


Abstract
Gramene (http://www.gramene.org), a knowledgebase founded on comparative functional analyses of genomic and pathway data for model plants and major crops, supports agricultural researchers worldwide. The resource is committed to open access and reproducible science based on the FAIR data principles. Since the last NAR update, we made nine releases; doubled the genome portal's content; expanded curated genes, pathways and expression sets; and implemented the Domain Informational Vocabulary Extraction (DIVE) algorithm for extracting gene function information from publications. The current release, #63 (October 2020), hosts 93 reference genomes—over 3.9 million genes in 122 947 families with orthologous and paralogous classifications. Plant Reactome portrays pathway networks using a combination of manual biocuration in rice (320 reference pathways) and orthology-based projections to 106 species. The Reactome platform facilitates comparison between reference and projected pathways, gene expression analyses and overlays of gene–gene interactions. Gramene integrates ontology-based protein structure–function annotation; information on genetic, epigenetic, expression, and phenotypic diversity; and gene functional annotations extracted from plant-focused journals using DIVE. We train plant researchers in biocuration of genes and pathways; host curated maize gene structures as tracks in the maize genome browser; and integrate curated rice genes and pathways in the Plant Reactome.
INTRODUCTION
Plants carry complex genomes in which gene duplication, polyploidy and transposons serve as the raw materials for adaptive responses to the environment, as well as agronomic improvement. Over the past five years, we have seen a large increase in high-quality reference genomes, supported by a reduction in sequencing costs, improvements in sequencing technology, and genome annotation workflows. These reference assemblies cover a wide phylogenetic range, from non-vascular plant moss to higher order flowering plants, and thus represent blueprints for functional diversity. Together, they provide a solid foundation for obtaining novel insights into plant gene function. However, a thorough exploitation of the genomic data requires systems-level frameworks, as well as analytic and visualization tools for comparative analysis of coding, noncoding and small molecule components across species.
Since its inception in 2001, Gramene has strived to provide a unique genome-scale data visualization platform and analytical tools to support the global community of plant genomics researchers, molecular biologists, geneticists, breeders, etc. (1,2). As the knowledgebase has evolved, we have included the Ensembl Genome Browser, which enables visualization of genomic data (molecular variation, gene expression, epigenomic) in the context of individual sequenced plant species and a BioCyc-based platform that provides cellular-level metabolic pathway models. In 2012, we created the Plant Reactome portal (https://plantreactome.gramene.org; (3), which enables enhanced visualization of plant pathways within the subcellular context; in addition, its data model allows depiction of various molecular events and macromolecular interactions, and is thus useful for visualizing important plant processes and genetic regulatory networks alongside the cellular-level plant metabolic network. In addition, Gramene's individual gene pages and Plant Reactome pathways link to other public resources to provide accessory information and display basal tissue-specific gene-expression profiles and anatomogram images, which are fetched programmatically in real time from the EMBL-EBI’s Expression Atlas (4). Gramene's multifaceted platform is built on a powerful and flexible document-based architecture that supports integrated search across all of its portals (Ensembl, Reactome, and Expression Atlas) and interactive views that graphically summarize the gene-centric results of a query in five categories: gene features and genomic context, phylogenetic trees, gene expression profiles, pathways and cross-references.
We also equip the plant biologist community with tools and recommendations for annotating genomic data sets supported by standards and ontology concepts. We support analysis and visualization of user-defined datasets in the context of species-specific Genome and Pathway Browsers. All data and results of user data analyses are downloadable in graphical and tabular formats and made accessible for bulk downloading via programmatic interfaces and FTP sites.
Since our last publication (5), we have added 49 new plant genome browsers (total 93); gene-orthology based pathway projections for 40 new species (total 107), including all 76 species hosted on the Genome portal and 30 additional species projections derived from sequence and transcriptome data from other sources; and gene-expression data for 10 new species in the Expression Atlas (total 941 experiments in 28 species) (Table 1 and Supplementary Table S1). Recently, we developed the Domain Informational Vocabulary Extraction (DIVE) algorithm for automated extraction of gene functional information from peer-reviewed articles in The Plant Cell and Plant Physiology, two plant-focused journals published by the American Society of Plant Biologists (ASPB). Compared to the post-publication processing of NLP-based data mining by numerous resources, the unique workflow that we developed is now part of the publication process and the publisher's manuscript approval and handling system, and directly engages authors in the data mining process. It has processed 1436 articles to date, of which 456 were from The Plant Cell, and 980 were from Plant Physiology. We have continued to engage our users at scientific conferences, a news blog and social media. We have also conducted live webinars and recorded video tutorials. We have also trained plant researchers in structural biocuration of maize genes, and rice genes and pathways by organizing onsite and online jamborees.
Table 1.
New additions and updates in data, analyses, tools and infrastructure at Gramene
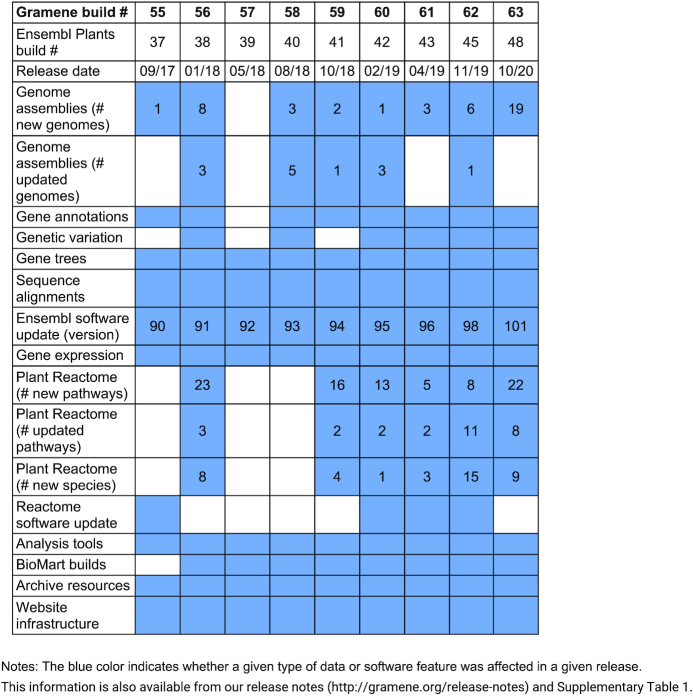
|
All data in Gramene are freely accessible through visual and programmatic interfaces at http://www.gramene.org.
