over the time and pay more attentions to newly emerging mutations.
Here we present the Global Evaluation of SARS-CoV-2/hCoV-19 Sequences (GESS), a database developed based on SNVs identified among over forty-two thousand high-quality and high-coverage SARS-CoV-2 complete genome sequences as of July 22nd, 2020. There will be more and more SARS-CoV-2 sequences collected by GESS with weekly updates. We do not include phylogenetic analysis in GESS, considering that many arguments have been raised about current phylogenetic network analysis due to unbalanced numbers of virus samples collected around the world that led to biased sampling (11). Particularly very few viral genomes during the early breakout of COVID-19 in Wuhan were available which makes the analysis more challenging. Also, lack of strong evidences on homologous recombination and horizontal gene transfer brought more questions about the results of phylogenetic network at this moment (12). Instead, the purpose of GESS is to provide multiple embedded functions and mutation results for users to quickly elucidate detailed information for SNVs without re-analyzing tens of thousands of whole genome sequences. This assists researchers who have no background or capacity for sequence analysis to find SNVs of their own interest, besides those mutation hotspots which have been published and discussed. The novel functions by the GESS can give users opportunities to study SARS-CoV-2 mutations from different angles. In general, GESS features several levels of evaluations on all SNVs. First, it allows user to achieve and download the SNV information, including amino acid (AA) changes caused by the SNVs on viral genes, through different options by either searching single or multiple viral genome positions, or selecting a specific genomic region or protein, or focusing on samples collected from a certain country/area. GESS features interactive maps of geographical distributions of selected SNVs, both worldwide and in the states of USA, in addition to the time-dependent patterns of SNV occurrence ratios. Moreover, the ‘genome region search’ on specific genomic location or protein has applications for downstream study, including assisting people on vaccine designs by examining the evolutionary conservation of viral sequences. Interestingly, many SNVs were simultaneously identified on the same genomes (13–15), suggesting co-occurrence or cooperation between those viral sequence variants. Hence, GESS highlights one function, ‘concurrence search’, to identify all SNVs that show significant concurrence ratios with specified SNV chosen by user. This may help elucidate how SARS-CoV-2 utilizes the sequence mutations to orchestrate its affiliation with potential receptors, while exploring specific viral strains in fewer population or within special geographical locations. Such results are also very useful for epistasis research in SARS-CoV-2. Another GESS function of ‘SNV birth query’ is a necessity to monitor SNVs newly detected in each month, which is important to uncover their original countries or areas and help virus prevention as well as potential treatments. Combining all these functions, GESS gives users comprehensive overviews and detailed information of SARS-CoV-2 SNVs, with distinct transmission at different countries/areas and evolution patterns over time.
MATERIALS AND METHODS
Data collection
The first version of GESS database contains 42 461 high-quality and high-coverage SARS-CoV-2/hCoV-19 genome sequences downloaded from GISAID as of 22 July 2020. Total 12 252 genomic locations were detected with SNVs by sequence alignments against the reference genome, Wuhan-Hu-1 (NCBI: NC_045512.2). Wuhan-Hu-1 genome is the full-length genome sequence of the SARS‐CoV‐2 detected in China in December 2019 (16). It has been collected in the NCBI GenBank as reference. To our knowledge, over hundreds SARS-CoV-2 genetic and genomic studies used Wuhan-Hu-1 as the reference genome (17,18). Many main vaccines, such as MRNA mRNA-1273 (19), BNTX BNT162b2 (20) and Ad5-nCoV (21,22) vaccines, were designed based on Wuhan-Hu-1 sequence as well. Another hCoV-19 genome, Wuhan-WIV04 (23), was identified in China in December 2019 as well, which has been used as the reference by GISAID and other studies. These two genomes, Wuhan-WIV04 and Wuhan-Hu1, are almost exactly the same, except that Wuhan-Hu-1 has 12 more poly-As at the end of the viral genome.
All results of SARS-CoV-2 single nucleotide mutation will be updated weekly and downloaded from GESS with technical support by GISAID.
