


Abstract
Proteins are intricate, dynamic structures, and small changes in their amino acid sequences can lead to large effects on their folding, stability and dynamics. To facilitate the further development and evaluation of methods to predict these changes, we have developed ThermoMutDB, a manually curated database containing >14,669 experimental data of thermodynamic parameters for wild type and mutant proteins. This represents an increase of 83% in unique mutations over previous databases and includes thermodynamic information on 204 new proteins. During manual curation we have also corrected annotation errors in previously curated entries. Associated with each entry, we have included information on the unfolding Gibbs free energy and melting temperature change, and have associated entries with available experimental structural information. ThermoMutDB supports users to contribute to new data points and programmatic access to the database via a RESTful API. ThermoMutDB is freely available at: http://biosig.unimelb.edu.au/thermomutdb.
Graphical Abstract
Graphical Abstract.
ThermoMutDB is an online resource associating effects of missense mutations on protein thermodynamics.
INTRODUCTION
Protein thermodynamic stability is a fundamental property of proteins that significantly influences their structure, function, expression, and solubility. Changes in protein stability have been shown to be a main driving molecular mechanism of genetic diseases (1–8) and even drug resistance (9–18). Small changes in the protein sequence can have significant consequences on their intricate structures, reflected in changes in their stability and ability to correctly fold (19). This is often a significant consideration whenever considering a new mutation, whether in the context of protein engineering or variant characterisation (20,21).
The accurate prediction of the effects of mutations on protein stability remains a complex and challenging problem. The development of computational approaches to tackle this have required large mutational datasets, however in turn have been limited by the quantity and quality of data available.
One of the first databases to collect information on the effects of mutations on protein stability, ProTherm (22), led to the exploration and rapid development of new computational approaches (23–28). However, this database has not been updated for 7 years and many errors have been identified previously (29,30), limiting both previous methods and future developments.
To overcome this, we have developed a new comprehensive and user-friendly resource for thermodynamic data from protein mutations, ThemoMutDB. Figure 1 depicts the database development workflow, which is divided into three main stages: (i) data acquisition and curation, (ii) mutation annotation and (iii) web-server development. By using a rigorous and careful data curation approach, ThemoMutDB represents a significant improvement in both the quantity and quality of data. This will not only enable the development of a new generation of methods but also an unbiased assessment of previously proposed ones.
Figure 1.
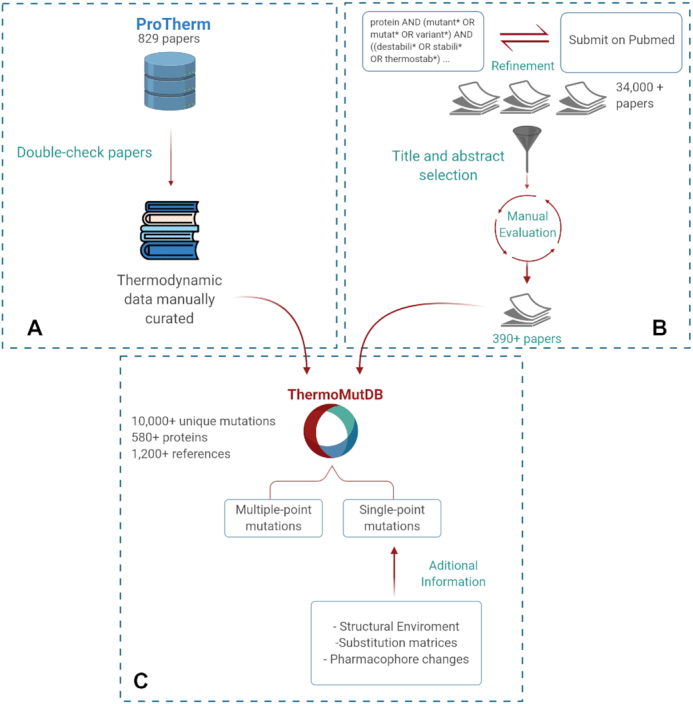
ThermoMutDB workflow for data acquisition and processing. The development workflow is divided into three steps: (A) verification of previously available mutation thermodynamics information (B) collection and manual curation of new data and (C) data aggregation and mutation annotation.
