


Abstract
TransCirc (https://www.biosino.org/transcirc/) is a specialized database that provide comprehensive evidences supporting the translation potential of circular RNAs (circRNAs). This database was generated by integrating various direct and indirect evidences to predict coding potential of each human circRNA and the putative translation products. Seven types of evidences for circRNA translation were included: (i) ribosome/polysome binding evidences supporting the occupancy of ribosomes onto circRNAs; (ii) experimentally mapped translation initiation sites on circRNAs; (iii) internal ribosome entry site on circRNAs; (iv) published N-6-methyladenosine modification data in circRNA that promote translation initiation; (v) lengths of the circRNA specific open reading frames; (vi) sequence composition scores from a machine learning prediction of all potential open reading frames; (vii) mass spectrometry data that directly support the circRNA encoded peptides across back-splice junctions. TransCirc provides a user-friendly searching/browsing interface and independent lines of evidences to predicte how likely a circRNA can be translated. In addition, several flexible tools have been developed to aid retrieval and analysis of the data. TransCirc can serve as an important resource for investigating the translation capacity of circRNAs and the potential circRNA-encoded peptides, and can be expanded to include new evidences or additional species in the future.
INTRODUCTION
Circular RNAs (circRNAs) have recently been demonstrated as a class of abundant and conserved RNAs in animals and plants (1–4). Most circRNAs are produced from a special type of alternative splicing known as back-splicing, and are predominantly localized in cytoplasm (3,5,6). Recent studies have revealed that circRNAs may play diverse biological roles by functioning as either no-coding or coding RNAs. Because circRNAs are more stable than their linear counterpart, they can naturally function as competitors of the linear RNAs to play regulatory roles in gene expression. For example, several circRNAs were found to function as miRNA sponges to sequestrate miRNAs (7,8), thus can increase the level of the cognate miRNA targets (4). In addition, circRNAs can also bind and sequestrate RNA binding proteins (RBPs) (4), and thus to modulate transcription or interfere with splicing regulation (9–11). Nevertheless, the functions of most circRNAs are still unclear and sometime controversial despite intensive studies (12).
Since most circRNAs contain exonic sequences and are localized in cytoplasm, many of these circRNAs may also function as mRNA to direct protein translation. Indeed, it has been previously reported that in vitro synthesized circRNAs can be translated in cap-independent fashion (13). Recent studies indicated that some cytoplasmic circRNAs can be effectively translated into detectable peptides, and many short sequences, including N-6-methyladenosine (m6A) sites, have been reported to function as IRES-like elements to drive circRNA translation (14–17). The translation of circRNA was up-regulated during cellular stresses (15), and some circRNA-encoded proteins were found to play key roles in regulating cancer cell growth (18–20).
However, the identification of circRNA-encoded protein has been a very difficult task, mainly because the sequences from circRNAs and their cognate linear mRNAs of host gene have a large overlap and differ only at the small window across back-splice junction. One of the common criteria used to identify translatable RNAs is to measure the association of the RNA with ribosomes using ribosome profiling (21), however it is very rare to find a ribo-seq read covering a back-splice junction of circRNAs (22,23). Similarly, the proteins translated from circRNAs often overlap with the known proteins translated from linear mRNA, except for the sequences near the back-splice junction. Therefore, it is also challenging to identify peptides encode by sequences at the back-splice junctions using Mass spectrometry. In addition, translation of a circRNA has to be initiated by an internal ribosome entry site (IRES) in a cap-independent fashion. However only a small fraction of IRESs in endogenous human genes have been identified. There are also other indirect evidences that can be used to support the coding potential of circRNAs, but none of these methods can make reliable prediction by themselves. As a result, while a large number of circRNAs (10 000–500 000) have been identified though high throughput transcriptome sequencing (24–26) and several databases for circRNA annotation have be developed (27–30), a specialized and comprehensive database for translatable circRNAs is still lacking. Although existing databases include the information on potential proteins encoded by circRNAs (31,32), these predicted ORFs may also be translated from linear mRNAs because they were not required to span the back-splice junction.
To meet this need, we describe a comprehensive database, TransCirc, which contains information of >300 000 circRNAs together with multi-omics evidence from published literatures to support circRNA translations. By integrating several direct and indirect evidences, we conducted an integrative analysis to predict the potential of all circRNAs in coding for functional peptides. This database provides an interactive data search engine and visualization interface for the translatable circRNAs and their translation products, as well as the regulatory elements that support its translation and analytic tools for potential function of circRNA encoded genes. Collectively, we expect the TransCirc database will facilitate further analysis of circRNA function, and streamline the identification of circRNA translation product. All of the information and data is freely available at https://www.biosino.org/transcirc.
