


Abstract
We present DescribePROT, the database of predicted amino acid-level descriptors of structure and function of proteins. DescribePROT delivers a comprehensive collection of 13 complementary descriptors predicted using 10 popular and accurate algorithms for 83 complete proteomes that cover key model organisms. The current version includes 7.8 billion predictions for close to 600 million amino acids in 1.4 million proteins. The descriptors encompass sequence conservation, position specific scoring matrix, secondary structure, solvent accessibility, intrinsic disorder, disordered linkers, signal peptides, MoRFs and interactions with proteins, DNA and RNAs. Users can search DescribePROT by the amino acid sequence and the UniProt accession number and entry name. The pre-computed results are made available instantaneously. The predictions can be accesses via an interactive graphical interface that allows simultaneous analysis of multiple descriptors and can be also downloaded in structured formats at the protein, proteome and whole database scale. The putative annotations included by DescriPROT are useful for a broad range of studies, including: investigations of protein function, applied projects focusing on therapeutics and diseases, and in the development of predictors for other protein sequence descriptors. Future releases will expand the coverage of DescribePROT. DescribePROT can be accessed at http://biomine.cs.vcu.edu/servers/DESCRIBEPROT/.
INTRODUCTION
As the amount of sequence data grows rapidly, currently including over 189 million protein coding regions in the UniProt release 2020_04 (1), scientists face the huge task to characterize novel proteins functionally and structurally. The functions and structures of proteins can be annotated at three levels of resolution: atomic, amino-acid (AA) and whole-protein. The primary repository of atomic-level information is the Protein Data Bank (PDB) (2), which currently houses ∼160 thousand protein structures. Protein-level data can be collected from several resources, including the manually reviewed Swiss-Prot and computationally annotated TrEMBL (1,3). Intermediate level annotations, also called 1D descriptors (4,5), describe structural and functional features of the AAs that compose protein chains. Popular AA-level structure descriptors include solvent accessibility, secondary structure, torsion angles, intrinsic disorder and flexibility. Common function descriptors at the AA level cover annotations of protein domains, catalytic residues and residues that interact with specific types of partners, such as proteins, RNA, DNA, membranes, nucleotides, and a variety of small ligands. While these AA-level annotations can be computed from PDB files and collected from Swiss-Prot/TrEMBL records, they cover a relatively small subset of proteins in the case of PDB and a small subset of AAs in the Swiss-Prot/TrEMBL annotated sequences.
The absence of the AA-level annotations can be remedied with the help of computational tools that predict them from protein sequence. Hundreds of these predictors have been developed over the last few decades (5–24). For instance, there are over 60 tools for predicting secondary structure (10,22,23), over 70 predictors for intrinsic disorder (14,16,20,21), and close to 40 predictors of AAs-nucleic acids interactions (24). Recent empirical assessments demonstrate that many of these tools provide accurate predictions (11,25–31). Many of them are also heavily used by the community, with examples that include SignalP (32–35) [21 941 citations in Google Scholar as of July 2020], PSIPRED (36,37) [9,050 citations] and IUPred (38–40) [2865 citations].
These tools can typically be used through web interfaces or downloadable programs provided by the authors. However, using tools directly can become complicated when collecting predictions for a large set of proteins and/or for multiple structural as well as functional characteristics. Web interfaces are typically ill-suited to running large numbers of predictions, and use of downloadable programs requires finding these resources, often time-consuming execution of the predictions, and assembling the results from outputs that rely on different formats. Several recently published prediction platforms, such as PSIPRED workbench (41), SCRATCH (42), PredictProtein (43), MULTICOM (44) and DEPICTER (45), alleviate some of these issues by providing integrated access to multiple predictors. However, these platforms use a substantial amount of time to complete the predictions and most of them focus on a specific category of the AA-level descriptors. For instance, PSIPRED workbench, SCRATCH and MULTICOM primarily focus on the structural descriptors while DEPICTER covers both structural and functional features but solely for the disordered regions. Two databases of pre-computed AA-level predictions, D2P2 (46) and MobiDB (47,48), offer an alternative solution. They provide fast and convenient access to results generated by multiple predictors. However, D2P2 was last updated in 2012 and both repositories cover a rather narrow set of putative structural and functional features, primarily focusing on disorder predictions (Table 1). More specifically, D2P2 covers only three descriptors (one structural and two functional) including the intrinsic disorder descriptor that is predicted by nine different methods. Similarly, MobiDB includes four putative descriptors (two structural and two functional) when using ten predictors of the intrinsic disorder.
Table 1.
Summary of the databases of predicted AA-level descriptors. The descriptors are categorized into three groups: structural descriptors (Str), functional descriptors (Fun), and sequence descriptors (Seq)
| Database | Last updated | No. of descriptors | List of descriptors | URL |
|---|---|---|---|---|
| MobiDB | 2019 | 4 | Intrinsic disorder (Str), Secondary structure (Str), Protein-binding (Fun), and Domains (Fun) | https://mobidb.bio.unipd.it/ |
| D2P2 | 2012 | 3 | Intrinsic disorder (Str), Disordered protein binding (Fun), and Domains (Fun) | http://d2p2.pro/ |
| DescribePROT | 2020 | 9 | Solvent accessibility (Str), Secondary structure (Str), Sequence conservation (Seq), Protein-binding (Fun), RNA-binding (Fun), DNA-binding (Fun), Intrinsic disorder (Str), Disordered linkers (Str), and Signal peptides (Seq) | http://biomine.cs.vcu.edu/servers/DESCRIBEPROT/ |
DescribePROT (Database of structure and function residue-based predictions of PROTeins) is a new resource that offers access to the predictions of nine key AA-level descriptors generated by 10 predictors for a collection of over 1.3 million proteins from 83 complete proteomes of popular organisms (Table 1). The current version of DescribePROT (v.1.1) provides a comprehensive collection of four structural descriptors, three functional descriptors and two sequence descriptors.
MATERIALS AND METHODS
Sequences
The AA-level predictions were processed on the sequence library of 83 complete proteomes selected from the UniProt's reference proteomes list in 2019_08 release of UniProt. We focused on selecting organisms that are popular research targets, such as human, mouse, rat, zebrafish, macaque, fruit fly, yeast, C. elegans, A. thaliana, E. coli, as well as prevalent viruses that include herpes, Ebola, HIV1, measles and mumps. The 83 proteomes contain 1.36 million proteins with close to 600 million AAs, and cover the four taxonomic kingdoms: Eukaryota (with multiple Animalia, Plantae, Fungi and Protista proteomes), Bacteria, Archaea and Viruses (Table 2). Figure 1 summarizes the taxonomic distribution of the proteins and proteomes included in DescribePROT. Figure 1B shows that 67% of proteomes are from Eukaryota, with the largest portion of 39% animal proteomes, while the remaining 33% are composed of 16% viral, 10% bacterial and 7% archaeal proteomes. Figure 1A reveals that DescribePROT includes about 2.3% bacterial proteins, 1.0% archaeal proteins, 0.1% viral proteins and 96.6% eukaryotic proteins. The latter is due to the relatively large sizes of the eukaryotic proteomes, particularly compared to the very small viral proteomes.
Table 2.