NEW AND UPDATED PLANT GENOMES
We currently host 93 plant reference genomes, including 49 new genomes (16 monocots, 31 dicots and 2 non-vascular plants; Supplementary Table S2), more than doubling the number since the last publication (5), and increasing the dicot-to-monocot ratio (Figure 1A).
Figure 1.
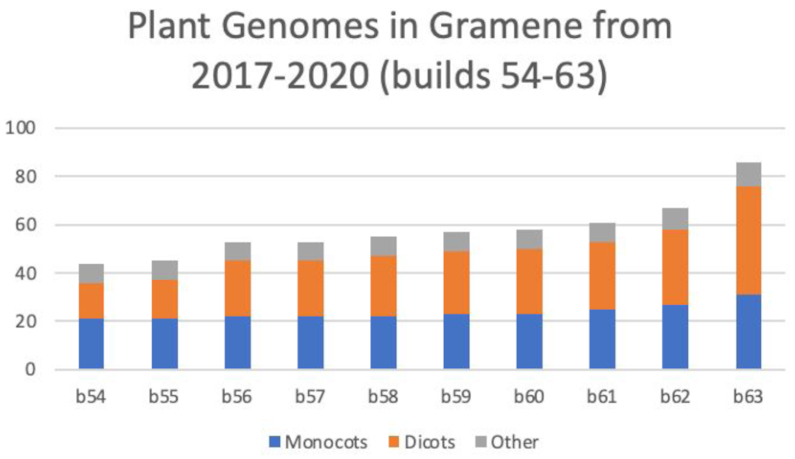
Plant genomes update in Gramene from release b54 to b63.
The new species include agronomically important cereals (durum wheat, emmer wheat, and teff), fruits and vegetables (apple, pineapple, watermelon, cantaloupe, clementine, cherry, kiwi, carrot, cucumber, cassava, hot pepper), specialty crops (coffee, olive, pistachio and almond), plants of special or emerging interest (cotton, jute and beans, a non-commercial tobacco and cannabis or hemp), and flowers (sunflower, rose, lily). In addition to five new varieties of bread wheat, and new varieties of barley and cacao (Supplementary Table S2). This release also features plant research models, including C4 warm-season grasses and brassicas, as well as other species that fill phylogenetic gaps for plant evolution studies.
In addition to the new species, genome assemblies and corresponding gene annotations were updated for 13 species (Supplementary Table S2). Two of the most significant assembly updates were for sorghum and wheat. Wheat is hexaploid, composed of three closely related and independently maintained genomes that resulted from a series of naturally occurring hybridization events. The ancestral wheat progenitor genomes are considered to be Triticum urartu (the A-genome donor) and an unknown grass thought to be related to Aegilops speltoides (the B-genome donor). This first hybridization event produced tetraploid emmer wheat (AABB, T. dicoccoides), which hybridized again with Aegilops tauschii (the D-genome donor) to yield modern bread wheat (AABBDD, T. aestivum). In release #58, the TGACv1 bread wheat assembly was replaced with the International Wheat Genome Sequencing Consortium version 1 (IWGSCv1). Gene ID mapping was provided between 98 270 high-confidence TGACv1 genes and the new IWGSC gene models. Another notable genome update was performed for sorghum (Sorbi1 → Sorghum_bicolor_NCBIv3), with the added capability of interconverting genomic coordinates to the intermediate Sorghum_bicolor_v2 assembly using Ensembl's Assembly Converter tool (http://ensembl.gramene.org/Sorghum_bicolor/Tools/AssemblyConverter). Similarly, the soybean genome assembly was updated twice (sequential updates from V1 to V3) (Supplementary Table S2).
The total number of genes has increased by 91% since the last NAR publication (5). All new and updated genes have been subjected to uniform functional annotations, such as protein domain mapping with InterProScan (6), and classified using ontology terms from the Gene Ontology Annotation (7).
NEW NATURAL AND INDUCED GENETIC SEQUENCE VARIATION
Genetic diversity is a major contributor to phenotypic variation between individuals. Characterization of genetic diversity, both natural and induced, provides insights into the life history of a species and serves as a resource for testing functional hypotheses. Gramene hosts variation data for 15 species (Supplementary Table S3) (8–32) comprising almost 238 million variants. Although the bulk of this variation is associated with short variants (i.e. single-nucleotide polymorphisms [SNPs] and indels), we also host nearly 30 000 structural variants in rice (from Gramene's legacy markers database; see http://archive.gramene.org/markers) and sorghum (from the Database of Genomic Variants [DGV] (33)).
