


Abstract
One of the most prominent topics in drug discovery is efficient exploration of the vast drug-like chemical space to find synthesizable and novel chemical structures with desired biological properties. To address this challenge, we created the DrugSpaceX (https://drugspacex.simm.ac.cn/) database based on expert-defined transformations of approved drug molecules. The current version of DrugSpaceX contains >100 million transformed chemical products for virtual screening, with outstanding characteristics in terms of structural novelty, diversity and large three-dimensional chemical space coverage. To illustrate its practical application in drug discovery, we used a case study of discoidin domain receptor 1 (DDR1), a kinase target implicated in fibrosis and other diseases, to show DrugSpaceX performing a quick search of initial hit compounds. Additionally, for ligand identification and optimization purposes, DrugSpaceX also provides several subsets for download, including a 10% diversity subset, an extended drug-like subset, a drug-like subset, a lead-like subset, and a fragment-like subset. In addition to chemical properties and transformation instructions, DrugSpaceX can locate the position of transformation, which will enable medicinal chemists to easily integrate strategy planning and protection design.
INTRODUCTION
For a long time, computational chemists have attempted to explore and generate drug-like ligands accurately and efficiently in large virtual chemistry spaces. Despite recognized pitfalls, virtual screening (1) is still a practical route in searching for novel bioactive compounds and pharmaceutical research. Traditionally, the compound sources for virtual screening are from either natural or commercial databases. The molecular structures from natural product libraries are diverse, but their source, isolation, identification and chemical modification are complicated. Commercial compound libraries are generally constructed with the same core scaffold and the introduction of various substituents, which leads to a lack of molecular diversity (2). Therefore, many compounds in these commercial libraries are not novel, which may generate intellectual property issues. Moreover, the same compounds can be ordered by competitors working on the same projects, which results in resupply issues for some vendors (3).
The solution to space searching is to expand the realms of possibility by using virtual molecules, and some novel virtual chemical libraries have been proposed by researchers. One of the most prominent examples is the generic database (GDB) approach conducted by the Reymond laboratory, with its current incarnation enumerating virtual molecules containing up to 11, 13 and 17 atoms formed by combining elements: C, N, O, S and halogen atoms (4,5). The types of molecules generated by this approach are of great novelty; however, their structures may be the major obstacle to the establishment of synthetic routes. Smaller subsets focusing on solving this issue have also been proposed, such as the fragment database (FDB17) (6), the Medicinal Chemistry Aware Database (GDBMedChem) (7), and the ChEMBL-Likeness Score and Database (GDBChEMBL) (8).
To solve the aforementioned issue, medicinal chemists have utilized chemoinformatics to design molecules that can be synthesized more conveniently. One approach is to use chemical reaction information to direct synthetic routes for compounds, which will make the use of virtual libraries more attractive (9). For example, the TIN database contains over 28 million product structures that are virtually novel and synthetically accessible. It is a combinatorial database built around the synthetic feasibility of multicomponent reactions (10). Similarly, based on the ‘click reactions’ of triazoles, the ZINClick database is composed of over 16 million of 1,4-disubstituted 1,2,3-triazoles, whose structures are novel, synthetically feasible and patentable (11). Another free virtual library is ‘Screenable Chemical Universe Based on Intuitive Data OrganizatiOn’ (SCUBIDOO). In SCUBIDOO, 58 robust reactions were applied to 18 561 common molecular building blocks to generate more than 21 million compounds (12). In addition, the REAL database, described in the VirtualFlow platform, contains more than 1.4 billion make-on-demand compounds. The REAL database has been built with 113 260 high-score qualified in-stock building blocks via 194 high-score validated reaction procedures and shows outstanding ease of synthesis (13). The successful applications of these platforms have demonstrated the importance of intelligent reaction knowledge in the field of exploiting chemical space.
It is true that the databases created by rule-based transformations may lack structural diversity due to the limited reaction rules. To address this issue, we chose to use Nova and BIOSTER from StarDrop, which has the most comprehensive collection of transformation rules currently available (14). There are a total of 29 218 reliable and hand-drawn rules collected from the literature, ranging from simple substitutions or bioisostere replacements to more dramatic modifications of the molecular framework, such as ring opening or closing, and this large collection of rules generates molecules with good structural novelty and diversity (15). Notably, starting from old drugs provides a more efficient method for the rapid identification and development of new pharmaceuticals. As pointed out in a recent review (16), the success rate of the drug repurposing approach can be up to 30%. In contrast, for a typical de novo drug discovery programme starting from the identification of lead molecules, it takes 10–15 years to bring a drug to market entry, and the probability of success is <10%. Therefore, structural modifications to some approved drugs can rediscover the potential value of these drugs, such as the repositioning of thalidomide and the target discovery of pomalidomide and lenalidomide (17).
In a multidimensional space, recognized reference points will be required to fulfil a traverse mission. Principal component analysis (PCA) is commonly utilized to visualize the chemical space, with the advantage of reducing the number of dimensions without causing unnecessary loss of information (9). This approach was adopted by the Reymond laboratory with a few different tools: MQN-mapplet (18), webDrugCS (19) and WebMolCS (20). In addition, the principal moments of inertia (PMI) analysis can be used to compare the shape space covered by different compound sets to rapidly assess and visualize the diversity of molecular shape (21). Meanwhile, in exploring the chemical space, some remarkable methods are also employed to evaluate the similarity between molecules, such as chemical space networks (CSNs) (22) and similarity maplets (23). There are also several other interactive tools to visualize chemical space, such as TMAP (24), ChemGPS-NPWeb (25) and ChemMaps.com (26).
