


Abstract
The Database of Antimicrobial Activity and Structure of Peptides (DBAASP) is an open-access, comprehensive database containing information on amino acid sequences, chemical modifications, 3D structures, bioactivities and toxicities of peptides that possess antimicrobial properties. DBAASP is updated continuously, and at present, version 3.0 (DBAASP v3) contains >15 700 entries (8000 more than the previous version), including >14 500 monomers and nearly 400 homo- and hetero-multimers. Of the monomeric antimicrobial peptides (AMPs), >12 000 are synthetic, about 2700 are ribosomally synthesized, and about 170 are non-ribosomally synthesized. Approximately 3/4 of the entries were added after the initial release of the database in 2014 reflecting the recent sharp increase in interest in AMPs. Despite the increased interest, adoption of peptide antimicrobials in clinical practice is still limited as a consequence of several factors including side effects, problems with bioavailability and high production costs. To assist in developing and optimizing de novo peptides with desired biological activities, DBAASP offers several tools including a sophisticated multifactor analysis of relevant physicochemical properties. Furthermore, DBAASP has implemented a structure modelling pipeline that automates the setup, execution and upload of molecular dynamics (MD) simulations of database peptides. At present, >3200 peptides have been populated with MD trajectories and related analyses that are both viewable within the web browser and available for download. More than 400 DBAASP entries also have links to experimentally determined structures in the Protein Data Bank. DBAASP v3 is freely accessible at http://dbaasp.org.
INTRODUCTION
Antimicrobial peptides (AMPs) have attracted growing attention for several decades due to their wide array of biological activities and the worldwide challenge of increasing resistance of pathogens to existing drugs. Although the field of AMPs is rapidly developing, only a few AMPs have advanced to the clinic. Of 35 FDA approved anti-infective peptides and proteins for which data are included in the THPdb database (1), only a few peptides are annotated as antimicrobial drugs. Another database, DRAMP (2), which derives its data largely from patents, has information on clinical trials of 76 AMPs. The limited success of AMPs at the clinical level is associated with susceptibility to proteolysis, toxicities and high costs of production. Limitations both in understanding of AMP modes of action and of existing in silico methods for targeted design further hinder the development of peptides for clinical use. To effectively leverage the fast growth in relevant AMP studies, data have to be collected and stored in modern databases and connected to pipelines for analysis, modelling and design of novel peptides (2–11).
The Database of Antimicrobial Activity and Structure of Peptides (DBAASP) was launched in 2014 as both the bibliographic collection of published information about AMPs and the resource aiming to facilitate structure activity relationship studies and the development of models for de novo design of peptide-based drugs (12). The first version of DBAASP had information on about 4000 AMPs. Between 2014 and 2016, the database was updated and expanded resulting in the release of the second version (DBAASP v2) with about 8000 entries (7). The expanded volume of data, including structural information, and new functionality made DBAASP v2 widely used in the research community, contributing to the development of several databases such as LAMP2 (8), dbAMP (10), PlantPepDB (13), starPepDB (14) and ADAPTABLE (15). DBAASP continues to expand through the addition of new content and additional user services, and from 2016 to 2019 the number of entries increased to >15 000. The DBAASP team continues to develop predictive tools for use in the de novo design of peptides having desired activities, and in vitro tests of several synthetic peptides have demonstrated that DBAASP predictions can be effectively used in the design of peptide-based antimicrobial agents against both gram-negative and gram-positive bacteria (16,17).
DBAASP’s ever-increasing volume of extensively annotated content is often used as a source of training set data for building statistical models for AMP design and prediction (18–20). Due to its high degree of curation and annotation of experimental information, Speck-Planche and co-authors (21) relied on DBAASP in developing a multitarget chemo-bioinformatic model for the discovery of anti-gram positive AMPs. Gull and Minhas used DBAASP in the development of a novel method that can extrapolate biological activity predictions to species that were not included in the training set (22). DBAASP was used as a source of positive antibacterial instances for the data used to train bidirectional long short-term memory recurrent neural networks for AMP classification (23). DBAASP peptides constituted the training data sets for machine learning approaches taken by two research groups in developing methods for the prediction of antitubercular peptides (24,25). Additionally, DBAASP information has been used to develop a predictive model of peptide toxicity that is one of major challenges for advancing AMPs into clinical use. Kleandrova and co-authors used DBAASP data to develop a multitasking computational model focused on performing simultaneous predictions of antibacterial activities and cytotoxicity. The authors noted that DBAASP annotates the particular strain of a given bacterial species against that AMPs are assayed when such information is available (26). HemoPred, a web-server developed to predict and analyze the hemolytic activity of peptides (27), uses data from both DBAASP and the Hemolytic database (28). DBAASP is not only used to study peptides with antimicrobial and/or cytotoxic potency; its data can also be used to predict sequences with other functions. mACPpred is a support vector machine-based meta-predictor for identification of anticancer peptides (29). During model development, the mACPpred team made use of information collected from different AMP databases including DBAASP. Taking into account the above-mentioned use cases and feedback from the research community, with significantly expanded content and newly introduced tools DBAASP v3 is aimed at becoming an indispensable tool for AMP study and design.