Summary and taxonomic classification of the protein data and predictions included in DescribePROT
| Taxonomic classification | No. of proteomes | No. of sequences | No. of AAs | No. of predictions | |
|---|---|---|---|---|---|
| Eukaryotes | Animalia | 33 | 790 891 | 373 185 044 | 4 851 405 572 |
| Plantae | 13 | 431 824 | 169 255 167 | 2 200 317 171 | |
| Fungi | 7 | 49 388 | 23 586 301 | 306 621 913 | |
| Protista | 3 | 48 395 | 19 407 557 | 252 298 241 | |
| Bacteria | 8 | 31 453 | 10 141 624 | 131 841 112 | |
| Archaea | 6 | 13 155 | 3 724 684 | 48 420 892 | |
| Virus | 13 | 840 | 214 886 | 2 793 518 | |
| Total | 83 | 1 365 946 | 599 515 263 | 7 793 698 419 | |
Figure 1.

Taxonomic distribution of the proteins (panel A) and proteomes (panel B) in DescribePROT.
Predictions
The predictive methods included in DescribePROT satisfy three key characteristics: (i) short runtime, which is necessary given the large scope of DescribePROT; (ii) complementary coverage of a comprehensive set of AA-level descriptors and (iii) strong predictive performance. Consequently, the current version of DescribePROT (v1.1) includes results generated by ten predictors (alphabetically): solvent accessibility by ASAquick (49,50), disordered linkers by DFLpred (51), disordered protein-, RNA-, and DNA-binding AAs by DisoRDPbind (52–54), structure-derived DNA- and RNA-binding AAs by DRNApred (55), multiple sequence alignment profiles by MMseqs2 (56,57), short disordered protein-binding regions by MoRFchibi (58), secondary structure by PSIPRED (36,59), structure-derived protein-binding AAs by SCRIBER (60), signal peptides by SignalP (34,61), and intrinsically disordered AAs by VSL2B (62,63). Table 3 summarizes these methods. Empirical measurements of the runtime conducted using proteins included in DescribePROT are shown in Table 3 and reveal that these predictors are indeed fast and require only between 0.07 s (for VSL2B) and 11 s (for both DRNApred's predictions) to make predictions for a single protein sequence. Each predictor produces different descriptors and they collectively cover four structural descriptors (solvent accessibility, secondary structure, intrinsic disorder and disordered linkers), three functional descriptors (protein-binding, RNA-binding and DNA-binding AAs), as well as two sequence descriptors (sequence conservation and signal peptides). Following, we briefly highlight key features of each tools.
Table 3.
Overview of the ten predictors that were used to derive data for DescribePROT. The runtime was measured using five batches with 100 proteins each on the Intel i7 CPU; we report averages and standard deviations over the five runs
| Predictor | Last updated | Version | Reference(s) | Runtime avg±stdev [s] per 100 proteins | Notes | URL |
|---|---|---|---|---|---|---|
| ASAquick | 2017 | 1.0 | (49,50) | 25.49±0.58 | Fast and accurate predictor of solvent accessibility. | http://mamiris.com/services.html |
| DFLpred | 2016 | 1.0 | (51) | 30.23±0.55 | Sole, fast and accurate predictor of disordered linkers. | http://biomine.cs.vcu.edu/servers/DFLpred/ |
| DisoRDPbind | 2015 | 1.0 | (52–54) | 30.15±1.12 | Sole, fast and accurate predictor of the disordered DNA-, RNA- and protein-binding AAs. | http://biomine.cs.vcu.edu/servers/DisoRDPbind/ |
| DRNApred | 2017 | 1.0 | (55) | 1094.03±79.89 | Accurate predictor of DNA- and RNA-binding AAs annotated from structure. | http://biomine.cs.vcu.edu/servers/DRNApred/ |
| MMseqs2 | 2019 | 2.0 | (56,57) | 932.99±4.04 | Fast and sensitive multiple sequence alignment. | https://search.mmseqs.com/search |
| MoRFchiBi | 2016 | 1.03 | (58) | 179.87±8.73 | Fast and accurate predictor of MoRF regions. | https://gsponerlab.msl.ubc.ca/software/morf_chibi/ |
| PSIPRED | 2019 | 4.01 | (36,59) | 90.91±3.44 | Popular and accurate predictor of secondary structure. | http://bioinf.cs.ucl.ac.uk/psipred/ |
| SCRIBER | 2019 | 1.0 | (60) | 770.56±40.81 | Accurate predictor of protein-binding AAs annotated from structure. | http://biomine.cs.vcu.edu/servers/SCRIBER/ |
| SignalP | 2019 | 5.0 | (34,61) | 492.80±0.12 | Popular, accurate and comprehensive predictor of signal peptides. | http://www.cbs.dtu.dk/services/SignalP/ |
| VSL2B | 2006 | N/A | (62,63) | 7.76±0.044 | Fast and accurate predictor of intrinsically disordered AAs. | http://www.dabi.temple.edu/disprot/predictor.php |
PSIPRED (36,59) is arguably the most popular predictor of secondary structure. It generates accurate three-state prediction of secondary structure, which includes numeric propensities for helix (H), strand (E) and coil (C) conformations and a predicted label corresponding to the secondary structure with the highest putative propensity. PSIPRED was ranked as one of the most accurate predictors in multiple comparative studies (28,64). We run the single-sequence version of PSIPRED that can scale to the size of DescribePROT.
ASAquick (49,50) is a very fast predictor of the AA-level accessible surface area (ASA). The quick runtime stems from the fact that ASAquick does not utilize the time-consuming multiple sequence alignments. However, its predictive performance is competitive with other methods that are much slower due to using the alignments (50). We convert the outputs produced by this tool into the relative solvent accessibility (RSA) by normalizing the putative ASA value by the AA-specific factors taken from (65). We also use the RSA values to annotate buried residues based on the approach described in (66,67), i.e. AAs with the putative RSA < 0.16 are assumed to be buried.
SignalP (34,61) is the most commonly used predictor of signal peptides. It generates numeric propensities for the presence of signal peptides and the corresponding binary labels (signal peptide versus no signal peptide) for the first 70 AAs in a given protein chain. We utilize the newest version 5.0 of SignalP that features very accurate predictions, works across all taxonomic kingdoms of life and differentiates between multiple types of the prokaryotic signal peptides (34). We set the organism groups parameter of SignalP to be compatible with the species of the query sequence.
MMseqs2 (56,57) is a very fast homology search tool that can produce multiple sequence alignments and position specific scoring matrices (PSSMs) from the search results. We utilize this tool to generate PSSMs using the reference proteomes set from the 2019_08 release of UniProt as the background set of sequences. We compute sequence conservation scores from PSSM using the relative entropy-based approach (68,69) where the background amino acid frequencies are from BLOSUM-62 (70). Moreover, we bin the conservation scores into decile intervals and provide binary annotation of the highly conserved AAs belonging to the top decile. MMseqs2 is two orders of magnitude faster than the popular PSI-BLAST while maintaining similar or better levels of sensitivity (57).
VSL2B (62,63) is a fast and popular predictor of intrinsic disorder (71–73). It generates numeric propensity for intrinsic disorder and a binary label (disordered vs. structured) for each AA in the protein sequence. It couples a short runtime with high levels of predictive performance. VSL2B was scored as the best disorder predictor in CASP6 (74) and was subsequently ranked among the top-performing methods in multiple other assessments (16,26,75).