To explore the space of drug-like compounds more efficiently, we constructed a virtual compound library called DrugSpaceX based on transformation rules with approved drug molecules as the starting points. The DrugSpaceX database is freely accessible online via our website (https://drugspacex.simm.ac.cn/) and contains more than 100 million chemical products for virtual screening at this release. Cheminformatics analyses show that the proposed database possesses significant novelty and diversity and covers a large three-dimensional chemical space. Moreover, the DrugSpaceX database not only provides the physicochemical and drug-likeness properties represented by radar charts but also displays transformation details to guide compound synthesis.
MATERIALS AND METHODS
The data sets
The drug data set
The set of 2215 approved small molecule drugs used in the validation of the transformations (the ‘Drug Set’) was derived as follows: version 5.1.4 of the DrugBank Small Molecule database (27) was obtained on 7 July 2019. The original set with 2617 approved small molecule drugs was further processed by removing molecules with ambiguous or untransformable SMILES, which produced a drug set consisting of 2215 compounds.
The transformation rules data set
The Nova and BIOSTER cheminformatics library was used within the StarDrop software platform to exponentially broaden the search by taking the ‘Drug Set’ molecules and creating new generations of related compounds. The module contains a unique compilation of 29 218 transformation rules encompassing a broad range of optimization strategies, such as bioisosteric replacements, linker replacements, homologization, introduction of conformational constraints and reversible derivatizations.
The reference databases
As a reference, we also collected existing databases dated 7 January 2020, e.g. DrugBank (10 666 compounds), PDB (28 987 compounds), BindingDB (757 467 compounds), ChEMBL (1 870 267 compounds) and CSD (1 055 799 compounds) (27–31). After standardizing the SMILES with the RDKit library, we compared the canonical SMILES from the external database and DrugSpaceX. Any two structures with the same canonical SMILES can be accessed from the external dataset using the matched ID tag as a reference.
Property rules
To represent the properties of molecules, we calculated some chemical descriptors for each molecular entity with the RDKit library. The set of descriptors were molecular weight (MW), octanol–water partition coefficien (logP) (32), number of hydrogen bond acceptors (HBA), number of hydrogen bond donors (HBD), total polar surface area (TPSA), number of rotatable bonds (RotB), the quantitative estimate of drug-likeness (QED) (33), and MCE-18 (34). The 752 molecules in the drug dataset did not comply with Lipinski's five rules (Ro5) (35). Nevertheless, there is no need to omit these drugs from DrugSpaceX owing to the application of a more appropriate modified version of Ro5 (36). The modified rules were as follows: MW ≤ 103 Da, –2 ≤ log P ≤ 10, HBD ≤ 6, HBA ≤ 15, TPSA ≤ 250 Å2 and RotB ≤20.
Chemical space visualization
The drug dataset and the DrugSpaceX database were compared using PCA (37). The descriptors were MW, log P, number of H-bond donors, number of H-bond acceptors, number of rotatable bonds and topological polar surface area, which provides an overall estimate of molecular complexity. The PMI analysis is a method to calculate the lowest energy conformation and PMI value of each molecule in the compound library using the molecular shape descriptors (21). Based on this approach, all of the molecules were classified as rods, discs or spheres to characterize the shape and distribution of the library around the triangle, which demonstrated the molecular shape diversity of the compound library.
Synthetic accessibility scores
The transformation rules used here do not necessarily correspond to specific chemical reactions or synthetic routes; rather, they are intended to describe changes to molecules that a medicinal chemist might consider in the course of an optimization project (38). A single transformation might require multiple synthetic steps or the synthesis of new building blocks, and thus, a synthetic accessibility scoring measure has been included for reference. The synthetic accessibility (SA) score was calculated for each of the molecules using an RDKit-based Python script (39). SA score estimation is based on fragment contributions and a complexity penalty (chiral centres, weight, large rings). The SA score ranges from 1 to 10, with a greater value representing a more difficult synthesis.
Structure-based virtual screening of DDR1 inhibitors
In the process of finding the DDR1 kinase inhibitors, we first downloaded the drug set as samples from DrugSpaceX, which provides the corresponding structure SMILES (.smi) and 2D and 3D structures (.sdf). Once downloaded, the samples were prepared via LigPrep (version 3.4; Schrödinger, LLC: New York, NY, 2015), which was used to generate stereoisomers and tautomers. The ligands were protonated at pH 7.0 ± 2.0 with Epik (40). For other parameters, the default values were used. Second, the crystal structure of DDR1 complexed with ponatinib (PDB accession code: 3ZOS) was selected for molecular docking (41). The structure was prepared with the Protein Preparation Wizard Workflow provided in the Maestro module of Schrödinger software (Schrödinger, LLC: New York, NY, 2015). The protein structure was first fixed by assigning bond orders, adding hydrogens, creating zero-order bonds to metals, filling in missing side chains using Prime, deleting water molecules >3 Å from the het group, removing waters with less than three H-bonds to nonwaters, and restraining the minimization to allow only hydrogen atoms to be freely minimized. Based on the optimized protein structures, the receptor grid of the complex was generated with the Glide module of Schrödinger software, and the grid files were defined as a 10 × 10 × 10 Å3 region centred at the original ligand of the complex structure. The prepared ligand conformations were docked to the corresponding target protein grid files by Glide with the SP precision mode. For the other parameters, the default values were used. Third, we identified promising drugs based on the docking scoring results and downloaded the drug analogues to reconstruct the dataset for another molecular docking process. Eventually, the hit compounds were determined via the comprehensive evaluation of docking scores, ligand efficiency (LE) (42) and SA.