MATERIALS AND METHODS
Data collection
The basic information including the genomic coordinates, host genes and full-length sequences of human circRNAs were downloaded from circAtlas database (31). In addition, we also imported intron-free full-length circRNAs detected by CIRI-AS and CIRI-full pipeline (26,33) from BioProject PRJCA000751 that encompasses normal 17 human tissue (34,35). For multiple circRNA isoforms sharing a common back-splicing junction, we treated them as different circRNAs with additional index appending to the cognate ID. Currently, 255 396 and 72 684 full-length exonic human circRNAs derived from circAtlas and RNA-seq data respectively are included in TransCirc. For all human circRNAs, we used available multi-omics data to derive a series of evidences that support their coding potentials presented with a relative score system in a radar chart.
Analysis of multi-omics evidence for circRNA translation
To detect ribosome- and polysome-associated circRNAs, we integrated data from several independent studies of transcriptome-wide ribosome profiling and polysome profiling (15,18,36,37). CIRCexplorer (38) and CIRI2 (34) were used to detect reads spanning back-splice junction in ribosome/polysome profiling. We merged the results (≥2 BSJ reads for each candidate detected by any pipeline) and further mapped to all known circRNAs with annotation of Refseq gene. We assign a relative score according to the percent of times the given circRNAs were detected in total datasets.
To identify IRES elements in circRNAs, we used the data from a systematic screening of IRES elements in human genome (39), which generated a series of statistically enriched hexamers (Z score > 7) with IRES-like activity. We mapped these enriched hexamers in circRNA sequence, and merged the overlapping hexamers into a single IRES fragment. We calculated the raw score of each IRES fragment using the sum of enrichment Z-scores of all overlapping hexamers divided by the length of the fragment. The relative score were computed by normalizing to the maximum raw score.
Previously m6A modification sites were reported to function as IRESs to drive circRNA translation (15). We used published m6A modification data from REPIC database (40), and mapped them back to circRNA sequences. Identification by at least three different tools (exomePeak, MACS2 and MeTPeak) and the presence of consensus modification motif (‘RAC’ sequence) are required for potential methylated adenosine. The experimentally validated m6A sites in circRNA also served as a predictor for translatable circRNA (15), which are integrated into this database.
To map the experimentally identified translation initiation site (TIS), we used the data from TISdb based on GTI-seq (42), and mapped to the circRNAs using annotations from GENCODE (43). For any circRNA, we scored the presence or absence of TIS and m6A with a score of 1 or 0, respectively.
The putative ORFs encoded by the circRNAs in three reading frames were predicted using several criteria. We considered potential ORFs with ‘NTG’ as start codon because these non-ATG start codons are common in IRES-mediated translation (41,42). We further required the ORFs to cross the back-splice junction at least once (i.e. circRNA-specific ORFs), and the number of times each ORF passed the back-splice junction were marked as ‘translation cycles’ in the database. We set a threshold of ORF length at ≥20aa, and used the longest ORFs when multiple ORFs overlap. If a particular ORF doesn’t contain a stop codon, it was defined as rolling circle ORF (rcORF, i.e. an infinite ORF translated in rolling circle fashion, translation cycle = 3). The presence or absence of ORF within a given circRNA was score scored as 1 or 0.
To directly identify circRNA-coded proteins with Mass spectrometry, we searched a published comprehensive human proteome dataset (44) against a combined database containing all UniProt human proteins and all potential circRNA-specific peptides using an open search engine pFind(v3.1.6) (45). We selected positive mass spectra across back splice junction using the following thresholds: q ≤0.01, peptides length ≥8 with a new sequence of at least 2 aa at either side of the back-splice junction, missed cleavage sites ≤3, allowing only common modifications (cysteine carbamidomethylation, oxidation of methionine, protein N-terminal acetylation, deamidation of asparagine, and phosphorylation of serine, threonine, and tyrosine). The presence or absence of MS-supported peptide across BSJ of each circRNA was score scored as 1 or 0. In addition, the Q values representing the confidence of BSJ-spanning peptide were also provided for each spectrum.
The detailed information of bioinformatic analysis on multi-omics evidence was also summarized as a supplementary table (Supplementary Table S1).
Sequence composition analysis using machine learning
To predict how likely a given sequence can encode for natural proteins, we tested several machine learning methods and chose sparse partial least squares discriminant analysis (sPLS-DA) to predict the translation ability of circRNAs. To train the classification model, we downloaded the RNA sequences of all protein coding genes of human genome (hg19 from UCSC) as the positive group, and compared with two negative non-coding groups including the randomly shuffled sequences and reversed coding sequences. We used the features of nucleotide composition from all sequence in these groups to train the PLS-DA model (Supplementary Figure S1a), and found that the coding and non-coding sequences can be clearly separated (Supplementary Figure S1b). We subsequently scored the distance of any given sequence to the coding group versus the non-coding group, and use their difference to predict the translation propensity. Using this predictive model, we ranked the translation propensity of all ORFs from circRNAs and gave a relative score to each ORF. The sPLS-DA model applied in our study is adopted from the R package ‘mixOmics’.