MATERIALS AND METHODS
Data collection
Peer-reviewed articles containing potentially relevant AMP data are identified through Google and PubMed (30) searches using the keywords ‘antimicrobial peptides’, ‘antibacterial peptides’, ‘antifungal peptides’, ‘antiviral peptides’, ‘antitumor peptides’, ‘anticancer peptides’ and ‘anti-parasitic peptides’. These articles are subsequently screened based on the presence of additional keywords such as ‘antimicrobial activity (activities)’, ‘antibacterial activity (activities)’, ‘antiviral activity (activities)’, ‘antifungal activity (activities)’, ‘anticancer activity (activities)’, ‘inhibitory concentration’, ‘susceptibility testing’, ‘susceptibility test’, ‘virus entry inhibition’, ‘fusion inhibition’, ‘replication inhibition’ and ‘viral protease inhibition’. Articles including such terms are then manually read and processed to extract peptide data concerning amino acid sequence, C- and N-terminal modifications, incorporation of unusual amino acids and/or post-translational modifications, source and target organisms, antimicrobial/anticancer activities and cytotoxicity. Data on each structurally unique peptide are used to create a ‘peptide card’, a database record containing a structured set of information about a particular peptide. The Protein Databank (PDB, www.rcsb.org) (31) is also queried to identify experimentally determined 3D structures of AMPs. Given that such structures are available for only a small fraction of DBAASP peptides, high-throughput molecular dynamics (MD) simulations are performed to make available structural information for additional peptides (see below).
Database technology stack
DBAASP v3 was built on Spring Boot 2.x with Docker-based Centos 8.x, Java8 and Swagger Framework to implement OpenAPI Specification. MariaDB was applied to manage the data as the back end. NGL Viewer (32) was integrated into the database in order to visualize experimental structures retrieved from the PDB or representative structures and trajectories generated from in-house MD simulations. DBAASP updates, backup, recovery and web optimization are performed regularly.
Database structure
Home page
The Home page provides an overview of DBAASP, highlights database tools and features, and provides DBAASP relevant references. This page also provides up-to-date information on the number of monomer, multimer and multi-peptide records in the database, and includes a link to allow quick access to all records populated with data from MD simulations. From this page, users can also access the function-specific pages of the database including Search, Property Calculation, Prediction, Statistics, Help and API pages.
Search page
DBAASP content can be searched using either a single keyword or a combination of terms from the database’s indexed fields. These allow for searches based on, for example, peptide ID, name, complexity, synthesis type, sequence, sequence length, N- and C- terminal modifications, unusual amino acids, presence and type of intrachain bonds, source organism, target species, target group, target object, UniProt ID, availability of 3D structural information, hemolytic/cytotoxic activities and bibliographic characteristics. Search results are presented as a table with each row corresponding to a chemically unique peptide (Supplementary Figure S1) and displaying the peptide ID, name, sequence and any N- and C- terminal modifications. Clicking on the ‘View’ link to the right of a row will open a new page displaying additional information available for that peptide.
Ranking search page
The Ranking Search function provides information on peptides that are active against target microbes or cell lines. The peptides are ranked by the numerical value of their reported activity. A target species or cell type, as well as activity measure, can be selected from a dropdown menu. Additional search options include sequence length, N- and C- terminal modifications, complexity, unusual amino acids, bond type, synthesis type, taxonomy of the source and experimental conditions such as assay medium or descriptors such as the colony forming unit (CFU) count of susceptibility tests.
Property calculation page
In the previous version of DBAASP, the Property Calculation tool allowed for the calculation of the following physiochemical properties of peptides: normalized hydrophobic moment, normalized hydrophobicity, net charge, isoelectric point, penetration depth, tilt angle, disordered conformation propensity, linear moment and propensity for in vitro aggregation. The methods used for calculating these properties are described in Vishnepolsky et al. (33). In DBAASP v3 the angle subtended by the hydrophobic residues, amphiphilicity index and propensity to form polyproline II (PPII) coil are additionally calculated as follows:
Angle Subtended by the Hydrophobic Residues is calculated based on a helical wheel representation of the polypeptide chain in the ideal α-helix approximation generated using in-house software. The definition of hydrophobic residues depends on the user-selected hydrophobicity scale (arXiv:1307.6160[q-bio.BM]).
Amphiphilicity Index is calculated as the sum of the amphiphilicity indices of the comprising amino acids (34) divided by the peptide sequence length.
Propensity for PPII coil is calculated as the sum of the PPII preferences (35) of the comprising amino acids divided by the peptide sequence length. PPII coil refers to coil structure that retains a degree of local order similar to the PPII helix, short stretches of which are interspersed with turns (36).
Prediction page
This page organizes the Prediction of general antibacterial activity (PGA) and Prediction of activity against specific microbial species (PAASS) tools:
PGA tool was introduced in the previous version of DBAASP to predict whether a peptide with a user-specified sequence has antimicrobial activity against microbial targets generally (33).
PAASS is a new tool introduced in DBAASP v3 that predicts whether a user-defined peptide would have activity against a particular microbial strain. A list of target strains for which predictions can be made is accessible via a drop-down menu.
The algorithm underlying the PAASS tool has been previously described (16–17,37). JavaScript was used to develop the front-end web interface for the prediction tools, and the main code was written in FORTRAN.
Statistics page
This page assembles the General Data, Compositional Data and Physicochemical Data subpages that together provide statistics calculated over the contents of the entire database or a subset of the database defined through user query.
General Data, as in DBAASP v2, present a compositional summary of peptides included in the database according to type of synthesis, complexity, target groups, etc. Summary data are updated concurrent with database expansion.
Compositional Data, newly introduced in DBAASP v3, allow users to perform assessments of amino acid composition (Supplementary Figure S2), including distribution of i-spaced amino acid pairs (DiSAAP), over a user-specified subset of peptides from the database defined by selecting options from a set of dropdown menus (Figure 1). The algorithm used to define the DiSAAP is described below.
Figure 1.