RESULTS
Creation of the data sets
Creation of the product database
Using the Drug Set as a starting point, the virtual chemical library DrugSpaceX was built based on transformation rules to explore the chemical space of drug-like molecules (Figure 1). In the first round, the 2215 approved drugs were transformed through the transformation rules on the StarDrop software platform, and the Sample Set including 937 230 products was generated after removing duplicate ones. The application of one generation of transformations produced 423 child compounds, suggesting that exhaustive enumeration through more than two generations would be intractable. Therefore, only two rounds of transformations were performed. Afterward, two rounds of transformations were performed within property rule limits, giving rise to a final database of 100 946 534 products. We also checked the novel structures in DrugSpaceX. After removing existing chemicals collected in various databases (including Zinc, ChEMBL, BindingDB, PDB, PubChem and CSD), the proportion of novel structures following the first round of transformations was 95.31%, and the proportion following the second round of transformations increased to 99.58%.
Figure 1.
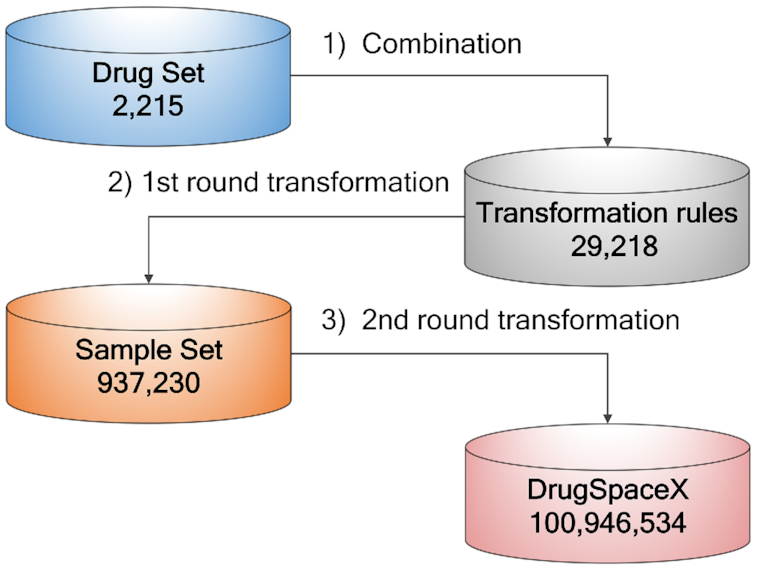
Assembly procedure for chemotype library DrugSpaceX.
Creation of representative samples
For the assembly procedure, the 100 million products were divided into three sets of representative samples with different sizes (D, S and A), which represent the Drug Set, the Sample Set and the All Data Set, respectively. In addition to these three, Supplementary Table S1 also describes five different collections of DrugSpaceX compounds that were prepared based on the following criteria: (i) an extended drug-like subset (DSX-EL) included compounds with MW ≤ 700 Da, 0 ≤ log P ≤ 7.5, HBD ≤ 5, TPSA ≤ 200 Å2 and RotB ≤ 20 (36). (ii) A drug-like subset (DSX-DL) was based on the following rules: MW ≤ 500 Da, log P ≤ 5, HBD ≤ 5, HBA ≤ 10, TPSA ≤ 150 Å2, RotB ≤ 7 (35). (iii) A lead-like subset (DSX-LL) was defined as follows: MW ≤ 350 Da and MW ≥ 250 Da, log P ≤ 3.5, and RotB ≤ 7 (43). (iv) A fragment-like subset (DSX-FL) was also defined with MW ≤ 250 Da, log P ≤ 3.5, and RotB ≤ 5 (44). (v) To reduce the number of DrugSpaceX compounds, a random selection (10%) of the compounds in DrugSpaceX (DSX-10%) was generated.
Analysis of the data sets
Chemical properties
The distributions for each descriptor of the compounds from DrugSpaceX and the Drug Set were compared using distribution histograms and probability density curves (Figure 2A). Based on these algorithms, the distributions of logP show Gaussian-shaped curves with a peak centred on 4 log P units, and the two datasets have similar logP values. Those clusters are equally populated between 1 and 5, while the Drug Set shows a preference for the region between 1 and 4. Similarly, the MW distributions also show Gaussian-shaped curves for the peak value between 450 and 550 Da. The MW space covered by DrugSpaceX has higher values than the Drug Set because larger starting compounds have more opportunities to match the transformation rules and produce more derivatives. The peaks for HBA and HBD are 8 and 2, and both curves fall off rapidly from their maxima of 20 and 12. The number of H-bond acceptors and donors in DrugSpaceX compounds is twice as high as they are in the Drug Set. The TPSA and RotB distributions have peaks at approximately 100 Å2 and 10, respectively.
