


Abstract
For over 10 years, ModelSEED has been a primary resource for the construction of draft genome-scale metabolic models based on annotated microbial or plant genomes. Now being released, the biochemistry database serves as the foundation of biochemical data underlying ModelSEED and KBase. The biochemistry database embodies several properties that, taken together, distinguish it from other published biochemistry resources by: (i) including compartmentalization, transport reactions, charged molecules and proton balancing on reactions; (ii) being extensible by the user community, with all data stored in GitHub; and (iii) design as a biochemical ‘Rosetta Stone’ to facilitate comparison and integration of annotations from many different tools and databases. The database was constructed by combining chemical data from many resources, applying standard transformations, identifying redundancies and computing thermodynamic properties. The ModelSEED biochemistry is continually tested using flux balance analysis to ensure the biochemical network is modeling-ready and capable of simulating diverse phenotypes. Ontologies can be designed to aid in comparing and reconciling metabolic reconstructions that differ in how they represent various metabolic pathways. ModelSEED now includes 33,978 compounds and 36,645 reactions, available as a set of extensible files on GitHub, and available to search at https://modelseed.org/biochem and KBase.
INTRODUCTION
Genome-scale metabolic reconstructions and models have become central tools for systems biology research. These models are valuable for their capacity to consolidate and represent the functional annotations of biology using the more concrete and universal language of biochemistry. By representing annotations with chemistry, we can move beyond a simple cataloguing of observed and predicted functions and begin to assemble those functions into the interconnected series of metabolic pathways that comprise the chemical foundation of any metabolic model. Models can then be applied to automatically identify any gaps that interrupt these pathways and suggest new hypothesis-driven experiments to fill these gaps (1).
Beyond the capacity of models to give structure and chemical meaning to functional annotations in biology, these models are also valuable for their predictive capacity. Today, models can be used to predict a wide range of biological phenotypes, including: (i) respiration, photosynthesis and fermentation types (2–11); (ii) feasible growth conditions and Biolog phenotype array profiles (12–15); (iii) essential genes and reactions (16–20); (iv) potential existing or engineerable by-product biosynthesis pathways (21–25); and (v) the yields and even titre available for those pathways (26–29).
Metabolic models are also now emerging as ideal tools for the integration of fluxomes, metabolomes, transcriptomes and proteomes. This capability has been applied to empower the development and parametrization of dynamic kinetic models (30–32); the reconstruction of tissue specific metabolic models (33–35); the discovery of new chemistry and pathways from metabolomes (36); and the simulation and analysis of interactions within a microbial community (37–39).
Given the rapid adoption of these models as tools in systems biology, the pace with which new models are produced has grown dramatically, particularly with the emergence of numerous automated model reconstruction pipelines (40). The diversity of resources now producing large numbers of these models has created new challenges due to a lack of standardization in models and their underlying biochemistry, assumptions and associated data. Tools like MEMOTE aid in improving standardization in metabolic models (41), but variations in how the same metabolic pathways are represented in different models remains a problem. The ability to rapidly map chemistry across different metabolic models is critical to facilitate the comparison and reconciliation of models or to permit models to interoperate within larger microbiome community models (42). It is also critical to support the integration of supplementary data for models, including thermodynamic properties (43), kinetic constants (44) and metabolomics data (45,46).
Here we present the ModelSEED biochemistry database, a transparent resource of biochemistry designed to support standardization and data integration. This database embodies several properties tailored to this objective. First, biochemistry data are unified and integrated from multiple major external sources, including KEGG (47,48), MetaCyc (49) and BiGG (50). All reactions and compounds from these sources are integrated and retained within the database to facilitate rapid automated mapping of new models to the database. Second, special attention and curation are performed to ensure that as many compounds in the database as possible have chemical structures associated with them, and compounds with identical structures are mapped together within the database. This facilitates the checking of reaction mass and charge balance, and the mapping of database metabolites to metabolomics data. Third, thermodynamic properties and pH-based molecular ion charges are computed consistently for compounds in the database, with these data being further used to compute reaction properties, including proton stoichiometry, Gibbs energy change of reaction, and predicted reversibility and directionality. These reaction and compound properties may then be mapped to models, where they can be used to evaluate thermodynamic feasibility of model output. Fourth, we applied flux balance analysis to explore how the connectivity of this new release of ModelSEED has improved in terms of activating diverse pathways and simulating biomass production in diverse media. This new release of the ModelSEED database also includes an ontology, which maps equivalent reactions from various data sources to each other. This ontology can be used to automatically convert a model to a standard biochemical representation to facilitate rapid comparison and integration. Finally, the database is encoded within GitHub, with a collection of testing scripts and a continuous integration environment, designed to facilitate the rapid extension of the ModelSEED database with community contributions, as well as providing an update and release mechanism enabling users to sync with database changes and see full details on how the database changes with each update cycle. Such extensibility is critical to keep pace with rapid discovery of new chemistry. Below we describe each of these capabilities in detail.
MATERIALS AND METHODS
Collation of biochemical data
We downloaded the molecular structures from KEGG and MetaCyc, and the compounds and reactions from >20 biochemistry databases and published metabolic models. See Supplemental Table S1 for a full list of sources.
Biochemical integration
We successively integrated the downloaded biochemistry in multiple layers (Table 1 and Figure 1), prioritizing first the KEGG and MetaCyc biochemistry as primary sources, merging metabolites if they shared the same molecular structure, then selected BioCyc databases and published models using KEGG/MetaCyc identifiers were integrated as secondary sources, wherein, the compounds were integrated with the database if the identifiers matched. The rest of the published models were treated as tertiary sources, and the compounds were integrated into the database if a synoym was matched.
Table 1.
The degree to which ModelSEED biochemistry was integrated from different data sources via different means: structure, identifier and synonym
| Sources | Integrated compounds | Integrated reactions | Unintegrated compounds | Unintegrated reactions |
|---|---|---|---|---|
| KEGG/MetaCyc | 6290 (6229) | 4889 (4777) | 24 318 (21 858) | 22 902 (19 251) |
| BioCyc (11 databases) | 6960 (6896) | 6736 (6599) | 997 | 1613 |
| Published models (34 models) | 3688 | 4332 | 1792 | 5406 |
It should be noted that the integration was done in stages (see ‘Biochemical integration’), so ‘unintegrated’ for KEGG and MetaCyc means not integrated with each other, but ‘unintegrated’ for published models means not integrated with either KEGG, MetaCyc or BioCyc. For KEGG/MetaCyc, the number in parentheses are for integration using structures only, and for BioCyc, the numbers in parentheses are for integration using BioCyc identifiers only.
Figure 1.

The growth of the ModelSEED biochemistry database. Since the release of the ModelSEED resource, along with its biochemistry, we have steadily updated the biochemistry database with the latest data in several public databases as well as integrated more published metabolic reconstructions. At the same time, we have refined our approach for integrating structural data, and so our database has grown not only in size, but also in quality: today, we have a biochemistry database of >20,000 mass-balanced reactions that can be utilized in metabolic reconstructions spanning the microbial, fungal and plant kingdoms.
At each stage, we integrated the compounds first, then integrated reactions based on whether they use the same reactants, products and stoichiometry (allowing for variations in proton stoichiometry). Crucially, this means we did not integrate reactions based on names or identifiers. For the integration of the primary sources, we used InChI and SMILES representations of the available molecular structures to match compounds from KEGG and MetaCyc. For the primary and secondary sources, if a new compound did not have an available structure, or a matching KEGG/MetaCyc identifier, we used the available synonyms to find matches if any. Finally, in order to provide external links with other databases known for use with metabolic models, we integrated the identifiers of Rhea and MetaNetX using matching KEGG and MetaCyc identifiers (Table 3).
Table 3.
A description of the biochemistry data that we have integrated from various sources
| ModelSEED | KEGG | MetaCyc | BiGG | MetaNetX | Rhea | |
|---|---|---|---|---|---|---|
| Compounds | 33 958 | 17 760 | 19 138 | 2704 | 30 858 | – |
| Structures | 28 120 | 16 198 | 18 118 | – | – | – |
| Reactions | 36 193 | 10 850 | 21 830 | 4306 | 23 758 | 8786 |
Only KEGG and MetaCyc were completely integrated so the numbers for the other databases may not reflect their published content.
Provenance
We do not exclude any compounds and reactions in the process of integrating the various biochemical sources. As such, a number of unconventional compounds and reactions, such as those that were manually created for individual published models, or incompletely curated reactions in some BioCyc databases, are included in the ModelSEED Biochemistry Database. We keep these in our database, and their external identifiers, so that any researcher can trace where they came from, for the twin purpose of transparency and reproducibility. We insist that their presence in our database does not mean that they are suitable for use in metabolic reconstructions, and as such, we direct users to use the ‘status’ field to find suitable reactions; a value of ‘OK’ indicates that the reaction is both mass-balanced and charge-balanced (see ‘Balancing of reactions in ModelSEED’).