DFLpred (51) is currently the only predictor of disordered linker regions, which are defined as intrinsically disordered regions that serve as linkers or spacers between domains in multi-domain proteins and between structured constituents within domains (76). DFLpred outputs the numeric propensity for the linkers and the corresponding binary label (disordered linker vs. non-linker) for each AA of the input sequence. This method was shown to produce accurate predictions in sub-second time for a single protein (51).
The functional descriptors that are included in DescribePROT focus on the annotations of interactions with proteins, DNA and RNA. The corresponding predictive models have been in development for well over a decade (7,8,11,13,30). The selection of the four functional predictors included in DescribePROT was informed by two observations. First, the two major classes of these predictors—ones that are trained using the intrinsically disordered AAs that bind proteins/DNA/RNA vs. ones that are trained using structured protein–protein, protein–DNA and protein–RNA complexes—were shown to provide complementary results (77,78). Second, multiple recent studies demonstrate that many of these methods cross-predict the three types of interacting AAs (11,30,31,79). This means that, for instance, predictors of protein-binding AAs would also incorrectly predict DNA- and RNA-binding AAs as protein-binding while predictors of DNA-binding residues would also incorrectly predict protein- and RNA-binding residues as DNA-binding. Correspondingly, we include both classes of predictors (disorder and structure trained) and we ensure that they were designed to minimize the amount of the cross-predictions.
DisoRDPbind (52–54) is the only currently available predictor of intrinsically disordered AAs that interact with DNA and RNA. This tool also provides predictions of disordered, protein-binding AAs. It generates three numeric propensities for protein-, DNA- and RNA-binding by disordered AAs and the corresponding three binary labels (protein/DNA/RNA binding versus non-binding) for each AAs of the input protein chain. DisoRDPbind excels through short runtime (the three types of interactions are predicted in under a second for a single protein), was ranked among the top predictors of disordered, protein-binding AAs (77), and generates low amounts of cross-predictions (52,77).
We also cover prediction of an abundant subclass of disordered, protein-binding AAs, called MoRFs (molecular recognition features) (80,81). MoRFs are short disordered protein regions (between 5 and 25 AAs in length) that undergo disorder-to-order transition upon binding the protein partner(s). A significant majority of functional predictors that address disordered AAs focus on this type interaction (16,18). We use a fast and accurate predictor, MoRFchibi (58), which outputs numeric propensities for MoRFs and binary labels (MoRF versus non-MoRF). This method was recently ranked among the most accurate predictors of MoRFs (18).
DRNApred (55) accurately predicts DNA and protein–RNA binding AAs that are annotated based on structured protein-nucleic acids complexes. It produces propensities for DNA-binding, propensities for RNA binding, and two corresponding binary labels (RNA binding versus non-RNA binding and DNA-binding versus non-DNA-binding) for each AA of the input sequence. This method is the sole predictor of nucleic-acid interacting AAs that was trained to specifically reduce cross-predictions (55,79).
SCRIBER (60) is an accurate predictor of protein-binding AAs annotated based on structured complexes. It outputs both the numeric propensities for protein-binding and the corresponding binary labels for each AA in the input protein sequence. Similar to DRNApred in the context of interactions with nucleic acids, this is the only method that was specifically designed to successfully minimize cross-predictions of protein binding residues (60,77).
The methods that we employ were shown to provide accurate predictions on the corresponding benchmark datasets (26,28,34,50,51,55,58,60,75,77). These datasets typically broadly cover the taxonomic space. However, only two of these methods, DisoRDPbind and SignalP, were comparatively evaluated across different species or domains of life to probe robustness of their predictions. DisoRDPbind demonstrates consistent levels of predictive performance across human, mouse, fruit fly and C. elegans proteomes (52). Similarly, SignalP provides comparable predictive quality across Archaea, gram-negative Bacteria, gram-positive Bacteria and Eukaryota (34). Availability of DescribePROT will facilitate future studies that provide analogous comparative analyses for the other methods.
DATABASE
The database is available at http://biomine.cs.vcu.edu/servers/DESCRIBEPROT/. DescribePROT’s backend is implemented with the MariaDB relational database. We use php and JavaScript to deliver the user interface and python to access database, parse data, and generate downloadable files. Following, we explain the data stored in DescribePROT, how to access these data, how to search the database, and how to use and understand the graphical interface.
Data
The data of DescribePROT include protein names, UniProt entry names, sequences, accession numbers that are used to link to the UniProt records, and 12 predictions that are provided as raw numeric propensities and propensity-derived labels. DescribePROT stores the numerical propensities for solvent accessibility, each of the three secondary structure states, signal peptides, intrinsic disorder, disordered linkers, MoRFs, disordered protein-, DNA- and RNA-binding and structure-annotated protein-, DNA- and RNA-binding. We also store the three-state secondary structure labels and the binary labels for buried AAs, signal peptides, intrinsically disordered AAs, disordered linkers, MoRFs, disordered protein-, DNA- and RNA-binding AAs, and structure-annotated protein-, DNA- and RNA-binding AAs. Finally, we include the PSSM, numeric conservation scores and the 10-state (decile-based) conservation level labels for each AA.
These data are available to the end user in multiple convenient and complementary ways. We provide the source data in JSON format for each of the 83 proteomes as well as for the entire database. This option is available under the ‘Download’ link on the main page of the database. We also provide access to the data for each individual protein via an interactive graphical interface and downloadable PNG file of this graphic, as well as a CSV-formatted file and a parsable JSON-formatted file with the raw predictions and binary results. We explain how to access this information in the ‘Results Page’ section.
Search types
Users can search DescribePROT in three ways: by the UniProt accession number, the UniProt entry name and the AA sequence. The AA sequence search generates a collection of proteins included in DescribePROT that are sorted by their similarity to the input protein chain. These proteins can be sorted by the E-value (by default), alignment coverage and identity values that are produced by BLAST (82,83). This information is accompanied by the corresponding accession numbers linking to UniProt records and the taxonomy IDs, to provide context for the selection of the most relevant protein. DescribePROT also provides direct access to the data for a particular protein using the UniProt accession number, e.g. users can fetch results for P04637 (p53 protein) using the following direct link: http://biomine.cs.vcu.edu/servers/DESCRIBEPROT/result.php?uniprot=P04637. This allows for direct cross-linking with other databases.
