


Abstract
With the advent of next-generation sequencing, large-scale initiatives for mining whole genomes and exomes have been employed to better understand global or population-level genetic architecture. India encompasses more than 17% of the world population with extensive genetic diversity, but is under-represented in the global sequencing datasets. This gave us the impetus to perform and analyze the whole genome sequencing of 1029 healthy Indian individuals under the pilot phase of the ‘IndiGen’ program. We generated a compendium of 55,898,122 single allelic genetic variants from geographically distinct Indian genomes and calculated the allele frequency, allele count, allele number, along with the number of heterozygous or homozygous individuals. In the present study, these variants were systematically annotated using publicly available population databases and can be accessed through a browsable online database named as ‘IndiGenomes’ http://clingen.igib.res.in/indigen/. The IndiGenomes database will help clinicians and researchers in exploring the genetic component underlying medical conditions. Till date, this is the most comprehensive genetic variant resource for the Indian population and is made freely available for academic utility. The resource has also been accessed extensively by the worldwide community since it's launch.
Graphical Abstract
Graphical Abstract.
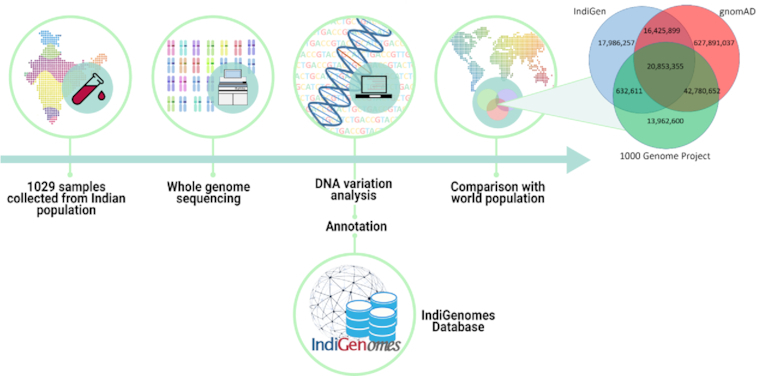
Overview of the Indigenomes sample collection and data analysis.
INTRODUCTION
India is the second largest country in terms of population density with more than 1.3 billion individuals encompassing 17% of the world population. The country is very diverse with more than 4500 anthropologically distinct populations (1). These populations have been segregated on the bases of caste, tribe and religious groups that differ in terms of cultural practices, geographical locations, climatic conditions, physical features, marriage practices, linguistics, as well as their genetic architecture (1,2). India is also considered as one of the major southern coastal routes for human migration out of Africa, and in the recent past was witness to multiple waves of migrations and invasions. These resulted in the enrichment of the genetic diversity of the Indian subcontinent (3). Despite having this rich genetic diversity, India has been under-represented in global genome studies (4). Furthermore, the population in India is also stratified into multiple large endogamous groups and is also characterized by consanguineous marriages. This has resulted in high prevalence of recessive alleles in the Indian population. In the absence of large-scale whole genome studies from India, these sub-population-specific genetic variants are also not adequately captured and catalogued in global medical literature (5).
Over the past decade, the advent of next generation sequencing and its increasing affordability has revolutionized the understanding of the genetic architecture of various populations across the globe (6–12). In this effort, multiple global population datasets including the 1000 genome project (7), ExAC (13), ESP6500 (https://evs.gs.washington.edu/EVS/) and gnomAD (14) have generated reference and patient genome datasets from populations across the continents. Although these datasets include the genomes from Indian individuals, the number is relatively small to represent the genetic diversity and heterogeneity of the Indian population (4,15). Apart from the global datasets, few Asian and Indian population specific studies have been conducted to understand the genetic architecture in this part of the world. The Indian Genome Variation (IGV) consortium study used SNP- based genotyping of 900 genes from over 1800 individuals across 55 sub-populations to underscore the heterogeneity of the Indian population (16). This study led to the discovery of unique founder mutations in the Indian subcontinent and better understanding of specific genetic markers establishing distinct genotype-phenotype correlations. Recently, the GenomeAsia100K study addressed a wide range of questions focusing on specific Asian population groups (17). This project included 598 samples from India, primarily tribal groups and specific castes majorly from the southern part of India. India being a country of more than one billion people, these datasets only represent a fraction of the genetic diversity. The sampling for genetic/ genomic studies from India needs to be performed extensively focusing on cultural, ethnic and geographical diversity. Population specific genome sequencing of Indian individuals can help in characterizing variants or polymorphisms associated with diseases, improving precision medicine outcomes, building population specific reference genome datasets, efficiently imputing genotype data and can be used for phasing and haplotype predictions (18–23).