Figure 2.

Analysis of chemical properties. (A) Comparison of the property distributions for the Drug Set (blue) and DrugSpaceX (red). Relative frequencies of the descriptors logP (upper left), molecular weight (upper right), H-bond acceptors (middle left), H-bond donors (middle right), rotatable bonds (lower left), and TPSA (lower right). (B) A principal component analysis (PCA) plot comparing the chemical space defined by the DrugSpaceX databases: all compounds (red), the Drug Set (blue). (C) A molecular 3D shape analysis of the diversity of DrugSpaceX (red) and Drug Set (blue) by the principal moments of inertia. (D) Distribution of SA scores for all of the products contained in DrugSpaceX (red) and Drug Set (blue).
Diversity and novelty
To generate a representation of the chemical space covered by DrugSpaceX, a PCA of the chemical descriptors represented by DrugSpaceX was performed (Figure 2B). Analysis of the space spanned by the aforementioned physicochemical properties using the two main principal components, accounting for 70.24% and 18.02% of the X-variance, shows that DrugSpaceX overlaps with the property regions of known drug compounds. This indicates that most of the generated products are in principle drug-like. It also shows that the two databases do not completely overlap, and DrugSpaceX has effectively explored new regions of chemical space.
Molecular shape
Drug-like molecules can be classified in terms of shape by analysing the PMI of their 3D structures, which allows the classification of molecules as rods (linear shape, e.g. propyne), discs (cyclic planar shape, e.g. benzene), or spheres (globular shape, e.g., adamantane). Normalized PMI ratios (NPRs) are plotted into two-dimensional triangular graphs and then used to compare the shape space covered by different compound sets to rapidly assess and visualize the diversity in molecular shape associated with a given compound set (21). As shown in Figure 2C, the compounds of DrugSpaceX were subjected to the above shape analysis, and the results were compared with the data from the Drug Set. This figure shows that the vast majority of approved small molecule drugs are either rodlike or disklike, while the DrugSpaceX database densely populates the third dimension in shape space, suggesting that it contains many more scaffold types and spherical molecules that are rare in conventional compound libraries (45). Due to the application of transformation rules, the shape diversity of compounds reflects the transformation of drugs at different positions. As a result, by comparison with the Drug Set, the DrugSpaceX compounds almost entirely cover the whole area of three-dimensional chemical space, highlighting the advantage of this database in terms of diversity.
Synthetic accessibility
To obtain a computational assessment of the ease of synthesis for all of the products within DrugSpaceX, the SA score was computed. The distribution of SA scores, plotted in Figure 2D, is centred around the value of 4.0, with the vast majority lying below an SA score of 6.0, which indicates that they are easily synthesized products rather than overcomplex molecules.
In addition, we compared the size and key properties of DrugSpaceX with those of all comparable resources, and Supplementary Table S2 summarizes the available resources attempting to define the unknown chemical space (left column) and the size of DrugSpaceX. As the second largest virtual chemical library, in contrast to GDB-17, DrugSpaceX mainly aims to explore the drug-like chemical space efficiently. Regarding the compounds in this database, we care more about their similarity to existing drugs (Figure 3A), feasibility of synthesis (Figure 3B), and structural diversity (Figure 3C). Based on these analyses, we may find that DrugSpaceX shows outstanding drug-likeness, synthesizability, and large three-dimensional chemical space coverage.
Figure 3.

Key property distributions of different chemical libraries. (A) Molecular fingerprint-based similarity to the approved drugs, which quantifies the closest distance of chemicals to existing drugs; (B) The synthetic accessibility (SA) score calculated by an RDKit-based Python script, where a lower value indicates easier synthesis; (C) Structural diversity measured by molecular 3D shape analysis based on the principal moment of inertia (PMI), which allows the classification of molecules as rods (linear shape, e.g. propyne), discs (cyclic planar shape, e.g., benzene), or spheres (globular shape, e.g. adamantane). Considering the large size of these databases, 100 000 samples were randomly selected from each for analysis of these properties.
Website interface
The DrugSpaceX database can be accessed for free via our website (https://drugspacex.simm.ac.cn/), and all of the structures are available to download in the canonical SMILES format. As shown in Figure 4A, several subsets have been created to narrow the number of tailored to specific applications, which are also available to download. Generally, the user can manually write the compound structure using the SMILES notation or draw the 2D chemical structure at the ‘Draw Molecule Interface’ (Figure 4B) webpage and click the ‘search’ button to obtain a results page, where all of the compounds found in DrugSpaceX are depicted (Figure 4C). In addition, our UI design aims to simplify the interface and keep it lightweight. Only minimal information has been provided on the query result page, but detailed information can be accessed via functions such as mouse hovering and clicking, and the hitlist can be downloaded via an autohide sidebar. For each result, five compound properties are available in a radar map: logP, MW, HBA, HBD and RotB. A details page (Figure 4D) shows the physicochemical parameters and is also represented by a radar map with the corresponding descriptions. All of the chemical descriptors are computed using the RDKit library. In addition to chemical properties, we have provided the related ID codes and cross-reference links to other publicly available databases whenever a compound is present in other resources, including DrugBank, ChEMBL, BindingDB, PDB, CCDC, Sure ChEMBL, and so on. Additionally, DrugSpaceX could also provide transformation instructions, such as the transformation type and the position of transformation. The functional groups involved in the transformation have been highlighted in both the parent and child compounds. An example is given below, showing the ‘benzamide to aminoquinoline’ transformation of the drug ponatinib. The transformation details enable medicinal chemists to easily make systematic decisions on strategy planning and protection design. Because of the relatively large size of the library, we do not provide a downloadable link for all the 3D format molecules in the DrugSpaceX database, but the compound structures can be downloaded as a single file in SMILES notation or as a 2D or 3D SDF file for user convenience.