Frequencies of occurrence of pairs of hydrophobic residues (Val, Phe, Ile, Leu, Trp) separated by i residues (i = 0,1,2,…,10) for (A) linear ribosomal AMPs and (B) cyclic ribosomal AMPs. Orange bars represent frequencies calculated over the respective sets of DBAASP peptides (fi). Green and purple bars are frequencies calculated after shuffling the sequences within the filtered sets (Fi) +/- 3σ, respectively. See ′Materials and Methods’ section for additional details.
Physicochemical Data, also newly introduced in the current release of the database, allow users to assess the distributions of various physicochemical properties, such as hydrophobicity or isoelectric point calculated, for a particular subset of database peptides which can be defined using dropdown menus (Supplementary Figure S3)
API page
This page describes how the database can be accessed using REST APIs. All resources, individual entries as well as sets of entries based on queries, are accessible using simple URLs.
Help page
This page offers users a detailed description of the various features and tools incorporated in the database, gives definitions of terms used and provides keys to the abbreviations of, for example, terminal modifications, unusual amino acids or media used in experimental assays that are commonly listed as options in dropdown menus across DBAASP.
Assessment of distributions of i-spaced amino acid pairs
The Compositional Data subpage accessible from the Statistics page allows users to calculate the distribution of i-spaced amino acid pairs (DiSAAP) (Figure 1). If the DiSAAP of a particular amino acid or group of amino acids is to be determined with itself, the option ‘X’ should be chosen from the ‘Type of Task’ dropdown (Supplementary Figure S4a). Membership in group X is specified through a row of ten dropdown menus labelled ‘Choose Symbols’ each allowing for selection of one of the 20 naturally occurring amino acids. Thus, X can comprise as few as one residue type or as many as ten. As an example, a user may wish to specify X to be the set of hydrophobic amino acids (Leu, Ile, Val, Phe, Ala, Trp) or the set of charged amino acids (Arg, Lys, Glu, Asp).
The DiSAAP is presented as a bar chart of the frequencies of occurrence (fi) of a pair of X type amino acids calculated over all DBAASP sequences or a subset thereof at a given separation i, the number of intervening residues. The frequencies are calculated as:
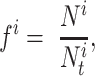 |
where 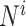 =
=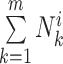 and
and 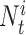 =
=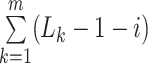 , m is the number of peptides in the subset, Nik is the total number of pairs of X type residues separated by i residues in the k-th peptide of the subset, and Lk is the length of the k-th peptide of the subset. In addition to the frequencies thus calculated (fi), frequencies estimated for random sequences (Fi) generated by shuffling the sequences of peptides satisfying the filter criteria are also calculated. Shuffling is repeated 500 times to assess the average and standard deviation (σi) of Fi. At each separation value, i, the chart shows bars corresponding to Fi - 3σi, fi, and Fi + 3σi.
, m is the number of peptides in the subset, Nik is the total number of pairs of X type residues separated by i residues in the k-th peptide of the subset, and Lk is the length of the k-th peptide of the subset. In addition to the frequencies thus calculated (fi), frequencies estimated for random sequences (Fi) generated by shuffling the sequences of peptides satisfying the filter criteria are also calculated. Shuffling is repeated 500 times to assess the average and standard deviation (σi) of Fi. At each separation value, i, the chart shows bars corresponding to Fi - 3σi, fi, and Fi + 3σi.
If instead the option ‘XY’ is selected from the ‘Type of Task’ dropdown, a DiSAAP of two different groups of amino acids X and Y will be constructed. Upon selecting this option two rows of menus labelled ‘Choose Symbols’ will appear that allow for the specification of the groups X and Y (Supplementary Figure S4b), respectively. Membership in group X excludes membership in group Y, and, in general, statistics calculated for the pair XY are different from those calculated for the pair YX. After specifying the X and Y groups and clicking the ‘Load’ button, the DiSAAP is presented as described above.
MD modelling
An automated pipeline has been written for generating structural information for the peptides present in DBAASP. The pipeline, consisting of a collection of bash, python and tcl scripts, is executed on NVIDIA Tesla K80 GPU-equipped nodes of the National Institute of Allergy and Infectious Diseases (NIAID) HPC cluster. The pipeline processes a set of DBAASP peptide IDs implementing lock files to allow multiple instances to execute in parallel. For each peptide, the corresponding peptide card is first downloaded from DBAASP, and the peptide card metadata are queried to filter out peptides that do not meet current processability criteria. The peptide must be monomeric and consist of fewer than 30 amino acids. As previously described (7), the starting models for simulations performed on peptides of length 30 amino acids or greater are generated through comparative modelling, a step that has not yet been incorporated into the pipeline. For pipeline processing, peptide composition is at present further restricted to the L-forms of the 20 naturally occurring amino acids; the only terminal modifications allowed are acetylation and carboxylation of the N- and C-termini, respectively; and disulfide bonds are the only intrachain bond type permitted. The starting coordinates for a peptide satisfying these criteria are generated with Chimera (38) assigning peptide ϕ and ψ values of 180°, and a protein structure file (PSF) is generated with the VMD (39) plugin psfgen. The peptide is then solvated with TIP3P waters using a 12 Å pad about the extent of the peptide in the x, y and z directions, and sodium and chloride ions are added to neutralize the system and adjust the salt concentration to 250 mM using the VMD packages solvate and autoionize, respectively. The VMD package chirality is then used to generate chiral restraints that are employed during an initial 2000 steps of conjugate gradient minimization of the system with NAMD (40). The system is next subjected to 1500 steps of unrestrained minimization followed by 100 ps of dynamics in the NVT ensemble and 2 ns in the NPT ensemble, all using AceMD (41). This is followed by 400 ns of production dynamics with AceMD using a 4 fs timestep with snapshots written out every 0.2 ns. Direct-space electrostatic interactions are truncated at 9 Å with a switching distance of 7.5 Å, and long range electrostatics are calculated using the PME method. Temperature is maintained at 310 K in the NVT ensemble using a Langevin thermostat with a damping constant of 0.1 ps−1. The CHARMM36m force field (42) is used throughout.