Database structure
The GESS database is built using R language with Shiny package, whose infrastructure is shown in Figure 1A. The complete genome sequences of SARS-CoV-2 were captured from the GISAID database and processed as in the paper (14). After filtering low-quality or low-coverage samples and removing white spaces within the sequences, we used a software, minimap2 (24), to pairwise align SARS-CoV-2 sequences with the reference genome Wuhan-Hu-1. Minimap2 is a fast and efficient pairwise aligner that can also handle alignment of long sequences in the fasta format. The results of pairwise alignment will not be affected by other sequences, which can avoid the interference caused by newly collected genome samples. Then a tool ANNOVAR (25) was adopted to annotate variants based on NCBI reference sequence: NC_045512.2. SNVs were identified and matched with corresponding sample information, including patients’ geographical locations and dates of sample collections, among others. In order to examine whether two SNVs arise simultaneously, a concurrence ratio, R, between pair of SNVs is calculated in the way,
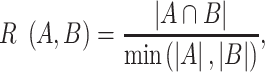 |
where A and B are any two SNVs detected from at least 0.1% of total viral genomes in the study, 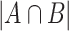 is the number of samples presenting both A and B, whereas
is the number of samples presenting both A and B, whereas 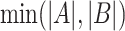 represents the minimum number of samples bearing either A or B. The larger the concurrence ratio, the more likely two SNVs coexist in the same viral genomes.
represents the minimum number of samples bearing either A or B. The larger the concurrence ratio, the more likely two SNVs coexist in the same viral genomes.
Figure 1.

Overview of GESS database. (A) The infrastructure of GESS database. The central part is individual SNV analysis and concurrence of SNVs. (B) Home page of GESS database. Besides a brief description of the website, the ‘Home’ page includes distributions of viral genome numbers around the world, or in the USA, or within each month. Several subpages contain different functions under the ‘Home’ page, in addition to the ‘Tutorial’ with the introduction to the use of GESS.
For any selected SNVs, GESS first shows their AA annotations, followed by interactive maps of SNVs’ distributions around the world and USA. The patterns of time-dependent occurrence ratios of SNVs reveal their frequencies in the populations for each month, suggesting the roles of SNVs during viral transmission and evolution. Indeed, all information involved in the result webpages can be downloaded directly from the website for further research and future publication.
RESULTS
The user interface of GESS database starts with a ‘Home’ page showing a brief summary of the number of high-quality and high-coverage samples collected from the world and the USA, separately (Figure 1B). The ‘SNV results download’ provides detailed information updated weekly for all SNVs. The profile up to 22 July 2020 can be found in Supplemental materials with this paper. GESS consists a set of ‘Search Functions’ pages, ‘Tutorial’, ‘Acknowledgement’, and information of the ‘Research Team’ (Figure 1B). Five search options are provided on separate sub-pages of GESS: SARS-CoV-2 ‘Genome position search’, ‘Genome region search’, ‘Country/Area search’, ‘Concurrence search’ and ‘SNV birth query’. The central part of all searches (Figure 1A) includes features of individual SNVs including corresponding sample information and concurrence correlations with each other. Here we present details of each search function provided by current version of GESS.
Genome position search
The page of ‘Genome position search’ contains two subset options: ‘Single position’ and ‘Multiple positions’ search. The ‘Single position’ search allows user to load only one SARS-CoV-2 genomic site at the select box. There are likely multiple different SNVs at the same genomic site. The select gear grants user to choose any SNVs of interest with corresponding mutation annotations. ‘All’ and ‘None’ buttons enables selection of all or none of SNVs at the input position for convenience. In ‘Multiple positions’ search, users can enter or select multiple genome positions in the selection box. Then the dropdown selection box shows SNVs at the input positions to let user select their interests. The relationship between selected SNVs can be determined as either ‘AND’ or ‘OR’ by ‘Relationship’ radio buttons. The ‘AND’ option reveals the viral genomes carrying all selected SNVs simultaneously, whereas the ‘OR’ option leads to samples with any one of SNVs chosen. The consequent results are exhibited in the tabs of ‘World-Wide Status’, ‘United States Status’ and ‘Time Series’ with statistics on input SNVs. The interactive maps in both ‘World-Wide Status’ and ‘United States Status’ allow user to get more detailed information on the countries/areas or the states of the USA by moving the mouse on the maps to any part of their specific interest. Additionally, a table within the ‘World-Wide Status’ tab can be downloaded showing the counts of SNVs in each country/area. By clicking specific country/area, user can get SNV distributions in different divisions or locations of selected country/area. The ‘Time Series’ tab displays bar plots of SNVs frequencies, indicating the temporal ratio of genomes bearing the mutations in each month.