Transport
We identify, parse and integrate all transport reactions from each of the BioCyc databases and published models. A transport reaction is identified as one where reagents are allocated to different compartments, regardless of which compartment. In order to best integrate transport reactions, and also to make them available for use with any combination of compartments in any metabolic reconstruction, we generalize the compartments as 0 or 1, thus removing their original identity. This means that, for example, a biochemical reaction catalyzed by ATP synthase can be used with a model abstracting a bacterial periplasmic membrane as well as a mitochondrial inner membrane, or a plastidial thylakoid membrane.
Protonation and conversion of ModelSEED compounds
Marvin from ChemAxon was used to protonate all molecular structures at a pH of 7 and to convert every molecular structure into InChI and SMILES format, Marvin 19.1, ChemAxon (https://www.chemaxon.com). Due to limitations in (i) the molecular structures, (ii) the InChI format and (iii) Marvin, we are unable to protonate or convert every molecular structure to InChI or SMILES format. Where possible, we defaulted to the InChI representation of the protonated structure and, failing that, the SMILES representation of the unprotonated structure.
Balancing of reactions in ModelSEED
A combination of RDKit 2020.03.1.0 (51) and OpenBabel 2.4.1 (52), two open source cheminformatics software packages, was used to derive the correct formula and charge from the molecular structures. We used two separate packages because each would fail or report errors on a small but differing set of molecular structures. We derived the formula and charge from each of the InChI and SMILES strings generated by Marvin after protonating the original structure. Using the stoichiometry and formula of each reagent and product in a reaction, we calculate how the elemental mass of a biochemical reaction is canceled out on each side of the equation. If the total number of each atomic element found in the reagents was equal to the number found in the products, we’d consider the reaction to be mass-balanced. The same process was carried out for the electronic charge of each reagent and product. We indicate the result of this balancing process in the ‘status’ field, where ‘OK’ indicates that a reaction is both mass-balanced and charge-balanced, and is appropriate for use in metabolic reconstructions.
As we use Marvin to protonate each molecular structure at a pH of 7, and thereby change the number of protons, we find that a significant number of reactions are mass-imbalanced, but only with protons. Therefore, we add, or remove, protons from the reactions in order to make sure they are completely mass and charge balanced. We only do this if we find we can balance out the reactions. We indicate for which reactions this was done by adding ‘HB’ in ‘notes’ field of the reaction. This was done for 7175 reactions.
Computation of thermodynamic properties of ModelSEED compounds and reactions
For previous releases of the ModelSEED biochemistry, the standard Gibbs energy of formation for each compound (ΔfG'o) and the standard Gibbs energy of reaction for each reaction (ΔrG'o) were estimated using a group contribution approach (53). For this new release, we recalculated these energies using eQuilibrator, a more recent approach developed by Noor et al., 0.2.5 (http://equilibrator.weizmann.ac.il) (54). All energies were calculated at pH 7.0, ionic strength of 0.25 M and temperature of 298.15 K. Data from these two methods were integrated in a complementary manner, giving precedence to the results from eQuilibrator.
The set of complete structures in the ModelSEED biochemistry from which energy of formation was computed using the group contribution approach, as integrated from KEGG and MetaCyc, only partially overlaps with the set of complete structures in MetaNetX, from which eQuilibrator computed pKa values. There was a total of 23 989 unique InChI structures in ModelSEED, and 465 752 unique InChI structures in MetaNetX, but only 19 479 of these structures were shared between the two databases. In addition, neither the group contribution method nor eQuilibrator was able to return an estimate for (ΔfG'o) for every structure, and as such, there were only 19 621 ModelSEED compounds with an estimate for (ΔfG'o) from the group contribution method and 17 510 ModelSEED compounds with an estimate for (ΔfG'o) from eQuilibrator. For each compound, we use the value from eQuilibrator in our database where possible, but we retain the values computed by the group contribution approach in the repository.
A reaction was considered to be ‘complete’ when every reactant had a defined structure and for which (ΔfG'o) was available via either the group contribution method or eQuilibrator. There were 18 930 ModelSEED reactions defined as complete by the group contribution method, and 15 574 ModelSEED reactions defined as complete by eQuilibrator, with 14 677 reactions shared between the two. For each of these reactions, we used the value of (ΔrG'o) computed by eQuilibrator, except when estimated error returned by eQuilibrator exceeds an arbitrary value of 100 kilocal.mole−1 (2459 reactions). For those, the value computed by the group contribution method was used. When computing ΔrG'o, we always used either eQuilibrator or group contribution values exclusively, and never mixed and matched ΔrG'o values from these two methods to compute a single ΔrG'o because they differ in their reference states, and thus would cause significant error. For each of the reactions with an estimated ΔrG'o, we applied a heuristic to estimate the thermodynamic reversibility of the reaction based on a set of rules developed in earlier work (55).
Undetermined compounds
Many compounds in the database, whether they were assigned a structure or not, were considered to be undetermined for a number of reasons. The compound may be ‘lumped’ if their structure was partially or wholly unknown and the ‘lumped’ structure was represented by an ‘R’ group. Where we could, we included the reactions containing such compounds while making sure that the ‘R’ groups balanced and represented the same substructure. The compound may have been generic or abstract, in that they were representative of a class of compounds; so, we include these compounds by generating hierarchical links between them and their structurally-specific representatives. Compounds that were partially or wholly determined can be found by searching for the ‘R’ group in the ‘Formula’ column on our website.
GitHub policies for community contributions
GitHub offers a valuable venue and toolkit to support community curation, and it has been applied to this purpose for the codevelopment of computer code by a vast user community. This community has demonstrated how a large group of individuals can work together on a single project and be effective. The tools that GitHub offers its developer-users, as well as the policies and practices it encourages, are critical components that make large-scale cooperative projects possible. With this release of the ModelSEED biochemistry database, we anticipate the same principles and methods apply and will ultimately support large-scale community-curation of biochemistry data in the ModelSEED. To accomplish this goal, we have adopted many of the same practices used by developers.
Use of branches
The ModelSEED repository includes a dev and master branch in GitHub. All releases will be deployed to the master branch, which will be tagged with release identifiers when the release is complete. All active new curation work will take place in the dev branch, where all external contributors are encouraged to submit their pull requests.
External user contributions
The ModelSEED database, despite years of development, is still far from being perfect or complete. We welcome contributions from external users, including: (i) new proposed compounds, reactions and pathways; (ii) curations to existing data including correcting reaction stoichiometry, aliases or molecular structures; and/or (iii) integrating new tools to support database maintenance, quality control and analysis. Users can propose changes by creating their own fork of the ModelSEED GitHub repository, implementing their changes within this fork, running ModelSEED test scripts to ensure that the proposed changes meet data quality and minimal information standards and submitting changes to the ModelSEED team for review by issuing a pull request in GitHub against the dev branch. Once the team has ensured that proposed changes meet all standards, pull requests can be merged. The pull request mechanism on GitHub includes a built-in discussion forum to permit interactive discussion of proposed changes. We utilize Travis CI (56) along with scripts for testing data immediately, and reporting whether or not data in the pull request is valid.
Release procedure
On a quarterly basis, we will release a new version of the ModelSEED database via GitHub. Releases will always be deployed from the master branch in GitHub, and each release will be tagged with a version in GitHub. Additionally, on release, updated data will be deployed into the ModelSEED modeling environment as well as the chemistry database in the U.S. Department of Energy (DOE) Systems Biology Knowledgebase, KBase (57).
RESULTS
Growth in the compounds and reactions included in the ModelSEED database
Development of the ModelSEED database began 10 years ago with the release of the first ModelSEED resource for microbial metabolic model reconstruction (58). The database was expanded in 2014 by integrating additional sources of plant biochemistry with the release of the PlantSEED (59). Here, for the first time, we are releasing the ModelSEED biochemistry database as a stand-alone resource. This new release includes expansions of the ModelSEED, updating data from our source databases, and adding additional sources. As expected, the ModelSEED database has expanded over time from 13 257 reactions in 2010 to 36 193 reactions today (Table 2).
Table 2.
The statistics of the ModelSEED biochemistry database over time (Figure 1)
| 2010 (ModelSEED) | 2014 (PlantSEED) | Current 2020 | |
|---|---|---|---|
| Compounds | 16 275 | 27 694 | 33 992 |
| Compounds with structures | 13 821 (85%) | 19 605 (71%) | 28 120 (83%) |
| Compounds with generic groups | 1261 (8%) | 1402 (5%) | 4416 (13%) |
| Reactions | 13 257 | 27 558 | 36 193 |
| Complete* reactions | 11 338 (86%) | 7898 (29%) | 27 991 (77%) |
| Balanced reactions | 10 263 (77%) | 17 264 (63%) | 25 457 (70%) |
| Reactions with generic groups | 1988 (15%) | 2939 (11%) | 9772 (27%) |
*A complete reaction is one where every reactant has a fully defined metabolic structure in our database. Balanced reactions were calculated to be mass- and charge-balanced, see ‘Balancing of reactions in ModelSEED’.