In order to better understand the genetic architecture of Indian population, whole-genome sequencing of 1029 healthy Indian individuals was performed under the ‘IndiGen’ program as a pilot exercise. Using these genomes, we have built one of the largest and well-annotated Indian population variant databases called IndiGenomes. The database contains information on variant allele frequency, allele number, allele count, number of heterozygous and homozygous individuals recorded in this study. This will help clinicians and researchers in querying variants for various medical applications and will enable them to differentiate the pathogenic variants from the benign ones in the context of Indian population. The IndiGenomes database is publicly available at the URL http://clingen.igib.res.in/indigen/.
MATERIALS AND METHODS
Participant selection
A total of 1029 self-declared healthy individuals were selected from a pool of volunteers. The volunteers were selected to represent the different states in India and their ancestries were also mapped to the geographical locations. Out of 1029 participants, 495 were males and 534 were females with mean age of 41.35 and 32.96 years respectively. The information regarding the number of participants selected from each state of India is tabulated in Supplementary Data 1. We have also marked on a map of India the geographical location of the sampled individuals (Supplementary Data 2). This study was approved by the Institutional Human Ethics Committee (IHEC) of CSIR-Institute of Genomics and Integrative Biology. The participants were explained about the informed consent process as per the approved IHEC guidelines.
Genome sequencing and analysis
After informed consent, 5 ml of blood was collected from each volunteer using venipuncture and genomic DNA was extracted using the salting out method (24). Whole-genome libraries were prepared using TruSeq DNA PCR free library preparation kit as per manufacturer's instructions (Illumina Inc. San Diego, CA, USA, Cat. no. FC-121-9006DOC). Following the library preparation, sequencing was performed on Illumina NovaSeq 6000 platform (Illumina Inc. San Diego, CA, USA) and data was generated as 150 × 2 bp paired-end reads. Alignment, post-processing and default quality filtered variant calling was performed on the Illumina DRAGEN v3.4 Bio-IT platform (Illumina Inc. San Diego, CA, USA) using GRCh38 as a human reference genome. The ‘Dynamic Read Analysis for GENomics’ (DRAGEN) pipeline functions on massively parallel highly reconfigurable field-programmable gate array (FPGA) logic that maximizes the speed without compromising the quality of the data (25). The joint variant calling was performed using Sentieon that mirrors the tools and protocol of GATK gold standard with a highly optimized back-end, which makes it faster with identical accuracy (26–28). Sentieon calculates the allele frequency, allele count and allele number for the variants while performing joint calling. Further analysis was carried out only on single allelic variants. We calculated the number of heterozygous and homozygous individuals in the joint variant call format file using bespoke shell script.
Annotation of variants
The variants were systematically annotated using ANNOVAR (v.2018-04-16) (29), which comprises of annotations from multiple databases such as RefGene (30), dbSNP (31), dbNSFP35a (32) and various global population databases (7,14,17). RefGene database contains a curated and non-redundant sequence of genomes, transcripts and proteins of 3774 organisms. This database was employed for annotating location of variants in the genome (30). dbSNP (avsnp150) serves as the largest repository for human genomic variations and was used to provide a unique identifier to the variants (31). In order to interpret the functional effect of the variants, dbNSFP35a database was used. It comprises of multiple variant pathogenicity prediction tools such as SIFT, Polyphen, LRT, MutationTaster, PROVEAN, MutationAccessor, FATHMM, RadialSVM, CADD, DANN and phylop among others (32). Allele frequencies of the variants were fetched from global population databases such as Genome Aggregation Database (gnomAD v3; repository of 71,702 whole genome sequenced individuals from eight populations) (14), 1000 Genome project (1000g2015aug_all; catalogue of 2504 whole genome sequence data of healthy individuals from five super populations including 26 subpopulations) (7), ESP6500 (esp6500siv2_all; comprising of 6503 whole exome data from 2203 African-American and 4300 European-American individuals), ExAC (exac03; exome data of 60 706 unrelated individuals from five ancestries) (33), and the Greater Middle East (GME) Variome Project (includes high-quality exome data of 1,111 unrelated individuals from six different regions) (11). To understand the clinical significance of the variants in Indian population, ClinVar, the repository of clinically interpreted human genetic variants, was used to identify variants known in disease contexts (34).
Comparison with the global datasets
The compendium of variants in IndiGenomes database was overlapped with the global population datasets to identify the common and unique variants. The publicly available VCF files without genotype information were downloaded from gnomAD v3 and 1000g2015aug_all followed by matching of the genomic location, reference, and alternate alleles of the variants using a bespoke shell script.