Genome region search
The ‘Genome region search’ allows user to explore SNVs in a special region of the viral genome (‘Region based search’) or on a specific protein (‘Protein based search’). User has an additional option to filter rare SNVs by choosing the count cut-off in the box. SNVs within selected region or protein are presented in the dot plot, where y-axis represents numbers of samples bearing the SNVs while nts and AAs are listed on corresponding genomic positions. The plot can be downloaded by clicking the button ‘camera’. Users are encouraged to check any individual SNV by moving the mouse. The detailed information for selected SNV is presented after being automatically redirected to the results in the page of ‘Genome position search’ by clicking it. For example, on spike protein (S), A23403G shows the highest incidence on 32 915 samples, leading to an AA change on the S:D614G, which was regarded as a viral strain with high infectivity (26). This mutation was found mostly in England and USA, followed by almost all other countries/areas in the world (Figure 2A and B). The prevailing ratios of A23403G increased from 2.51% in January 2020 up to 96.52% in June and 99.23% in July 2020 (Figure 2C), indicating a dominant role of this mutation in driving the evolution and spreading of SARS-CoV-2 (13).
Figure 2.

An example to show the process of data mining via GESS. (A) World map of distribution of the mutation A23403G (S:D614G). (B) Table including event counts of A23403G in different counties/areas. (C) Temporal occurrence ratios of A23403G along the time. (D) SNVs in concurrence with A23403G (identified in at least 1,000 samples with the ratio larger than 0.9).
Country/area search
The function of ‘Country/Area search’ helps user to focus on SNVs identified in a certain country or area. The summary of SNVs in the country/area includes names of SNVs, genomic positions and annotations, numbers and percentages of viral genomes carrying the SNV in selected country/area. User can also filter or do a quick search on SNVs given interesting attributes by adding the key word in the search box. The summary table can be downloaded by user as either CSV format or Excel format. With curiosity of learning more details about any specific SNV, user may click on the SNV and then will be directed to the results on the webpage of ‘Genome position search’.
Concurrence search
Among over forty thousand SARS-CoV-2 genomes, SNVs were observed on approximately 29% of genome positions. Some SNVs are considered as random mutations, which might have no biological significance on viral infection, migration, evolution, and mortality (27), while others could be important drivers of virus genetic diversity in populations. It has already been noticed that some of SARS-CoV-2 SNVs were detected in the same patients simultaneously, suggesting potential functional interplay and cooperation among the mutations on these genomic sites. The concurrence ratio (see Methods) was calculated for each pair of SNVs that were identified in at least 0.1% of all viral genomes. The webpage of ‘Concurrence search’ allows user to choose any one of SNVs from the full list. After clicking the search button, user may browse, search, and sort all other SNVs with the information about SNV annotations, counts, and concurrence ratios. The filter options under each column can help user concentrate on SNVs of their interests. The tables containing all details about the SNV results can be downloaded from GESS. Take A23403G as an example here, we identified 13 other SNVs in at least 1000 viral genomes which had concurrence ratios larger than 0.9 with A23403G (Figure 3D). These co-occurrent SNVs with A23403G includes nonsynonymous mutations C14408T leading to an AA change on ORF1ab (Genbank, 2020) (13), G28881A, G28882A, and G28883C on ORF9 (Genbank, 2020) among others. Another interesting nonsynonymous mutation is G10097A on ORF1a/1ab, which was detected first time in March 2020 mainly from the viral genomes of England and Scotland collected in the study. These SNVs also showed a similar trend of increased incidences together with A23403G.