Figure 4.

Detailed view of the DrugSpaceX webpage: (A) Details of the subsets available to download; (B) Details of the search section; (C) Example of a search results page; (D) Details page for a DrugSpaceX molecule.
Case studies
Identification of Discoidin domain receptor 1 kinase inhibitors
Discoidin domain receptor 1 (DDR1) is a collagen-activated receptor tyrosine kinase implicated in fibrosis and other diseases that has attracted significant attention as a therapeutic target (46). Since 2013, at least eight chemotypes have been released as selective small molecule inhibitors of DDR1 (or DDR1 and DDR2), some of which have been modified from multitarget inhibitors (47). In this case study, the Drug Set of DrugSpaceX (https://drugspacex.simm.ac.cn/download/) was used to reposition known drugs to DDR1. Structure-based virtual screening was performed with Schrödinger's GLIDE docking program, and the top 10 drugs with the lowest docking scores were selected (Figure 5A). Among them, imatinib, nilotinib and ponatinib have been reported to show cross-reactivity to DDR1 (47). The first round of transformation products of these top 10 drugs were retrieved from DrugSpaceX as Set1. By searching available resources, including BindingDB, ChEMBL, PubChem, and SureChEMBL databases, we found that 152 drug analogues in Set1 (6.08%) are known, among which 16 compounds have reported kinase activity, and one compound has reported DDR1 activity. The Set1 compounds were further tested by docking. Figure 5A shows a t-SNE structural diversity distribution map of these drugs and analogues, among which the top 10 analogues (Table S3 in the Supporting Information) highlighted in blue are mainly located near ponatinib, indicating that transformations around ponatinib may be more promising for developing DDR1 inhibitors. Interestingly, a literature survey suggests that one of the top 10 analogues, i.e. the third-ranked compound DE209841, has been covered by a DDR1 inhibitor patent recently reported by Insilico Medicine (NO. WO2020079652A1). Figure 5B shows the putative binding mode of DE209841 (DDR1 docking score = –16.44 kcal/mol and ligand efficiency = –0.411 kcal/mol), which perfectly overlaps with cocrystallized ponatinib within the active site of DDR1 (PDB id: 3ZOS) and agrees well with the binding model proposed by Zhavoronkov et al (48). In the hinge-binding region, the imidazo[1,2-b]pyridazine group establishes a hydrogen bond with the backbone amide nitrogen atom of M704, which has proven to be essential in the binding of DDR1 inhibitors (41). Moreover, extensive hydrogen bonding interactions are formed with E672, V763, H764 and D784, resulting in a very low docking score. Similarly, the second round of transformation products of the top 10 analogues can be retrieved and screened by following the same procedures, leading to Set2. As shown in Figure 5C, the points on the heat map represent the compounds after two rounds of transformations. Among them, compound DE50204704, obtained by sequential modification to the highlighted red ‘tail’ and green ‘linker’ regions of ponatinib, yields a compound with a docking score and ligand efficiency score even higher than those of the first round of transformation analogues.
Figure 5.

Docking-based virtual screening of DDR1 inhibitors against DrugSpaceX compounds. (A) The t-SNE projection of the compounds in Set1, including the top 10 drugs repositioned in relation to DDR1 and their first round of transformation products. The chemical structure features were encoded as an ECFP4 512-bit vector for t-SNE analysis. (B) The putative binding mode of DE209841 (cyan carbons) derived from docking simulations compared to the crystallized ligand ponatinib (salmon carbons) in DDR1 kinase (PDB code: 3ZOS). (C) The t-SNE projection of the compounds in Set2 coloured by docking scores ranging from the lowest in orange to the highest in yellow. The compound DE50204704, showing the lowest docking score, can be traced back to ponatinib in two rounds of transformation.
CONCLUSION AND DISCUSSION
One of the most prominent problems in small molecule drug discovery is to efficiently explore the vast drug-like chemical space to find synthesizable and novel chemical structures with desired biological properties. In this study, with approved drugs as the starting point, we created the DrugSpaceX (https://drugspacex.simm.ac.cn) database based on expert-defined transformation rules to facilitate the reuse of drug molecules. The current release of DrugSpaceX contains >100 million chemical products for virtual screening. Cheminformatics analyses show that the database possesses outstanding structural novelty and diversity as well as large three-dimensional chemical space coverage. A case study illustrates that DrugSpaceX offers a viable alternative for rapid lead identification. Moreover, DrugSpaceX also provides good annotations and display functions, such as radar charts of physicochemical and drug-likeness properties and information on the transformation pathway from known parent drugs, which will be useful to guide the subsequent lead compound optimization process.