Database and web server
A web-based search tool was integrated with the IndiGenomes database to allow the users to easily explore the genes and variants found in the Indian population. The database was converted into JavaScript Object Notation (JSON). All variants were stored into MongoDB v3.4.10 and the server was hosted using Apache HTTP server. Access to the data was provided through a web interface running on the Apache HTTP server and PHP 7.0. MongoDB v3.4.10 was used to keep track of data processing and database through the web interface. The search query was optimized using indexing in MongoDB, with most search terms, including gene names, variant and dbSNP ID populating the search bar. Optimization of the search query in MongoDB allows faster loading. The web interface was coded in HTML5, CSS3, Bootstrap version 4.0 (Material Design) and AngularJS. Bootstrap is the world's most popular framework for building responsive, mobile-first sites that enable database web-based system optimizations feasible for mobile browsing. The web-based system is fully responsive and compatible with all devices like desktops, tablets and smartphones. The back-end of the database was constructed using PHP 7.0. AngularJS gives users a rich and responsive experience. AngularJS and PHP 7.0 scripts were used to retrieve and bind the data from the database.
RESULTS
Genome sequencing, analysis and genetic variant classification
Whole genome sequencing was undertaken for 1029 self-declared healthy individuals with an average genome coverage of ∼30×. Analysis of the sequenced genomes led to the identification of 55,898,122 single allelic genetic variants that mapped to the human reference genome GRCh38. The genetic variants were annotated using ANNOVAR, which provides genomic location and functionality based on the RefGene database (30). In context of the genomic location, 3,952,209 variants (7.07%) mapped to exonic and ncRNA exonic region, 31,134,798 variants (55.69%) mapped to intergenic region, 19,455,670 variants (34.80%) mapped to intronic region and 4,147 variants (0.007%) mapped to splicing region in the genome. The remaining 1,351 298 variants (2.41%) mapped to the untranslated region (UTR), downstream, upstream and undetermined regions in the genome. The variant classification has been depicted in the pie-chart in Figure 1A.
Figure 1.

Classification of the variants: (A) Based on the genomic location; (B) Based on type of variant in the exonic region.
Further, 497,390 exonic variants were classified on the basis of variant type. There were 287,289 (57.75%) nonsynonymous variants, 182,694 (36.73%) synonymous variants, 9,373 (1.88%) frameshift variants, 7,174 (1.44%) non-frameshift variants, 6,996 stop-gain (1.40%) and 343 (0.068%) stop-loss variants. The remaining 3,521 (0.70%) variants were classified under unknown variant type. The classification of exonic variants has been depicted in Figure 1B.
Variant comparison with the global datasets
We compared the variants of IndiGenomes database with the global population databases gnomAD and 1000 Genomes project. We found 37,249,254 (66.63%) variants in the IndiGenomes common with the gnomAD database and 21,485,966 (38.43%) variants common with the 1000 Genomes project. There were 20,853,355 (37.30%) variants common in all the three databases whereas 18,016,257 (32.23%) variants were unique to only IndiGenomes database. A Venn diagram representing variant numbers in each database is shown in Figure 2.
Figure 2.
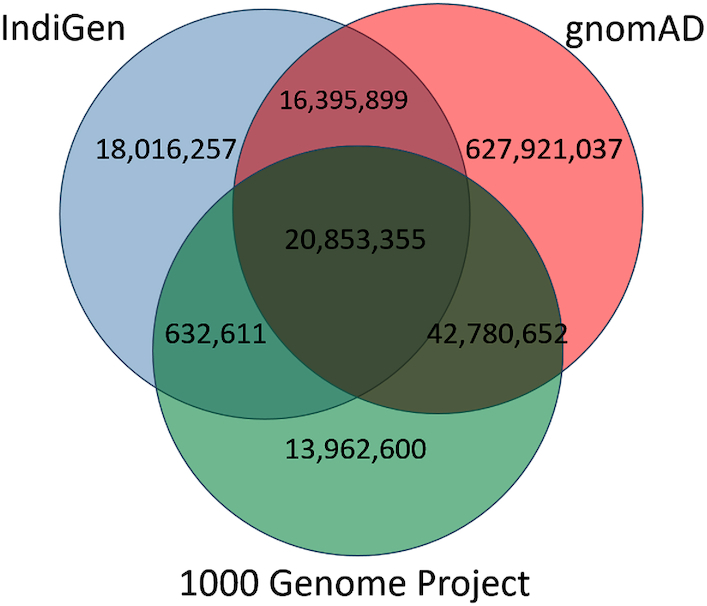
IndiGenomes variant number comparison with the 1000 Genomes Project and gnomAD.