Since our last NAR update, we have integrated new variation data for three species (Supplementary Table S3): Malus domestica (apple, 10.6 million SNPs; (26)), Helianthus annuus (sunflower, 11 834 SNPs; (27,28), and T. turgidum (durum wheat, 1.9 million SNPs; (29)). For apple, SNP variants with minor allele frequency (MAF) ≧ 0.05 were called on 70 varieties and lines through the FruitBreedomics project (26). Sunflower SNPs were called in three sunflower pre-breeding collections belonging to INTA (Argentina), INRA (France), and USDA-UBC (USA and Canada) to estimate the distribution pattern of global genetic diversity (27,28). The variants for durum wheat from the 35K (60 391 variants), 820K (1 496 076 variants), 90K (182 507 variants) and TabW280K (24 969 variants) SNP arrays (29) were imported from CerealsDB (30).
Most notable was the near doubling of bread wheat variation data since the previous update, reaching over 18 million short variants. New additions include the Axiom 35K (31 774 variants) and 820K (768 664 variants) SNP arrays from CerealsDB (30); 3.6 million inter-homeologous variants (IHVs) between the A, B and D genome components (31); 710 chromosome-specific KASP markers (32); and additional ethyl methanesulfonate (EMS)-induced mutations from the Kronos and Cadenza TILLING populations (4 827 302 and 8 900 333 variants, respectively) (25). The wheat EMS data set grew by 85%, representing novel genetic variation that can be used to improve precompetitive breeding germplasm; arguably, this expansion constitutes the most agronomically important addition to our collection of variation data. Overall, there are nearly 13.7 million and 1.5 million EMS variants in bread wheat and sorghum, respectively.
Genetic and structural variants for sorghum, Brachypodium, tomato and bread wheat were also remapped to newer assemblies, yielding small changes in the number of variants. We continued to routinely run all new and updated variants through Ensembl's variant effect prediction (VEP) workflow (34). Users can carry out the VEP analysis of their own datasets online (limited data allowed/upload) or in bulk by downloading the tool as previously described (35). Another new addition is the linkage disequilibrium (LD) display for the Axiom and KASP wheat variants, available in the Location view of the bread wheat genome browser.
NEW COMPARATIVE GENOMICS DATA AND DISPLAYS
Gene Tree families are based on the Ensembl Compara pipeline (36,37), which recently incorporated a step that consists of a Hidden Markov Model (HMM) search on the TreeFam HMM library (see https://www.ensembl.org/info/genome/compara/hmm_lib.html), based on the Panther database (http://www.pantherdb.org, version 9; (38)), to classify protein sequences into families. Currently, there are 122 947 gene family trees constructed with 3 169 866 input proteins, representing a 97% increase in the number of trees since the previous update (5). Gene trees are in turn used to determine orthologs, paralogs, predict split genes and uncover potential structural annotation errors. Split gene models might result from a legitimate evolutionary process, but most commonly they are related to an annotation artifact in which a single gene is annotated as two or more genes due to incomplete evidence. Therefore, at every release, we provide lists of potential split genes (see ftp://ftp.gramene.org/pub/gramene/CURRENT_RELEASE/split_genes) predicted using the Ensembl Compara pipeline.
The phylogenetic trees (Figure 2) provide a standard way for the community to infer gene function across species, as well as insights into the fluid nature of gene gain and loss across the evolutionary spectrum, as shown in the less conventional radial tree views (Figure 2A), which are scored by CAFE, a computational tool for the study of gene family evolution (39,40). The region comparison view (Figure 2B) allows users to compare a region of up to 100 000 Mb across multiple species, simultaneously. The gene tree views of the Gramene search (Figure 2D and E) are highly customizable, highlight gene structure, amino acid sequence, and splice junction conservation in a phylogenetic context; support pruning of the number of species on display; and offer the ability to zoom in and out from an amino acid–level view to entire gene neighborhoods (up to 10 genes flanking each side). We have leveraged the flexibility of our gene tree views into a Gene Tree Visualizer Tool for our community annotation efforts (41); see also the Biocuration section below).
Figure 2.

Comparative genomic visualizations in Gramene. Four ways to graphically determine whether a gene is present or absent relative to other species in Gramene. (A) Circular gene family tree for a rice repressor protein, OsDrAp1 (Os11g0544700) showing significant gene gain (red lines; e.g. Brassica napus with 8 paralogs), and gene loss (zero paralogs, and species names grayed out; e.g., Oryza meridionalis and Triticum urartu). An apparent gene loss could be evolutionary important for a species, but could also indicate a gap in the gene annotation. (B) Region comparison view of the syntenic segment surrounding the OsDrAp1 gene. The gene is conserved in the monocot model Brachypodium, but absent in O. meridionalis and T. urartu. (C) Synteny map between the model plant Brachypodium (chromosome 2) and Japonica rice (chromosome 1) in the region surrounding the rice auxin efflux carrier, OsPIN9, and a table listing the corresponding syntelogs in this region. (D and E). Gene neighborhood views for the ZmPIN9 (blue arrow) gene tree. ZmPIN9 is the maize ortholog of OsPIN9. The view shows (D) a high level of microsynteny in monocots (sorghum and wheat, including the new emmer wheat genome; brachy and rice, shown in a green rectangle), and (E) microsynteny among dicots (Arabidopsis species marked with a red rectangle; and three new genomes: tobacco, hot pepper and coffee).
In addition to the gene tree protein alignments, we provide 382 pairwise whole-genome sequence alignments (WGAs; see http://ensembl.gramene.org/compara_analyses.html) across 93 species in Gramene. At a minimum, each species was aligned against the monocot reference O. sativa japonica, and dicot references A. thaliana and V. vinifera. The DNA alignments complement the gene trees and provide insights into conserved non-coding regions. All alignments can be viewed in sections from 50 bp to 500 Mb and downloaded as images or in text form (aligned DNA sequences).