Figure 3.

Function of ‘SNV birth query’. (A) Word cloud of new SNVs in March 2020. (B) Table listing corresponding information for new SNVs in March 2020. (C) Word cloud of new SNVs and (D) information of new SNVs in June 2020.
SNV birth query
Similar to other viruses, SARS-CoV-2 have created genetic diversity via temporally accumulated mutations. In order to uncover the connections between featured mutations and viral migrations at a specific time point, ‘SNV birth query’ is set up for users to overview new SNVs which were discovered for the first time in each month. After the time selected, a word cloud plot is popped out with at most top 100 SNVs which had highest occurrence ratios in selected month (Figure 3A). The larger size of the SNV in the word cloud, the higher frequency of the SNV. More detailed information for all newly emerging SNVs is provided in the table on the webpage. In addition to SNVs, their counts, and percentage of samples with the SNVs in total genomes collected in the month of query, the table lists the country/area which had the most number of corresponding SNVs, and other information in the top country/area during the same time period, e.g. numbers of the genomes with the SNVs, numbers of total samples, and their ratios in percentages (Figure 3B). The table can be downloaded from the webpage in either excel or csv format. When clicking the SNV in the word cloud plot or in the table, user is automatically redirected to the page of ‘Genome position search’ with details about the SNV. For example, the mutation of C27046T had the highest incident number 469 (2.67% of all samples) that was identified for the first time in March 2020 (Figure 3A). C27046T contributing to ORF5:T175M was mainly found in Netherlands in March, where 100 out of 589 (∼16.98%) viral genomes bore this new SNV (Figure 3B). Several other European countries, e.g. England, Iceland, and Sweden, followed Netherlands in terms of numbers of genomes identified to carry C27046T, suggesting that the mutation spread quickly in March, or the mutation had already existed before March but was not discovered from our database due to limited numbers of samples or biased sampling at the early stage of COVID-19 breakout. As comparison, we checked the GESS with more recent dates. There were 569 newly emerging SNVs (Figure 3C) uncovered in June 2020, majority of which were specific to one country/area only (Figure 3D). For example, all 58 genomes carrying A2292C in June were from India, which led the AA mutation on ORF1a:Q676P or Nsp2:Q496P. Another nonsynonymous SNV, G2036T, causing alteration on ORF1ab:A591S, or ORF1a:A591S, or Nsp2:A411S, was found only in USA in June (Figure 3D).
DISCUSSION
Comparing to existing databases of SARS-CoV-2 mutations, GESS provides a more convenient and straightforward way to obtain the comprehensive overview on SARS-CoV-2 SNVs by easy operations. The summary pages of GESS contain the information about numbers of samples collected by date around the world and in the USA, with details for each month. Multiple search functions on GESS offer users a larger flexibility to browse and search SNV patterns from different aspects, while keeping the focus on SNV characteristic. Notably, one important feature of GESS is usage of correlation function on SNVs, where a parameter, concurrence ratio R, is adopted to identify SNVs cooccurred simultaneously. GESS also provides a novel function for SNV birth query to assist monitoring newly occurred SNVs each month. This may help user to better understand the migration, transmission, spread and evolution of SARS-CoV-2 via featured sequence mutations.