Database interface and features
A web-based search tool has been created for querying genes and their variants from IndiGenomes. The home page of the search tool has been made user-friendly and contains the search bar as well as a downloadable user manual. The VCF file containing variants unique to IndiGenomes has been made available for download. The home page also includes the graphical representation of variant number comparison with different datasets, variant classification based on genomic location, and variant type for all the exonic variants.
The search bar option has been designed in such a way that a user can simply enter the query using either the gene name, dbSNP ID or variant ID in the given format as chromosome number-position-reference-alternate or chromosome number:position:reference:alternate. The search by variant (GRCh38) and search by dbSNP ID are case sensitive, while the search by gene name is case insensitive. When a query term is entered in the search bar, the list of all the matching entries from the database will be retrieved. The query search results will be displayed below the example search box in the form of a table, constructed by bootstrap 4 data-tables with the basic information summarized, including gene, chromosome, position, ref, alt, dbSNP ID, gene function and exonic function as shown in Figure 3. The query search can be further customized using the search bar present in the ‘Search results’ box. Additional detailed annotation information about a specific variant can be obtained by selecting the variant entry. Upon selecting a specific variant from the search results table, a new page will open with all the information about the queried variant. The additional results page contains two panels, as shown in Figure 4.
Figure 3.

Result display for the search query.
Figure 4.

Left and right panels showing the detailed annotation of the variant.
The left panel of the variant search page consists of three windows. The first window ‘Gene’ provides the basic information about the variant, i.e. gene name, chromosome number, chromosomal position, reference allele, alternate allele, amino acid change, UCSC variant location (35), OMIM ID (36), dbSNP ID (31), gene function, exonic function, including links to various external resources. The second window ‘Clinical Annotations and Linkouts’, contains details about the disease condition(s) and clinical significance of the variant as mentioned in the ClinVar database. The third window ‘Computational prediction and annotation’ provides the predictions of pathogenicity of the variant from various tools.
The right panel of the variant search page consists of three windows. The first window ‘IndiGenome allele frequency’ provides the information about the allele count, allele number, allele frequency as well as number of individuals being heterozygous or homozygous for the variant queried in the IndiGenomes dataset. The second window ‘Global allele frequencies’ provides the frequency of the variant in various publicly available global population databases that includes 1000 Genomes Project (7), gnomAD (14), GME (11) and Esp6500 (https://evs.gs.washington.edu/EVS/). The third window ‘Update on clinical annotations’ provides an option to the users to submit a variant for retrieving the variant annotation according to the American College of Medical Genetics and Genomics or Association of Molecular Pathology (ACMG/AMP) guidelines (37). To obtain this annotation, the user needs to register using email id and contact telephone number. After successful registration, the user will be able to submit variants by logging on to the account using login credentials. Once variant annotation based on automated ACMG/AMP criteria is updated, the user will be notified via email.
DISCUSSION
It is well documented in literature that the Indian population comprises of multiple endogamous groups. These communities widely practice consanguineous marriage traditions, leading to accumulation of rare deleterious variants (38). These rare/unique variations amplify as founder mutations within specific endogamous populations, which in due course translates to population specific genetic diseases. It is estimated that ∼70 million people in India are affected by genetic diseases with about 64 out of 1000 live births carrying disorders such as primary immunodeficiencies, lysosomal storage disorders, mitochondrial diseases, cardiac disorders and muscle-related disorders (5). The government of India is now focusing on effective cataloging of the genomic variants specific for the Indian population so as to create a blueprint for identification of disease epidemiology, population-specific variants and pharmacogenomic markers ultimately leading to the development of effective therapies for treating genetic disorders (https://pib.gov.in/PressReleseDetailm.aspx?PRID=1605509). In this context, the IndiGen programme aims to create a compendium of genetic variants representing the contemporary Indian population with an objective to classify variants involved in mendelian disorders and improve precision medicine outcomes. The resource can also enable the identification of markers for carrier screening, prevention of adverse events and provide better diagnosis and optimal therapy through mining data of clinically actionable pharmacogenetic variants. The phased data will allow researchers to build Indian-specific reference genome dataset and efficiently impute haplotype information.