The updated ModelSEED database now contains reactions from KEGG, MetaCyc, BiGG, MetaNetX and Rhea, but it is important to note that only the KEGG and MetaCyc databases have been integrated in their entirety (Table 3).
Molecular structure is very important in the ModelSEED because we use structure as our primary tool to map together identical compounds from our source databases and because we apply structures with thermodynamic property estimation tools to predict Gibbs free energy change for compounds and reactions. Currently, 84% of compounds in the ModelSEED have specified structure, which translates to 81% of reactions defined as complete, meaning the structure is defined for every reactant involved in the reaction (Table 2).
One significant area of improvement for this new release of the ModelSEED was the identification and correction of redundant copies of various compounds and reactions that were previously added to the database in error due to a failure to match identical compounds. We previously failed to match identical compounds based on three problems: (i) no associated molecular structure, making it impossible to automatically match these compounds to other compounds in our database based on structure; (ii) errors or inconsistencies in compound structures that prevented a match from being made; or (iii) missing stereochemistry information in compound structures. We identified and corrected some of these issues in this latest release by reviewing and correcting many problems. For example, we reviewed 47 cases where sets of two or more compounds in our database appeared to have completely identical structures, involving 3320 reactions. Ultimately 32 compounds were consolidated, which led to the correction of 1301 reactions in our database that involved one or more of these compounds as reactants. This subsequently led to the consolidation of 7577 reactions. We were also able to identify and correct previously automated consolidations of compounds and reactions that turned out to be erroneous. These cases primarily consisted of stereochemically generic compounds being consolidated with stereochemically specific versions. In the majority of cases, we only had to ensure the correct structure was used. We corrected the remainder of cases by disambiguating 39 compounds, leading to the correction of 20 reactions. We note that this is an ongoing curation effort and our data is by no means completely ‘fixed’ or perfect but a work in progress. This is why we made the decision to officially distribute the ModelSEED database via GitHub (see a detailed discussion later). GitHub enables users to easily clone our database in a form that can also be easily updated as the database improves overtime (as well as showing users detailed information about every aspect of the data that have changed).
Thermodynamics
For this release of the ModelSEED database, eQuilibrator (54) was applied to update 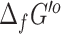 for the compounds in the database.
for the compounds in the database. 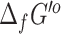 could be computed by eQuilibrator for 17 510 (73%) of the compounds with assigned structures in the database. The remaining 10 885 compounds had structures representing abstract molecules, macromolecules, or in some cases compounds containing functional groups with no associated energy contribution in eQuilibrator. About 9884 (56%) of the successful
could be computed by eQuilibrator for 17 510 (73%) of the compounds with assigned structures in the database. The remaining 10 885 compounds had structures representing abstract molecules, macromolecules, or in some cases compounds containing functional groups with no associated energy contribution in eQuilibrator. About 9884 (56%) of the successful 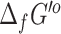 predictions from eQuilibrator had very low uncertainty (<5%), but 6930 (40%) had a high uncertainty of over 100%. Given the large disparity in uncertainty values between eQuilibrator and our previously applied group contribution method (53) for computing
predictions from eQuilibrator had very low uncertainty (<5%), but 6930 (40%) had a high uncertainty of over 100%. Given the large disparity in uncertainty values between eQuilibrator and our previously applied group contribution method (53) for computing 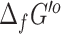 , we retain the
, we retain the 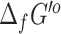 values from both methods in our repository, with the source and uncertainty of each value labeled.
values from both methods in our repository, with the source and uncertainty of each value labeled.
We similarly applied eQuilibrator to predict new 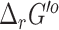 for the reactions in the ModelSEED database, where we observed similar results with 13 042 (84%) reactions having an uncertainty below 5% and 2459 (16%) reactions having an uncertainty over 100%. As with the
for the reactions in the ModelSEED database, where we observed similar results with 13 042 (84%) reactions having an uncertainty below 5% and 2459 (16%) reactions having an uncertainty over 100%. As with the 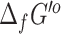 predictions, we retained both our original
predictions, we retained both our original 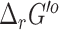 and the eQuilibrator
and the eQuilibrator 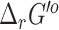 values in our repository, with the source and uncertainty of each value labeled. Unlike with the
values in our repository, with the source and uncertainty of each value labeled. Unlike with the 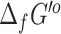 values, it may be possible to mix and match
values, it may be possible to mix and match 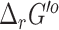 values from these competing methods. The results from our recomputation of
values from these competing methods. The results from our recomputation of 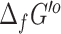 and
and 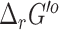 using eQuilibrator and our original group contribution method are highlighted in Table 4.
using eQuilibrator and our original group contribution method are highlighted in Table 4.
Table 4.
Integration of thermodynamics data from the Group Contribution approach (GC) and eQuilibrator (eQ)
| Compounds | Reactions | |
|---|---|---|
| All | 33 958 | 36 193 |
| Structures (GC) | 28 087 (100%) | 36 193 (100%) |
| Complete (GC) | – | 18 930 (52%) |
| Accepted (GC) | 9208 (33%) | 6141 (17%) |
| Structures (eQ) | 17 510 (62%) | 36 193 (100%) |
| Complete (eQ) | – | 17 440 (48%) |
| Accepted (eQ) | 10 580 (37%) | 13 298 (37%) |
| Final | 19 788 (70%) | 19 439 (54%) |
The two approaches can only be applied to compounds for which structures are available. The number of reactions in rows denoted ‘Structures’ includes any reactions for which a reagent has associated thermodynamics data, and the number of reactions in rows denoted ‘Complete’ includes any reactions for which all reagents have associated thermodynamics data. The ‘Final’ number of reactions includes the total number of reactions for which the thermodynamics data were accepted for GC approach and eQ. “Accepted" means the data were utilized if it passed several basic tests, but precedent was placed on data from eQuilibrator (see the main text).
When comparing the newly calculated 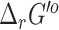 computed by eQuilibrator to that computed using the group contribution method, we find that most (65%) of the reactions had a difference of <5 kcal/mol (Figure 2). As the difference increases, the number of reactions dropped significantly, only 9% of the reactions had a difference above 15 kcal/mol. The uncertainty of the
computed by eQuilibrator to that computed using the group contribution method, we find that most (65%) of the reactions had a difference of <5 kcal/mol (Figure 2). As the difference increases, the number of reactions dropped significantly, only 9% of the reactions had a difference above 15 kcal/mol. The uncertainty of the 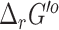 computed by eQuilibrator for reactions with a difference >15 kcal/mol was also high, at least 80% of these reactions had >100% uncertainty, which implies a low confidence in the eQuilibrator energies for these reactions with high deviations in their
computed by eQuilibrator for reactions with a difference >15 kcal/mol was also high, at least 80% of these reactions had >100% uncertainty, which implies a low confidence in the eQuilibrator energies for these reactions with high deviations in their 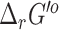 values. When directly comparing the uncertainty between the two approaches, 43% of the compared reactions exhibited >100% uncertainty for the group contribution method and only 10% of the compared reactions exhibited >100% uncertainty for eQuilibrator (Figure 2). For the purpose of this analysis, we only compared nontransport and mass-balanced reactions
values. When directly comparing the uncertainty between the two approaches, 43% of the compared reactions exhibited >100% uncertainty for the group contribution method and only 10% of the compared reactions exhibited >100% uncertainty for eQuilibrator (Figure 2). For the purpose of this analysis, we only compared nontransport and mass-balanced reactions
Figure 2.

Thermodynamics in ModelSEED and eQuilibrator. We integrated the results calculated by eQuilibrator, a more recently developed approach, to estimate the Gibbs energy of formation for >17K compounds and 17K reactions in the database. In comparing the results for reactions to those of the group contribution approach used in this and prior releases of ModelSEED, we find that there is very little change. In a very small percentage of cases, the error reported for values computed by eQuilibrator is much higher than that reported for the group contribution approach (inset). The integration of data computed by eQuilibrator led to the adjustment of thermodynamic reversibility for roughly ∼5% of the reactions in our database (see ‘Thermodynamics’ section).
We apply these newly calculated reaction energies, using the eQuilibrator values when the uncertainty was low and using the group contribution method values otherwise, to determine the reversibility of all reactions in the ModelSEED (see ‘Materials and Methods’ section). Based on this analysis, we find that we were able to determine the reversibility of 3894 reactions that had not been determined before. For the reactions for which reversibility had been determined before, we compared the reversibility if we were to use the group contribution method alone versus if we were to apply eQuilibrator values (Figure 2). We find that 85% of the compared reactions exhibited no change. Having adopted the approach integrating eQuilibrator values, we double-checked the subset of reactions that we use in our automated metabolic reconstructions in ModelSEED and in KBase, to ensure that they did not disrupt the behavior of the drafted reconstructions, and we integrated the updated reversibility constraints into the gap-filling reactions used by the reconstruction process.