Following simulation, the structure undergoes a quality control assessment that evaluates bond lengths, chirality, and the integrity of the dcd file. For passing peptides, a representative structure is selected and self-consistency heatmaps and secondary structure plots are generated as previously described (7). The peptide trajectory (.dcd) and protein structure file (.psf), a PDB file of the representative structure, and analysis files are then uploaded to the database for viewing or download.
RESULTS AND DISCUSSION
Data update
The current version of DBAASP includes information extracted from about 3000 articles published by about 10 000 authors. DBAASP is updated continuously, and at present, DBAASP v3 contains >15 700 entries. The set of target organisms and cell types include bacteria, fungi, viruses, insects, molicutes, nematodes, protists, cancer cells and mammalian cells.
Adding to the expanded number of peptide entries, DBAASP v3 features an increased number of entries that include structural information from modelling, new annotation types and tools, and an updated, more user-friendly interface. About 3/4 of the entries included in the database at present were added after DBAASP was initially described (12). MD simulations have been performed for >3200 peptides, and >400 DBAASP entries have links to experimentally determined structures in the Protein Data Bank (31). The set of calculated physicochemical values for each peptide is expanded to include the angle subtended by the hydrophobic residues, an amphiphilicity index and a propensity for PPII coil (36). An updated Statistics page allows for data to be summarized over a user-specified subset of DBAASP peptides, and a new version of the API allows users to retrieve DBAASP data more efficiently.
New statistical analysis tools
Due to the significant variation in both sequence and structure of AMPs present in DBAASP, when performing structure activity relationship studies it might be desirable to first cluster the peptides and to consider the features of a particular cluster. Sequence-based characteristics such as amino acid composition or the pairwise distribution of residues along the peptide chain might be used as valuable descriptors and lead to the identification of hallmarks of a particular subset of AMPs. The updated version of DBAASP offers a new tool located on the Statistics page that enables users to assess both amino acid composition and pairwise distribution of residues for AMP sequences.
To demonstrate the use of this tool, we looked for compositional biases among three structurally distinct groups of AMPs present in DBAASP, namely linear peptides, cyclic peptides having a covalent link between N- and C- termini, and peptides with intramolecular disulfide bonds. To identify patterns among these groups, it is reasonable to consider only sequence data of ribosomal peptides that are evolved sequences. To reveal any potentially functionally relevant signatures based on amino acid composition, comparisons should be made to a set of proteins of general function for which the net impact of evolutionary pressure can be assumed to be neutral. The new tool labelled ‘Comparative analysis of AMP composition’ reports the difference in amino acid composition of a selected set of DBAASP peptides relative to all proteins in the UniProt database (43), a repository of proteins having many different functions. Applying the appropriate filters within the new search tool, we determined that in DBAASP there were 1443 ribosomal linear peptides, 123 ribosomal cyclic peptides with a linkage between N- and C-termini, and 1095 ribosomal peptides containing one or more disulfide bonds. As shown in Figure 2, the amino acid composition of the three structural groups of ribosomal peptides are distinct from each other. At the same time, shared features are clearly seen between linear and disulfide-bonded peptides. An abundance of Lys and Gly and a lower percentage acidic amino acids appear as a general property.
Figure 2.

Amino acid compositions of (A) linear, (B) disulfide bonded and (C) cyclic ribosomal peptides presented as differences from a set of UniProt sequences.
In contrast, an abundance of bulky hydrophobic and/or aromatic amino acids (Phe, Ile, Leu, Trp and His) appears as a feature unique to linear AMPs. Furthermore, cyclic and disulfide-bonded peptides are found to be rich in Cys, Lys and Gly, whereas a hallmark of cyclic peptides is an elevated level of Pro, Ser and Thr. The latter is consistent with the requirement of turns and bends necessary to form a cyclic structure. It is worth noting that in contrast to the other structural classes, ribosomal cyclic AMPs are not enriched in positively charged amino acids. This suggests that structurally constrained cyclic peptides having fewer positively charged residues function through different mechanisms of action than linear peptides enriched in positively charged residues that lack cyclizing constraints. At the membrane, cyclic peptides maintain their structure and tend to self-aggregate (44). In contrast, the majority of linear peptides, which tend to be disordered in an aqueous environment, adopt regular secondary structure, mainly alpha helical, at the membrane–water interface (45). For such peptides, the precise order and periodicity of the amino acid character result in amphipathic structures that project hydrophobic amino acids toward the membrane and polar, and charged amino acids towards the aqueous environment. This is consistent with the results of an analysis of the distribution of i-spaced hydrophobic residues pairs for the different structural classes (Figure 1). For the set of linear peptides (Figure 1A) hydrophobic residues are distributed with a periodicity expected for an amphipathic alpha helix. The same is not true for the cyclic peptides (Figure 1B).
New prediction tools
Application of in silico methods represents a comparatively simple, fast and cost-effective approach to the design of new AMPs. Most in silico design methods are based on predictive models trained on data obtained from different AMP databases, and many extant AMP databases make available such prediction tools (2–5,7). However, as a rule, these tools only offer predictions of whether a peptide has general antimicrobial activity, and do not predict antimicrobial activity against particular strains (46). This is unsurprising when one considers that the mechanical and biophysical properties of membranes with which AMPs interact vary considerably with composition and organization which in turn vary considerably among microbial species or strains (47). Thus, one of the main obstacles to the in silico design of new AMPs is the lack of predictive models developed to design new amino acid sequences with a high therapeutic effect against particular microbial strains.