Results page
The putative structural, functional, and sequence descriptors for a given protein are available in an interactive graphical format, which utilizes the ‘Feature Viewer’ software (DOI: 10.5281/zenodo.345324), and parsable structured format on the results page (Figure 2). The top of the page includes the accession number (linked to the corresponding UniProt record), protein name, taxonomy ID and length of the sequence. The red marker 1 in Figure 2 points the question mark icon that links to the help and tutorial videos. The JSON and CSV-formatted putative annotations can be downloaded by clicking the arrow icons indicated by the red markers 2 and 3, respectively. The graphical view shown at the bottom of Figure 2 is available for download as an image in the PNG format by clicking the arrow icon identified by the red marker 4. The results are divided into three sections (Figure 2): (a) putative structural descriptors that include predictions from VSL2B (intrinsic disorder), ASAquick (solvent accessibility) and PSIPRED (secondary structure); (b) putative functional descriptors that cover predictions from DisoRDPbind (disordered protein-, DNA- and RNA-binding binding), MoRFchibi (MoRF regions), DRNApred (structure-derived DNA- and RNA-binding) and SCRIBER (structure-derived protein-binding) and (c) other descriptors that feature results from MMseqs2 (PSSM and sequence conservation) and predictions from DFLpred (disordered linkers) and SignalP (signal peptides). The predictions are displayed using a graphical report that summarizes the numeric propensities and labels. The red oval marker at the top of Figure 2 identifies the checkbox that opens graphical reports for specific predictions. The graphical reports can be scaled (zoomed in and out) and offer functionality to highlight regions of predicted labels and to display the boundaries of these regions and the underlying propensities on the mouse over. Examples of the latter features are shown using the red oval markers in the middle of Figure 2.
Figure 2.
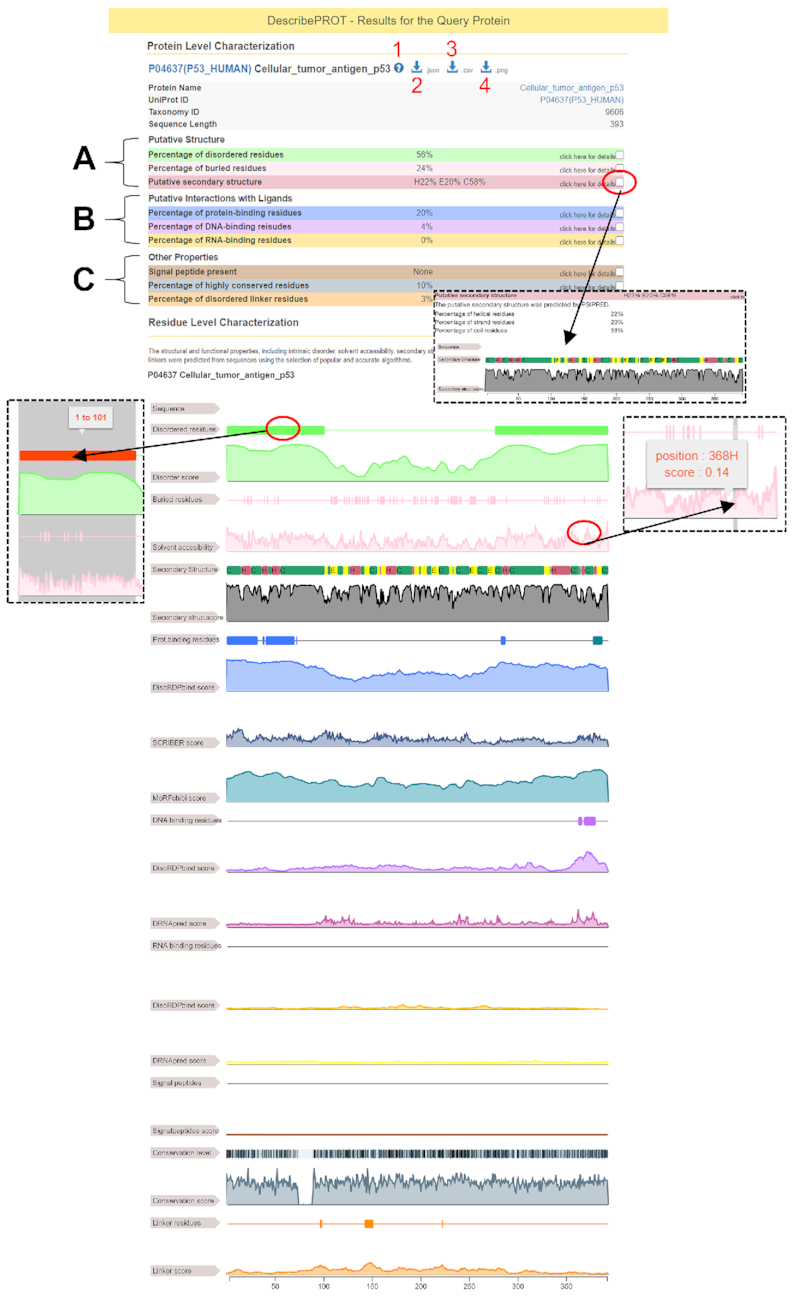
An example results page generated by DescribePROT database for the human p53 protein (UniProt ID: P04637).
We explain how to interpret the data from the results page using an example analysis of the human p53 protein shown in Figure 2. The p53 protein is involved in several key cellular processes, such as apoptosis and DNA repair (84). Studies have shown that p53 is an intrinsically disordered proteins that carries out its functions by interacting with a large numbers of protein (85–91) and DNA (92,93) partners. According to the results from VSL2B shown in the light green color in Figure 2, DescribePROT suggests that 56% of the p53 sequence is disordered, with two long disordered regions at the N-terminus (positions 1–101) and the C-terminus (positions 277–393). This is in good agreement with the experimentally annotated disordered regions that are localized at the N-terminus (positions 1–92) and the C-terminus (positions 293–393 AAs) (94). Moreover, DescribePROT suggests that 20% of AAs bind protein partners (blue highlights in Figure 2). This prediction combines together, using union operation, the results produced by the relevant methods that include DisoRDPbind, MoRFchibi and SCRIBER. Detailed analysis reveals that in this case the interactions are predicted by DisoRDPbind (regions 1–32, 41–70 and 283–287) and MoRFchibi (region 378–387). Their predictions are in line with the experimental data (88). For instance, p53 was shown to interact with several protein partners, such as p300 and CBP, via the transactivation domain (region 1–61) (89), and with another group of proteins, including sirtuin and CBP, in the 374–388 region (90,91). Moreover, research shows that the central structured domain of p53 is highly conserved (95) while the flanking disordered regions have diversified during the evolution (96). Correspondingly, the gray-colored results in Figure 2 show that highly conserved residues (darker grays) are primarily located in the structured domain. This example demonstrates the richness of the information that can be gleaned from the results reported by DescribePROT.
Global analysis of the putative descriptors
Figure 3 visualizes color-coded Spearman correlation coefficients (SCCs) between each pair of the 14 AA-level putative propensities for the protein structure and function generated by nine predictive tools. We exclude SignalP from this analysis since its predictions concern only the 70 AAs at the N-terminus of the protein chain. The majority of the propensities are not correlated (SCC < 0.2), which confirms that they characterize distinct descriptors of AAs. The few correlated descriptors include the PSIPRED-predicted secondary structures, where propensity for the helical conformation is negatively correlated with the propensities for strands and coils (SCC < –0.6) and where propensities for strands and coils are weakly correlated (SCC = 0.2). The DRNApred-generated propensities for DNA-binding and RNA-binding are negatively correlated (SCC = –0.54), and this stems from the fact that DRNApred was designed to minimize cross-prediction between DNA and RNA binding AAs (55,79). Similar observation is true for DisoRDPbind's predictions of protein-binding and RNA-binding that are also slightly negatively correlated (SCC = –0.24) (52,77). Finally, the modestly correlated predictions from SCRIBER and MoRFchibi (SCC = 0.25) can be explained by the fact that both methods predict protein-binding AAs. SCRIBER predicts protein-binding residues that form structured complexes while MoRFchibi focuses on MoRFs (short disordered protein-binding regions that fold upon binding).