Database construction
The TransCirc web server was built with a mature and convention-over-configuration Model-View-Controller (MVC) framework, Spring boot (http://spring.io), which is embedded on Tomcat service (https://tomcat.apache.org) in a CentOS Linux 7.8 environment. The circRNA data and associated with evidence data were stored in MongoDB (https://www.mongodb.com), which is a general purpose, document-based distributed database. The user interfaces were built using JavaScript (https://www.javascript.com) to provide responsive and user-friendly web pages. All modules are packaged into docker to ensure flexibility in website deployment.
DATABASE CONTENT
Through data collection and integrative analysis, we summarized information of 328,080 human circRNAs together with seven types of evidences that support their translation potential (Figure 1). Each class of evidence provides a direct or indirect support for the coding capacity of circRNA, which can be used in combination for a more reliable prediction. For convenience, we converted all evidences into a normalized score ranging from 0 to 1, and use the sum of scores as a general predictor for coding propensity.
Figure 1.

Schematic of overall design of TransCirc. The combined circRNA sequences, seven types of supporting evidence for coding potentials and their associated statistic scores are integrated into an interactive database TransCirc, which also provides user-friendly interface for data accessing, evidence searching and downloading.
circRNA-specific ORFs
We require the circRNA-specific ORFs to span the back-splice junction for at least 6-nt at both ends and consider all possible reading frames (see Materials and Methods). The length of potential ORF is a common predictor for coding RNA versus non-coding RNAs, as a long ORF is unusual in a non-coding RNA. We used the ORF length ≥20 aa as a minimal requirement, which is a relatively weak predictor as many small peptides were found to be coded by ‘non-coding’ RNAs (46).
Sequences that drive cap-independent translation
Since circRNAs are covalently closed, their translation must use an unconventional initiation mechanism known as cap-independent translation. Such pathway has to be driven by IRESs that are typically short fragments with specialized secondary structure (47). Although most well-studied IRESs were found in viral RNAs, there are several cases where the endogenous genes contain IRES to drive translation. Recently, we and other group conducted a systematic screen for IRES elements in human genome or from random sequences (39,48), and thus all the available IRES information was used in TransCirc as evidence.
The N-6-methyladenosine is the most common base modification found in many types of non-coding and coding RNAs. We have recently found that circRNAs undergo extensive modification m6A, which can drive circRNA translation through recruiting reader protein YTHDF3 that interacts with translation initiation factors (most notably eIF4G2) (15).
Direct evidences for translation and translation products
The translation of mRNAs is carried out by ribosomes, which usually form polysomes in actively translated mRNAs. The association with ribosomes/polysomes can serve as a strong predictor of the potential for translatable circRNA, which is usually supported by transcriptome-wide mapping of ribosomes. TransCirc uses several high-quality ribo-seq datasets with additional manual curation.
Mass spectrometry is an important method to directly identify and characterize proteins in proteomic scale. Several large scale MS experiments have been conducted to study human proteome (44,49), however only ∼50% of mass spectra can be reliably assigned to known peptides coded by canonical mRNAs even considering the post-translational modifications, suggesting a large fraction of ‘hidden proteome’ encoded by non-canonical mRNA. We have defined a set of empirical rules and rigorous filters to search circRNA-encoded peptide from MS dataset, and use the search results as a strong evidence for circRNA translation. TransCirc also provides all the raw spectra supporting the circRNA encoded peptides that cross the back-splice junctions.
Sequence composition of translation products
The amino acid sequences of natural proteins only occupy a very small fraction of the possible sequence space, mostly because only a small fraction of sequences can form stable proteins. The protein with ‘unnatural’ sequence tends to be mis-folded and degraded rapidly, and thus the similarity to all natural protein sequences can serve as an independent predictor to identify authentic proteins in random strings of amino acid sequences. Such similarity was measured using a new machine learning approach (see method).
USER INTERFACE
Data access and download
TransCirc provides a user-friendly interface and flexible routes to visualize or mine the data. The users can access the data through several common routes: (i) TransCirc provides a quick navigation and search engine at the front page, where users can choose different types of translatable evidences to retrieve a list of circRNAs. Users can also search the entire database with a keyword, and the optional keywords include the TransCirc ID, IDs from other circRNA databases, symbols of the host genes, GENECODE/Ensembl gene ID, or a specific genomic location. (ii) For users who only have circRNA or peptide sequences (e.g., raw reads from experiments without gene name or genomic location), TransCirc provides a direct tool, TransCirc-BLAT, to conduct customized sequence search that allows identification of relevant circRNAs with associated translatable evidences. (iii) All evidences can be filtered in the search result list, enabling the users to combine different evidences for a customized ranking of circRNA candidates. This feature will be helpful to prioritize candidates for functional studies.