Previously, we developed an algorithm that can distinguish peptides that are active from those that are inactive against select microbial strains (37). This algorithm has been incorporated into a tool that is newly available in DBAASP v3. In addition to antimicrobial activity, the tool can predict activity against human erythrocytes. Its utility was previously demonstrated when it was used to predict peptides active against Escherichia coli strain ATCC 25922 and Staphylococcus aureus strain ATCC 25923 but not against red blood cells. The results of subsequent in vitro characterization of the designed peptides justify the tool’s use in the design of new AMPs (16,17). At present the tool is capable of providing predictions of activity against seven microbial species or strains (Escherichia coli ATCC 25922, Pseudomonas aeruginosa ATCC 27853, Staphylococcus aureus ATCC 25923, Klebsiella pneumoniae, Bacillus subtilis, Candida albicans and Saccharomyces cerevisiae) in addition to human erythrocytes. Inclusion of additional microbes is hindered at present due to an insufficient amount of data required for model training available for most microbial strains within DBAASP. Through database expansion and the use of methods to evaluate similarity between microbial organisms, we aim to markedly expand the applicability of this tool.
MD models
Since the release of DBAASP v2, a pipeline has been written to fully automate the approach to peptide modelling described previously for peptides of fewer than 30 amino acids (7). Commonly between 16 and 32 instances of the pipeline are run concurrently, each processing peptides that satisfy current processability criteria (see ‘Materials and Methods’ section) at a rate of one peptide per 1.75 days on average.
Two modifications have been introduced to the modelling protocol previously described (7). First and most significantly, in order to avoid the bias toward alpha-helical structure that has been noted for short peptides simulated using the CHARMM22 protein force field (48), a switch was made to the use of the CHARMM36m force field that has been shown to more accurately model the secondary structure of intrinsically disordered proteins and peptides (42). It is likely that many peptides adopt stable secondary structure only at the membrane–water interface (45), and, consistently, we have noted an increase in the number of peptides that remain largely disordered over the course of the 400 ns simulations relative to previous simulations performed using the CHARMM22 force field.
A second change that has been made to the modelling protocol relative to that used in the previous release is the introduction of a step in which the system is pre-minimized with chiral restraints applied to the peptide. As the peptides are generated initially in an extended conformation, those peptides for which disulfide bonds are specified between distant residues experience extremely large forces at the outset of minimization, with the result that, in the absence of chiral restraints, these residues can in some cases be ‘pulled’ into their D-forms. To prevent this, a minimization step with chiral restraints is now performed in NAMD ahead of unrestrained minimization in the program AceMD as previously described. At the time of writing, structural data have been generated for >3200 peptides; data for all but approximately 400 of which were generated using the pipeline protocol described here. Peptides modelled with the updated protocol have a value of 10 assigned to the structure protocol Id key present within the peptide card that can be accessed using the DBAASP API.
A method for extracting a representative structure from the MD trajectory was previously described (7). This model along with an MP4 formatted movie of the simulation trajectory was made available for download. While the representative structure is still selected and made available for download or viewing in an instance of the newly implemented NGL Viewer (32), in place of the movie, the full 400 ns production trajectories are now made available for download or for viewing directly in the web-browser (Figure 3). This allows the end-user to either use tools made available in the NGL Viewer instance to interrogate the simulation or to download the data for offline processing. For the displayed trajectory, solvent is removed and the peptides are aligned to the first frame. The raw, unaligned trajectory with solvent and accompanying protein structure file are made available for offline processing.
Figure 3.

MD trajectory displayed within NGL Viewer instance. A screen capture showing the 101st frame from the MD trajectory for the Brevinin-2 related peptide (DBAASP peptide ID 14) displayed in-browser in an instance of NGL Viewer (32). Side chain heavy atoms are represented in ball-and-stick, and the backbone is represented in cartoon coloured by residue index. The distance between the OD2 atom of residue D4 and the NZ atom of residue K16 has been measured and is indicated with a dashed green line and distance label. This measurement is dynamically updated with trajectory playback.
Distinction from other databases of the general type
In our previous publication describing DBAASP v2 (7), we presented a comparison of DBAASP to other general-type AMP databases including APD (3), CAMP3 (4), YADAMP (49) and LAMP (50). Each of these databases has been created with a unique purpose that is reflected in the differences in data content and structure among them. For example, APD focuses on natural AMPs with defined sequence and activity (3). CAMP aims to expand and accelerate antimicrobial peptide family-based studies (4). YADAMP focuses on short alpha-helical peptides interacting with cell membranes (49), and LAMP has been created as a tool to supply information on AMPs within a single platform (50). One of DBAASP’s conceptual aims is to facilitate the de novo design of AMPs with desired properties. This goal has defined DBAASP’s focus on peptides for which experimentally determined, comprehensive data on susceptibility testing are available, regardless of whether the peptides are determined to be active or inactive. In contrast, the other AMP databases referenced above do not require that such data be available. Thus, while the overlap among these databases in terms of peptide sequences may be significant, the overlap in terms of the published and derived metadata made accessible to the end-user is much smaller.
To the best of our knowledge, LAMP2 (8), dbAMP (10), ADAPTABLE (15) and starPepDB (14) are the only general-type AMP databases to have come online since the time of our previous publication. ADAPTABLE is a web platform that aims to create families of property- and sequence-related peptides (15). starPepDB was created to solve the problem of data redundancy and duplication by standardizing it (14). The goals of these two databases, therefore, have little in common with those of DBAASP. On the other hand, the intent of dbAMP (10) to provide functional and physicochemical information on AMPs in order to facilitate drug discovery, does partly overlap with the aims of DBAASP. Consistently, dbAMP includes a section called ’Against Target Species’ containing data on antimicrobial/cytotoxic activities; however, it should be noted that to a large extent these data were derived from DBAASP.