One of most important ways to combat viral pandemic is to develop effective vaccines. Much effort has been devoted to design vaccines against SARS-CoV2 globally. Until now, there are more than 100 planned and ongoing clinical studies of vaccines for COVID-19, several of which have entered Phase II or III clinical trial. Ideal vaccine must have strong immunogenicity with useful T lymphocyte or B lymphocyte response via their specific epitopes, and less immunotoxicity. For this purpose, several groups designed and optimized different HLA-based cytotoxic T-lymphocyte, helper T-lymphocyte and B-lymphocyte epitopes, and linked the selected epitopes together and clonal to expression vectors (28–31). In order to produce a universal antibody to neutralize different viral strains, we must also consider the conservation of targeted sequences. The database of GESS can be of great help to calibrate mutation rates for specially designed regions. For example, Helper T lymphocyte-based epitope LLLQYGSFCTQLNRA is located on the Spike protein within the genomic region: 23 816 – 23 860 (30). Using ‘Genome Region Search’, we found that the sequences within 23 816 – 23 848 were very conserved (Figure 4A), where very few mutation events occurred among over forty thousand viral genomes. However, about twenty samples bore mutations on the genomic positions 23 849, 23 854 and 23 856, suggesting possible limitations of this vaccine design due to mutations on these sites. Another example is B lymphocyte epitope located on ORF3a, QGEIKDATPSDF, within the viral genome: 25 441 – 25 476 (28), where the mutation levels seemed higher than those of previous design on Spike protein, indicating potential restrictions or reduced effectiveness of the vaccine for those mutated viruses.
Figure 4.

Examples to use GESS to calibrate sequence conservation for vaccine design. (A) Helper T lymphocyte-based epitope LLLQYGSFCTQLNRA on the genomic region: 23 816 – 23 860 (Spike). The y-axis in the dot plot represents the numbers (base-10 log scale) of viral genomes with point mutations at each position. Nucleotides and amino acids are marked, respectively, under the dot plot at corresponding genomic locations. (B) B lymphocyte epitope, QGEIKDATPSDF, within the viral genome: 25 441 – 25 476 (ORF3a). The screenshots were taken from GESS.
The example results from GESS presented in the paper was based on metadata provided by GISAID as of 22 July 2020. The subsequent updates will be performed weekly given newly submitted and collected whole genome sequencing data passing the quality control. The goal of GESS is to provide a user-friendly, state-of-art database to explore the associations and interactions between SNVs. In general, by fetching the data of SNVs and using functions embedded in GESS to analyze their significant features, users may gain new insights into the molecular drivers of SARS-CoV-2 transmission, migration, and evolution.
DATA AVAILABILITY
GESS is published webpage served by R shiny and available at address (https://wan-bioinfo.shinyapps.io/GESS/). GISAID data provided on GESS are subject to GISAID'S Terms and Conditions (https://www.gisaid.org/registration/terms-of-use/).
Supplementary Material
ACKNOWLEDGEMENTS
We are very grateful to GISAID Initiative and all its data contributors, i.e. the Authors from the Originating laboratories responsible for obtaining the specimens and the Submitting laboratories where genetic sequence data were generated and shared via the GISAID Initiative, on which this research is based. Special thanks to Tobias Gattermayer, a software engineer at GISAID, for technical support. We appreciate Dr Daniel J. Rigden from Liverpool University for suggestive discussion about GESS development.
Contributor Information
Shuyi Fang, Department of BioHealth Informatics, School of Informatics and Computing, Indiana University–Purdue University Indianapolis, Indianapolis, IN, USA.
Kailing Li, Department of BioHealth Informatics, School of Informatics and Computing, Indiana University–Purdue University Indianapolis, Indianapolis, IN, USA.
Jikui Shen, Wilmer Eye Institute, Johns Hopkins University School of Medicine, Baltimore, MD, USA.
Sheng Liu, Department of Medical and Molecular Genetics, Indiana University School of Medicine, Indianapolis, IN, USA; Collaborative Core for Cancer Bioinformatics (C3B) shared by Indiana University Simon Comprehensive Cancer Center and Purdue University Center for Cancer Research, Indianapolis, IN, USA.
Juli Liu, Department of Pediatrics, Indiana University School of Medicine, Indianapolis, IN, USA.
Lei Yang, Department of Pediatrics, Indiana University School of Medicine, Indianapolis, IN, USA.
Chang-Deng Hu, Department of Medicinal Chemistry and Molecular Pharmacology, Purdue University, West Lafayette, IN, USA; Purdue University Center for Cancer Research, Purdue University, West Lafayette, IN, USA.