Once in the detailed page for each circRNA, the users can visualize extensive information of sequences and supporting evidences using clickable tabs, and a radar chart is also generated to facilitate the visualization and comparison of the strength for translation evidences. All the sequence-based information in circRNAs, including the exon sequences, IRESs, TISs, and m6As, were highlighted on an interactive diagram to help selection of the corresponding sequences (an example showed in Figure 2). All information of the circRNA-specific ORFs, including the diagrams of circular translation, the corresponding nucleic acid and amino acid sequences, and MS evidence from peptides spanning back-splice junction, are listed in a specific row of ORF table (Figure 2).
Figure 2.
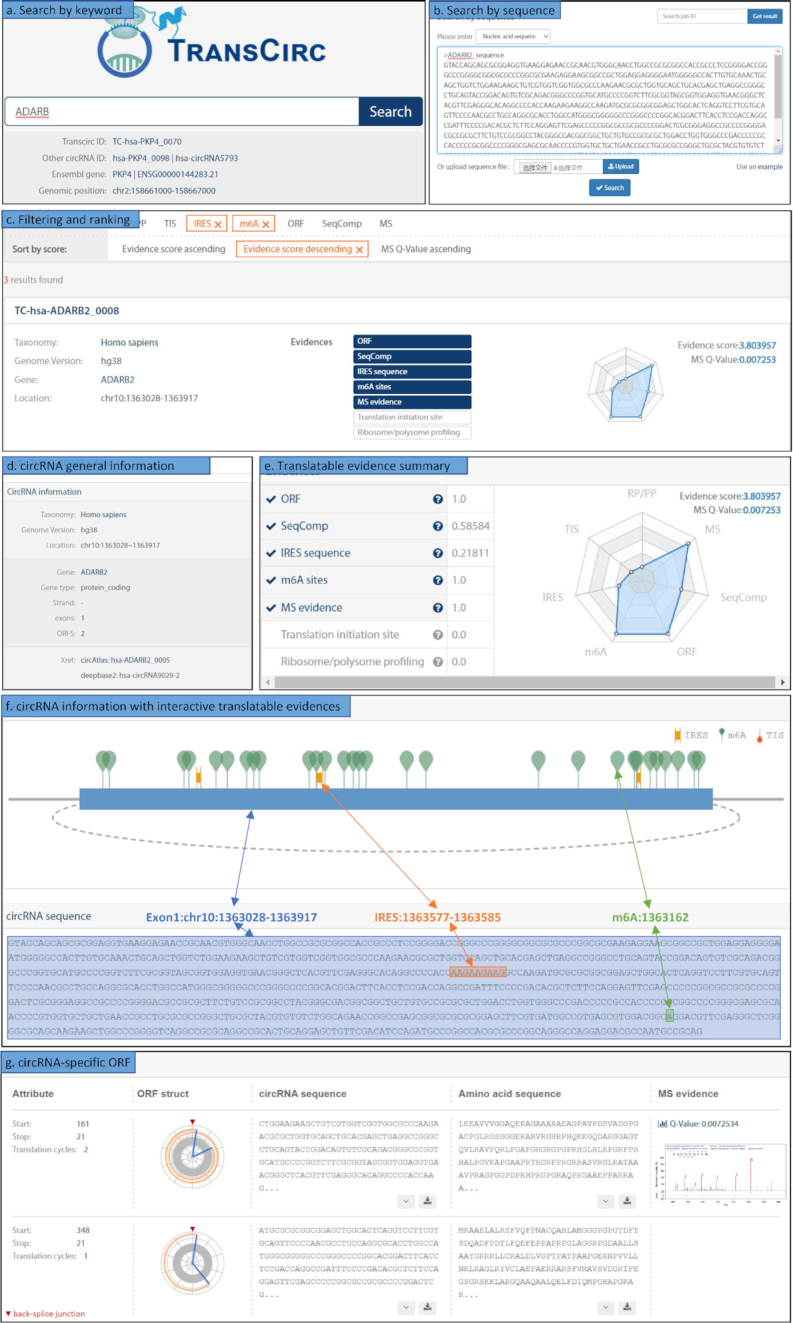
TransCirc user interface using the circRNA from ADARB2 as an example. (A) Searching TransCirc using keywords; (B) searching TransCirc with nucleic acid or amino acid sequences using TransCirc-BLAT; (C) an example of search result list, which can be filtered by evidence types and sorted by scores; (D) circRNA general information, including species information, genome region, gene information, exon/ORF number and cross-reference information; (E) scoring of seven types of translatable evidence, total score and visualization by radar chart; (F) display of circRNA information and translatable evidences; (G) display of circRNA-specific ORF information.