Figure 3.

Spearman correlation coefficients (SCCs) between each pair of the numeric propensities produced by the 14 AA-level predictions of protein structure and function. The color-coded SCCs were computed over the AAs included in DescribePROT. The structure predictions include RSA by ASAquick, disordered linkers by DFLpred, helix, strand and coil conformations by PSIPRED, and intrinsic disorder by VSL2B. The function predictions cover disordered RNA-binding, DNA-binding and protein-binding by DisoRDPbind, MoRFs by MoRFchibi, structure-annotated DNA-binding and RNA-binding by DRNApred and structure-annotated protein-binding by SCRIBER. We also include sequence conservation computed from the profiles generated by MMSeqs2.
Figure 4 shows distributions of the protein-level content values that are aggregated from the AA-level labels predicted by the ten methods. Content is defined as the fraction of AAs with a given label in the protein sequence, e.g. fraction of buried AAs is computed as the number of buried AAs divided by the sequence length. We cover the content of highly conserved residues (AAs in the top docile of the database-wide conservation scores), content of helix (H), strand (E) and coil (C) conformations, content of buried AAs (RSA < 0.16 (66,67)), and contents of the disordered AAs, disordered linkers, as well as protein-binding, RNA-binding and DNA-binding AAs. Several interesting observations can be gleaned from these data. For instance, the content of highly conserved AAs ranges between 0.03 and 0.24, which suggests that sequence-level conservation can vary by as much as an order of magnitude. The median content of helical AAs is at about 0.4, which is slightly lower than the median content of coils at 0.45, and substantially higher that the median content of strands that is at ∼0.15. The median content of buried AAs is 0.3, but the fraction of buried residues can vary widely between nearly zero and half the sequence. The median content of intrinsic disorder is at around 0.1 while about 35% of proteins have majority of their AAs disordered, and some proteins are fully disordered. These observations are in agreement with past studies of the abundance of the intrinsic disorder (97,98).
Figure 4.

Distributions of the putative protein-level content of the structural, functional and sequence-derived descriptors included in DescribePROT. The boxplots represent the following 12 intervals, where the consecutive rectangles corresponding to 5–12.5, 12.5–20, 20–27.5, 27.5–35, 35–42.5, 42.5–50, 50–57.5, 57.5–65, 65–72.5, 72.5–80, 80–87.5, 87.5–95 percentile ranges. The black horizontal lines represent medians.
DISCUSSION
DescribePROT provides convenient access to a variety of AA-level descriptors of protein structure and function for a collection of complete proteomes that cover popular model organisms. It includes predictions of intrinsic disorder, secondary structure, solvent accessibility, RNA-, DNA- and protein-binding, MoRFs, disordered linkers and signal peptides. It also offers access to the pre-computed PSSM and sequence conservation values. This resource complements the current databases of AA-level predictions, D2P2 (46) and MobiDB (47,48), that primarily focus on the intrinsic disorder. The putative annotations included by DescriPROT are useful for a wide range of studies, spanning from basic investigations of protein function, through applied projects that focus on diseases and therapeutics, to projects that design and test novel methods for the prediction of other characteristics of protein sequences. For instance, just recently, VSL2B was used to characterize function and structure of the EZH2 protein (99), DisoRDPbind was used to analyze the SARS-CoV-2 proteome (100), and PSIPRED and ASAquick were applied to devise a deep-learning predictor of caspase and matrix metalloprotease cleavage sites (101).
DescribePROT provides multiple ways to access the data. It features an interactive graphical interface that offers the opportunity to simultaneously explore multiple structural and functional descriptors. It also provides parsable downloads of the source data at the protein, proteome and whole database scales. Moreover, DescriPROT website features help and tutorial videos that explain how to search the database and how to use and understand the graphical interface.
Future work will primarily concentrate on expanding the coverage of the database, with the long-term goal to cover the entire content of UniProt. Our high-priority short-term objective is to include experimental annotations available in several relevant reference databases, such as PDB (2) and DisProt (102). We intend to add additional and complementary functional and structural descriptors, with examples being putative domain boundaries, post-translational modifications, and interactions with small molecule ligands. We plan to provide access to the underlying data programmatically via API, to supplement the multitude of the currently available downloadable file formats. Overall, we aim to update the DescribePROT resource quarterly. We are also eager to hear and consider suggestions concerning the future developments from the community of users.
ACKNOWLEDGEMENTS
We gratefully acknowledge contributions of the authors of the predictive tools that were used to develop this resource, which were developed by the labs of Drs Keith A Dunker, Jörg Gspooner, David T Jones, Andrzej Kloczkowski, Lukasz Kurgan, Henrik Nielsen, Zoran Obradovic and Johannes Söding.
Contributor Information
Bi Zhao, Department of Computer Science, Virginia Commonwealth University, Richmond, VA, USA.
Akila Katuwawala, Department of Computer Science, Virginia Commonwealth University, Richmond, VA, USA.
Christopher J Oldfield, Department of Computer Science, Virginia Commonwealth University, Richmond, VA, USA.
A Keith Dunker, Center for Computational Biology and Bioinformatics, Indiana University School of Medicine, Indianapolis, IN, USA.
Eshel Faraggi, Battelle Center for Mathematical Medicine at the Nationwide Children's Hospital, and Department of Pediatrics, The Ohio State University, Columbus, OH, USA.
Jörg Gsponer, Michael Smith Laboratories, University of British Columbia, Vancouver, BC, Canada.
Andrzej Kloczkowski, Battelle Center for Mathematical Medicine at the Nationwide Children's Hospital, and Department of Pediatrics, The Ohio State University, Columbus, OH, USA.
Nawar Malhis, Michael Smith Laboratories, University of British Columbia, Vancouver, BC, Canada.
Milot Mirdita, Quantitative and Computational Biology, Max Planck Institute for Biophysical Chemistry, Göttingen, Germany.
Zoran Obradovic, Department of Computer and Information Sciences, Temple University, Philadelphia, PA, USA.
Johannes Söding, Quantitative and Computational Biology, Max Planck Institute for Biophysical Chemistry, Göttingen, Germany.
Martin Steinegger, School of Biological Sciences and Institute of Molecular Biology & Genetics, Seoul National University, Seoul, Republic of Korea.
Yaoqi Zhou, Institute for Glycomics, Griffith University, Gold Coast, Queensland, Australia.
Lukasz Kurgan, Department of Computer Science, Virginia Commonwealth University, Richmond, VA, USA.
FUNDING
National Science Foundation [1617369, 1661391]; National Institutes of Health [R01 GM127701]; Robert J. Mattauch Endowment funds. Funding for open access charge: Endowment funds.
Conflict of interest statement. None declared.