MATERIALS AND METHODS
Data acquisition and curation
Data acquisition for ThermoMutDB was divided into two steps: manual checking of previously mined data available in other resources (Figure 1A) and manual literature curation of new thermodynamic data (Figure 1B). Within ThermoMutDB we captured thermodynamic information, experimental conditions, and literature citations. We also standardized measurements and calculations across the data entries, including temperature in Kelvin, energy in kcal/mol, and Gibbs free energy (ΔΔG) as in the formula:
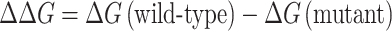 |
where negative ΔΔG values indicate that the mutation has destabilized the protein and positive ΔΔG values that the mutant protein is more stable.
On the first data acquisition stage, all 1,902 references in ProTherm were manually checked and validated. References that did not contain data about missense mutations were removed, leaving 829 papers. During this process, errors in data fields were corrected, duplicate entries were removed, and 329 new data-points not previously captured, but present in the original papers, were included.
New data were identified through manual literature curation. Optimized search terms (Supplementary Figure S1) were used to identify an initial pool of over 34,000 manuscripts available on PubMed. These were further narrowed down to those that contained experimental thermodynamic results for missense mutations. In total, 393 papers were analyzed and 5,654 new data points obtained, which were confirmed by at least two independent curators. Supplementary Figure S2 shows the distribution of unique mutations collected per year.
Mutation annotation
Collected mutations were mapped to protein structures available at the Protein Data Bank using (31). Different characteristics of the wild-type residue environment were calculated, including secondary structure, torsional angles, relative solvent accessibility (32) and residue depth (33). Additional residue-level information used to annotate the mutations included different substitution matrix scores. Mutation annotations were calculated using the Biopython (34). Mutation effects are also depicted via pharmacophore modeling (23). Pharmacophore modeling has been introduced in the context of mutation analysis in a previous work (23) to characterise the effect of mutations based on the differences in atom counts per pharmacophore type. Mutations that do not map to any available experimental structures are still listed but without any structure-based features calculated.
Database and web interface implementation
The database architecture was developed using SQLAlchemy, a database toolkit for Python (version 2.7). All data is stored in an SQLite database and available to download at http://biosig.unimelb.edu.au/thermomutdb/downloads. The backend system was developed using the Flask Python module (version 1.0.2) and the REStful API uses RestX extension for Flask (version 0.2.0). The web interface was implemented using the Bootstrap (version 4) framework. It also uses HTML5, CSS, JavaScript, and JQuery. JINJA2 templating language for Python was used to dynamically generate HTML templates.
RESULTS
Web interface and usage
ThermoMutDB contains information of the protein, mutational information, experimental methods and conditions, thermodynamic parameters, derived data, and literature information (details are available in Supplementary Table S1 and Figure S2). The database provides a user-friendly web interface that contains five modules: Explore and Browse, Contribute, Downloads, API and a detailed tutorial.
Explore and browse
In order to access the data, a search can be performed. This can be done either by selecting the ‘Browse’ page from the navigation bar or by writing the desired words on search input available on the ‘Home’ page. In both cases, users can use different filter combinations (Figure 2A), include or exclude columns, and download selected results in several formats (JSON, XML, CSV, TXT, SQL, MS-Excel and PDF).
Figure 2.