Improvements to database connectivity
One key purpose driving the development of the ModelSEED biochemistry database was to serve as the underlying chemistry source for the reconstruction of metabolic models in the ModelSEED (58), PlantSEED (58,60) and KBase (57). As such, we needed to evaluate how our overall database performs in flux balance analysis. Metabolic modeling and flux balance analysis both place very specific demands on a biochemistry database. First and foremost, FBA was designed to be applied to reaction networks comprised of mass- and charge-balanced reactions, so imbalanced reactions should be filtered out of any network that is to be leveraged for FBA. This does not mean that imbalanced reactions are excluded from the ModelSEED database, as it was also important to have as complete a database as possible to support annotation comparison, which is discussed later. However, all imbalanced reactions were identified and filtered from the database prior to the use of data in any FBA-based approach. Currently, of the 36K reactions in the ModelSEED database, 25 457 (70%) are charge and mass balanced, 2424 (7%) are mass balanced but not charge balanced and 8338 (23%) are not mass balanced. This represents a significant improvement over previous releases which included only 22 421 mass balanced reactions.
Secondly, FBA also depends on having constraints on the reversibility of reactions to ensure that reactions have the flexibility needed to replicate true biological behavior without having too much flexibility leading to thermodynamically unrealistic flux profiles. For this, the improved 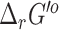 estimates in this release of the ModelSEED lead to an adjustment of the reversibility rules associated with 2783 reactions as described in the previous section. These changes impact how every reaction in the ModelSEED network is connected to every other reaction and the number of functional reactions when flux balance analysis is applied. When constructing an FBA model of the ModelSEED biochemistry, we included all balanced single-compartment reactions as intracellular, while transport reactions were used to define the metabolites that are allowed to move from the extracellular space into the intracellular compartment. The SEED bacterial biomass reaction was added as a sink for biomass metabolites, as many biomass components would otherwise become dead-ends in the metabolic network.
estimates in this release of the ModelSEED lead to an adjustment of the reversibility rules associated with 2783 reactions as described in the previous section. These changes impact how every reaction in the ModelSEED network is connected to every other reaction and the number of functional reactions when flux balance analysis is applied. When constructing an FBA model of the ModelSEED biochemistry, we included all balanced single-compartment reactions as intracellular, while transport reactions were used to define the metabolites that are allowed to move from the extracellular space into the intracellular compartment. The SEED bacterial biomass reaction was added as a sink for biomass metabolites, as many biomass components would otherwise become dead-ends in the metabolic network.
We used this whole-biochemistry model to study how our updates to the ModelSEED biochemistry affected the connectivity and flux profile of our reaction network when used with flux balance analysis (Table 5). First, we applied flux variability analysis assuming every metabolite in the extracellular space is available for uptake. This enabled us to determine how many reactions in our database are functional, meaning they are capable of carrying a nonzero mass-balanced flux from one set of transported metabolite inputs to another set of transported metabolite outputs (61). We actually saw a very slight decline in the number of functional reactions versus our previous release with the PlantSEED. This is primarily a result of our efforts to combine and eliminate redundant reactions in our database (described earlier). Additionally, some reactions are characterized to be nonfunctional because they lead exclusively to the production of terminal compounds that lack transport reactions and are not currently included in our biomass composition. To test the extent to which this new release has improved capabilities in simulating phenotypes, we applied our database to simulate biomass production using an example bacterial biomass objective function in 390 Biolog growth conditions (62). In this study, our database was capable of successfully producing biomass for 355 (91%) of the Biolog conditions, which was an improvement over previous releases.
Table 5.
Results from running flux balance analysis on ModelSEED database
| Release | Total reactions | Mass balanced | Reversible | Functional reactions* | Functional growth conditions |
|---|---|---|---|---|---|
| Current (2020) | 36 193 | 25 457 | 18 399 | 21 403 (22 530) | 355 (91%) |
| PlantSEED (2014) | 27 558 | 17 264 | 8906 | 21 917 (23 461) | 337 (86%) |
| ModelSEED (2010) | 13 257 | 10 263 | 6195 | 8505 (9073) | 330 (85%) |
*A reaction is considered functional if it were determined to be capable of carrying nonzero mass-balanced flux (see the main text). Values in parentheses are without thermodynamic constraints (i.e., all reactions reversible).
Leveraging ModelSEED biochemistry to map annotation ontologies and support comparison
The field of biology has long struggled to arrive at a standardized controlled way of describing the function of genes and their products. A variety of controlled vocabularies do exist (e.g. UniRef (63), Enzyme Classification numbers (64), Gene Ontology (65), KEGG orthology (48) and SEED (66)), but many annotation platforms do not use these controlled vocabularies. Additionally, in order to compare the annotations of one platform with those of another, it is critically important to map together equivalent ontology terms. This enables one to differentiate cases where a difference in the function assigned to a gene by two platforms represents an actual disagreement in the function of the gene rather than a difference in nomenclature. Unfortunately, in the absence of any other abstraction, this mapping of functional descriptions becomes a largely manual exercise in syntactic interpretation (e.g., is the function described by these words equivalent to the function described by these other words). Fortunately, in the case of metabolic functions, we have another abstraction: biochemistry. Biochemistry is distinctive in that, if one knows the stoichiometry of a reaction and the molecular structure of the metabolites involved in the reaction, one does not need to rely on alias mapping or syntactic interpretation to determine if one reaction is equivalent to another. Instead, it is possible to computationally encode the compound structure and associated reactions into unique strings that make it possible to automatically compare reactions from different databases to one another. This makes biochemistry a valuable tool that may be leveraged to automatically map between functional ontologies where those ontologies have been associated with some kind of biochemistry database (which is increasingly becoming the case for most annotation ontologies).
Extensive effort has already been applied to exploit biochemistry structure codes (e.g., InChI and SMARTS) data to automatically generate translation tables among the reaction and compound identifiers in various biochemistry databases (e.g., MetRxn (67) and MetaNetX (68)). Indeed, this semi-automated mapping is a major component of our own ongoing curation of the ModelSEED database, as previously discussed. We already highlighted one mechanism by which this automated mapping procedure may fail (e.g., if some compounds have missing or erroneous structures associated with them). The other significant reason why this mapping process may fail is that, in many cases, different biochemistry databases will represent the same biochemical pathway using different reactions (e.g., Figure 3B is an example of this in lipid metabolism). This issue can lead to significant apparent disagreement between the chemistry assigned by different resources to the same genes. For example, a comparison of the ModelSEED model of Escherichia coli with the earliest manually curated model of E. coli, the iJR904 (69) revealed extensive apparent differences between these models. Of the 625 distinct compounds included in the iJR904 model, only 547 overlapped perfectly with the ModelSEED. However, many of these apparent miss-matching compounds were due to differences in representation of the same biochemistry between BiGG and ModelSEED.
Figure 3.

Directed acyclic graph representation of compound classes. Hierarchy is defined by their functional relationship in metabolism. (A) Example of DAG representation of a few electron transfer compounds. (B) Parallel representation of compound and reaction hierarchy; both rxn06138 and rxn09551 are abstract representations of rxn09067. However, rxn09551 is a context adapted version for the yeast, thus it has a different stoichiometry weight.
We have now developed a mechanism within the ModelSEED database for identifying and accounting for these differences in representation when comparing models and genome annotations. This approach begins with a general policy applied by the ModelSEED when we integrate chemistry from other resources into our own database. Unlike other biochemistry databases (e.g., KEGG and MetaCyc), the chemistry in the ModelSEED is not necessarily nonredundant. The same biochemistry may occur multiple times in the ModelSEED with differing representations. This is an explicit design decision in the ModelSEED made to facilitate the creation and maintenance of an ontological map between these various representations. The creation of this map begins with a semi-automated process of mapping together compounds in the database that are structurally different but chemically equivalent. Some associations can be made automatically (e.g., mapping α-D-glucose and β-D-glucose to D-glucose and mapping D-glucose and L-glucose to generic glucose). Note, other frameworks like ChEBI support this type of automated mapping as well (Hastings et al., 2009). Other associations must be made manually (e.g., mapping three different chain-length representations of fatty acid together as done in Figure 3B). Once these compound associations have been created, we then have an automated mechanism for the creation of a directed acyclic graph (DAG) connecting equivalent reactions to one another based on the associations among reactants. Once constructed, this DAG can be used to automatically support the translation of chemistry from one mapping to another, which in turn enables the automated comparison of metabolic annotations between resources. Applying our current DAG to our example comparison of the ModelSEED and iJR904 model of E. coli, this translation process reduced the number of mismatching compounds from 78 to 31. The impact of the reactions (Figure 4) is also significant, the number of uniques detected in iJR904 is reduced from 258 to 159, the usage of different isomers had the highest impact, followed by abstract representation of phospholipids and lumped fatty acid metabolism.