The IndiGenomes database includes over 18 million unique variants. This can provide useful insights for clinicians and researchers in comprehending genetics not only at the population level but at the individual level. As of 8 August 2020, there are ∼200,000 pageviews on the IndiGenomes web page from users spanning 27 countries. The highest number of searches in the database till date correspond to the genes ACE2, MVK, BRCA1, AGRN, SCO2, DSP, SIRT2, TRDN, ITGB3, LHB, NDUFB3, and SNTA1. It is intriguing to note that during the COVID-19 pandemic in the year 2020, ACE2 variants have been widely queried using the database. It has been predicted that ACE2 variants can potentially alter host susceptibility to SARS-CoV2 virus (39,40). This depicts the utility of such population-scale databases in understanding the genetics of specific genes and variants of human interest in the Indian population. Currently, the IndiGenomes database enables the search of only single-allelic variants. However, we envision addition of multiallelic variations and structural variations in the future updates of the database. To the best of our knowledge, this is the most comprehensive large-scale database of genetic variations specific to the Indian population.
DATA AVAILABILITY
The variants unique to IndiGenomes have been submitted to the NCBI Variation Submission Portal with submission ID: SUB8153462 and also made available for download on the IndiGenomes database website http://clingen.igib.res.in/indigen/.
Supplementary Material
ACKNOWLEDGEMENTS
Authors acknowledge the volunteers who participated in this study. Authors acknowledge Mukta Poojary for constructive suggestions and Sr. B. Velangini, Principal, St. Pious X Degree & PG College for Women, Hyderabad for encouragement.
Contributor Information
Abhinav Jain, CSIR-Institute of Genomics and Integrative Biology, New Delhi 110025, India; Academy of Scientific and Innovative Research (AcSIR), Ghaziabad, Uttar Pradesh 201002, India.
Rahul C Bhoyar, CSIR-Institute of Genomics and Integrative Biology, New Delhi 110025, India.
Kavita Pandhare, CSIR-Institute of Genomics and Integrative Biology, New Delhi 110025, India.
Anushree Mishra, CSIR-Institute of Genomics and Integrative Biology, New Delhi 110025, India.
Disha Sharma, CSIR-Institute of Genomics and Integrative Biology, New Delhi 110025, India.
Mohamed Imran, CSIR-Institute of Genomics and Integrative Biology, New Delhi 110025, India; Academy of Scientific and Innovative Research (AcSIR), Ghaziabad, Uttar Pradesh 201002, India.
Vigneshwar Senthivel, CSIR-Institute of Genomics and Integrative Biology, New Delhi 110025, India; Academy of Scientific and Innovative Research (AcSIR), Ghaziabad, Uttar Pradesh 201002, India.
Mohit Kumar Divakar, CSIR-Institute of Genomics and Integrative Biology, New Delhi 110025, India; Academy of Scientific and Innovative Research (AcSIR), Ghaziabad, Uttar Pradesh 201002, India.
Mercy Rophina, CSIR-Institute of Genomics and Integrative Biology, New Delhi 110025, India; Academy of Scientific and Innovative Research (AcSIR), Ghaziabad, Uttar Pradesh 201002, India.
Bani Jolly, CSIR-Institute of Genomics and Integrative Biology, New Delhi 110025, India; Academy of Scientific and Innovative Research (AcSIR), Ghaziabad, Uttar Pradesh 201002, India.
Arushi Batra, CSIR-Institute of Genomics and Integrative Biology, New Delhi 110025, India; Academy of Scientific and Innovative Research (AcSIR), Ghaziabad, Uttar Pradesh 201002, India.
Sumit Sharma, CSIR-Institute of Genomics and Integrative Biology, New Delhi 110025, India.
Sanjay Siwach, CSIR-Institute of Genomics and Integrative Biology, New Delhi 110025, India.
Arun G Jadhao, Department of Zoology, RTM Nagpur University, Nagpur, Maharashtra 440033, India.
Nikhil V Palande, Department of Zoology, Shri Mathuradas Mohota College of Science, Nagpur, Maharashtra 440009, India.
Ganga Nath Jha, Department of Anthropology, Vinoba Bhave University, Hazaribag, Jharkhand 825301, India.
Nishat Ashrafi, Department of Anthropology, Vinoba Bhave University, Hazaribag, Jharkhand 825301, India.
Prashant Kumar Mishra, Department of Biotechnology, Vinoba Bhave University, Hazaribag, Jharkhand 825301, India.
Vidhya A. K., Department of Biochemistry, Dr. Kongu Science and Art College, Erode, Tamil Nadu 638107, India.
Suman Jain, Thalassemia and Sickle cell Society, Hyderabad, Telangana 500052, India.
Debasis Dash, CSIR-Institute of Genomics and Integrative Biology, New Delhi 110025, India; Academy of Scientific and Innovative Research (AcSIR), Ghaziabad, Uttar Pradesh 201002, India.
Nachimuthu Senthil Kumar, Department of Biotechnology, Mizoram University, Aizawl, Mizoram 796004, India.