REFERENCES
- 1. UniProt, C. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019; 47:D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. ww, P.D.B.c. Protein Data Bank: the single global archive for 3D macromolecular structure data. Nucleic Acids Res. 2019; 47:D520–D528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Boutet E., Lieberherr D., Tognolli M., Schneider M., Bansal P., Bridge A.J., Poux S., Bougueleret L., Xenarios I.. UniProtKB/Swiss-Prot, the manually annotated section of the UniProt KnowledgeBase: how to use the entry view. Methods Mol. Biol. 2016; 1374:23–54. [DOI] [PubMed] [Google Scholar]
- 4. Rost B. Prediction in 1D: secondary structure, membrane helices, and accessibility. Methods Biochem. Anal. 2003; 44:559–587. [PubMed] [Google Scholar]
- 5. Kurgan L., Disfani F.M.. Structural protein descriptors in 1-dimension and their sequence-based predictions. Curr. Protein Pept. Sci. 2011; 12:470–489. [DOI] [PubMed] [Google Scholar]
- 6. Si J., Cui J., Cheng J., Wu R.. Computational prediction of RNA-binding proteins and binding sites. Int. J. Mol. Sci. 2015; 16:26303–26317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Si J., Zhao R., Wu R.. An overview of the prediction of protein DNA-binding sites. Int. J. Mol. Sci. 2015; 16:5194–5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhao H., Yang Y., Zhou Y.. Prediction of RNA binding proteins comes of age from low resolution to high resolution. Mol. Biosyst. 2013; 9:2417–2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fernandez-Recio J. Prediction of protein binding sites and hot spots. Wires Comput. Mol. Sci. 2011; 1:680–698. [Google Scholar]
- 10. Oldfield C.J., Chen K., Kurgan L.. Computational prediction of secondary and supersecondary structures from protein sequences. Methods Mol. Biol. 2019; 1958:73–100. [DOI] [PubMed] [Google Scholar]
- 11. Zhang J., Kurgan L.. Review and comparative assessment of sequence-based predictors of protein-binding residues. Brief. Bioinform. 2018; 19:821–837. [DOI] [PubMed] [Google Scholar]
- 12. Roche D., Brackenridge D.A., McGuffin L.J.. Proteins and their interacting partners: an introduction to protein-ligand binding site prediction methods. Int. J. Mol. Sci. 2015; 16:29829–29842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Maheshwari S., Brylinski M.. Predicting protein interface residues using easily accessible on-line resources. Brief. Bioinform. 2015; 16:1025–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. He B., Wang K., Liu Y., Xue B., Uversky V.N., Dunker A.K.. Predicting intrinsic disorder in proteins: an overview. Cell Res. 2009; 19:929–949. [DOI] [PubMed] [Google Scholar]
- 15. Puton T., Kozlowski L., Tuszynska I., Rother K., Bujnicki J.M.. Computational methods for prediction of protein-RNA interactions. J. Struct. Biol. 2012; 179:261–268. [DOI] [PubMed] [Google Scholar]
- 16. Meng F., Uversky V.N., Kurgan L.. Comprehensive review of methods for prediction of intrinsic disorder and its molecular functions. Cell. Mol. Life Sci. 2017; 74:3069–3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jiang Q., Jin X., Lee S.J., Yao S.. Protein secondary structure prediction: a survey of the state of the art. J. Mol. Graph. Model. 2017; 76:379–402. [DOI] [PubMed] [Google Scholar]
- 18. Katuwawala A., Peng Z., Yang J., Kurgan L.. Computational prediction of MoRFs, short disorder-to-order transitioning protein binding regions. Comput Struct Biotechnol J. 2019; 17:454–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xie J., Ding W., Chen L., Guo Q., Zhang W.. Advances in protein contact map prediction based on machine learning. Med. Chem. 2015; 11:265–270. [DOI] [PubMed] [Google Scholar]
- 20. Lieutaud P., Ferron F., Uversky A.V., Kurgan L., Uversky V.N., Longhi S.. How disordered is my protein and what is its disorder for? A guide through the “Dark Side” of the protein universe. Intrinsic. Disord. Proteins. 2016; 4:e1259708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Meng F., Uversky V., Kurgan L.. Computational prediction of intrinsic disorder in proteins. Curr. Protoc. Protein Sci. 2017; 88:2.16.11–12.16.14. [DOI] [PubMed] [Google Scholar]
- 22. Kashani-Amin E., Tabatabaei-Malazy O., Sakhteman A., Larijani B., Ebrahim-Habibi A.. A systematic review on popularity, application and characteristics of protein secondary structure prediction tools. Curr. Drug Discov. Technol. 2018; 16:159–172. [DOI] [PubMed] [Google Scholar]
- 23. Meng F., Kurgan L.. Computational prediction of protein secondary structure from sequence. Curr. Protoc. Protein Sci. 2016; 86:2.3.1–2.3.10. [DOI] [PubMed] [Google Scholar]
- 24. Zhang J., Ma Z., Kurgan L.. Comprehensive review and empirical analysis of hallmarks of DNA-, RNA- and protein-binding residues in protein chains. Brief. Bioinform. 2019; 20:1250–1268. [DOI] [PubMed] [Google Scholar]
- 25. Monastyrskyy B., Kryshtafovych A., Moult J., Tramontano A., Fidelis K.. Assessment of protein disorder region predictions in CASP10. Proteins. 2014; 82:127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Walsh I., Giollo M., Di Domenico T., Ferrari C., Zimmermann O., Tosatto S.C.. Comprehensive large-scale assessment of intrinsic protein disorder. Bioinformatics. 2015; 31:201–208. [DOI] [PubMed] [Google Scholar]
- 27. Katuwawala A., Oldfield C.J., Kurgan L.. Accuracy of protein-level disorder predictions. Brief. Bioinform. 2020; 21:1509–522. [DOI] [PubMed] [Google Scholar]
- 28. Zhang H., Zhang T., Chen K., Kedarisetti K.D., Mizianty M.J., Bao Q., Stach W., Kurgan L.. Critical assessment of high-throughput standalone methods for secondary structure prediction. Brief. Bioinform. 2011; 12:672–688. [DOI] [PubMed] [Google Scholar]
- 29. Schaarschmidt J., Monastyrskyy B., Kryshtafovych A., Bonvin A.. Assessment of contact predictions in CASP12: Co-evolution and deep learning coming of age. Proteins. 2018; 86:51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yan J., Friedrich S., Kurgan L.. A comprehensive comparative review of sequence-based predictors of DNA- and RNA-binding residues. Brief. Bioinform. 2016; 17:88–105. [DOI] [PubMed] [Google Scholar]
- 31. Miao Z., Westhof E.. A large-scale assessment of nucleic acids binding site prediction programs. PLoS Comput. Biol. 2015; 11:e1004639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nielsen H., Engelbrecht J., Brunak S., vonHeijne G.. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 1997; 10:1–6. [DOI] [PubMed] [Google Scholar]
- 33. Bendtsen J.D., Nielsen H., von Heijne G., Brunak S.. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 2004; 340:783–795. [DOI] [PubMed] [Google Scholar]
- 34. Almagro Armenteros J.J., Tsirigos K.D., Sonderby C.K., Petersen T.N., Winther O., Brunak S., von Heijne G., Nielsen H.. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019; 37:420–423. [DOI] [PubMed] [Google Scholar]
- 35. Petersen T.N., Brunak S., von Heijne G., Nielsen H.. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods. 2011; 8:785–786. [DOI] [PubMed] [Google Scholar]