Furthermore, DrugSpaceX uses a concept that efficiently explores the vast drug-like chemical space, incorporating intelligent reaction knowledge into key considerations so that we can readily deliver available molecules with a desirable biological effect. These vast resources enable medicinal chemists to execute rapid scaffold-hopping experiments and rapid hit expansion in intellectual property-free territory and at low cost. Ultra-large-scale screening could improve the true positive rate. If the DrugSpaceX database can be combined with an ultra-large-scale screening platform similar to VirtualFlow, the quality of hits could be improved by expanding the initial screening scale, such as screening from a larger subset or even the entire library. Moreover, it is possible to consider adding clinical-stage compounds and corresponding analogues to the database in the same way to further expand the size of the database. In addition, since DrugSpaceX is a Web-based application, feedback from the scientific community can be gathered to update the products in future versions for improved utility.
DATA AVAILABILITY
DrugSapceX is freely available online at https://drugspacex.simm.ac.cn and can be accessed with a JavaScript-enabled browser.
Supplementary Material
ACKNOWLEDGEMENTS
The DrugSpaceX website is licensed under a Creative Commons Attribution-NonCommercial 4.0 International (CC BY-NC 4.0), which permits reusers to distribute, remix, adapt, and build upon the material in any medium or format, for noncommercial purposes only. If material is not included in the article's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit https://creativecommons.org/licenses/by-nc/4.0/. The authors would like to thank ChemAxon and StarDrop™ for their support as well as all users of DrugSpaceX for their valuable feedback and suggestions.
Contributor Information
Tianbiao Yang, Drug Discovery and Design Center, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, China; Department of Pharmacy, University of Chinese Academy of Sciences, No.19A Yuquan Road, Beijing 100049, China; School of Pharmaceutical Science and Technology, Hangzhou Institute for Advanced Study, UCAS, Hangzhou 310024, China.
Zhaojun Li, School of Information Management, Dezhou University, No. 566 University Rd. West, Dezhou 253023, Shandong, China.
Yingjia Chen, Drug Discovery and Design Center, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, China; Department of Pharmacy, University of Chinese Academy of Sciences, No.19A Yuquan Road, Beijing 100049, China.
Dan Feng, Drug Discovery and Design Center, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, China; Department of Chemistry, College of Sciences, Shanghai University, Shanghai, China.
Guangchao Wang, School of Information Management, Dezhou University, No. 566 University Rd. West, Dezhou 253023, Shandong, China.
Zunyun Fu, Drug Discovery and Design Center, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, China; Nanjing University of Chinese Medicine, 138 Xianlin Road, Jiangsu, Nanjing 210023, China.
Xiaoyu Ding, Drug Discovery and Design Center, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, China; Department of Pharmacy, University of Chinese Academy of Sciences, No.19A Yuquan Road, Beijing 100049, China.
Xiaoqin Tan, Drug Discovery and Design Center, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, China; Department of Pharmacy, University of Chinese Academy of Sciences, No.19A Yuquan Road, Beijing 100049, China.
Jihui Zhao, Drug Discovery and Design Center, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, China; Department of Pharmacy, University of Chinese Academy of Sciences, No.19A Yuquan Road, Beijing 100049, China.
Xiaomin Luo, Drug Discovery and Design Center, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, China; Department of Pharmacy, University of Chinese Academy of Sciences, No.19A Yuquan Road, Beijing 100049, China.
Kaixian Chen, Drug Discovery and Design Center, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, China; Department of Pharmacy, University of Chinese Academy of Sciences, No.19A Yuquan Road, Beijing 100049, China.
Hualiang Jiang, Drug Discovery and Design Center, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, China; Department of Pharmacy, University of Chinese Academy of Sciences, No.19A Yuquan Road, Beijing 100049, China; School of Pharmaceutical Science and Technology, Hangzhou Institute for Advanced Study, UCAS, Hangzhou 310024, China; School of Life Science and Technology, ShanghaiTech University, 393 Huaxiazhong Road, Shanghai 200031, China.