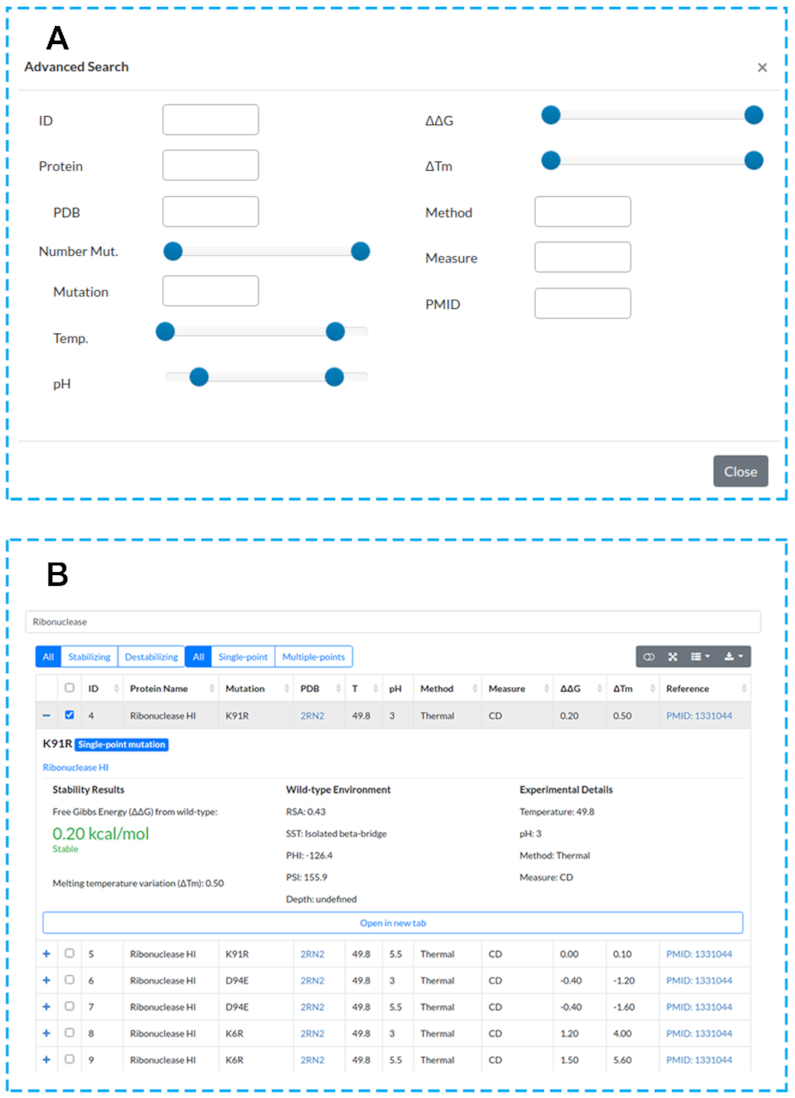
ThermoMutDB web interface search and results pages. (A) ThermoMutDB offers 12 query modes, with detailed information available about each query type through the ‘Help’ page at the top navigation bar and through on-page help in the form of question mark tooltips. (B) The general layout of the result page, showing a summary of information for each entry as well as detailed view.
The search results are shown in an interactive table, with columns providing experimental information recovered from literature and also derived properties (Figure 2B). Aiming to improve user experience, it is possible to visualize a summary for each entry by clicking on the ‘+’ icon. This option can lead to a ‘Details’ page that shows all information about the mutation and provides related files to download (Supplementary Figure S3).
User contributions
To facilitate a continuous database update, we have implemented a user's contribution section (Supplementary Figure S4), which allows the scientific community to share new data or identify potential errors that will be manually checked by our team. To submit contributions it is just required to fill the form with mutation and thermodynamics data, to inform a contact email and a reference (paper published, accepted, or pre-print). Although significant effort has been devoted to ensure high quality data curation, users have the option to report any issues with the data to our team. These are important efforts to further expand and improve the database.
Downloads
All data in the database can be downloaded from the ‘Download’ page in CSV or JSON formats. It is also possible to download the protein structure files related to data available.
Programmatic access via an API
ThermoMutDB supports programmatic access via a RESTful API to allow other services to harness our data easily. The ‘API’ page provides documentation of all endpoints available and allows users to execute queries using provided fields. Other queries can be performed by passing parameters through the URL (Supplementary Figure S5).
Data statistics
Examining the distribution of mutations in the ThermoMutDB reveals a number of natural biases that need to be taken into consideration when developing, or evaluating, new predictive tools. ThermoMutDB contains thermodynamic information on 14,669 mutations across 588 proteins. This represents a significant increase over ProTherm, with a 83% increase in unique mutations and over 300 new proteins. Supplementary Figure S6 shows the distribution of unique mutations collected per year. The majority of these are single-point mutations (82.8%), with mutations to alanine being over-represented (Figure 3D). This becomes evident when we look at the distribution of wild-type and mutant amino acid residues within the database (Supplementary Figure S7). The most frequent mutations were from Leucine and Valine to Alanine, while 10 mutations were not present in the dataset, including W→G, W→P and C→K among others, which seem to denote large changes in residue physicochemical properties.