Since the previous update, the recently optimized Ensembl Compara Enredo–Pecan–Ortheus (EPO) pipeline (42,43) was applied to compute a multiple genome alignment for 11 Oryza species. Another achievement was the release of a polyploid genome view, displaying aligned segments of homologous wheat chromosomes from the three subgenomes of allohexaploid wheat (AABBDD), simultaneously.
Gene trees and WGA data have been used to build synteny maps for a limited number of species (see http://ensembl.gramene.org/compara_analyses.html). Currently, we have 84 synteny maps; O. sativa Japonica holds the record with 21 maps, of which nine are against other rice genomes. All synteny maps can be downloaded as images, and a list of corresponding syntelogs can be downloaded for up to 15 genes in a given region (Figure 2C).
PLANT GENE EXPRESSION IN ATLAS
Curation of plant gene expression data continued, with a 29% increase in the number of curated experiments and a 56% expansion in the number of plant species (from 731 experiments in 18 plant species (5) to 941 experiments in 28 species (Expression Atlas, August 2020) (Supplementary Table S4). The new plant species with curated gene expression include Trifolium pratense, Setaria italica, Prunus persica, Musa acuminata, Brassica napus and Beta vulgaris subsp. vulgaris. In addition, previously curated datasets were reanalyzed against the updated genomes, such as wheat IWGSCv1. Other analyses and visualizations included ‘enrichment’ in each differential comparison of GO terms, Plant Reactome pathways, and InterPro domains, as well as a functionality to find genes with similar expression profiles in a baseline experiment page, and another to visualize the distribution of baseline expression across biological replicates via box plots. The expression data is available to users via the EMBL-EBI’s Expression Atlas database site, as well as via widgets and APIs. The views also allow importing the expression data for individual samples as an on-demand track on the species-specific genome browser.
In the past year, a new workflow for analyzing single-cell expression (SCE) data was developed and applied to five SCE experiments in A. thaliana (44–47).
PLANT REACTOME: A PLANT PATHWAY KNOWLEDGEBASE
The Plant Reactome knowledgebase is the only plant pathway platform that provides visualization of various types of molecular events, reactions and macromolecular interactions, in the context of subcellular architecture of the plant cell. It currently hosts pathways associated with plant metabolism, hormone signaling, genetic regulation, plant organ development, and differentiation, as well as responses to biotic and abiotic stresses. To build a repertoire of plant pathways, we utilize a combination of manual biocuration in the reference species, rice (O. sativa subsp. japonica), and automated gene orthology projections. As of Gramene release #63, the knowledgebase contains 320 rice reference pathways (>27 000 projected pathways) mapped onto 1887 reactions (>69 000 projected reactions) including 2170 gene products (>149 000 projected orthologs), 267 transcripts, 36 small RNAs and 1018 small molecules and metabolites. Since our last report (5), the total number of species, including the Japonica rice reference, has increased from 66 to 107. Orthology data for 76 species are from the reference genomes in Gramene, and the additional 30 species are from other sequenced genomes or transcriptomes provided by the Planteome project (48). Users can access pathway data in graphical and tabular format for any member species and compare the projected pathways with the reference pathways in rice.
Plant Reactome has adopted the Reactome data model for biocuration of rice pathways. Since our last report (5), 64 new pathways have been added, and 28 pathways were updated (Supplementary Table S5). Previously, the Plant Reactome database contained 11 evolutionarily conserved pathways (including DNA replication and repair, transcription, translation, and vesicle transport) that were projected from humans to rice (49). Recently, we replaced the projected pathway for DNA synthesis with the curated rice pathway (Figure 3). Projections of the curated rice pathways are available for other species hosted by the Plant Reactome (Figure 3B). For instance, a projected pathway for A. thaliana can be compared with the reference rice pathway (Figure 3C). The pathway browser also displays anatomogram images and RNA-seq baseline expression data, fetched in real time from the Expression Atlas (4) (Figure 3D). A similar level of expression data can be accessed for the 28 plant species mentioned in the Plant Gene Expression in ATLAS section. Recently, we added the gene–gene interaction overlay feature, which allows users to select and display gene–gene interaction data from the BioAnalytic Resource (BAR) (50), the IntAct database (51), our own plant interactome, and several other sources via PSICQUIC web services (52). Users can also upload interaction data from comma- and/or tab-delimited files, or by providing a URL for locally stored data. For genes for which interaction data are available, users can filter the number of interactors by adjusting the confidence score. An example overlay of interactome data from the BAR on an A. thaliana pathway is shown in Figure 3E. The interactor count appears next to the proteins on the pathway diagram; clicking on it opens a spoke-and-wheel diagram showing the identities of the interactors (Figure 3E). A detailed description of the interactome overlay feature that allows users to extend the nodes of interactors to fine-tune their research hypothesis has been published (3).
Figure 3.

Plant Reactome pathway features and functionalities. (A) Pathway hierarchy navigation (upper panel) and pathway diagram view (lower panel); (B) Species selector for viewing pathway projections in any of the available 106 projected plant species. (C) Species comparison feature shows curated DNA synthesis pathway in the reference species (blue color) and overlay of projected orthologous A. thaliana matches (yellow colored blocks; the complete and partial overlap respectively suggest same of different number of homologs between two species, whereas, blue boxes suggest absence of Arabidopsis homologs. (D) Baseline gene expression data for selected entity in pathway diagram served by EMBL-EBI Gene Expression Atlas (lower panel); (E) A gene-gene interaction from Bio Analytical Resource (BAR) is displayed over the A. thaliana DNA synthesis pathway that includes 247 unique interactors for five genes (upper panel) and an enlarged view of 107 interactors associated with the gene AT5G26680 is shown as spoke-and-wheel overlay (lower panel).