Figure 4.
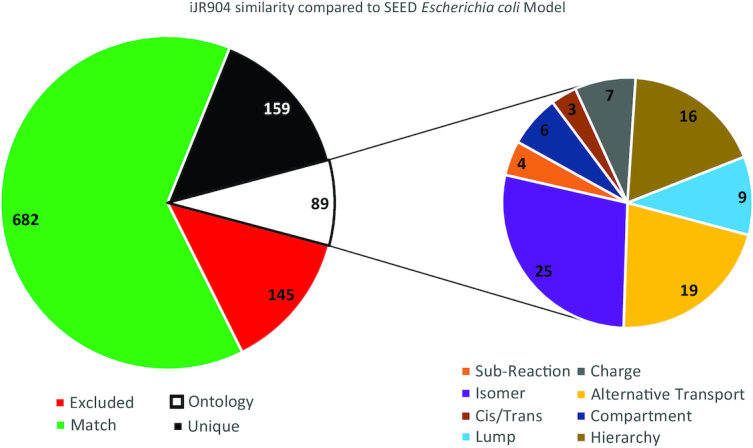
Reaction comparison between iJR904 and ModelSEED biochemistry of the Escherichia coli genome-scale model. Excluded - Exchange reactions, Biomass, maintenance ATP (ATPM); Match - Traditional matching approach (identity matching) with protonation comparison. Unique - Reactions that are not present in the ModelSEED model. Subchart: reactions otherwise marked as unique but with alternative representation in ModelSEED. Isomer and Cis/Trans: reaction present but utilizing different isomer or cis/trans metabolite; Subreaction/Lump: reaction present but is a merge or split version of a ModelSEED reaction; Charge: reaction matches but compounds carry different charge; Hierarchy: reaction matches ModelSEED reaction but utilizing an abstract representation of the compound; Compartment: reaction matches exact stoichiometry but utilizing different compartment configuration; Alternative transport: transport of the compound present but using different mechanism or co-substrate. The submitted manuscript has been created by UChicago Argonne, LLC as Operator of Argonne National Laboratory (‘Argonne’) under Contract No. DE-AC02–06CH11357 with the U.S. Department of Energy. The U.S. Government retains for itself, and others acting on its behalf, a paid-up, nonexclusive, irrevocable worldwide license in said article to reproduce, prepare derivative works, distribute copies to the public, and perform publicly and display publicly, by or on behalf of the Government. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan.
The current ontological mappings established in the ModelSEED represent three different types of relationships: (i) equivalent compound sets, (ii) lumped reaction sets and (iii) context-specific reaction sets. We stored each of these sets in separate DAGs that connect together ModelSEED compound and reaction entities. The equivalent compound and reaction set relationships expose compounds and reactions that are abstract/generic representations of other compounds and reactions. For example, in many electron transfer reactions (Figure 3A) when the co-substrate is unknown, both KEGG and MetaCyc use a pair of abstract compounds to act as placeholders (acceptor/donor). These reactions must be adapted when used for modelling purposes. KEGG and MetaCyc currently contain 235 and 644 reactions respectively that are represented with generic acceptors. Using the ontological relationships implemented in the ModelSEED, we identified 104 clusters of reactions with specific cosubstrates that represent instantiations of generic acceptor/donor reactions. Now if two models from two different sources use different members of these clusters to represent the same overall reaction, we can automatically determine that these models at least agree that the generic reaction is happening. Other common problematic abstractions include: sugar isomers, and pathways that apply the same chemical actions on multiple substrates (i.e., pathways that are replicated for multiple representations of the participating substrate, such as phospholipids, quinones and fatty acid chain lengths, etc). The lumped reaction set relationship connects lumped versions of reactions to the series of substep reactions that have been lumped. Examples of these reactions include composite reactions in MetaCyc or multi-step reactions in KEGG. However, metabolic models may also merge reactions for other purposes (e.g., modeling mechanism, hypothesizing unknown pathways and avoiding poorly defined intermediates). We use these relationships to automatically convert lumped reactions into their component reactions or vice versa. The organism/context specific relationship connects entities that were modified to fit a certain context (e.g., adaptation for modeling purposes) to their standard representation (Figure 3B). In general, these reactions are unfit for modelling purposes, but they might contribute knowledge for the database (e.g., related genes).
We note that the ModelSEED is far from the first biochemistry database to apply ontologies to metabolites and biochemical reactions. ChEBI, MetaCyc and KEGG all do this to varying extents (70). In general, most of the existing ontologies represent identity (e.g., cross-references) or structural relationships (e.g., alpha/beta glucose); the ModelSEED ontology is designed to add additional relationships required to translate among the representational heterogeneity that exists between various metabolic databases and modeling approaches. For example, we add relationships to represent compound adaptations often used in modeling, such as using FAD instead of electron transfer flavoprotein (Figure 3A) or using an adapted version of a generic compound (Figure 3B). However, other database ontologies have focused on classifying and categorizing these entities, whereas our focus is on mapping data from disparate sources to better facilitate comparison, reconciliation and integration of annotation information.
Using GitHub as a tool for community contributions and maintenance
One major goal for this release of the ModelSEED biochemistry database is to become a community-driven resource, meaning that contributions, updates and corrections could be rapidly integrated from the research community. We also want the database to evolve over time in as transparent a manner as possible. To accomplish these goals, we have released the ModelSEED database to the public, using the Creative Commons Attribution License, in the Git repository: https://github.com/ModelSEED/ModelSEEDDatabase. Note, data directly derived from KEGG and MetaCyc are still subject to licenses from these resources. Using Git to store the changes made to the data and to the underlying scripts inherently maintains provenance of the data and scripts.
All the main datasets in the repository are well-formatted, and accompanied by instructions and a library of scripts for loading and handling the data across several folders. The main compound and reaction databases were formatted as tables that can be exported to Excel and as structured JSON objects that can be directly imported into any scripting language and web application. For example, a researcher can load the data directly into a local Solr instance (as described below). These files are accompanied by the data and scripts we use to maintain metabolic structure, thermodynamics, and external identifiers and synonyms.
Crucially, we expect the ModelSEED database to grow and to be improved over time, and invite researchers to collaborate with us. The use of a Git repository in GitHub provides the means by which we can interact with researchers and include changes from external teams with the accompanying provenance. Researchers will be able to submit edits, additions, and changes to the current data via use of Git and GitHub Pull Requests. We particularly welcome any new metabolic pathways relevant to the microbial, fungal, and plant kingdoms, as well as any new metabolic structures or thermodynamic data that would improve the process of reconstructing metabolism. We will review these submissions, and interact with the wider community to merge the new data and maintain the repository at a high standard. Policies for community contributions to the ModelSEED Github are described in the ‘Materials and Methods’ section. Finally, we will release new changes and data on a quarterly basis.
DISCUSSION
Currently in the field of bioinformatics, there are many powerful techniques for predicting gene function, including numerous homology methods like BLAST, Hidden Markov Models and k-mer indexing. There are also numerous nonhomology methods exploiting chromosomal context, coexpression, gene fitness data, co-occurrence and protein structure. No single approach is a panacea. Rather, it has been demonstrated numerous times that optimal results in bioinformatics are obtained by combining many different approaches and data sources together to obtain a consensus result (71). One of the biggest impediments to building such a consensus approach for biology today is the lack of a single standard ontology for describing gene functions. Another impediment is the need for a mechanism to be able to test predicting gene functions for consistency with available phenotypic evidence (e.g., growth conditions and gene fitness data). A final impediment is the need for a streamlined mechanism for the research community to rapidly integrate new annotations and pathways into these chemistry databases, as well as track full provenance on changes in those databases over time.
The ModelSEED biochemistry database was designed to address these challenges. By integrating together diverse chemistry databases and building and maintaining mappings to those databases based on structure and ontology, we provide a resource that can automatically translate many different annotation ontologies into a single chemical representation. This in turn facilitates the rapid comparison and reconciliation of annotations. By also making that single chemical representation ‘modelable’ and integrating deeply with model reconstruction platforms like the ModelSEED, PlantSEED and KBase, we offer a means of converting that single chemical representation into mechanistic models. Mechanistic models can then apply functional annotations to predict conditional phenotypes like gene fitness or growth conditions so that competing annotations may be tested and reconciled to maximize consistency between phenotype predictions and experimental data. Finally, by deploying our database on GitHub, we provide an easy, trackable method for rapidly accepting contributions of new chemistry and data from the research community. GitHub also provides an excellent built in system for tracking database changes over time, as well as tracking who is responsible for each change.