Figure 3.
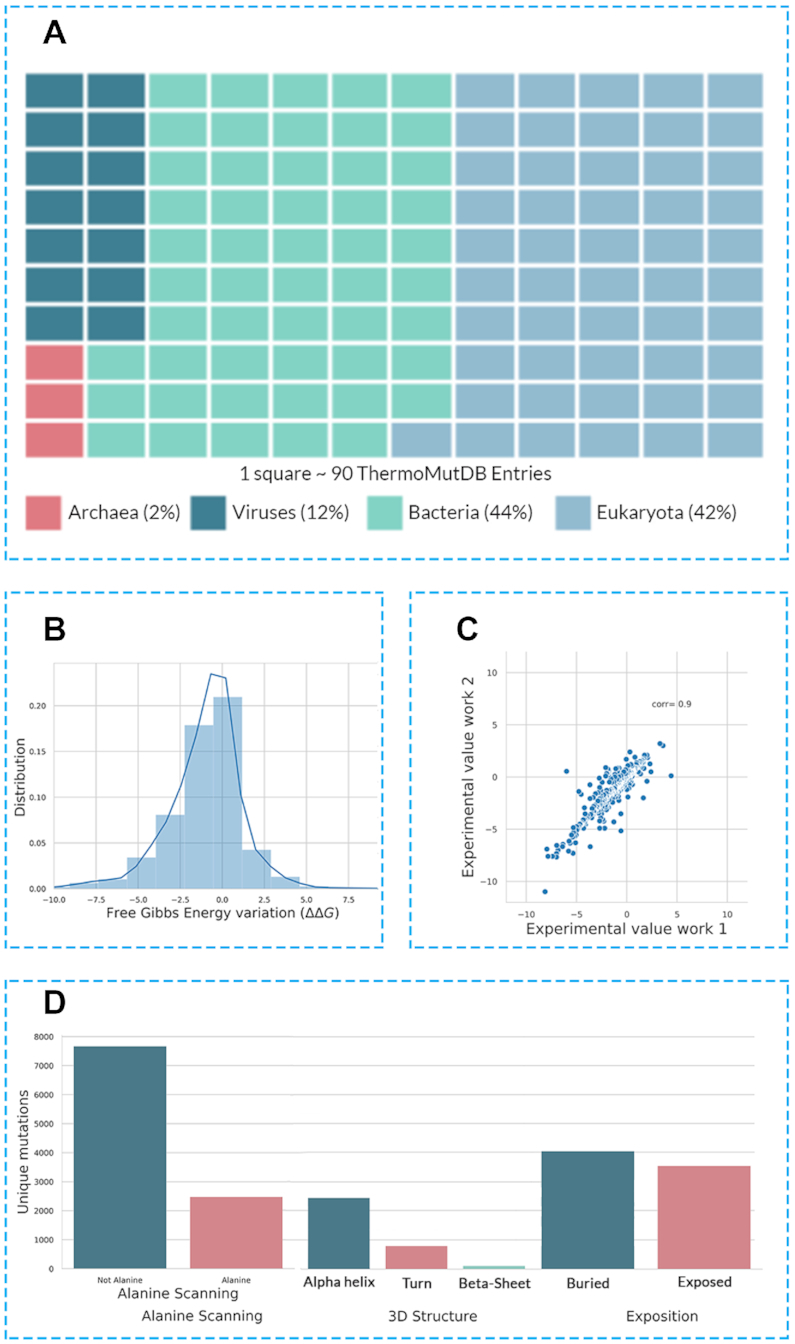
Composition of ThermoMutDB entries. (A) depicts the distribution of phylogenetic kingdoms of proteins in the database. (B) highlights the distribution of thermodynamic effects of mutation in the database, given as the variation in Gibbs Free Energy (ΔΔG). (C) Experimental variability of mutation assessed under different conditions and groups. (D) Distribution of mutations in ThermoMutDB based on type (mutation to alanine/non-alanine), their location and residue environment.