For specific circRNAs, all the sequence information (RNA fragments and ORFs) can be downloaded using the clickable exporting tabs. In addition, the packed metadata of all circRNAs is available for public download.
An example of TransCirc use
ADARB2 (adenosine deaminase RNA specific B2) is a gene coding for a catalytically inactive RNA deaminase expressed mainly in neuronal cells (50,51). The exact activity of ADARB2 is unclear but its mutations were found to be associated with neuronal diseases including dyschromatosis symmetrica hereditaria and juvenile astrocytoma (51). If a user wants to find a translatable circRNA associated with ADARB2, he/she can use ADARB as a keyword to search the TransCirc (Figure 2A and B). The search engine returned 68 records in the result page, and the user would use evidence filters to select the circRNAs with IRESs and/or m6A sites assuming he/she was interested in its translation initiation pathways. Three potential translatable circRNAs passed this filter (Figure 2C). Sorting by the evidence score, the user would find TC-hsa-ADARB2_0008 interesting as it has the highest translation propensity (Figure 2C).
Intrigued by this candidate, the user may click into the circRNA detail page, where the basic information such as genomic region, species information, gene information, and cross-references related to TC-hsa-ADARB2_0008 were displayed (Figure 2D) accompanied by the detailed scores of each evidence in a radar chart (Figure 2E). Using the information table of the candidate circRNA, the user can inspect the exonic configuration and sequences, as well as the positions of various sequence features such as putative IRESs and m6A sites. Each piece of information can also be highlighted using a clickable icon (Figure 2F). Scrolling down to the ORF table, two circRNA-specific ORFs were presented by a diagram of ORF structure and the associated RNA or peptide sequences (Figure 2G). The mass spectra associated with the peptide spanning back-splice junction were also provided as a link of a image (Figure 2G). The user can download any of these data for further study of this specific candidate.
CONCLUSIONS AND FUTURE DEVELOPMENT
Increasing evidences suggest that the circRNA-encoded proteins play critical roles in regulating various cellular events, however identification of translatable circRNAs and circRNA-specific proteins is lagging due to technical difficulties. Here we report an interactive database where multi-omics evidence were integrated to predict circRNA-specific ORFs, some of which were also supported by peptides with mass spectra across back-splice junction. The database is easy to navigate with an interactive interface, and all data can be downloaded in batch for additional offline analysis. In addition, this database provides a standard format that can accommodate the accumulation of additional multi-omics data (e.g. Ribo-seq or MS), as well as an expandable platform where new types of evidences can be integrated. Our initial efforts were focused on the human circRNAs because of the vast amount of existing transcriptome data and the best genome annotation, and the database will be expanded to other organisms with the available multi-omics data in future versions. We expect the database will facilitate circRNA related research from a broad range of users by saving their computational efforts in mining multi-omics data.
Supplementary Material
ACKNOWLEDGEMENTS
The authors want to thank Dr Fangqing Zhao for useful discussion and suggestions in using the data from circAtlas.
Contributor Information
Wendi Huang, Bio-Med Big Data Center, CAS Key Laboratory of Computational Biology, CAS-MPG Partner Institute for Computational Biology, Shanghai Institute of Nutrition and Health, Chinese Academy of Sciences, Shanghai 200031, China; University of Chinese Academy of Sciences, Beijing, China.
Yunchao Ling, Bio-Med Big Data Center, CAS Key Laboratory of Computational Biology, CAS-MPG Partner Institute for Computational Biology, Shanghai Institute of Nutrition and Health, Chinese Academy of Sciences, Shanghai 200031, China.
Sirui Zhang, Bio-Med Big Data Center, CAS Key Laboratory of Computational Biology, CAS-MPG Partner Institute for Computational Biology, Shanghai Institute of Nutrition and Health, Chinese Academy of Sciences, Shanghai 200031, China; University of Chinese Academy of Sciences, Beijing, China.
Qiguang Xia, Bio-Med Big Data Center, CAS Key Laboratory of Computational Biology, CAS-MPG Partner Institute for Computational Biology, Shanghai Institute of Nutrition and Health, Chinese Academy of Sciences, Shanghai 200031, China; University of Chinese Academy of Sciences, Beijing, China.