Consequently, it is fair to say that DBAASP is the most comprehensive repository of experimental data from in vitro tests assessing antimicrobial/cytotoxic activities of peptides; it provides users with complete information on the chemical and 3D structure of peptides including making available MD models for >3200 AMPs; and it provides a unique set of calculated, sequence-based physicochemical properties of the peptides it contains. These and many other properties of the database make it a comprehensive resource to perform structure–activity relationship studies and to develop models for the de novo design of peptides with desired antimicrobial properties (18–29). For this purpose, we note that the DBAASP incorporates experimental data on peptide activity regardless of whether a peptide is shown to be highly active in a particular assay. Data on ‘inactive’ peptides are important because both positive and negative examples are essential for studying AMP structure–activity relationships and for generating predictive models. To our knowledge, it is not possible to compile a set of empirically determined, inactive peptides from any other peptide database. For this reason, the majority of machine learning-based predictive models have been developed using peptides that have not been empirically shown to be inactive as the negative training data.
Among the new tools introduced in DBAASP v3 that differentiate it from other AMP databases is an application to make predictions of peptide activity against select microbial strains and tools that allow users to perform their own statistical analyses on subsets of DBAASP data to reveal new sequence signals that might be used as descriptors to improve predictive models. Furthermore, a reworked API page describes how users can retrieve any data from DBAASP by REST API.
CONCLUSION
AMPs have great potential to be used as an alternative to conventional antibiotics. As antimicrobial drug design may benefit from knowledge of the mechanisms and evolutionary ‘discoveries’ that AMPs employ for their activities, well-annotated AMP databases are helpful to researchers in this field. DBAASP aims to be a comprehensive resource and collection of tools for structure–activity studies and the de novo design of AMPs with desired biological functions. The database is a repository of data associated with >15 700 peptides. These data include amino acid sequence, C- and N-terminal modifications, incorporation of unusual amino acids and/or post-translational modifications, peptide source and target organisms, antimicrobial/anticancer activities, cytotoxicity and bibliographic characteristics. The database offers high quality MD models for >3200 peptides. All these data are easily retrieved by user-friendly search engines. Additionally, the database makes available a unique set of calculated, sequence-based physicochemical properties of relevance to many biologically active peptides. Users can perform their own analyses to reveal new signatures in the AMP sequences. DBAASP’s sequence-based prediction services allow end-users to perform de novo design of peptides with activity against particular microbial strains. These features have made DBAASP a widely used resource to develop predictive models of AMPs and to facilitate the de novo design of novel bioactive peptides (18–29). DBAASP is continuously expanding both in terms of data and functionality with the aim of maintaining it as the comprehensive resource to support the field of AMP exploration and design.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Ramandeep Kaur for her work with transitioning the DBAASP website to the National Institute of Allergy and Infectious Diseases (NIAID) Monarch Environment. This study used the Office of Cyber Infrastructure and Computational Biology (OCICB) High Performance Computing (HPC) cluster at NIAID, Bethesda, MD.
Contributor Information
Malak Pirtskhalava, Ivane Beritashvili Center of Experimental Biomedicine, Tbilisi 0160, Georgia.
Anthony A Amstrong, Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD 20892, USA.
Maia Grigolava, Ivane Beritashvili Center of Experimental Biomedicine, Tbilisi 0160, Georgia.
Mindia Chubinidze, Ivane Beritashvili Center of Experimental Biomedicine, Tbilisi 0160, Georgia.
Evgenia Alimbarashvili, Ivane Beritashvili Center of Experimental Biomedicine, Tbilisi 0160, Georgia.
Boris Vishnepolsky, Ivane Beritashvili Center of Experimental Biomedicine, Tbilisi 0160, Georgia.
Andrei Gabrielian, Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD 20892, USA.
Alex Rosenthal, Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD 20892, USA.
Darrell E Hurt, Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD 20892, USA.
Michael Tartakovsky, Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD 20892, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institute of Allergy and Infectious Diseases (NIAID) under BCBB Support Services Contract [HHSN316201300006W/HHSN27200002] to MSC; International Science and Technology Center [G-2102]. Funding for open access charge: International Science and Technology Center [G-2102].
Conflict of interest statementек. None declared.