As would be expected by chance, two thirds of mutations within the database are destabilising (Supplementary Figure S8). This natural bias creates an extra challenge for computational methods built using this information, in particular those based on machine learning approaches, regarding the prediction of stabilising mutations, which are less well represented. It is important to note, however, that the data on ThermoMutDB represents an increase of over 100% in stabilising mutations in comparison with previous resources. No apparent correlation was identified between the mutation effects and their location within protein structures, with mutations leading to increased and decreased stability similarly distributed across protein structures when looking at residue depth (Supplementary Figure S9). Mutations in ThermoMutDB are spread across different protein classes (Supplementary Figure S10) and diverse in terms of secondary structure (Supplementary Figure S11).
Within ThermoMutDB, we identified mutations that had been experimentally measured at least twice and, by comparing the variance between these replicate results (Figure 3C), we identified a Pearson's correlation of 0.9. This provides a measure of the intrinsic noise in the data, and suggests a theoretical maximum performance that should be expected for predictive stability tools built using this data.
DISCUSSION
ThermoMutDB represents a significant increase in availability, reliability and diversity of thermodynamics data linking effects of mutations to protein stability. We believe this resource will have a significant impact on understanding the effects of mutations on protein structure and stability. It will enable experimental scientists to identify previously characterised mutations in proteins of interest, and provide computational scientists with a comprehensive and refined set of experimental data to query the relationship between changes in protein sequence and stability, facilitating the development of new computational tools to analyse these relationships and develop prediction algorithms.
New mutation thermodynamics data collected and compiled in ThermoMutDB will also allow for more robust, comprehensive and independent validation of currently available computational predictors. The database will be continuously maintained and updated, enabling submission of user contributions and data access through an intuitive web-based interface (http://biosig.unimelb.edu.au/thermomutdb) as well as programmatic access through an API.
Supplementary Material
Contributor Information
Joicymara S Xavier, Institute of Agricultural Sciences, Universidade Federal dos Vales do Jequitinhonha e Mucuri; Instituto René Rachou, Fundação Oswaldo Cruz.
Thanh-Binh Nguyen, Bio 21 Institute, University of Melbourne.
Malancha Karmarkar, Bio 21 Institute, University of Melbourne; Computational Biology and Clinical Informatics, Baker Heart and Diabetes Institute.
Stephanie Portelli, Bio 21 Institute, University of Melbourne; Computational Biology and Clinical Informatics, Baker Heart and Diabetes Institute.
Pâmela M Rezende, Instituto René Rachou, Fundação Oswaldo Cruz.
João P L Velloso, Instituto René Rachou, Fundação Oswaldo Cruz.
David B Ascher, Bio 21 Institute, University of Melbourne; Computational Biology and Clinical Informatics, Baker Heart and Diabetes Institute; Department of Biochemistry, University of Cambridge.
Douglas E V Pires, Bio 21 Institute, University of Melbourne; Computational Biology and Clinical Informatics, Baker Heart and Diabetes Institute; School of Computing and Information Systems, University of Melbourne.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
D.B.A. and D.E.V.P. were funded by a Newton Fund RCUK-CONFAP Grant awarded by the Medical Research Council (MRC) and Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG) [MR/M026302/1]; Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq); Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES); Jack Brockhoff Foundation [JBF 4186, 2016]; Wellcome Trust [200814/Z/16/Z]; Investigator Grant from the National Health and Medical Research Council (NHMRC) of Australia [GNT1174405]. Funding for open access charge: Wellcome Trust.
Conflict of interest statement. None declared.