Competing biochemistry resources do exist that meet one of these challenges. MetaCyc and BIGG are both top tier resources for supporting metabolic model reconstruction, but neither of these databases supports direct community contributions, and of this pair, only MetaCyc offers significant ontology support. Even in MetaCyc, the ontology support is directed more at classification rather than mapping between databases. Other resources exist that focus more specifically on supporting database mapping and ontology, including Rhea, which is integrated with gene ontology, and MetaNetX, which maintains mappings of identical compounds and reactions from numerous data sources. However, again neither of these resources supports direct community contribution or model reconstruction.
Mapping, reconciling, testing and integrating knowledge of gene function in biological systems is one of the primary driving missions of KBase. The ModelSEED biochemistry database is an important part of the KBase platform. The tools presented here significantly advance that mission by providing a structured, extensible framework, with provenance, to support all of these activities for metabolism. As implemented in KBase, the ModelSEED biochemistry database exemplifies how a specialized, independently curated resource that provides valuable integration of multidimensional omics data can significantly enrich the available data content and structure in KBase, and thereby further empower a systems biological analytical approach for all KBase users.
DATA AVAILABILITY
We have released the ModelSEED biochemistry database to the public, using the Creative Commons Attribution License, in the Git repository: https://github.com/ModelSEED/ModelSEEDDatabase. The release of data will be not only in the repository but also deployed to several key resources: ModelSEED (https://modelseed.org) and its accompanying SOLR database (https://modelseed.org/solr) and KBase (https://kbase.us) by way of inclusion in all of our metabolic modeling Apps in KBase narratives, and also via the KBase search interface (Figure 5; https://narrative.kbase.us/#biochem-search).
Figure 5.

Biochemistry in DOE Systems Biology Knowledgebase (KBase). The ModelSEED biochemistry database is widely used for a range of metabolic modeling Apps in KBase Narratives (https://kbase.us) and there is also an interface for searching the entire biochemistry. The screenshot shows an example of a search result where ‘pyruvate’ is used for the search term, and the first returned hit is expanded (by clicking in the first column).
In addition to our establishment of a GitHub repository for the ModelSEED data, we have also created a web interface in both the ModelSEED and KBase environments to search and browse this data. The ModelSEED interface to the biochemistry data is available at http://modelseed.org/biochem/reactions. This interface includes a compound and reaction table, fully searchable by column, including supporting search by aliases from other databases. The interface also includes compound and reaction landing pages showing a more detailed view of these entities. We also updated this interface to ensure that all biochemistry data are accessible without logging in. Finally, we added an interface to KBase for browsing this biochemistry data: https://narrative.kbase.us/#biochem-search. Like the ModelSEED interface, this tabular view enables users to search for reactions and compounds by a variety of terms, including aliases, and redirects compound and reaction views to the landing pages in the ModelSEED.
In addition to these web interfaces for manually browsing the ModelSEED biochemistry data, we have also created programmatic APIs. All data can be loaded into an Apache Solr (https://lucene.apache.org/solr/) database, which offers a publically accessible REST API for accessing the data. In the Solr folder of the ModelSEED GitHub, we have several examples of how a researcher can fetch the data directly via https, or via a python script. If researchers wish to setup their own Solr endpoint to serve their own biochemistry data, formatted in the same manner as our public one, a set of instructions for how to do that is in the same folder.
Supplementary Material
Contributor Information
Samuel M D Seaver, Computing, Environment, and Life Sciences Division, Argonne National Laboratory, Lemont, IL 60439, USA.
Filipe Liu, Computing, Environment, and Life Sciences Division, Argonne National Laboratory, Lemont, IL 60439, USA.
Qizhi Zhang, Computing, Environment, and Life Sciences Division, Argonne National Laboratory, Lemont, IL 60439, USA.
James Jeffryes, Computing, Environment, and Life Sciences Division, Argonne National Laboratory, Lemont, IL 60439, USA.
José P Faria, Computing, Environment, and Life Sciences Division, Argonne National Laboratory, Lemont, IL 60439, USA.
Janaka N Edirisinghe, Computing, Environment, and Life Sciences Division, Argonne National Laboratory, Lemont, IL 60439, USA.
Michael Mundy, Center for Individualized Medicine, Mayo Clinic, Rochester, MN 55905, USA.
Nicholas Chia, Center for Individualized Medicine, Mayo Clinic, Rochester, MN 55905, USA.
Elad Noor, Department of Biology, Institute of Molecular Systems Biology, Eidgenössische Technische Hochschule Zürich, CH-8093 Zürich, Switzerland.
Moritz E Beber, Novo Nordisk Foundation Center for Biosustainability, Technical University of Denmark, Kongens Lyngby, 2800, Denmark.
Aaron A Best, Department of Biology, Hope College, Holland, MI 49423, USA.
Matthew DeJongh, Department of Computer Science, Hope College, Holland, MI 49423, USA.
Jeffrey A Kimbrel, Biosciences and Biotechnology Division, Lawrence Livermore National Laboratory, Livermore, CA 94550, USA.
Patrik D’haeseleer, Biosciences and Biotechnology Division, Lawrence Livermore National Laboratory, Livermore, CA 94550, USA.
Sean R McCorkle, Computational Science Initiative, Brookhaven National Laboratory, Upton, NY 11973, USA.
Jay R Bolton, Environmental Genomics and Systems Biology Division, E.O. Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA.
Erik Pearson, Environmental Genomics and Systems Biology Division, E.O. Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA.
Shane Canon, Environmental Genomics and Systems Biology Division, E.O. Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA.
Elisha M Wood-Charlson, Environmental Genomics and Systems Biology Division, E.O. Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA.
Robert W Cottingham, Biosciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37830, USA.
Adam P Arkin, Environmental Genomics and Systems Biology Division, E.O. Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA.
Christopher S Henry, Computing, Environment, and Life Sciences Division, Argonne National Laboratory, Lemont, IL 60439, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
U.S. Department of Energy [DE-AC02–06CH11357, DE-AC02–05CH11231, DE-AC05–00OR22725 to C.S.H., S.S., J.P.F., J.J., J.E., Q.Z., F.L., E.P., S.C., E.M.W.C., R.W.C., A.A.; DE-AC52–07NA27344 to J.A.K., P.D.]; National Cancer Institute [R01CA179243 to N.C., M.M.]; National Science Foundation [GEPR-1444202 to C.S.H., S.S., Q.Z.; MCB-1716285 to A.A.B., M.D.J.]; Horizon 2020 - Research and Innovation Framework Programme [686070 (DD-DeCaF) to M.E.B.]; Center for Individualized Medicine, Mayo Clinic [to M.M., N.C.]. Funding for open access charge: U.S. Department of Energy.
Conflict of interest statement. None declared.