Ruifang Cao, Bio-Med Big Data Center, CAS Key Laboratory of Computational Biology, CAS-MPG Partner Institute for Computational Biology, Shanghai Institute of Nutrition and Health, Chinese Academy of Sciences, Shanghai 200031, China.
Xiaojuan Fan, Bio-Med Big Data Center, CAS Key Laboratory of Computational Biology, CAS-MPG Partner Institute for Computational Biology, Shanghai Institute of Nutrition and Health, Chinese Academy of Sciences, Shanghai 200031, China.
Zhaoyuan Fang, Bio-Med Big Data Center, CAS Key Laboratory of Computational Biology, CAS-MPG Partner Institute for Computational Biology, Shanghai Institute of Nutrition and Health, Chinese Academy of Sciences, Shanghai 200031, China; CAS Center for Excellence in Molecular Cell Science, Chinese Academy of Sciences, Shanghai 200031, China.
Zefeng Wang, Bio-Med Big Data Center, CAS Key Laboratory of Computational Biology, CAS-MPG Partner Institute for Computational Biology, Shanghai Institute of Nutrition and Health, Chinese Academy of Sciences, Shanghai 200031, China; University of Chinese Academy of Sciences, Beijing, China; CAS Center for Excellence in Molecular Cell Science, Chinese Academy of Sciences, Shanghai 200031, China.
Guoqing Zhang, Bio-Med Big Data Center, CAS Key Laboratory of Computational Biology, CAS-MPG Partner Institute for Computational Biology, Shanghai Institute of Nutrition and Health, Chinese Academy of Sciences, Shanghai 200031, China; University of Chinese Academy of Sciences, Beijing, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Key Research and Development Program of China [2016YFC0901904, 2017YFC1201200]; Special Project on National Science and Technology Basic Resources Investigation [2019FY100102]; Strategic Priority Research Program of Chinese Academy of Sciences [XDB38040100]; National Natural Science Foundation of China [91940303, 31661143031, 31730110]; Science and Technology Commission of Shanghai Municipality [17JC1404900]; Z.W. is also supported by the type A CAS Pioneer 100-Talent program. Funding for open access charge: National Key Research and Development Program of China [2016YFC0901904, 2017YFC1201200]; Special Project on National Science and Technology Basic Resources Investigation [2019FY100102]; National Natural Science Foundation of China [91940303, 31661143031, 31730110]; Science and Technology Commission of Shanghai Municipality [17JC1404900]; Z.W. is also supported by the type A CAS Pioneer 100-Talent program.
Conflict of interest statement. None declared.
REFERENCES
- 1. Chen L.L. The biogenesis and emerging roles of circular RNAs. Nat. Rev. Mol. Cell Biol. 2016; 17:205–211. [DOI] [PubMed] [Google Scholar]
- 2. Barrett S.P., Salzman J.. Circular RNAs: analysis, expression and potential functions. Development. 2016; 143:1838–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li X., Yang L., Chen L.L.. The biogenesis, functions, and challenges of circular RNAs. Mol. Cell. 2018; 71:428–442. [DOI] [PubMed] [Google Scholar]
- 4. Kristensen L.S., Andersen M.S., Stagsted L.V.W., Ebbesen K.K., Hansen T.B., Kjems J.. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 2019; 20:675–691. [DOI] [PubMed] [Google Scholar]
- 5. Jeck W.R., Sorrentino J.A., Wang K., Slevin M.K., Burd C.E., Liu J., Marzluff W.F., Sharpless N.E.. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013; 19:141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Salzman J., Gawad C., Wang P.L., Lacayo N., Brown P.O.. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One. 2012; 7:e30733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hansen T.B., Jensen T.I., Clausen B.H., Bramsen J.B., Finsen B., Damgaard C.K., Kjems J.. Natural RNA circles function as efficient microRNA sponges. Nature. 2013; 495:384–388. [DOI] [PubMed] [Google Scholar]
- 8. Memczak S., Jens M., Elefsinioti A., Torti F., Krueger J., Rybak A., Maier L., Mackowiak S.D., Gregersen L.H., Munschauer M. et al.. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013; 495:333–338. [DOI] [PubMed] [Google Scholar]
- 9. Li Z., Huang C., Bao C., Chen L., Lin M., Wang X., Zhong G., Yu B., Hu W., Dai L. et al.. Exon-intron circular RNAs regulate transcription in the nucleus. Nat. Struct. Mol. Biol. 2015; 22:256–264. [DOI] [PubMed] [Google Scholar]
- 10. Zhang Y., Zhang X.O., Chen T., Xiang J.F., Yin Q.F., Xing Y.H., Zhu S., Yang L., Chen L.L.. Circular intronic long noncoding RNAs. Mol. Cell. 2013; 51:792–806. [DOI] [PubMed] [Google Scholar]