Andrew Vanlallawma, Department of Biotechnology, Mizoram University, Aizawl, Mizoram 796004, India.
Ranjan Jyoti Sarma, Department of Biotechnology, Mizoram University, Aizawl, Mizoram 796004, India.
Lalchhandama Chhakchhuak, Department of Pathology, Civil Hospital Aizawl, Mizoram 796001, India.
Shantaraman Kalyanaraman, TVMC, Tirunelveli Medical College, Tirunelveli, Tamil Nadu 627011, India.
Radha Mahadevan, TVMC, Tirunelveli Medical College, Tirunelveli, Tamil Nadu 627011, India.
Sunitha Kandasamy, TVMC, Tirunelveli Medical College, Tirunelveli, Tamil Nadu 627011, India.
Pabitha B. M., TVMC, Tirunelveli Medical College, Tirunelveli, Tamil Nadu 627011, India.
Raskin Erusan Rajagopal, TVMC, Tirunelveli Medical College, Tirunelveli, Tamil Nadu 627011, India.
Ezhil Ramya J., TVMC, Tirunelveli Medical College, Tirunelveli, Tamil Nadu 627011, India.
Nirmala Devi P., TVMC, Tirunelveli Medical College, Tirunelveli, Tamil Nadu 627011, India.
Anjali Bajaj, CSIR-Institute of Genomics and Integrative Biology, New Delhi 110025, India; Academy of Scientific and Innovative Research (AcSIR), Ghaziabad, Uttar Pradesh 201002, India.
Vishu Gupta, CSIR-Institute of Genomics and Integrative Biology, New Delhi 110025, India; Academy of Scientific and Innovative Research (AcSIR), Ghaziabad, Uttar Pradesh 201002, India.
Samatha Mathew, CSIR-Institute of Genomics and Integrative Biology, New Delhi 110025, India; Academy of Scientific and Innovative Research (AcSIR), Ghaziabad, Uttar Pradesh 201002, India.
Sangam Goswami, CSIR-Institute of Genomics and Integrative Biology, New Delhi 110025, India; Academy of Scientific and Innovative Research (AcSIR), Ghaziabad, Uttar Pradesh 201002, India.
Mohit Mangla, CSIR-Institute of Genomics and Integrative Biology, New Delhi 110025, India; Academy of Scientific and Innovative Research (AcSIR), Ghaziabad, Uttar Pradesh 201002, India.
Savinitha Prakash, CSIR-Institute of Genomics and Integrative Biology, New Delhi 110025, India.
Kandarp Joshi, CSIR-Institute of Genomics and Integrative Biology, New Delhi 110025, India.
Meyakumla, CSIR-Institute of Genomics and Integrative Biology, New Delhi 110025, India.
Sreedevi S., Department of Microbiology, St.Pious X Degree & PG College for Women, Hyderabad, Telangana 500076, India.
Devarshi Gajjar, Department of Microbiology, The Maharaja Sayajirao University of Baroda, Vadodara, Gujarat 390002, India.
Ronibala Soraisham, Department of Dermatology, Venereology and Leprology, Regional Institute of Medical Sciences, Imphal, Manipur 795004, India.
Rohit Yadav, CSIR-Institute of Genomics and Integrative Biology, New Delhi 110025, India; Academy of Scientific and Innovative Research (AcSIR), Ghaziabad, Uttar Pradesh 201002, India.
Yumnam Silla Devi, CSIR- North East Institute of Science and Technology, Jorhat, Assam 785006, India.
Aayush Gupta, Department of Dermatology, Dr. D.Y. Patil Medical College, Pune, Maharashtra 411018, India.
Mitali Mukerji, CSIR-Institute of Genomics and Integrative Biology, New Delhi 110025, India; Academy of Scientific and Innovative Research (AcSIR), Ghaziabad, Uttar Pradesh 201002, India.
Sivaprakash Ramalingam, CSIR-Institute of Genomics and Integrative Biology, New Delhi 110025, India; Academy of Scientific and Innovative Research (AcSIR), Ghaziabad, Uttar Pradesh 201002, India.
Binukumar B. K., CSIR-Institute of Genomics and Integrative Biology, New Delhi 110025, India; Academy of Scientific and Innovative Research (AcSIR), Ghaziabad, Uttar Pradesh 201002, India.
Vinod Scaria, CSIR-Institute of Genomics and Integrative Biology, New Delhi 110025, India; Academy of Scientific and Innovative Research (AcSIR), Ghaziabad, Uttar Pradesh 201002, India.