REFERENCES
- 1. Usmani S.S., Bedi G., Samuel J.S., Singh S., Kalra S., Kumar P., Ahuja A.A., Sharma M., Gautam A., Raghava G.P.S.. THPdb: Database of FDA approved peptide and protein therapeutics. PLoS One. 2017; 12:e0181748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kang X., Dong F., Shi C., Liu S., Sun J., Chen J., Li H., Xu H., Lao X., Zheng H.. DRAMP 2.0, an updated data repository of antimicrobial peptides. Sci. Data. 2019; 6:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang G., Li X., Wang Z.. APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016; 44:D1087–D1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Waghu F.H., Gurung P., Barai R.S., Idicula-Thomas S.. CAMPR3: a database on sequences, structures and signatures of antimicrobial peptides. Nucleic Acids Res. 2016; 44:D1094–D1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vijayaraghava V.S., Gabere M.N., Pretorius A., Adam S., Christoffels A., Lehväslaiho M., Archer J.A.C., Bajic V.B.. DAMPD: a manually curated antimicrobial peptide database. Nucleic Acids Res. 2012; 40:D1108–D1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Piotto S.P., Sessa L., Concilio S., Iannelli P.. YADAMP: yet another database of antimicrobial peptides. Int. J. Antimicrob. Agents. 2012; 39:346–351. [DOI] [PubMed] [Google Scholar]
- 7. Pirtskhalava M., Gabrielian A., Cruz P., Griggs H.L., Squires R.B., Hurt D.E., Grigolava M., Chubinidze M., Gogoladze G., Vishnepolsky B. et al.. DBAASP v.2: an enhanced database of structure and antimicrobial/cytotoxic activity of natural and synthetic peptides. Nucleic Acids Res. 2016; 44:D1104–D1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ye G., Wu H., Wang W., Ge K., Li, Zhong J., Huang Q.. LAMP2: a major update of the database linking antimicrobial peptides. Database. 2020; 2020:baaa061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee H.T., Lee C.C., Yang J.R., Lai J.Z.C., Chang K.Y.. A large-scale structural classification of antimicrobial peptides. BioMed Res. Int. Gale OneFile: Health Med. 2015; 2015:475062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chi Y.H., Li W.C., Lin T.H., Huang K.Y., Lee T.Y.. dbAMP: an integrated resource for exploring antimicrobial peptides with functional activities and physicochemical properties on transcriptome and proteome data. Nucleic Acids Res. 2019; 47:D285–D297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Singh S., Chaudhary K., Dhanda S.K., Bhalla S., Usmani S.S., Gautam A., Tuknait A., Agrawal P., Mathur D., Raghava GP.. SATPdb: a database of structurally annotated therapeutic peptides. Nucleic Acids Res. 2016; 44:D1119–D1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gogoladze G., Grigolava M., Vishnepolsky B., Chubinidze M., Duroux P., Lefranc M.P., Pirtskhalava M.. DBAASP: Database of antimicrobial activity and structure of peptides. FEMS Microbiol. Lett. 2014; 357:63–68. [DOI] [PubMed] [Google Scholar]
- 13. Das D., Jaiswal M., Khan F.N., Ahamad S., Kumar S.. PlantPepDB: A manually curated plant peptide database. Sci. Rep. 2020; 10:2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aguilera-Mendoza L., Marrero-Ponce Y., Beltran J.A., Tellez Ibarra R., Guillen-Ramirez H.A., Brizuela C.A.. Graph-based data integration from bioactive peptide databases of pharmaceutical interest: toward an organized collection enabling visual network analysis. Bioinformatics. 2019; 35:4739–4747. [DOI] [PubMed] [Google Scholar]
- 15. Ramos-Martin F., Annaval T., Buchoux S., Sarazin C., D’Amelio N.. ADAPTABLE: a comprehensive web platform of antimicrobial peptides tailored to the user's research. Life Sci. Alliance. 2019; 2:e201900512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vishnepolsky B., Zaalishvili G., Karapetian M., Nasrashvili T., Kuljanishvili N., Gabrielian A., Rosenthal A., Hurt D E., Tartakovsky M., Grigolava M. et al.. De Novo design and in vitro testing of antimicrobial peptides against Gram-Negative bacteria. Pharmaceuticals. 2019; 12:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vishnepolsky B., Zaalishvili G., Karapetian M., Gabrielian A., Rosenthal A., Hurt D E., Tartakovsky M., Grigolava M., Pirtskhalava M.. Development of the model of in silico design of AMPs active against Staphylococcus aureus 25923. 2019; 5th International Electronic Conference on Medicinal Chemistry session ECMC-5, 01/11/2019 - 30/11/2019, doi:10.3390/ECMC2019-06359.
- 18. Armas F., Pacor S., Ferrari E., Guida F., Pertinhez T.A., Romani A.A., Scocchi M., Benincasa M.. Design, antimicrobial activity and mechanism of action of Arg-rich ultra-short cationic lipopeptides. PLoS One. 2019; 14:e0212447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tucs A., Phuoc Tran D., Yumoto A., Ito Y., Uzawa T., Tsuda K.. Generating ampicillin-level antimicrobial peptides with activity-aware generative adversarial networks. ACS Omega. 2020; 5:22847–22851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nava Lara R.A., Aguilera-Mendoza L., Brizuela C.A., Peña A., Del Rio G.. Heterologous machine learning for the identification of antimicrobial activity in Human-Targeted drugs. Molecules. 2019; 24:E1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Speck-Planche A., Kleandrova V.V., Ruso J.M., Cordeiro M.N.. First multitarget chemo bioinformatic model to enable the discovery of antibacterial peptides against multiple gram-positive pathogens. J. Chem. Inf. Model. 2016; 56:588–598. [DOI] [PubMed] [Google Scholar]
- 22. Gull S., Minhas Z.. AMP0: Species-Specific Prediction of Antimicrobial Peptides using Zero and Few Shot Learning. IEEA/ACM Trans. Comput. Biol. Bioinform. 2019; doi:10.1109/TCBB.2020.2999399. [DOI] [PubMed] [Google Scholar]
- 23. Youmans M., Spainhour J.C.G., Qiu P.. Classification of antibacterial peptides using long short-term memory recurrent neural networks. IEEA/ACM Trans. Comput. Biol. Bioinform. 2019; 17:1134–1140. [DOI] [PubMed] [Google Scholar]