Mingyue Zheng, Drug Discovery and Design Center, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, China; Department of Pharmacy, University of Chinese Academy of Sciences, No.19A Yuquan Road, Beijing 100049, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [81773634 to M.Z.]; National Science & Technology Major Project ‘Key New Drug Creation and Manufacturing Program’, China [2018ZX09711002 to H.J.]; ‘Personalized Medicines—Molecular Signature-based Drug Discovery and Development’, Strategic Priority Research Program of the Chinese Academy of Sciences [XDA12050201 to M.Z.]. Funding for open access charge: National Natural Science Foundation of China [81773634 to M.Z.]; National Science & Technology Major Project ‘Key New Drug Creation and Manufacturing Program’, China [2018ZX09711002 to H.J.]; ‘Personalized Medicines—Molecular Signature-based Drug Discovery and Development’, Strategic Priority Research Program of the Chinese Academy of Sciences [XDA12050201 to M.Z.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Shoichet B.K. Virtual screening of chemical libraries. Nature. 2004; 432:862–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhang L., Zheng M.Y., Liu H.. Diversity-oriented synthesis and its application in drug discovery. Yao Xue Xue Bao. 2015; 50:419–433. [PubMed] [Google Scholar]
- 3. Chuprina A., Lukin O., Demoiseaux R., Buzko A., Shivanyuk A.. Drug- and lead-likeness, target class, and molecular diversity analysis of 7.9 million commercially available organic compounds provided by 29 suppliers. J. Chem. Inf. Model. 2010; 50:470–479. [DOI] [PubMed] [Google Scholar]
- 4. Reymond J.L. The chemical space project. Acc. Chem. Res. 2015; 48:722–730. [DOI] [PubMed] [Google Scholar]
- 5. Ruddigkeit L., van Deursen R., Blum L.C., Reymond J.L.. Enumeration of 166 billion organic small molecules in the chemical universe database GDB-17. J. Chem. Inf. Model. 2012; 52:2864–2875. [DOI] [PubMed] [Google Scholar]
- 6. Visini R., Awale M., Reymond J.L.. Fragment Database FDB-17. J. Chem. Inf. Model. 2017; 57:700–709. [DOI] [PubMed] [Google Scholar]
- 7. Awale M., Sirockin F., Stiefl N., Reymond J.L.. Medicinal chemistry aware database GDBMedChem. Mol. Inf. 2019; 38:e1900031. [DOI] [PubMed] [Google Scholar]
- 8. Buhlmann S., Reymond J.L.. ChEMBL-likeness score and database GDBChEMBL. Front. Chem. 2020; 8:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Opassi G., Gesu A., Massarotti A.. The hitchhiker's guide to the chemical-biological galaxy. Drug Discov. Today. 2018; 23:565–574. [DOI] [PubMed] [Google Scholar]
- 10. Dorschner K.V., Toomey D., Brennan M.P., Heinemann T., Duffy F.J., Nolan K.B., Cox D., Adamo M.F.A., Chubb A.J.. TIN − a combinatorial compound collection of synthetically feasible multicomponent synthesis products. J. Chem. Inf. Model. 2011; 51:986–995. [DOI] [PubMed] [Google Scholar]
- 11. Levré D., Arcisto C., Mercalli V., Massarotti A.. ZINClick v.18: expanding chemical space of 1,2,3-triazoles. J. Chem. Inf. Model. 2019; 59:1697–1702. [DOI] [PubMed] [Google Scholar]
- 12. Chevillard F., Kolb P.. SCUBIDOO: a large yet screenable and easily searchable database of computationally created chemical compounds optimized toward high likelihood of synthetic tractability. J. Chem. Inf. Model. 2015; 55:1824–1835. [DOI] [PubMed] [Google Scholar]
- 13. Gorgulla C., Boeszoermenyi A., Wang Z.F., Fischer P.D., Coote P.W., Padmanabha Das K.M., Malets Y.S., Radchenko D.S., Moroz Y.S., Scott D.A. et al.. An open-source drug discovery platform enables ultra-large virtual screens. Nature. 2020; 580:663–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Optibrium StarDrop. 2019; Optibrium Ltd; version 6.6. [Google Scholar]
- 15. Ujváry I., Hayward J.. Brown N. BIOSTER: a database of bioisosteres and bioanalogues. Bioisosteres in Medicinal Chemistry. 2012; Wiley VCH; 53–74. [Google Scholar]
- 16. Pillaiyar T., Meenakshisundaram S., Manickam M., Sankaranarayanan M.. A medicinal chemistry perspective of drug repositioning: Recent advances and challenges in drug discovery. Eur. J. Med. Chem. 2020; 195:112275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ashburn T.T., Thor K.B.. Drug repositioning: identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004; 3:673–683. [DOI] [PubMed] [Google Scholar]
- 18. Awale M., van Deursen R., Reymond J.-L.. MQN-Mapplet: visualization of chemical space with interactive maps of DrugBank, ChEMBL, PubChem, GDB-11, and GDB-13. J. Chem. Inf. Model. 2013; 53:509–518. [DOI] [PubMed] [Google Scholar]
- 19. Awale M., Reymond J.L.. Web-based 3D-visualization of the DrugBank chemical space. J. Cheminform. 2016; 8:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Awale M., Probst D., Reymond J.-L.. WebMolCS: a web-based interface for visualizing molecules in three-dimensional chemical spaces. J. Chem. Inf. Model. 2017; 57:643–649. [DOI] [PubMed] [Google Scholar]
- 21. Sauer W.H.B., Schwarz M.K.. Molecular shape diversity of combinatorial libraries: a prerequisite for broad bioactivity. J. Chem. Inf. Comput. Sci. 2003; 43:987–1003. [DOI] [PubMed] [Google Scholar]
- 22. Wu M., Vogt M., Maggiora G.M., Bajorath J.. Design of chemical space networks on the basis of Tversky similarity. J. Comput.-Aided Mol. Des. 2016; 30:1–12. [DOI] [PubMed] [Google Scholar]