Jun Wan, Department of BioHealth Informatics, School of Informatics and Computing, Indiana University–Purdue University Indianapolis, Indianapolis, IN, USA; Department of Medical and Molecular Genetics, Indiana University School of Medicine, Indianapolis, IN, USA; Collaborative Core for Cancer Bioinformatics (C3B) shared by Indiana University Simon Comprehensive Cancer Center and Purdue University Center for Cancer Research, Indianapolis, IN, USA; Center for Computational Biology and Bioinformatics, Indiana University School of Medicine, Indianapolis, IN 46202, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [P30CA082709 to J.W.]; Walther Cancer Foundation. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1. Poh C.M., Carissimo G., Wang B., Amrun S.N., Lee C.Y., Chee R.S., Fong S.W., Yeo N.K., Lee W.H., Torres-Ruesta A. et al.. Two linear epitopes on the SARS-CoV-2 spike protein that elicit neutralising antibodies in COVID-19 patients. Nat. Commun. 2020; 11:2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yang X., Dong N., Chan E.W., Chen S.. Genetic cluster analysis of SARS-CoV-2 and the identification of those responsible for the major outbreaks in various countries. Emerg. Microbes Infect. 2020; 9:1287–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hu Y., Riley L.W.. Dissemination and co-circulation of SARS-CoV2 subclades exhibiting enhanced transmission associated with increased mortality in Western Europe and the United States. 2020; medRxiv doi:15 July 2020, preprint: not peer reviewed 10.1101/2020.07.13.20152959. [DOI]
- 4. Kim S.J., Nguyen V.G., Park Y.H., Park B.K., Chung H.C.. A novel synonymous mutation of SARS-CoV-2: is this possible to affect their antigenicity and immunogenicity. Vaccines (Basel). 2020; 8:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Daniloski Z., Guo X., Sanjana N.E.. The D614G mutation in SARS-CoV-2 Spike increases transduction of multiple human cell types. 2020; bioRxiv doi:15 June 2020, preprint: not peer reviewed 10.1101/2020.06.14.151357. [DOI] [PMC free article] [PubMed]
- 6. Benvenuto D., Angeletti S., Giovanetti M., Bianchi M., Pascarella S., Cauda R., Ciccozzi M., Cassone A.. Evolutionary analysis of SARS-CoV-2: how mutation of Non-Structural Protein 6 (NSP6) could affect viral autophagy. J. Infect. 2020; 81:e24–e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Elbe S., Buckland-Merrett G.. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob Chall. 2017; 1:33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shu Y., McCauley J.. GISAID: Global initiative on sharing all influenza data - from vision to reality. Euro Surveill. 2017; 22:30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pickett B.E., Sadat E.L., Zhang Y., Noronha J.M., Squires R.B., Hunt V., Liu M., Kumar S., Zaremba S., Gu Z. et al.. ViPR: an open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012; 40:D593–D598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hadfield J., Megill C., Bell S.M., Huddleston J., Potter B., Callender C., Sagulenko P., Bedford T., Neher R.A.. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics. 2018; 34:4121–4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mavian C., Pond S.K., Marini S., Magalis B.R., Vandamme A.M., Dellicour S., Scarpino S.V., Houldcroft C., Villabona-Arenas J., Paisie T.K. et al.. Sampling bias and incorrect rooting make phylogenetic network tracing of SARS-COV-2 infections unreliable. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:12522–12523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sanchez-Pacheco S.J., Kong S., Pulido-Santacruz P., Murphy R.W., Kubatko L.. Median-joining network analysis of SARS-CoV-2 genomes is neither phylogenetic nor evolutionary. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:12518–12519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ugurel O.M., Ata O., Turgut-Balik D.. An updated analysis of variations in SARS-CoV-2 genome. Turkish journal of biology = Turk biyoloji dergisi. 2020; 44:157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu S., Shen J., Fang S., Li K., Liu J., Yang L., Hu C.D., Wan J.. Genetic spectrum and distinct evolution patterns of SARS-CoV-2. Front. Microbiol. 2020; doi:10.3389/fmicb.2020.593548. [DOI] [PMC free article] [PubMed]