The Plant Reactome pathway browser uses a dynamic ‘Fireworks' platform to display hierarchical organization of pathways, the interrelationships among them, and links to other public resources providing accessory information on biochemicals (PubChem: ChEBI), genes (Gramene), proteins (UniProt; (53), gene expression (Expression Atlas; (4), and publications (PubMed). We host a DiagramJs widget, which can be embedded at other sites to display a dynamic, interactive Plant Reactome pathway viewer (available at Ensembl Plants and Gramene gene pages, PubChem and Expression Atlas). We provide this feature to other databases upon request; and continue to support upload, visualization, and analysis of user-defined gene-expression (54), and gene–gene interaction data (3).
BIOCURATING GENE STRUCTURE AND FUNCTION WHILE SCALING
Accurate gene structure has always been important for biological interpretation and proper experimental design. Missing exons and incorrect splice sites directly impact interpretations of gene function, development of genetic markers, and predictions of the impact of natural and induced genetic variation. Following recent advances in plant transformation (55,56), coupled with advanced genomics and gene-editing breakthroughs such as the CRISPR technologies (57), it is now possible to make targeted modifications in many plant species. However, the lack of robust, accurate gene structure and functional regulatory information could negatively impact these efforts. For annotation of human gene structure and function, expert manual annotations serve as the gold standard (58). In plants, however, resources to support manual biocuration of genes have been limited, creating an obstacle to the assignment of accurate structural or functional annotations. To address this issue, Gramene has adopted a community curation approach for crowdsourcing biocuration of gene structure by recruiting undergraduate students and plant researchers to triage suspect gene models (Figure 4), evaluate the experimental evidence to support the existence of a transcript, and curate the gene's structure in the Apollo gene editor (59). Maize served as the initial prototype for these efforts (41). Gramene established a network of researchers and teaching faculty to support and implement genome annotation projects as Course-based Undergraduate Research Experiences (CUREs). In addition, we have hosted several in-person and virtual jamborees, trained over 50 participants, developed training materials and curation tools based on the gene trees to support triage of suspect models, and set up Apollo instances of reference assemblies for maize and sorghum. Importantly, the virtual jamborees highlighted methods for supporting a diverse collaborative environment for remote learning and provided a proof-of-concept for future community curation efforts to improve genomic annotations in maize and other important crops. Curated maize gene models are available as a track on Gramene's Maize Genome Browser (see the table with links to their corresponding genomic location at http://www.gramene.org/curated_maize_v4_gene_models, and Figure 4B).
Figure 4.

Community biocuration. (A) Gene tree visualizer tool with a multiple (amino acid) sequence alignment highlighting potential structural annotation artifacts at the 3′-end of the auxin efflux carrier ZmPIN9 gene (Zm00001d043179). (B) Maize genome track showing a curated gene structure (shorter internal exons) for ZmPIN9.
In addition, we focused biocuration on rice genes and pathways, based on evaluation of peer-reviewed published scientific literature, analysis of expression patterns of homologous genes, and evidence from gene–gene interaction data sets. Recent efforts accomplished (i) revision and enrichment of metabolic and transport pathways created during the very early stages of Plant Reactome; (ii) depiction of processes associated with plant development (organ formation, tissue differentiation, cell division, etc.); and (iii) integration of transcription networks. To engage the plant genomics community in such biocuration efforts, we hosted two jamborees in the autumns of 2017 and 2018 (60). In addition, we recruited community collaborators and leveraged individuals involved in data science courses, thesis projects, and experiential summer learning internships at Oregon State University (OSU), and trained seven undergraduate students in small biocuration research projects.
The availability of a large number of genomes, along with an extensive array of peer-reviewed literature and meeting abstracts reporting on gene function and phenotype, represents a gold mine for training Machine Learning methods for knowledge extraction. This is an important activity because even though biocuration is of extremely high quality, it is time-consuming, challenging, and constrained by limitations on resources, experts, and funding models for various plant biology databases. Moreover, the rate of reporting new data in the literature has substantially outpaced quality manual biocuration, making it practically impossible to capture everything manually. Therefore, in a pilot program, we implemented Domain Informational Vocabulary Extraction (DIVE) as a web service for the publication pipeline used by the two ASPB journals, The Plant Cell and Plant Physiology. In March 2018, the DIVE tool was integrated with The Plant Cell using ArticleExpress, a proprietary tool of Sheridan Journal Services. ArticleExpress is an XML-based author proofing system that allows authors to make changes directly to the article's XML, rather than the traditional method of marking up a PDF. The integration of DIVE into ArticleExpress added a step to an author's proofing process by presenting curated entities to confirm, edit, delete, or add. DIVE extracts a list of potentially important words and phrases from manuscripts and provides a web-based interactive user interface for biocuration by authors (61). An author is unable to submit the manuscript proofs for composition until the data mined by DIVE are approved by the authors. In April 2018, Plant Physiology began requiring authors to use ArticleExpress with the integrated DIVE tool. Thus, DIVE enriches digital publications by first detecting biological entities and key informational words, and then adding additional annotations to the published article. The system implements multiple strategies for biological entity detection, including the use of regular expression rules, ontology, and a keyword dictionary. These extracted entities are then stored in a database and made accessible for curation and evaluation by authors. The author approved edits and updates to the mined data can then be used to improve entity importance prediction for future article processing (62). For this work, we have used gene nomenclature data from Gramene, ontologies from the Planteome database (48) as our word bank, and ontology-based knowledgegraphs, to drive entity recognition and data mining. Other projects have performed post-publication implementation of natural language processing (NLP) (63–66); by contrast, DIVE implementation has processed 1436 accepted manuscripts (The Plant Cell: 456 and Plant Physiology: 980) by directly engaging authors as expert biocurators and seeking validation of text-mining data. To date (early September 2020), the system has mined 59005 entities, including 17317 genes from 112 unique species, and authors have made 3127 edits. We are in the process of integrating mined data sets into our Genome and Pathway portals.