REFERENCES
- 1. Satish Kumar V., Dasika M.S., Maranas C.D.. Optimization based automated curation of metabolic reconstructions. BMC Bioinform. 2007; 8:212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lieven C., Petersen L.A.H., Jørgensen S.B., Gernaey K.V., Herrgard M.J., Sonnenschein N.. A Genome-Scale Metabolic Model for Methylococcus capsulatus (Bath) Suggests Reduced Efficiency Electron Transfer to the Particulate Methane Monooxygenase. Front. Microbiol. 2018; 9:2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. de la Torre A., Metivier A., Chu F., Laurens L.M.L., Beck D.A.C., Pienkos P.T., Lidstrom M.E., Kalyuzhnaya M.G.. Genome-scale metabolic reconstructions and theoretical investigation of methane conversion in Methylomicrobium buryatense strain 5G(B1). Microb. Cell Fact. 2015; 14:188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Meadows A.L., Hawkins K.M., Tsegaye Y., Antipov E., Kim Y., Raetz L., Dahl R.H., Tai A., Mahatdejkul-Meadows T., Xu L. et al.. Rewriting yeast central carbon metabolism for industrial isoprenoid production. Nature. 2016; 537:694–697. [DOI] [PubMed] [Google Scholar]
- 5. Chen J., Daniell J., Griffin D., Li X., Henson M.A.. Experimental testing of a spatiotemporal metabolic model for carbon monoxide fermentation with Clostridium autoethanogenum. Biochem. Eng. J. 2018; 129:64–73. [Google Scholar]
- 6. Mendoza S.N., Cañón P.M., Contreras Á., Ribbeck M., Agosín E.. Genome-Scale Reconstruction of the Metabolic Network in Oenococcus oeni to Assess Wine Malolactic Fermentation. Front. Microbiol. 2017; 8:534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Marcellin E., Behrendorff J.B., Nagaraju S., DeTissera S., Segovia S., Palfreyman R.W., Daniell J., Licona-Cassani C., Quek L.-E., Speight R. et al.. Low carbon fuels and commodity chemicals from waste gases – systematic approach to understand energy metabolism in a model acetogen. Green Chem. 2016; 18:3020–3028. [Google Scholar]
- 8. Marshall C.W., Ross D.E., Handley K.M., Weisenhorn P.B., Edirisinghe J.N., Henry C.S., Gilbert J.A., May H.D., Norman R.S.. Metabolic Reconstruction and Modeling Microbial Electrosynthesis. Sci. Rep. 2017; 7:8391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Edirisinghe J.N., Weisenhorn P., Conrad N., Xia F., Overbeek R., Stevens R.L., Henry C.S.. Modeling central metabolism and energy biosynthesis across microbial life. BMC Genomics. 2016; 17:568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cheung C.Y.M., Poolman M.G., Fell D.A., Ratcliffe R.G., Sweetlove L.J.. A Diel Flux Balance Model Captures Interactions between Light and Dark Metabolism during Day-Night Cycles in C3 and Crassulacean Acid Metabolism Leaves. Plant Physiol. 2014; 165:917–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shameer S., Baghalian K., Cheung C.Y.M., Ratcliffe R.G., Sweetlove L.J.. Computational analysis of the productivity potential of CAM. Nat. Plants. 2018; 4:165–171. [DOI] [PubMed] [Google Scholar]
- 12. Plata G., Henry C.S., Vitkup D.. Long-term phenotypic evolution of bacteria. Nature. 2015; 517:369–372. [DOI] [PubMed] [Google Scholar]
- 13. diCenzo G.C., Checcucci A., Bazzicalupo M., Mengoni A., Viti C., Dziewit L., Finan T.M., Galardini M., Fondi M.. Metabolic modelling reveals the specialization of secondary replicons for niche adaptation in Sinorhizobium meliloti. Nat. Commun. 2016; 7:12219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bosi E., Monk J.M., Aziz R.K., Fondi M., Nizet V., Palsson B.Ø. Comparative genome-scale modelling of Staphylococcus aureus strains identifies strain-specific metabolic capabilities linked to pathogenicity. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:E3801–E3809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hartleb D., Jarre F., Lercher M.J.. Improved Metabolic Models for E. coli and Mycoplasma genitalium from GlobalFit, an Algorithm That Simultaneously Matches Growth and Non-Growth Data Sets. PLoS Comput. Biol. 2016; 12:e1005036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xavier J.C., Patil K.R., Rocha I.. Metabolic models and gene essentiality data reveal essential and conserved metabolism in prokaryotes. PLoS Comput. Biol. 2018; 14:e1006556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ding T., Case K.A., Omolo M.A., Reiland H.A., Metz Z.P., Diao X., Baumler D.J.. Predicting Essential Metabolic Genome Content of Niche-Specific Enterobacterial Human Pathogens during Simulation of Host Environments. PLoS One. 2016; 11:e0149423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Khodayari A., Maranas C.D.. A genome-scale Escherichia coli kinetic metabolic model k-ecoli457 satisfying flux data for multiple mutant strains. Nat. Commun. 2016; 7:13806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang C., Bidkhori G., Benfeitas R., Lee S., Arif M., Uhlén M., Mardinoglu A.. ESS: A Tool for Genome-Scale Quantification of Essentiality Score for Reaction/Genes in Constraint-Based Modeling. Front. Physiol. 2018; 9:1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guzmán G.I., Olson C.A., Hefner Y., Phaneuf P.V., Catoiu E., Crepaldi L.B., Micas L.G., Palsson B.O., Feist A.M.. Reframing gene essentiality in terms of adaptive flexibility. BMC Syst. Biol. 2018; 12:143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Harder B.-J., Bettenbrock K., Klamt S.. Model-based metabolic engineering enables high yield itaconic acid production by Escherichia coli. Metab. Eng. 2016; 38:29–37. [DOI] [PubMed] [Google Scholar]
- 22. Park J.M., Song H., Lee H.J., Seung D.. In silico aided metabolic engineering of Klebsiella oxytoca and fermentation optimization for enhanced 2, 3-butanediol production. J. Ind. Microbiol. Biotechnol. 2013; 40:1057–1066. [DOI] [PubMed] [Google Scholar]
- 23. Chen J., Henson M.A.. In silico metabolic engineering of Clostridium ljungdahlii for synthesis gas fermentation. Metab. Eng. 2016; 38:389–400. [DOI] [PubMed] [Google Scholar]
- 24. Alper H., Jin Y.-S., Moxley J.F., Stephanopoulos G.. Identifying gene targets for the metabolic engineering of lycopene biosynthesis in Escherichia coli. Metab. Eng. 2005; 7:155–164. [DOI] [PubMed] [Google Scholar]
- 25. Milne C.B., Eddy J.A., Raju R., Ardekani S., Kim P.-J., Senger R.S., Jin Y.-S., Blaschek H.P., Price N.D.. Metabolic network reconstruction and genome-scale model of butanol-producing strain Clostridium beijerinckii NCIMB 8052. BMC Syst. Biol. 2011; 5:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang J., Wang C., Song K., Wen J.. Metabolic network model guided engineering ethylmalonyl-CoA pathway to improve ascomycin production in Streptomyces hygroscopicus var. ascomyceticus. Microb. Cell Fact. 2017; 16:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li J., Sun R., Ning X., Wang X., Wang Z.. Genome-Scale Metabolic Model of Actinosynnema pretiosum ATCC 31280 and Its Application for Ansamitocin P-3 Production Improvement. Genes. 2018; 9:364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Niu W., Kramer L., Mueller J., Liu K., Guo J.. Metabolic engineering of Escherichia coli for the de novo stereospecific biosynthesis of 1, 2-propanediol through lactic acid. Metab. Eng. Commun. 2019; 8:e00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zuñiga C., Li C.-T., Huelsman T., Levering J., Zielinski D.C., McConnell B.O., Long C.P., Knoshaug E.P., Guarnieri M.T., Antoniewicz M.R. et al.. Genome-Scale Metabolic Model for the Green Alga Chlorella vulgaris UTEX 395 Accurately Predicts Phenotypes under Autotrophic, Heterotrophic, and Mixotrophic Growth Conditions. Plant Physiol. 2016; 172:589–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chakrabarti A., Miskovic L., Soh K.C., Hatzimanikatis V.. Towards kinetic modeling of genome-scale metabolic networks without sacrificing stoichiometric, thermodynamic and physiological constraints. Biotechnol. J. 2013; 8:1043–1057. [DOI] [PubMed] [Google Scholar]
- 31. Smallbone K., Simeonidis E., Swainston N., Mendes P.. Towards a genome-scale kinetic model of cellular metabolism. BMC Syst. Biol. 2010; 4:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stanford N.J., Lubitz T., Smallbone K., Klipp E., Mendes P., Liebermeister W.. Systematic construction of kinetic models from genome-scale metabolic networks. PLoS One. 2013; 8:e79195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Seaver S.M.D., Bradbury L.M.T., Frelin O., Zarecki R., Ruppin E., Hanson A.D., Henry C.S.. Improved evidence-based genome-scale metabolic models for maize leaf, embryo, and endosperm. Front. Plant Sci. 2015; 6:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jerby L., Shlomi T., Ruppin E.. Computational reconstruction of tissue-specific metabolic models: application to human liver metabolism. Mol. Syst. Biol. 2010; 6:401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bordbar A., Feist A.M., Usaite-Black R., Woodcock J., Palsson B.O., Famili I.. A multi-tissue type genome-scale metabolic network for analysis of whole-body systems physiology. BMC Syst. Biol. 2011; 5:180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jeffryes J.G., Colastani R.L., Elbadawi-Sidhu M., Kind T., Niehaus T.D., Broadbelt L.J., Hanson A.D., Fiehn O., Tyo K.E.J., Henry C.S.. MINEs: open access databases of computationally predicted enzyme promiscuity products for untargeted metabolomics. J. Cheminform. 2015; 7:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Witherden E.A., Moyes D.L., Bruce K.D., Ehrlich S.D., Shoaie S.. Using systems biology approaches to elucidate cause and effect in host–microbiome interactions. Curr. Opin. Syst. Biol. 2017; 3:141–146. [Google Scholar]
- 38. Ruiz-Moreno H.A., López-Tamayo A.M., Caro-Quintero A., Husserl J., González Barrios A.F.. Metagenome level metabolic network reconstruction analysis reveals the microbiome in the Bogotá River is functionally close to the microbiome in produced water. Ecol. Model. 2019; 399:1–12. [Google Scholar]
- 39. Cardona C., Weisenhorn P., Henry C., Gilbert J.A.. Network-based metabolic analysis and microbial community modeling. Curr. Opin. Microbiol. 2016; 31:124–131. [DOI] [PubMed] [Google Scholar]
- 40. Mendoza S.N., Olivier B.G., Molenaar D., Teusink B.. A systematic assessment of current genome-scale metabolic reconstruction tools. Genome Biol. 2019; 20:158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lieven C., Beber M.E., Olivier B.G., Bergmann F.T., Ataman M., Babaei P., Bartell J.A., Blank L.M., Chauhan S., Correia K. et al.. MEMOTE for standardized genome-scale metabolic model testing. Nat. Biotechnol. 2020; 38:272–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Henry C.S., Bernstein H.C., Weisenhorn P., Taylor R.C., Lee J.-Y., Zucker J., Song H.-S.. Microbial Community Metabolic Modeling: A Community Data-Driven Network Reconstruction. J. Cell. Physiol. 2016; 231:2339–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Soh K.C., Hatzimanikatis V.. Krömer J.O., Nielsen L.K., Blank L.M.. Constraining the Flux Space Using Thermodynamics and Integration of Metabolomics Data. Metabolic Flux Analysis: Methods and Protocols. 2014; NY: Springer; 49–63. [DOI] [PubMed] [Google Scholar]
- 44. Chowdhury A., Khodayari A., Maranas C.D.. Improving prediction fidelity of cellular metabolism with kinetic descriptions. Curr. Opin. Biotechnol. 2015; 36:57–64. [DOI] [PubMed] [Google Scholar]
- 45. Töpfer N., Seaver S.M.D., Aharoni A.. António C. Integration of Plant Metabolomics Data with Metabolic Networks: Progresses and Challenges. Plant Metabolomics: Methods and Protocols. 2018; NY: Springer; 297–310. [DOI] [PubMed] [Google Scholar]
- 46. Bordbar A., Yurkovich J.T., Paglia G., Rolfsson O., Sigurjónsson Ó.E., Palsson B.O.. Elucidating dynamic metabolic physiology through network integration of quantitative time-course metabolomics. Sci. Rep. 2017; 7:46249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kanehisa M., Goto S., Sato Y., Kawashima M., Furumichi M., Tanabe M.. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014; 42:D199–D205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kanehisa M., Sato Y., Kawashima M., Furumichi M., Tanabe M.. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016; 44:D457–D462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Caspi R., Billington R., Ferrer L., Foerster H., Fulcher C.A., Keseler I.M., Kothari A., Krummenacker M., Latendresse M., Mueller L.A. et al.. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2016; 44:D471–D480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. King Z.A., Lu J., Dräger A., Miller P., Federowicz S., Lerman J.A., Ebrahim A., Palsson B.O., Lewis N.E.. BiGG Models: A platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Res. 2016; 44:D515–D522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Landrum G. RDKit: Open-Source Cheminformatics Software. 2016; (01 August 2020, date last accessed)http://rdkit.org/.