REFERENCES
- 1. Jafri M., Wake N.C., Ascher D.B., Pires D.E., Gentle D., Morris M.R., Rattenberry E., Simpson M.A., Trembath R.C., Weber A. et al.. Germline mutations in the CDKN2B tumor suppressor gene predispose to renal cell carcinoma. Cancer Discov. 2015; 5:723–729. [DOI] [PubMed] [Google Scholar]
- 2. Ramdzan Y.M., Trubetskov M.M., Ormsby A.R., Newcombe E.A., Sui X., Tobin M.J., Bongiovanni M.N., Gras S.L., Dewson G., Miller J.M.L. et al.. Huntingtin inclusions trigger cellular quiescence, deactivate apoptosis, and lead to delayed necrosis. Cell Rep. 2017; 19:919–927. [DOI] [PubMed] [Google Scholar]
- 3. Soardi F.C., Machado-Silva A., Linhares N.D., Zheng G., Qu Q., Pena H.B., Martins T.M.M., Vieira H.G.S., Pereira N.B., Melo-Minardi R.C. et al.. Familial STAG2 germline mutation defines a new human cohesinopathy. NPJ Genom Med. 2017; 2:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Andrews K.A., Ascher D.B., Pires D.E.V., Barnes D.R., Vialard L., Casey R.T., Bradshaw N., Adlard J., Aylwin S., Brennan P. et al.. Tumour risks and genotype-phenotype correlations associated with germline variants in succinate dehydrogenase subunit genes SDHB, SDHC and SDHD. J. Med. Genet. 2018; 55:384–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ascher D.B., Spiga O., Sekelska M., Pires D.E.V., Bernini A., Tiezzi M., Kralovicova J., Borovska I., Soltysova A., Olsson B. et al.. Homogentisate 1,2-dioxygenase (HGD) gene variants, their analysis and genotype-phenotype correlations in the largest cohort of patients with AKU. Eur. J. Hum. Genet. 2019; 27:888–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hildebrand J.M., Kauppi M., Majewski I.J., Liu Z., Cox A.J., Miyake S., Petrie E.J., Silk M.A., Li Z., Tanzer M.C. et al.. A missense mutation in the MLKL brace region promotes lethal neonatal inflammation and hematopoietic dysfunction. Nat. Commun. 2020; 11:3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pires D.E.V., Rodrigues C.H.M., Ascher D.B.. mCSM-membrane: predicting the effects of mutations on transmembrane proteins. Nucleic Acids Res. 2020; 48:W147–W153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Trezza A., Bernini A., Langella A., Ascher D.B., Pires D.E.V., Sodi A., Passerini I., Pelo E., Rizzo S., Niccolai N. et al.. A computational approach from gene to structure analysis of the human ABCA4 transporter involved in genetic retinal diseases. Invest. Ophthalmol. Vis. Sci. 2017; 58:5320–5328. [DOI] [PubMed] [Google Scholar]
- 9. Ascher D.B., Wielens J., Nero T.L., Doughty L., Morton C.J., Parker M.W.. Potent hepatitis C inhibitors bind directly to NS5A and reduce its affinity for RNA. Sci. Rep. 2014; 4:4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Phelan J., Coll F., McNerney R., Ascher D.B., Pires D.E., Furnham N., Coeck N., Hill-Cawthorne G.A., Nair M.B., Mallard K. et al.. Mycobacterium tuberculosis whole genome sequencing and protein structure modelling provides insights into anti-tuberculosis drug resistance. BMC Med. 2016; 14:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Karmakar M., Globan M., Fyfe J.A.M., Stinear T.P., Johnson P.D.R., Holmes N.E., Denholm J.T., Ascher D.B.. Analysis of a novel pncA mutation for susceptibility to pyrazinamide therapy. Am. J. Respir. Crit. Care Med. 2018; 198:541–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Portelli S., Phelan J.E., Ascher D.B., Clark T.G., Furnham N.. Understanding molecular consequences of putative drug resistant mutations in Mycobacterium tuberculosis. Sci. Rep. 2018; 8:15356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Karmakar M., Rodrigues C.H.M., Holt K.E., Dunstan S.J., Denholm J., Ascher D.B.. Empirical ways to identify novel Bedaquiline resistance mutations in AtpE. PLoS One. 2019; 14:e0217169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hawkey J., Ascher D.B., Judd L.M., Wick R.R., Kostoulias X., Cleland H., Spelman D.W., Padiglione A., Peleg A.Y., Holt K.E.. Evolution of carbapenem resistance in Acinetobacter baumannii during a prolonged infection. Microbial Genomics. 2018; 4:e000165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Holt K.E., McAdam P., Thai P.V.K., Thuong N.T.T., Ha D.T.M., Lan N.N., Lan N.H., Nhu N.T.Q., Hai H.T., Ha V.T.N. et al.. Frequent transmission of the Mycobacterium tuberculosis Beijing lineage and positive selection for the EsxW Beijing variant in Vietnam. Nat. Genet. 