Sridhar Sivasubbu, CSIR-Institute of Genomics and Integrative Biology, New Delhi 110025, India; Academy of Scientific and Innovative Research (AcSIR), Ghaziabad, Uttar Pradesh 201002, India.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Council of Scientific and Industrial Research (CSIR), India [MLP1809, MLP2001]; CSIR fellowship (to A.J., M.K.D., B.J., A.B., S.M., R.Y.); Intel Research Fellowship (to D.S.); UGC fellowship (to V.G., S.G.). Funding for open access charge: Council of Scientific and Industrial Research, India [MLP1809, MLP2001].
Conflict of interest statement. None declared.
REFERENCES
- 1. Mastana S.S. Unity in diversity: an overview of the genomic anthropology of India. Ann. Hum. Biol. 2014; 41:287–299. [DOI] [PubMed] [Google Scholar]
- 2. Chaubey G., Metspalu M., Kivisild T., Villems R.. Peopling of South Asia: investigating the caste-tribe continuum in India. Bioessays. 2007; 29:91–100. [DOI] [PubMed] [Google Scholar]
- 3. Kivisild T., Rootsi S., Metspalu M., Mastana S., Kaldma K., Parik J., Metspalu E., Adojaan M., Tolk H.-V., Stepanov V. et al.. The genetic heritage of the earliest settlers persists both in Indian tribal and caste populations. Am. J. Hum. Genet. 2003; 72:313–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Popejoy A.B., Fullerton S.M.. Genomics is failing on diversity. Nature. 2016; 538:161–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. GUaRDIAN Consortium Sivasubbu S., Scaria V.. Genomics of rare genetic diseases-experiences from India. Hum. Genomics. 2019; 14:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Genome of the Netherlands Consortium Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat. Genet. 2014; 46:818–825. [DOI] [PubMed] [Google Scholar]
- 7. 1000 Genomes Project Consortium Auton A., Brooks L.D., Durbin R.M., Garrison E.P., Kang H.M., Korbel J.O., Marchini J.L., McCarthy S., McVean G.A. et al.. A global reference for human genetic variation. Nature. 2015; 526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gurdasani D., Carstensen T., Tekola-Ayele F., Pagani L., Tachmazidou I., Hatzikotoulas K., Karthikeyan S., Iles L., Pollard M.O., Choudhury A. et al.. The African Genome Variation Project shapes medical genetics in Africa. Nature. 2015; 517:327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mallick S., Li H., Lipson M., Mathieson I., Gymrek M., Racimo F., Zhao M., Chennagiri N., Nordenfelt S., Tandon A. et al.. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature. 2016; 538:201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nagasaki M., Yasuda J., Katsuoka F., Nariai N., Kojima K., Kawai Y., Yamaguchi-Kabata Y., Yokozawa J., Danjoh I., Saito S. et al.. Rare variant discovery by deep whole-genome sequencing of 1, 070 Japanese individuals. Nat. Commun. 2015; 6:8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Scott E.M., Halees A., Itan Y., Spencer E.G., He Y., Azab M.A., Gabriel S.B., Belkadi A., Boisson B., Abel L. et al.. Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat. Genet. 2016; 48:1071–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fakhro K.A., Staudt M.R., Ramstetter M.D., Robay A., Malek J.A., Badii R., Al-Marri A.A.-N., Abi Khalil C., Al-Shakaki A., Chidiac O. et al.. The Qatar genome: a population-specific tool for precision medicine in the Middle East. Hum Genome Var. 2016; 3:16016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O’Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B. et al.. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016; 536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P. et al.. The mutational constraint spectrum quantified from variation in 141, 456 humans. Nature. 2020; 581:434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sengupta D., Choudhury A., Basu A., Ramsay M.. Population stratification and underrepresentation of indian subcontinent genetic diversity in the 1000 Genomes Project Dataset. Genome Biol. Evol. 2016; 8:3460–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Indian Genome Variation Consortium Genetic landscape of the people of India: a canvas for disease gene exploration. J. Genet. 2008; 87:3–20. [DOI] [PubMed] [Google Scholar]
- 17. GenomeAsia100K Consortium The GenomeAsia 100K Project enables genetic discoveries across Asia. Nature. 2019; 576:106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mitt M., Kals M., Pärn K., Gabriel S.B., Lander E.S., Palotie A., Ripatti S., Morris A.P., Metspalu A., Esko T. et al.. Improved imputation accuracy of rare and low-frequency variants using population-specific high-coverage WGS-based imputation reference panel. Eur. J. Hum. Genet. 2017; 25:869–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hou L., Kember R.L., Roach J.C., O’Connell J.R., Craig D.W., Bucan M., Scott W.K., Pericak-Vance M., Haines J.L., Crawford M.H. et al.. A population-specific reference panel empowers genetic studies of Anabaptist populations. Sci. Rep. 2017; 7:6079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ahmad M., Sinha A., Ghosh S., Kumar V., Davila S., Yajnik C.S., Chandak G.R.. Inclusion of population-specific reference panel from India to the 1000 genomes phase 3 panel improves imputation accuracy. Sci. Rep. 2017; 7:6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Feero W.G., Wicklund C.A., Veenstra D.. Precision medicine, genome sequencing, and improved population health. JAMA. 2018; 319:1979–1980. [DOI] [PubMed] [Google Scholar]