- 24. Usmani S.S., Bhalla S., Raghava G.P.S.. Prediction of antitubercular peptides from sequence information using ensemble classifier and hybrid features. Front. Pharmacol. 2018; 9:954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Khatun S., Hasan M., Kurat H.. Efficient computational model for identification of antitubercular peptides by integrating amino acid patterns and properties. FEBS Letter. 2019; 593:3029–3039. [DOI] [PubMed] [Google Scholar]
- 26. Kleandrova V.V., Ruso J.M., Speck-Planche A., Dias Soeiro Cordeiro M.N.. Enabling the discovery and virtual screening of potent and safe antimicrobial peptides. Simultaneous prediction of antibacterial activity and cytotoxicity. ACS Comb. Sci. 2016; 18:490–498. [DOI] [PubMed] [Google Scholar]
- 27. Win T.S., Malik A.A., Prachayasittikul V., S Wikberg J.E., Nantasenamat C., Shoombuatong W.. HemoPred: a web server for predicting the hemolytic activity of peptides. Fut. Med. Chem. 2017; 9:275–291. [DOI] [PubMed] [Google Scholar]
- 28. Gautam A., Chaudhary K., Singh S., Joshi A., Anand P., Tuknait A., Mathur D., Varshney G.C., Raghava G.P.. Hemolytik: a database of experimentally determined hemolytic and non-hemolytic peptides. Nucleic Acids Res. 2014; 42:D444–D449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Boopathi V., Subramaniyam S Malik A., Lee G., Manavalan Band Yang D.C.. mACPpred: A support vector machine-based meta-predictor for identification of anticancer peptides. Int. J. Mol. Sci. 2019; 20:E1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. NCBI Resource Coordinators Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2015; 43:D6–D17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rose P.W., Prlic A., Bi C., Bluhm W.F., Christie C.H., Dutta S., Green R.K., Goodsell D.S., Westbrook J.D., Woo J. et al.. The RCSB Protein Data Bank: views of structural biology for basic and applied research and education. Nucleic Acids Res. 2015; 43:D345–D356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rose A.S., Bradley A.R., Valasatava Y., Duarte J.M., Prlic A., Rose P.W.. NGL viewer: web-based molecular graphics for large complexes. Bioinformatics. 2018; 34:3755–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vishnepolsky B., Pirtskhalava M.. Prediction of linear cationic antimicrobial peptides based on characteristics responsible for their interaction with the membranes. J. Chem. Inf. Model. 2014; 54:1512–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mitaku S., Hirokawa T., Tsuji T.. Amphiphilicity index of polar amino acids as an aid in the characterization of amino acid preference at membrane-water interfaces. Bioinformatics. 2002; 18:608–616. [DOI] [PubMed] [Google Scholar]
- 35. Adzhubei A.A., Eisenmenger F., Tumanyan V.G., Zinke M., Brodzinski S., Esipova N.G. Third type of secondary structure: noncooperative mobile conformation. Protein Data Bank analysis. Biochem. Biophys. Res. Commun. 1987; 146:934–938. [DOI] [PubMed] [Google Scholar]
- 36. Tiffany M.L, Krimm S.. New chain conformations of poly(glutamic acid) and polylysine. Biopolymers. 1968; 6:1379–1382. [DOI] [PubMed] [Google Scholar]
- 37. Vishnepolsky B., Gabrielian A., Rosenthal A., Hurt D.E., Tartakovsky M., Managadze G., Grigolava M., Makhatadze G.I., Pirtskhalava M.. Predictive model of linear antimicrobial peptides active against gram-negative bacteria. J. Chem. Inf. Model. 2018; 58:1141–1151. [DOI] [PubMed] [Google Scholar]
- 38. Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E.. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput. Chem. 2004; 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 39. Humphrey W., Dalke A., Schulten K.. VMD - Visual molecular dynamics. J. Mol. Graph. 1996; 14:33–38. [DOI] [PubMed] [Google Scholar]
- 40. Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kalé L., Schulten K.. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005; 26:1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Harvey M.J., Giupponi G., Fabritiis G.D.. ACEMD: Accelerating biomolecular dynamics in the microsecond time scale. J. Chem. Theory Comput. 2009; 5:1632–1639. [DOI] [PubMed] [Google Scholar]
- 42. Huang J., Rauscher S., Nawrocki G., Ran T., Feig M., de Groot B.L., Grubmüller H., MacKerell A.D. Jr. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods. 2017; 14:71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. The UniProt Consortium UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019; 47:D506–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Llamas-Saiz A.L., Grotenbreg G.M., Overhand M., van Raaij M.J.. Double-stranded helical twisted beta-sheet channels in crystals of gramicidin S grown in the presence of trifluoroacetic and hydrochloric acids. Acta Crystallogr. D. Biol. Crystallogr. 2007; 63:401–407. [DOI] [PubMed] [Google Scholar]
- 45. Blondelle S.E., Lohner K., Aguilar M.. Lipid-induced conformation and lipid-binding properties of cytolytic and antimicrobial peptides: determination and biological specificity. Biochim. Biophys. Acta. 1999; 1462:89–108. [DOI] [PubMed] [Google Scholar]
- 46. Vishnepolsky B., Pirtskhalava M.. Comment on: ‘Empirical comparison of web-based antimicrobial peptide prediction tools’. Bioinformatics. 2019; 35:2692–2694. [DOI] [PubMed] [Google Scholar]
- 47. Sohlenkamp C., Geiger O.. Bacterial membrane lipids: diversity in structures and pathways. FEMS Microbiol. Rev. 2016; 40:133–159. [DOI] [PubMed] [Google Scholar]
- 48. Freddolino P.L., Park S., Roux B., Schulten K.. Force field bias in protein folding simulations. Biophys. J. 2009; 96:3772–3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Piotto S.P., Sessa L., Concilio S., Iannelli P.. YADAMP: yet another database of antimicrobial peptides. Int. J. Antimicrob. Agents. 2012; 39:346–351. [DOI] [PubMed] [Google Scholar]
- 50. Zhao X., Wu H., Lu H., Li G., Huang Q.. LAMP: A database linking antimicrobial peptides. PLoS One. 2013; 8:e66557. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.