In addition to the above mentioned community biocuration activities, we engaged plant scientists, breeders, and budding biologists at annual conferences including the American Society of Plant Biology, Maize Genetics, Plant and Animal Genomes, and Plant Genome Evolution. We maintain a news blog (https://news.gramene.org/blog) and a social media presence (https://twitter.com/GrameneDatabase and https://www.facebook.com/Gramene) to provide information on the most recent releases, and events at scientific meetings. We continue to organize live webinars and recordings, which serve as online tutorials for users and are available in Gramene's YouTube channel (https://goo.gl/ln9RLD).
FUTURE DIRECTIONS
Major challenges for the future include keeping up with the increasing volume of data and the development of high-throughput approaches to support functional annotations of both transcribed and regulatory regions of the genome. Examples include single-cell sequencing and image-based phenotypes. Our continued collaborations with EMBL-EBI’s Expression Atlas will support integration of single-cell RNA-seq data (67), which will provide high spatial and temporal resolution of genes, in order to improve information about gene function. In recent years, the Expression Atlas’ infrastructure has been scaled up to accommodate large datasets from projects such as the Human Cell Atlas. In addition, analysis pipelines have been reviewed to increase their scalability and flexibility across different platforms. We will continue our collaboration with Ensembl Genomes (31) to address both scaling of the number of reference genomes and updates to our genome browser. The developers of the Ensembl platform are currently working on an update to their technology stack (68), and this will require an update to the Gramene infrastructure in the next cycle. Development of new content for Plant Reactome is centered on molecular details of the development of specialized plant tissues and responses to external stimuli and stresses. We will continue our work to integrate DIVE with Gramene, and to provide links back to the relevant publications from our gene and pathway views.
Over the past 15 years, Gramene has evolved from a genome database hosting the first crop reference assembly, rice (69), into the preeminent plant genomics data warehouse and knowledgebase. Phylogenomics provides insights into gene evolution and the fluid nature of the plant genomes. To date, Gramene's existing infrastructure and data acquisition has focused on cross-species comparisons, but an important future target will be storing and providing access to species pan-genomes. This effort will initially target the research communities for four species—rice, maize, sorghum and grapevine—to develop species-specific sites with a minimum of 20 high-quality reference genomes, and extend the existing workflows to support characterization of copy-number variations (CNVs) and core gene sets. We will work with EnsemblGenomes and the European Variation Archive to build upon the scalable workflows for ingestion of genome assemblies, gene annotations, and genetic variation. In addition, we will review ongoing activities with the Alliance of Genome Resources (70), and where appropriate, align those with the needs of the plant species’ communities to adopt new standards. Each species’ community will also provide requirements and usability testing for new phenotype search and views. In addition, the communities will have the opportunity to extend Gramene's community curation network to support expert review of gene structure annotations.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are grateful to Gramene's users, researchers, and numerous collaborators for sharing datasets generated by their projects, and for providing valuable suggestions and feedback that have helped us to improve the overall quality of Gramene as a community resource. We thank Cold Spring Harbor Laboratory (CSHL) and the Dolan DNA Learning Center, the Center for Genome Research and Biocomputing (CGRB) at Oregon State University (OSU), and the Ontario Institute for Cancer Research (OICR) for infrastructure support. We also thank Peter van Buren from CSHL for system administration support, and David Magda, Gino Yearwood and Robin Haw (OICR) for technical support on Plant Reactome development. In addition, we thank CyVerse for providing a mirror Plant Reactome server under the Powered-by-CyVerse program. We acknowledge the assistance of OSU undergraduate students Shayla Rao and Callan Stowell with biocuration of rice reference pathways. We thank Cristina F. Marco and David Micklos (MaizeCode Project) for jointly running the maize gene structural annotation jamborees. The funders had no role in study design, data analysis or preparation of this manuscript.
Contributor Information
Marcela K Tello-Ruiz, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY 11724, USA.
Sushma Naithani, Department of Botany and Plant Pathology, Oregon State University, Corvallis, OR 97331, USA.
Parul Gupta, Department of Botany and Plant Pathology, Oregon State University, Corvallis, OR 97331, USA.
Andrew Olson, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY 11724, USA.
Sharon Wei, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY 11724, USA.
Justin Preece, Department of Botany and Plant Pathology, Oregon State University, Corvallis, OR 97331, USA.
Yinping Jiao, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY 11724, USA.
Bo Wang, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY 11724, USA.
Kapeel Chougule, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY 11724, USA.
Priyanka Garg, Department of Botany and Plant Pathology, Oregon State University, Corvallis, OR 97331, USA.
Justin Elser, Department of Botany and Plant Pathology, Oregon State University, Corvallis, OR 97331, USA.
Sunita Kumari, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY 11724, USA.
Vivek Kumar, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY 11724, USA.
Bruno Contreras-Moreira, European Molecular Biology Laboratory, European Bioinformatics Institute, Wellcome Genome Campus, Hinxton CB10 1SD, UK.
Guy Naamati, European Molecular Biology Laboratory, European Bioinformatics Institute, Wellcome Genome Campus, Hinxton CB10 1SD, UK.