- 36. McGuffin L.J., Bryson K., Jones D.T.. The PSIPRED protein structure prediction server. Bioinformatics. 2000; 16:404–405. [DOI] [PubMed] [Google Scholar]
- 37. Jones D.T. Protein secondary structure prediction based on position-specific scoring matrices1. J. Mol. Biol. 1999; 292:195–202. [DOI] [PubMed] [Google Scholar]
- 38. Dosztányi Z., Csizmok V., Tompa P., Simon I.. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics. 2005; 21:3433–3434. [DOI] [PubMed] [Google Scholar]
- 39. Dosztányi Z., Csizmók V., Tompa P., Simon I.. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J. Mol. Biol. 2005; 347:827–839. [DOI] [PubMed] [Google Scholar]
- 40. Meszaros B., Erdos G., Dosztanyi Z.. IUPred2A: context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018; 46:W329–W337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Buchan D.W.A., Jones D.T.. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res. 2019; 47:W402–W407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cheng J., Randall A.Z., Sweredoski M.J., Baldi P.. SCRATCH: a protein structure and structural feature prediction server. Nucleic Acids Res. 2005; 33:W72–W76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yachdav G., Kloppmann E., Kajan L., Hecht M., Goldberg T., Hamp T., Hönigschmid P., Schafferhans A., Roos M., Bernhofer M. et al.. PredictProtein—an open resource for online prediction of protein structural and functional features. Nucleic Acids Res. 2014; 42:W337–W343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cheng J., Li J., Wang Z., Eickholt J., Deng X.. The MULTICOM toolbox for protein structure prediction. BMC Bioinformatics. 2012; 13:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Barik A., Katuwawala A., Hanson J., Paliwal K., Zhou Y., Kurgan L.. DEPICTER: intrinsic disorder and disorder function prediction server. J. Mol. Biol. 2020; 432:3379–3387. [DOI] [PubMed] [Google Scholar]
- 46. Oates M.E., Romero P., Ishida T., Ghalwash M., Mizianty M.J., Xue B., Dosztanyi Z., Uversky V.N., Obradovic Z., Kurgan L. et al.. D(2)P(2): database of disordered protein predictions. Nucleic Acids Res. 2013; 41:D508–D516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Piovesan D., Tabaro F., Paladin L., Necci M., Micetic I., Camilloni C., Davey N., Dosztanyi Z., Meszaros B., Monzon A.M. et al.. MobiDB 3.0: more annotations for intrinsic disorder, conformational diversity and interactions in proteins. Nucleic Acids Res. 2018; 46:D471–D476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Di Domenico T., Walsh I., Martin A.J.M., Tosatto S.C.E.. MobiDB: a comprehensive database of intrinsic protein disorder annotations. Bioinformatics. 2012; 28:2080–2081. [DOI] [PubMed] [Google Scholar]
- 49. Faraggi E., Zhou Y., Kloczkowski A.. Accurate single-sequence prediction of solvent accessible surface area using local and global features. Proteins. 2014; 82:3170–3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Faraggi E., Kouza M., Zhou Y., Kloczkowski A.. Fast and accurate accessible surface area prediction without a sequence profile. Methods Mol. Biol. 2017; 1484:127–136. [DOI] [PubMed] [Google Scholar]
- 51. Meng F., Kurgan L.. DFLpred: High-throughput prediction of disordered flexible linker regions in protein sequences. Bioinformatics. 2016; 32:i341–i350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Peng Z., Kurgan L.. High-throughput prediction of RNA, DNA and protein binding regions mediated by intrinsic disorder. Nucleic Acids Res. 2015; 43:e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Peng Z., Wang C., Uversky V.N., Kurgan L.. Prediction of disordered RNA, DNA, and protein binding regions using DisoRDPbind. Methods Mol. Biol. 2017; 1484:187–203. [DOI] [PubMed] [Google Scholar]
- 54. Oldfield C.J., Peng Z., Kurgan L.. Disordered RNA-binding region prediction with DisoRDPbind. Methods Mol. Biol. 2020; 2106:225–239. [DOI] [PubMed] [Google Scholar]
- 55. Yan J., Kurgan L.. DRNApred, fast sequence-based method that accurately predicts and discriminates DNA- and RNA-binding residues. Nucleic Acids Res. 2017; 45:e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mirdita M., Steinegger M., Soding J.. MMseqs2 desktop and local web server app for fast, interactive sequence searches. Bioinformatics. 2019; 35:2856–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Steinegger M., Soding J.. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017; 35:1026–1028. [DOI] [PubMed] [Google Scholar]
- 58. Malhis N., Jacobson M., Gsponer J.. MoRFchibi SYSTEM: software tools for the identification of MoRFs in protein sequences. Nucleic Acids Res. 2016; 44:W488–W493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jones D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999; 292:195–202. [DOI] [PubMed] [Google Scholar]
- 60. Zhang J., Kurgan L.. SCRIBER: accurate and partner type-specific prediction of protein-binding residues from proteins sequences. Bioinformatics. 2019; 35:i343–i353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nielsen H. Predicting secretory proteins with SignalP. Methods Mol. Biol. 2017; 1611:59–73. [DOI] [PubMed] [Google Scholar]
- 62. Obradovic Z., Peng K., Vucetic S., Radivojac P., Dunker A.K.. Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins. 2005; 61:176–182. [DOI] [PubMed] [Google Scholar]
- 63. Peng K., Radivojac P., Vucetic S., Dunker A.K., Obradovic Z.. Length-dependent prediction of protein intrinsic disorder. BMC Bioinformatics. 2006; 7:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rost B. Review: protein secondary structure prediction continues to rise. J. Struct. Biol. 2001; 134:204–218. [DOI] [PubMed] [Google Scholar]
- 65. Tien M.Z., Meyer A.G., Sydykova D.K., Spielman S.J., Wilke C.O.. Maximum allowed solvent accessibilites of residues in proteins. PLoS One. 2013; 8:e80635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kim H., Park H.. Prediction of protein relative solvent accessibility with support vector machines and long-range interaction 3D local descriptor. Proteins Struct. Funct. Bioinf. 2004; 54:557–562. [DOI] [PubMed] [Google Scholar]
- 67. Pollastri G., Baldi P., Fariselli P., Casadio R.. Prediction of coordination number and relative solvent accessibility in proteins. Proteins. 2002; 47:142–153. [DOI] [PubMed] [Google Scholar]
- 68. Fischer J.D., Mayer C.E., Soding J.. Prediction of protein functional residues from sequence by probability density estimation. Bioinformatics. 2008; 24:613–620. [DOI] [PubMed] [Google Scholar]
- 69. Wang K., Samudrala R.. Incorporating background frequency improves entropy-based residue conservation measures. BMC Bioinformatics. 2006; 7:385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Styczynski M.P., Jensen K.L., Rigoutsos I., Stephanopoulos G.. BLOSUM62 miscalculations improve search performance. Nat. Biotechnol. 2008; 26:274–275. [DOI] [PubMed] [Google Scholar]