2018; 50:849–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vedithi S.C., Malhotra S., Das M., Daniel S., Kishore N., George A., Arumugam S., Rajan L., Ebenezer M., Ascher D.B. et al.. Structural implications of mutations conferring rifampin resistance in Mycobacterium leprae. Sci. Rep. 2018; 8:5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Karmakar M., Rodrigues C.H.M., Horan K., Denholm J.T., Ascher D.B.. Structure guided prediction of Pyrazinamide resistance mutations in pncA. Sci. Rep. 2020; 10:1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Portelli S., Olshansky M., Rodrigues C.H.M., D'Souza E.N., Myung Y., Silk M., Alavi A., Pires D.E.V., Ascher D.B.. Exploring the structural distribution of genetic variation in SARS-CoV-2 with the COVID-3D online resource. Nat. Genet. 2020; 52:999–1001. [DOI] [PubMed] [Google Scholar]
- 19. Pandurangan A.P., Ascher D.B., Thomas S.E., Blundell T.L.. Genomes, structural biology and drug discovery: combating the impacts of mutations in genetic disease and antibiotic resistance. Biochem. Soc. Trans. 2017; 45:303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wijma H.J., Floor R.J., Janssen D.B.. Structure- and sequence-analysis inspired engineering of proteins for enhanced thermostability. Curr. Opin. Struct. Biol. 2013; 23:588–594. [DOI] [PubMed] [Google Scholar]
- 21. Pires D.E., Chen J., Blundell T.L., Ascher D.B.. In silico functional dissection of saturation mutagenesis: interpreting the relationship between phenotypes and changes in protein stability, interactions and activity. Sci. Rep. 2016; 6:19848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gromiha M.M., An J., Kono H., Oobatake M., Uedaira H., Sarai A.. ProTherm: thermodynamic database for proteins and mutants. Nucleic Acids Res. 1999; 27:286–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pires D.E., Ascher D.B., Blundell T.L.. mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics. 2014; 30:335–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pires D.E., Ascher D.B., Blundell T.L.. DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 2014; 42:W314–W319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pandurangan A.P., Ochoa-Montano B., Ascher D.B., Blundell T.L.. SDM: a server for predicting effects of mutations on protein stability. Nucleic Acids Res. 2017; 45:W229–W235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rodrigues C.H., Pires D.E., Ascher D.B.. DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 2018; 46:W350–W355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Laimer J., Hiebl-Flach J., Lengauer D., Lackner P.. MAESTROweb: a web server for structure-based protein stability prediction. Bioinformatics. 2016; 32:1414–1416. [DOI] [PubMed] [Google Scholar]
- 28. Quan L., Lv Q., Zhang Y.. STRUM: structure-based prediction of protein stability changes upon single-point mutation. Bioinformatics. 2016; 32:2936–2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang Y., Urolagin S., Niroula A., Ding X., Shen B., Vihinen M.. PON-tstab: protein variant stability predictor. Importance of training data quality. Int. J. Mol. Sci. 2018; 19:1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fang J. A critical review of five machine learning-based algorithms for predicting protein stability changes upon mutation. Brief. Bioinform. 2020; 21:1285–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Martin A.C. Mapping PDB chains to UniProtKB entries. Bioinformatics. 2005; 21:4297–4301. [DOI] [PubMed] [Google Scholar]
- 32. Gilis D., Rooman M.. Predicting protein stability changes upon mutation using database-derived potentials: solvent accessibility determines the importance of local versus non-local interactions along the sequence. J. Mol. Biol. 1997; 272:276–290. [DOI] [PubMed] [Google Scholar]
- 33. Chakravarty S., Varadarajan R.. Residue depth: a novel parameter for the analysis of protein structure and stability. Structure. 1999; 7:723–732. [DOI] [PubMed] [Google Scholar]
- 34. Cock P.J., Antao T., Chang J.T., Chapman B.A., Cox C.J., Dalke A., Friedberg I., Hamelryck T., Kauff F., Wilczynski B. et al.. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009; 25:1422–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.