- 11. Ashwal-Fluss R., Meyer M., Pamudurti N.R., Ivanov A., Bartok O., Hanan M., Evantal N., Memczak S., Rajewsky N., Kadener S.. circRNA biogenesis competes with pre-mRNA splicing. Mol. Cell. 2014; 56:55–66. [DOI] [PubMed] [Google Scholar]
- 12. Li H.M., Ma X.L., Li H.G.. Intriguing circles: conflicts and controversies in circular RNA research. Wiley Interdiscip Rev RNA. 2019; 10:e1538. [DOI] [PubMed] [Google Scholar]
- 13. Chen C.Y., Sarnow P.. Initiation of protein synthesis by the eukaryotic translational apparatus on circular RNAs. Science. 1995; 268:415–417. [DOI] [PubMed] [Google Scholar]
- 14. Wang Y., Wang Z.. Efficient backsplicing produces translatable circular mRNAs. RNA. 2015; 21:172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang Y., Fan X., Mao M., Song X., Wu P., Zhang Y., Jin Y., Yang Y., Chen L.L., Wang Y. et al.. Extensive translation of circular RNAs driven by N(6)-methyladenosine. Cell Res. 2017; 27:626–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Legnini I., Di Timoteo G., Rossi F., Morlando M., Briganti F., Sthandier O., Fatica A., Santini T., Andronache A., Wade M. et al.. Circ-ZNF609 is a circular RNA that can be translated and functions in myogenesis. Mol. Cell. 2017; 66:22–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pamudurti N.R., Bartok O., Jens M., Ashwal-Fluss R., Stottmeister C., Ruhe L., Hanan M., Wyler E., Perez-Hernandez D., Ramberger E. et al.. Translation of CircRNAs. Mol. Cell. 2017; 66:9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang M., Zhao K., Xu X., Yang Y., Yan S., Wei P., Liu H., Xu J., Xiao F., Zhou H. et al.. A peptide encoded by circular form of LINC-PINT suppresses oncogenic transcriptional elongation in glioblastoma. Nat. Commun. 2018; 9:4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xia X., Li X., Li F., Wu X., Zhang M., Zhou H., Huang N., Yang X., Xiao F., Liu D. et al.. A novel tumor suppressor protein encoded by circular AKT3 RNA inhibits glioblastoma tumorigenicity by competing with active phosphoinositide-dependent Kinase-1. Mol. Cancer. 2019; 18:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang M., Huang N., Yang X., Luo J., Yan S., Xiao F., Chen W., Gao X., Zhao K., Zhou H. et al.. A novel protein encoded by the circular form of the SHPRH gene suppresses glioma tumorigenesis. Oncogene. 2018; 37:1805–1814. [DOI] [PubMed] [Google Scholar]
- 21. Ingolia N.T., Hussmann J.A., Weissman J.S.. Ribosome profiling: global views of translation. Cold Spring Harb. Perspect. Biol. 2019; 11:a032698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. You X., Vlatkovic I., Babic A., Will T., Epstein I., Tushev G., Akbalik G., Wang M., Glock C., Quedenau C. et al.. Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat. Neurosci. 2015; 18:603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rybak-Wolf A., Stottmeister C., Glazar P., Jens M., Pino N., Giusti S., Hanan M., Behm M., Bartok O., Ashwal-Fluss R. et al.. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol. Cell. 2015; 58:870–885. [DOI] [PubMed] [Google Scholar]
- 24. Wang P.L., Bao Y., Yee M.C., Barrett S.P., Hogan G.J., Olsen M.N., Dinneny J.R., Brown P.O., Salzman J.. Circular RNA is expressed across the eukaryotic tree of life. PLoS One. 2014; 9:e90859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Szabo L., Salzman J.. Detecting circular RNAs: bioinformatic and experimental challenges. Nat. Rev. Genet. 2016; 17:679–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gao Y., Wang J., Zheng Y., Zhang J., Chen S., Zhao F.. Comprehensive identification of internal structure and alternative splicing events in circular RNAs. Nat. Commun. 2016; 7:12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vromman M., Vandesompele J., Volders P.J.. Closing the circle: current state and perspectives of circular RNA databases. Brief. Bioinform. 2020; doi:10.1093/bib/bbz175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen X., Han P., Zhou T., Guo X., Song X., Li Y.. circRNADb: A comprehensive database for human circular RNAs with protein-coding annotations. Sci. Rep. 2016; 6:34985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Glazar P., Papavasileiou P., Rajewsky N.. circBase: a database for circular RNAs. RNA. 2014; 20:1666–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dong R., Ma X.K., Li G.W., Yang L.. CIRCpedia v2: an updated database for comprehensive circular RNA annotation and expression comparison. Genomics Proteomics Bioinformatics. 