- 22. Lencz T., Yu J., Palmer C., Carmi S., Ben-Avraham D., Barzilai N., Bressman S., Darvasi A., Cho J.H., Clark L.N. et al.. High-depth whole genome sequencing of an Ashkenazi Jewish reference panel: enhancing sensitivity, accuracy, and imputation. Hum. Genet. 2018; 137:343–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jain A., Gandhi S., Koshy R., Scaria V.. Incidental and clinically actionable genetic variants in 1005 whole exomes and genomes from Qatar. Mol. Genet. Genomics. 2018; 293:919–929. [DOI] [PubMed] [Google Scholar]
- 24. Miller S.A., Dykes D.D., Polesky H.F.. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988; 16:1215–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Miller N.A., Farrow E.G., Gibson M., Willig L.K., Twist G., Yoo B., Marrs T., Corder S., Krivohlavek L., Walter A. et al.. A 26-hour system of highly sensitive whole genome sequencing for emergency management of genetic diseases. Genome Med. 2015; 7:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Freed D., Aldana R., Weber J.A., Edwards J.S.. The Sentieon Genomics Tools - a fast and accurate solution to variant calling from next-generation sequence data. 2017; bioRxiv doi:12 May 2020, preprint: not peer reviewed 10.1101/115717. [DOI]
- 27. Kendig K.I., Baheti S., Bockol M.A., Drucker T.M., Hart S.N., Heldenbrand J.R., Hernaez M., Hudson M.E., Kalmbach M.T., Klee E.W. et al.. Sentieon DNASeq variant calling workflow demonstrates strong computational performance and accuracy. Front. Genet. 2019; 10:736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. et al.. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011; 43:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang K., Li M., Hakonarson H.. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pruitt K.D., Tatusova T., Maglott D.R.. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007; 35:D61–D65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sherry S.T., Ward M.H., Kholodov M., Baker J., Phan L., Smigielski E.M., Sirotkin K.. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001; 29:308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu X., Wu C., Li C., Boerwinkle E.. dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016; 37:235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Karczewski K.J., Weisburd B., Thomas B., Solomonson M., Ruderfer D.M., Kavanagh D., Hamamsy T., Lek M., Samocha K.E., Cummings B.B. et al.. The ExAC browser: displaying reference data information from over 60 000 exomes. Nucleic Acids Res. 2017; 45:D840–D845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Landrum M.J., Lee J.M., Benson M., Brown G.R., Chao C., Chitipiralla S., Gu B., Hart J., Hoffman D., Jang W. et al.. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018; 46:D1062–D1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Karolchik D., Baertsch R., Diekhans M., Furey T.S., Hinrichs A., Lu Y.T., Roskin K.M., Schwartz M., Sugnet C.W., Thomas D.J. et al.. The UCSC Genome Browser Database. Nucleic Acids Res. 2003; 31:51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hamosh A., Scott A.F., Amberger J., Bocchini C., Valle D., McKusick V.A.. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2002; 30:52–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E. et al.. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015; 17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Reich D., Thangaraj K., Patterson N., Price A.L., Singh L.. Reconstructing Indian population history. Nature. 2009; 461:489–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stawiski E.W., Diwanji D., Suryamohan K., Gupta R., Fellouse F.A., Fah Sathirapongsasuti J., Liu J., Jiang Y.-P., Ratan A., Mis M. et al.. Human ACE2 receptor polymorphisms predict SARS-CoV-2 susceptibility. 2020; bioRxiv doi:10 April 2020, preprint: not peer reviewed 10.1101/2020.04.07.024752. [DOI] [PMC free article] [PubMed]
- 40. Hussain M., Jabeen N., Raza F., Shabbir S., Baig A.A., Amanullah A., Aziz B.. Structural variations in human ACE2 may influence its binding with SARS-CoV-2 spike protein. J. Med. Virol. 2020; doi:10.1002/jmv.25832. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The variants unique to IndiGenomes have been submitted to the NCBI Variation Submission Portal with submission ID: SUB8153462 and also made available for download on the IndiGenomes database website http://clingen.igib.res.in/indigen/.