- 23. Awale M., Reymond J.-L.. Similarity Mapplet: Interactive visualization of the directory of useful decoys and ChEMBL in high dimensional chemical spaces. J. Chem. Inf. Model. 2015; 55:1509–1516. [DOI] [PubMed] [Google Scholar]
- 24. Probst D., Reymond J.-L.. Visualization of very large high-dimensional data sets as minimum spanning trees. J. Cheminform. 2020; 12:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rosen J., Lovgren A., Kogej T., Muresan S., Gottfries J., Backlund A.. ChemGPS-NP(Web): chemical space navigation online. J. Comput.-Aided Mol. Des. 2009; 23:253–259. [DOI] [PubMed] [Google Scholar]
- 26. Borrel A., Kleinstreuer N.C., Fourches D.. Exploring drug space with ChemMaps.com. Bioinformatics. 2018; 34:3773–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wishart D.S., Feunang Y.D., Guo A.C., Lo E.J., Marcu A., Grant J.R., Sajed T., Johnson D., Li C., Sayeeda Z. et al.. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic. Acids. Res. 2018; 46:D1074–D1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E.. The Protein Data Bank. Nucleic. Acids. Res. 2000; 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gilson M.K., Liu T., Baitaluk M., Nicola G., Hwang L., Chong J.. BindingDB in 2015: a public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016; 44:D1045–D1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mendez D., Gaulton A., Bento A.P., Chambers J., De Veij M., Felix E., Magarinos M.P., Mosquera J.F., Mutowo P., Nowotka M et al.. ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res. 2019; 47:D930–D940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Groom C.R., Bruno I.J., Lightfoot M.P., Ward S.C.. The cambridge structural database. Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater. 2016; 72:171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wildman S.A., Crippen G.M.. Prediction of physicochemical parameters by atomic contributions. J. Chem. Inf. Comput. Sci. 1999; 39:868–873. [Google Scholar]
- 33. Bickerton G.R., Paolini G.V., Besnard J., Muresan S., Hopkins A.L.. Quantifying the chemical beauty of drugs. Nat. Chem. 2012; 4:90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ivanenkov Y.A., Zagribelnyy B.A., Aladinskiy V.A.. Are we opening the door to a new era of medicinal chemistry or being collapsed to a chemical singularity. J. Med. Chem. 2019; 62:10026–10043. [DOI] [PubMed] [Google Scholar]
- 35. Lipinski C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods. 2000; 44:235–249. [DOI] [PubMed] [Google Scholar]
- 36. Doak B.C., Over B., Giordanetto F., Kihlberg J.. Oral druggable space beyond the rule of 5: insights from drugs and clinical candidates. Chem. Biol. 2014; 21:1115–1142. [DOI] [PubMed] [Google Scholar]
- 37. Abdi H., Williams L.J.. Principal component analysis. WIREs Comput. Stat. 2010; 2:433–459. [Google Scholar]
- 38. Segall M., Champness E., Leeding C., Lilien R., Mettu R., Stevens B.. Applying medicinal chemistry transformations and multiparameter optimization to guide the search for high-quality leads and candidates. J. Chem. Inf. Model. 2011; 51:2967–2976. [DOI] [PubMed] [Google Scholar]
- 39. Ertl P., Schuffenhauer A.. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 2009; 1:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shelley J.C., Cholleti A., Frye L.L., Greenwood J.R., Timlin M.R., Uchimaya M.. Epik: a software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput.-Aided Mol. Des. 2007; 21:681–691. [DOI] [PubMed] [Google Scholar]
- 41. Canning P., Tan L., Chu K., Lee S.W., Gray N.S., Bullock A.N.. Structural mechanisms determining inhibition of the collagen receptor DDR1 by selective and multi-targeted type II kinase inhibitors. J. Mol. Biol. 2014; 426:2457–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Reynolds C.H., Bembenek S.D., Tounge B.A.. The role of molecular size in ligand efficiency. Bioorg. Med. Chem. Lett. 2007; 17:4258–4261. [DOI] [PubMed] [Google Scholar]
- 43. Teague S.J., Davis A.M., Leeson P.D., Oprea T.. The design of leadlike combinatorial libraries. Angew. Chem. Int. Ed. 1999; 38:3743–3748. [DOI] [PubMed] [Google Scholar]
- 44. Carr R.A.E., Congreve M., Murray C.W., Rees D.C.. Fragment-based lead discovery: leads by design. Drug Discov. Today. 2005; 10:987–992. [DOI] [PubMed] [Google Scholar]
- 45. Prosser K.E., Stokes R.W., Cohen S.M.. Evaluation of 3-dimensionality in approved and experimental drug space. ACS Med. Chem. Lett. 2020; 11:1292–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Moll S., Desmoulière A., Moeller M.J., Pache J.-C., Badi L., Arcadu F., Richter H., Satz A., Uhles S., Cavalli A. et al.. DDR1 role in fibrosis and its pharmacological targeting. Biochim. Biophys. Acta (BBA) - Mol. Cell Res. 2019; 1866:118474. [DOI] [PubMed] [Google Scholar]
- 47. Li Y., Lu X., Ren X., Ding K.. Small molecule discoidin domain receptor kinase inhibitors and potential medical applications. J. Med. Chem. 2015; 58:3287–3301. [DOI] [PubMed] [Google Scholar]
- 48. Zhavoronkov A., Ivanenkov Y.A., Aliper A., Veselov M.S., Aladinskiy V.A., Aladinskaya A.V., Terentiev V.A., Polykovskiy D.A., Kuznetsov M.D., Asadulaev A. et al.. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 2019; 37:1038–1040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
DrugSapceX is freely available online at https://drugspacex.simm.ac.cn and can be accessed with a JavaScript-enabled browser.