- 15. Khailany R.A., Safdar M., Ozaslan M.. Genomic characterization of a novel SARS-CoV-2. Gene Rep. 2020; 19:100682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wu F., Zhao S., Yu B., Chen Y.M., Wang W., Song Z.G., Hu Y., Tao Z.W., Tian J.H., Pei Y.Y. et al.. A new coronavirus associated with human respiratory disease in China. Nature. 2020; 579:265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Forster P., Forster L., Renfrew C., Forster M.. Phylogenetic network analysis of SARS-CoV-2 genomes. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:9241–9243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tang X., Wu C., Li X., Song Y., Yao X., Wu X., Duan Y., Zhang H., Wang Y., Qian Z. et al.. On the origin and continuing evolution of SARS-CoV-2. Natl. Sci. Rev. 2020; 7:1012–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Corbett K.S., Edwards D., Leist S.R., Abiona O.M., Boyoglu-Barnum S., Gillespie R.A., Himansu S., Schäfer A., Ziwawo C.T., DiPiazza A.T. et al.. SARS-CoV-2 mRNA vaccine development enabled by prototype pathogen preparedness. Nature. 2020; doi:10.1038/s41586-020-2622-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Walsh E.E., Frenck R., Falsey A.R., Kitchin N., Absalon J., Gurtman A., Lockhart S., Neuzil K., Mulligan M.J., Bailey R. et al.. RNA-based COVID-19 vaccine BNT162b2 selected for a pivotal efficacy study. 2020; medRxiv doi:28 August 2020, preprint: not peer reviewed 10.1101/2020.08.17.20176651. [DOI] [PMC free article] [PubMed]
- 21. Wu S., Zhong G., Zhang J., Shuai L., Zhang Z., Wen Z., Wang B., Zhao Z., Song X., Chen Y. et al.. A single dose of an adenovirus-vectored vaccine provides protection against SARS-CoV-2 challenge. Nat. Commun. 2020; 11:4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhu F.C., Guan X.H., Li Y.H., Huang J.Y., Jiang T., Hou L.H., Li J.X., Yang B.F., Wang L., Wang W.J. et al.. Immunogenicity and safety of a recombinant adenovirus type-5-vectored COVID-19 vaccine in healthy adults aged 18 years or older: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. 2020; 396:479–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W., Si H.-R., Zhu Y., Li B., Huang C.-L. et al.. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020; 579:270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018; 34:3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang K., Li M., Hakonarson H.. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Korber B., Fischer W.M., Gnanakaran S., Yoon H., Theiler J., Abfalterer W., Hengartner N., Giorgi E.E., Bhattacharya T., Foley B. et al.. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. 2020; 182:812–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chisholm P.J., Busch J.W., Crowder D.W.. Effects of life history and ecology on virus evolutionary potential. Virus Res. 2019; 265:1–9. [DOI] [PubMed] [Google Scholar]
- 28. Lin L., Ting S., Yufei H., Wendong L., Yubo F., Jing Z.. Epitope-based peptide vaccines predicted against novel coronavirus disease caused by SARS-CoV-2. Virus Res. 2020; 288:198082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kar T., Narsaria U., Basak S., Deb D., Castiglione F., Mueller D.M., Srivastava A.P.. A candidate multi-epitope vaccine against SARS-CoV-2. Sci. Rep. 2020; 10:10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Samad A., Ahammad F., Nain Z., Alam R., Imon R.R., Hasan M., Rahman M.S.. Designing a multi-epitope vaccine against SARS-CoV-2: an immunoinformatics approach. J. Biomol. Struct. Dyn. 2020; doi:10.1080/07391102.2020.1792347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lizbeth R.G., Jazmín G.M., José C.B., Marlet M.A.. Immunoinformatics study to search epitopes of spike glycoprotein from SARS-CoV-2 as potential vaccine. J. Biomol. Struct. Dyn. 2020; doi:10.1080/07391102.2020.1780944. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
GESS is published webpage served by R shiny and available at address (https://wan-bioinfo.shinyapps.io/GESS/). GISAID data provided on GESS are subject to GISAID'S Terms and Conditions (https://www.gisaid.org/registration/terms-of-use/).