- 71. van der Lee R., Buljan M., Lang B., Weatheritt R.J., Daughdrill G.W., Dunker A.K., Fuxreiter M., Gough J., Gsponer J., Jones D.T. et al.. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014; 114:6589–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Oldfield C.J., Uversky V.N., Dunker A.K., Kurgan L.. Salvi N. Introduction to intrinsically disordered proteins and regions. Intrinsically Disordered Proteins. 2019; Academic Press; 1–34. [Google Scholar]
- 73. Zhou J., Oldfield C.J., Yan W., Shen B., Dunker A.K.. Identification of intrinsic disorder in complexes from the Protein Data Bank. ACS Omega. 2020; 5:17883–17891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Jin Y., Dunbrack R.L. Jr. Assessment of disorder predictions in CASP6. Proteins. 2005; 61:167–175. [DOI] [PubMed] [Google Scholar]
- 75. Peng Z.L., Kurgan L.. Comprehensive comparative assessment of in-silico predictors of disordered regions. Curr. Protein Pept. Sci. 2012; 13:6–18. [DOI] [PubMed] [Google Scholar]
- 76. Dunker A.K., Brown C.J., Lawson J.D., Iakoucheva L.M., Obradovic Z.. Intrinsic disorder and protein function. Biochemistry. 2002; 41:6573–6582. [DOI] [PubMed] [Google Scholar]
- 77. Zhang J., Ghadermarzi S., Kurgan L.. Prediction of protein-binding residues: dichotomy of sequence-based methods developed using structured complexes vs. disordered proteins. Bioinformatics. 2020; 10.1093/bioinformatics/btaa573. [DOI] [PubMed] [Google Scholar]
- 78. Chowdhury S., Zhang J., Kurgan L.. In silico prediction and validation of novel RNA binding proteins and residues in the human proteome. Proteomics. 2018; 18:e1800064. [DOI] [PubMed] [Google Scholar]
- 79. Su H., Liu M., Sun S., Peng Z., Yang J.. Improving the prediction of protein-nucleic acids binding residues via multiple sequence profiles and the consensus of complementary methods. Bioinformatics. 2019; 35:930–936. [DOI] [PubMed] [Google Scholar]
- 80. Mohan A., Oldfield C.J., Radivojac P., Vacic V., Cortese M.S., Dunker A.K., Uversky V.N.. Analysis of molecular recognition features (MoRFs). J. Mol. Biol. 2006; 362:1043–1059. [DOI] [PubMed] [Google Scholar]
- 81. Yan J., Dunker A.K., Uversky V.N., Kurgan L.. Molecular recognition features (MoRFs) in three domains of life. Mol. Biosyst. 2016; 12:697–710. [DOI] [PubMed] [Google Scholar]
- 82. Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., Madden T.L.. BLAST+: architecture and applications. BMC Bioinformatics. 2009; 10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hu G., Kurgan L.. Sequence similarity searching. Curr. Protoc. Protein Sci. 2019; 95:e71. [DOI] [PubMed] [Google Scholar]
- 84. Toufektchan E., Toledo F.. The guardian of the genome revisited: p53 downregulates genes required for telomere maintenance, DNA repair, and centromere structure. Cancers (Basel). 2018; 10:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Bischoff J.R., Friedman P.N., Marshak D.R., Prives C., Beach D.. Human P53 is phosphorylated by P60-Cdc2 and Cyclin-B-Cdc2. Proc. Natl. Acad. Sci. U.S.A. 1990; 87:4766–4770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ferreon J.C., Lee C.W., Arai M., Martinez-Yamout M.A., Dyson H.J., Wright P.E.. Cooperative regulation of p53 by modulation of ternary complex formation with CBP/p300 and HDM2. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:6591–6596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wells M., Tidow H., Rutherford T.J., Markwick P., Jensen M.R., Mylonas E., Svergun D.I., Blackledge M., Fersht A.R.. Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:5762–5767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Oldfield C.J., Meng J., Yang J.Y., Yang M.Q., Uversky V.N., Dunker A.K.. Flexible nets: disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genomics. 2008; 9:S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Feng H., Jenkins L.M., Durell S.R., Hayashi R., Mazur S.J., Cherry S., Tropea J.E., Miller M., Wlodawer A., Appella E. et al.. Structural basis for p300 Taz2-p53 TAD1 binding and modulation by phosphorylation. Structure. 2009; 17:202–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Avalos J.L., Celic I., Muhammad S., Cosgrove M.S., Boeke J.D., Wolberger C.. Structure of a Sir2 enzyme bound to an acetylated p53 peptide. Mol. Cell. 2002; 10:523–535. [DOI] [PubMed] [Google Scholar]
- 91. Mujtaba S., He Y., Zeng L., Yan S., Plotnikova O., Sachchidanand Sanchez R., Zeleznik-Le N.J., Ronai Z., Zhou M.M.. Structural mechanism of the bromodomain of the coactivator CBP in p53 transcriptional activation. Mol. Cell. 2004; 13:251–263. [DOI] [PubMed] [Google Scholar]
- 92. Lidor Nili E., Field Y., Lubling Y., Widom J., Oren M., Segal E.. p53 binds preferentially to genomic regions with high DNA-encoded nucleosome occupancy. Genome Res. 2010; 20:1361–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. McLure K.G., Lee P.W.. How p53 binds DNA as a tetramer. EMBO J. 1998; 17:3342–3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Uversky V.N. p53 proteoforms and intrinsic disorder: an Illustration of the protein structure-function continuum concept. Int. J. Mol. Sci. 2016; 17:1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Soussi T., Beroud C.. Assessing TP53 status in human tumours to evaluate clinical outcome. Nat. Rev. Cancer. 2001; 1:233–240. [DOI] [PubMed] [Google Scholar]
- 96. Xue B., Brown C.J., Dunker A.K., Uversky V.N.. Intrinsically disordered regions of p53 family are highly diversified in evolution. Biochim. Biophys. Acta. 2013; 1834:725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Peng Z., Yan J., Fan X., Mizianty M.J., Xue B., Wang K., Hu G., Uversky V.N., Kurgan L.. Exceptionally abundant exceptions: comprehensive characterization of intrinsic disorder in all domains of life. Cell. Mol. Life Sci. 2015; 72:137–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Uversky V.N. Paradoxes and wonders of intrinsic disorder: Prevalence of exceptionality. Intrinsic. Disord Proteins. 2015; 3:e1065029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Jiao L., Shubbar M., Yang X., Zhang Q., Chen S., Wu Q., Chen Z., Rizo J., Liu X.. A partially disordered region connects gene repression and activation functions of EZH2. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:16992–17002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Giri R., Bhardwaj T., Shegane M., Gehi B.R., Kumar P., Gadhave K., Oldfield C.J., Uversky V.N.. Understanding COVID-19 via comparative analysis of dark proteomes of SARS-CoV-2, human SARS and bat SARS-like coronaviruses. Cell. Mol. Life Sci. 2020; 10.1007/s00018-020-03603-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Li F., Chen J., Leier A., Marquez-Lago T., Liu Q., Wang Y., Revote J., Smith A.I., Akutsu T., Webb G.I. et al.. DeepCleave: a deep learning predictor for caspase and matrix metalloprotease substrates and cleavage sites. Bioinformatics. 2020; 36:1057–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Hatos A., Hajdu-Soltesz B., Monzon A.M., Palopoli N., Alvarez L., Aykac-Fas B., Bassot C., Benitez G.I., Bevilacqua M., Chasapi A. et al.. DisProt: intrinsic protein disorder annotation in 2020. Nucleic Acids Res. 2020; 48:D269–D276. [DOI] [PMC free article] [PubMed] [Google Scholar]