- 52. O’Boyle N.M., Banck M., James C.A., Morley C., Vandermeersch T., Hutchison G.R.. Open Babel: An open chemical toolbox. J. Cheminform. 2011; 3:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jankowski M.D., Henry C.S., Broadbelt L.J., Hatzimanikatis V.. Group contribution method for thermodynamic analysis of complex metabolic networks. Biophys. J. 2008; 95:1487–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Noor E., Bar-Even A., Flamholz A., Lubling Y., Davidi D., Milo R.. An integrated open framework for thermodynamics of reactions that combines accuracy and coverage. Bioinformatics. 2012; 28:2037–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Henry C.S., Zinner J.F., Cohoon M.P., Stevens R.L.. iBsu1103: a new genome-scale metabolic model of Bacillus subtilis based on SEED annotations. Genome Biol. 2009; 10:R69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Travis C.I. 2020; (01 August 2020, date last accessed)https://travis-ci.org/.
- 57. Arkin A.P., Cottingham R.W., Henry C.S., Harris N.L., Stevens R.L., Maslov S., Dehal P., Ware D., Perez F., Canon S. et al.. KBase: The United States Department of Energy Systems Biology Knowledgebase. Nat. Biotechnol. 2018; 36:566–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Henry C.S., DeJongh M., Best A.A., Frybarger P.M., Linsay B., Stevens R.L.. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat. Biotechnol. 2010; 28:977–982. [DOI] [PubMed] [Google Scholar]
- 59. Seaver S.M.D., Lerma-Ortiz C., Conrad N., Mikaili A., Sreedasyam A., Hanson A.D., Henry C.S.. PlantSEED enables automated annotation and reconstruction of plant primary metabolism with improved compartmentalization and comparative consistency. Plant J. 2018; 95:1102–1113. [DOI] [PubMed] [Google Scholar]
- 60. Seaver S.M.D., Gerdes S., Frelin O., Lerma-Ortiz C., Bradbury L.M.T., Zallot R., Hasnain G., Niehaus T.D., El Yacoubi B., Pasternak S. et al.. High-throughput comparison, functional annotation, and metabolic modeling of plant genomes using the PlantSEED resource. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:9645–9650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mahadevan R., Schilling C.H.. The effects of alternate optimal solutions in constraint-based genome-scale metabolic models. Metab. Eng. 2003; 5:264–276. [DOI] [PubMed] [Google Scholar]
- 62. Bochner B.R. Global phenotypic characterization of bacteria. FEMS Microbiol. Rev. 2009; 33:191–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Suzek B.E., Wang Y., Huang H., McGarvey P.B., Wu C.H., Consortium U.P.. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics. 2015; 31:926–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. McDonald A.G., Boyce S., Tipton K.F.. ExplorEnz: the primary source of the IUBMB enzyme list. Nucleic Acids Res. 2009; 37:D593–D597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T. et al.. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000; 25:25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Overbeek R. The Subsystems Approach to Genome Annotation and its Use in the Project to Annotate 1000 Genomes. Nucleic Acids Res. 2005; 33:5691–5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kumar A., Suthers P.F., Maranas C.D.. MetRxn: a knowledgebase of metabolites and reactions spanning metabolic models and databases. BMC Bioinformatics. 2012; 13:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ganter M., Bernard T., Moretti S., Stelling J., Pagni M.. MetaNetX.org: a website and repository for accessing, analysing and manipulating metabolic networks. Bioinformatics. 2013; 29:815–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Reed J.L., Vo T.D., Schilling C.H., Palsson B.O.. An expanded genome-scale model of Escherichia coli K-12 (iJR904 GSM/GPR). Genome Biol. 2003; 4:R54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hastings J., de Matos P., Ennis M., Steinbeck C.. Towards automatic classification within the ChEBI ontology. Nat. Preced. 2009; doi:10.1038/npre.2009.3525.1. [Google Scholar]
- 71. Griesemer M., Kimbrel J.A., Zhou C.E., Navid A., D’haeseleer P.. Combining multiple functional annotation tools increases coverage of metabolic annotation. BMC Genomics. 2018; 19:948. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We have released the ModelSEED biochemistry database to the public, using the Creative Commons Attribution License, in the Git repository: https://github.com/ModelSEED/ModelSEEDDatabase. The release of data will be not only in the repository but also deployed to several key resources: ModelSEED (https://modelseed.org) and its accompanying SOLR database (https://modelseed.org/solr) and KBase (https://kbase.us) by way of inclusion in all of our metabolic modeling Apps in KBase narratives, and also via the KBase search interface (Figure 5; https://narrative.kbase.us/#biochem-search).
Figure 5.
Biochemistry in DOE Systems Biology Knowledgebase (KBase). The ModelSEED biochemistry database is widely used for a range of metabolic modeling Apps in KBase Narratives (https://kbase.us) and there is also an interface for searching the entire biochemistry. The screenshot shows an example of a search result where ‘pyruvate’ is used for the search term, and the first returned hit is expanded (by clicking in the first column).
In addition to our establishment of a GitHub repository for the ModelSEED data, we have also created a web interface in both the ModelSEED and KBase environments to search and browse this data. The ModelSEED interface to the biochemistry data is available at http://modelseed.org/biochem/reactions. This interface includes a compound and reaction table, fully searchable by column, including supporting search by aliases from other databases. The interface also includes compound and reaction landing pages showing a more detailed view of these entities. We also updated this interface to ensure that all biochemistry data are accessible without logging in. Finally, we added an interface to KBase for browsing this biochemistry data: https://narrative.kbase.us/#biochem-search. Like the ModelSEED interface, this tabular view enables users to search for reactions and compounds by a variety of terms, including aliases, and redirects compound and reaction views to the landing pages in the ModelSEED.
In addition to these web interfaces for manually browsing the ModelSEED biochemistry data, we have also created programmatic APIs. All data can be loaded into an Apache Solr (https://lucene.apache.org/solr/) database, which offers a publically accessible REST API for accessing the data. In the Solr folder of the ModelSEED GitHub, we have several examples of how a researcher can fetch the data directly via https, or via a python script. If researchers wish to setup their own Solr endpoint to serve their own biochemistry data, formatted in the same manner as our public one, a set of instructions for how to do that is in the same folder.