Nancy George, European Molecular Biology Laboratory, European Bioinformatics Institute, Wellcome Genome Campus, Hinxton CB10 1SD, UK.
Justin Cook, Informatics and Bio-computing Program, Ontario Institute of Cancer Research, Toronto M5G 1L7, Canada.
Daniel Bolser, European Molecular Biology Laboratory, European Bioinformatics Institute, Wellcome Genome Campus, Hinxton CB10 1SD, UK; Current affiliation: Geromics Inc., Cambridge CB1 3NF, UK.
Peter D’Eustachio, Department of Biochemistry and Molecular Pharmacology, New York University Grossman School of Medicine, New York, NY 10016, USA.
Lincoln D Stein, Adaptive Oncology Program, Ontario Institute for Cancer Research, Toronto M5G 0A3, Canada; Department of Molecular Genetics, University of Toronto, Toronto, ON M5S 1A8, Canada.
Amit Gupta, Texas Advanced Computing Center, University of Texas at Austin, Austin, TX 78758, USA.
Weijia Xu, Texas Advanced Computing Center, University of Texas at Austin, Austin, TX 78758, USA.
Jennifer Regala, American Society of Plant Biologists, Rockville, MD 20855-2768, USA; Current affiliation: American Urological Association, Linthicum, MD 21090, USA.
Irene Papatheodorou, European Molecular Biology Laboratory, European Bioinformatics Institute, Wellcome Genome Campus, Hinxton CB10 1SD, UK.
Paul J Kersey, European Molecular Biology Laboratory, European Bioinformatics Institute, Wellcome Genome Campus, Hinxton CB10 1SD, UK; Current affiliation: Royal Botanic Gardens, Kew Richmond, Surrey TW9 3AE, UK.
Paul Flicek, European Molecular Biology Laboratory, European Bioinformatics Institute, Wellcome Genome Campus, Hinxton CB10 1SD, UK.
Crispin Taylor, American Society of Plant Biologists, Rockville, MD 20855-2768, USA.
Pankaj Jaiswal, Department of Botany and Plant Pathology, Oregon State University, Corvallis, OR 97331, USA.
Doreen Ware, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY 11724, USA; USDA ARS NAA Robert W. Holley Center for Agriculture and Health, Ithaca, NY 14853, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Foundation (NSF) [IOS-1127112, IOS-1340112]; United States Department of Agriculture—Agricultural Research Service [58-1907-4-030, 8062-21000-041-00D to D.W., 52930113 to P.J.]; United Kingdom Biotechnology and Biosciences Research Council [BB/P016855/1 and Ensembl-4-Breeders workshop support]; European Molecular Biology Laboratory; The in-kind infrastructure and intellectual support for the development and running of the Plant Reactome is supported by the Reactome database project via a grant from the US National Institutes of Health [U41 HG003751 to L.S.]; EU grant [LSHG-CT-2005-518254]; ‘ENFIN’, the Ontario Research Fund; EMBL-EBI Industry Programme; Hosting of the Plant Reactome website on Cyverse was also supported by the NSF [IOS-1743442]; The Expression ATLAS received additional funding form the Single-Cell Gene Expression Atlas and ‘PRIDE Atlas’ grants from Wellcome [108437/Z/15/Z, WT101477MA to I.P.]; Open Targets; Chan-Zuckerberg Initiative; Ensembl Plants received additional funding from ELIXIR implementation studies FONDUE and ‘Apple as a Model for Genomic Information Exchange’; OSU provided partial support for trainee undergraduate students and project personnel at the university. Funding for open access charge: NSF [IOS-1127112].
Conflict of interest statement. None declared.
REFERENCES
- 1. Ware D., Jaiswal P., Ni J., Pan X., Chang K., Clark K., Teytelman L., Schmidt S., Zhao W., Cartinhour S. et al.. Gramene: a resource for comparative grass genomics. Nucleic Acids Res. 2002; 30:103–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jaiswal P., Ware D., Ni J., Chang K., Zhao W., Schmidt S., Pan X., Clark K., Teytelman L., Cartinhour S. et al.. Gramene: development and integration of trait and gene ontologies for rice. Comp. Funct. Genomics. 2002; 3:132–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Naithani S., Gupta P., Preece J., D’Eustachio P., Elser J.L., Garg P., Dikeman D.A., Kiff J., Cook J., Olson A. et al.. Plant Reactome: a knowledgebase and resource for comparative pathway analysis. Nucleic Acids Res. 2020; 48:D1093–D1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Papatheodorou I., Moreno P., Manning J., Fuentes A.M.-P., George N., Fexova S., Fonseca N.A., Füllgrabe A., Green M., Huang N. et al.. Expression Atlas update: from tissues to single cells. Nucleic Acids Res. 2020; 48:D77–D83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tello-Ruiz M.K., Naithani S., Stein J.C., Gupta P., Campbell M., Olson A., Wei S., Preece J., Geniza M.J., Jiao Y. et al.. Gramene 2018: unifying comparative genomics and pathway resources for plant research. Nucleic Acids Res. 2018; 46:D1181–D1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jones P., Binns D., Chang H.-Y., Fraser M., Li W., McAnulla C., McWilliam H., Maslen J., Mitchell A., Nuka G. et al.. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014; 30:1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huntley R.P., Sawford T., Mutowo-Meullenet P., Shypitsyna A., Bonilla C., Martin M.J., O’Donovan C.. The GOA database: gene Ontology annotation updates for 2015. Nucleic Acids Res. 2015; 43:D1057–D1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Atwell S., Huang Y.S., Vilhjalmsson B.J., Willems G., Horton M., Li Y., Meng D., Platt A., Tarone A.M., Hu T.T. et al.. Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature. 2010; 465:627–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Clark R.M., Schweikert G., Toomajian C., Ossowski S., Zeller G., Shinn P., Warthmann N., Hu T.T., Fu G., Hinds D.A. et al.. Common sequence polymorphisms shaping genetic diversity in Arabidopsis thaliana. Science. 2007; 317:338–342. [DOI] [PubMed] [Google Scholar]