2018; 16:226–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu W., Ji P., Zhao F.. CircAtlas: an integrated resource of one million highly accurate circular RNAs from 1070 vertebrate transcriptomes. Genome Biol. 2020; 21:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Xia S., Feng J., Chen K., Ma Y., Gong J., Cai F., Jin Y., Gao Y., Xia L., Chang H. et al.. CSCD: a database for cancer-specific circular RNAs. Nucleic Acids Res. 2018; 46:D925–D929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zheng Y., Ji P., Chen S., Hou L., Zhao F.. Reconstruction of full-length circular RNAs enables isoform-level quantification. Genome Med. 2019; 11:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gao Y., Zhang J., Zhao F.. Circular RNA identification based on multiple seed matching. Brief. Bioinform. 2018; 19:803–810. [DOI] [PubMed] [Google Scholar]
- 35. Ji P., Wu W., Chen S., Zheng Y., Zhou L., Zhang J., Cheng H., Yan J., Zhang S., Yang P. et al.. Expanded expression landscape and prioritization of circular RNAs in mammals. Cell Rep. 2019; 26:3444–3460. [DOI] [PubMed] [Google Scholar]
- 36. Ragan C., Goodall G.J., Shirokikh N.E., Preiss T.. Insights into the biogenesis and potential functions of exonic circular RNA. Sci. Rep. 2019; 9:2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Floor S.N., Doudna J.A.. Tunable protein synthesis by transcript isoforms in human cells. Elife. 2016; 5:e10921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang X.O., Wang H.B., Zhang Y., Lu X., Chen L.L., Yang L.. Complementary sequence-mediated exon circularization. Cell. 2014; 159:134–147. [DOI] [PubMed] [Google Scholar]
- 39. Fan X., Yang Y., Chen C., Wang Z.. Pervasive translation of circular RNAs driven by short IRES-like elements. 2020; bioRxiv doi:28 November 2018, preprint: not peer reviewed 10.1101/473207. [DOI] [PMC free article] [PubMed]
- 40. Liu S., Zhu A., He C., Chen M.. REPIC: a database for exploring the N(6)-methyladenosine methylome. Genome Biol. 2020; 21:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ingolia N.T., Lareau L.F., Weissman J.S.. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011; 147:789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee S., Liu B., Huang S.X., Shen B., Qian S.B.. Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:E2424–E2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Frankish A., Diekhans M., Ferreira A.M., Johnson R., Jungreis I., Loveland J., Mudge J.M., Sisu C., Wright J., Armstrong J. et al.. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019; 47:D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim M.S., Pinto S.M., Getnet D., Nirujogi R.S., Manda S.S., Chaerkady R., Madugundu A.K., Kelkar D.S., Isserlin R., Jain S. et al.. A draft map of the human proteome. Nature. 2014; 509:575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chi H., Liu C., Yang H., Zeng W.F., Wu L., Zhou W.J., Wang R.M., Niu X.N., Ding Y.H., Zhang Y. et al.. Comprehensive identification of peptides in tandem mass spectra using an efficient open search engine. Nat. Biotechnol. 2018; 36:1059–1061. [DOI] [PubMed] [Google Scholar]
- 46. Choi S.W., Kim H.W., Nam J.W.. The small peptide world in long noncoding RNAs. Brief. Bioinform. 2019; 20:1853–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yang Y., Wang Z.. IRES-mediated cap-independent translation, a path leading to hidden proteome. J. Mol. Cell Biol. 2019; 11:911–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Weingarten-Gabbay S., Elias-Kirma S., Nir R., Gritsenko A.A., Stern-Ginossar N., Yakhini Z., Weinberger A., Segal E.. Systematic discovery of cap-independent translation sequences in human and viral genomes. Science. 2016; 351:aad4939. [DOI] [PubMed] [Google Scholar]
- 49. Bekker-Jensen D.B., Kelstrup C.D., Batth T.S., Larsen S.C., Haldrup C., Bramsen J.B., Sorensen K.D., Hoyer S., Orntoft T.F., Andersen C.L. et al.. An optimized shotgun strategy for the rapid generation of comprehensive human proteomes. Cell Syst. 2017; 4:587–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fagerberg L., Hallstrom B.M., Oksvold P., Kampf C., Djureinovic D., Odeberg J., Habuka M., Tahmasebpoor S., Danielsson A., Edlund K. et al.. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteomics. 2014; 13:397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang Y., Chung D.H., Monteleone L.R., Li J., Chiang Y., Toney M.D., Beal P.A.. RNA binding candidates for human ADAR3 from substrates of a gain of function mutant expressed in neuronal cells. Nucleic Acids Res. 2019; 47:10801–10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.