- 10. Fox S.E., Preece J., Kimbrel J.A., Marchini G.L., Sage A., Youens-Clark K., Cruzan M.B., Jaiswal P.. Sequencing and de novo transcriptome assembly of Brachypodium sylvaticum (Poaceae). Appl. Plant Sci. 2013; 1:1200011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li Z.-M., Zheng X.-M., Ge S.. Genetic diversity and domestication history of African rice (Oryza glaberrima) as inferred from multiple gene sequences. Theor. Appl. Genet. 2011; 123:21–31. [DOI] [PubMed] [Google Scholar]
- 12. 3,000 rice genomes project The 3,000 rice genomes project. Gigascience. 2014; 3:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gan X., Stegle O., Behr J., Steffen J.G., Drewe P., Hildebrand K.L., Lyngsoe R., Schultheiss S.J., Osborne E.J., Sreedharan V.T. et al.. Multiple reference genomes and transcriptomes for Arabidopsis thaliana. Nature. 2011; 477:419–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. International Barley Genome Sequencing Consortium Mayer K.F.X., Waugh R., Brown J.W.S., Schulman A., Langridge P., Platzer M., Fincher G.B., Muehlbauer G.J., Sato K. et al.. A physical, genetic and functional sequence assembly of the barley genome. Nature. 2012; 491:711–716. [DOI] [PubMed] [Google Scholar]
- 15. Ariyadasa R., Mascher M., Nussbaumer T., Schulte D., Frenkel Z., Poursarebani N., Zhou R., Steuernagel B., Gundlach H., Taudien S. et al.. A sequence-ready physical map of barley anchored genetically by two million single-nucleotide polymorphisms. Plant Physiol. 2014; 164:412–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mace E.S., Tai S., Gilding E.K., Li Y., Prentis P.J., Bian L., Campbell B.C., Hu W., Innes D.J., Han X. et al.. Whole-genome sequencing reveals untapped genetic potential in Africa's indigenous cereal crop sorghum. Nat. Commun. 2013; 4:2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McNally K.L., Childs K.L., Bohnert R., Davidson R.M., Zhao K., Ulat V.J., Zeller G., Clark R.M., Hoen D.R., Bureau T.E. et al.. Genomewide SNP variation reveals relationships among landraces and modern varieties of rice. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:12273–12278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Morris G.P., Ramu P., Deshpande S.P., Hash C.T., Shah T., Upadhyaya H.D., Riera-Lizarazu O., Brown P.J., Acharya C.B., Mitchell S.E. et al.. Population genomic and genome-wide association studies of agroclimatic traits in sorghum. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:453–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Myles S., Chia J.-M., Hurwitz B., Simon C., Zhong G.Y., Buckler E., Ware D.. Rapid genomic characterization of the genus vitis. PLoS One. 2010; 5:e8219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhao K., Wright M., Kimball J., Eizenga G., McClung A.. Genomic diversity and introgression in O. sativa reveal the impact of domestication and breeding on the rice genome. PLoS One. 2010; 5:e10780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zheng L.-Y., Guo X.-S., He B., Sun L.-J., Peng Y., Dong S.-S., Liu T.-F., Jiang S., Ramachandran S., Liu C.-M. et al.. Genome-wide patterns of genetic variation in sweet and grain sorghum (Sorghum bicolor). Genome Biol. 2011; 12:R114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Consortium, 100 Tomato Genome Sequencing Aflitos S., Schijlen E., de Jong H., de Ridder D., Smit S., Finkers R., Wang J., Zhang G., Li N. et al.. Exploring genetic variation in the tomato (Solanum section Lycopersicon) clade by whole-genome sequencing. Plant J. 2014; 80:136–148. [DOI] [PubMed] [Google Scholar]
- 23. Chia J.M., Song C., Bradbury P., Costich D., de Leon N., Doebley J.C., Elshire R.J., Gaunt B.S., Geller L., Glaubitz J.C. et al.. Capturing extant variation from a genome in flux: maize HapMap II. Nat. Genet. 2012; 44:803–807. [DOI] [PubMed] [Google Scholar]
- 24. Jiao Y., Burke J., Chopra R., Burow G., Chen J., Wang B., Hayes C., Emendack Y., Ware D., Xin Z.. A sorghum mutant resource as an efficient platform for gene discovery in grasses. Plant Cell. 2016; 28:1551–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Krasileva K.V., Vasquez-Gross H.A., Howell T., Bailey P., Paraiso F., Clissold L., Simmonds J., Ramirez-Gonzalez R.H., Wang X., Borrill P. et al.. Uncovering hidden variation in polyploid wheat. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:E913–E921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bianco L., Cestaro A., Linsmith G., Muranty H., Denance C., Theron A., Poncet C., Micheletti D., Kerschbamer E., Di Pierro E.A. et al.. Development and validation of the Axiom® Apple480K SNP genotyping array. Plant J. 2016; 86:62–74. [DOI] [PubMed] [Google Scholar]
- 27. Filippi C.V., Aguirre N., Rivas J.G., Zubrzycki J., Puebla A., Cordes D., Moreno M.V., Fusari C.M., Alvarez D., Heinz R.A. et al.. Population structure and genetic diversity characterization of a sunflower association mapping population using SSR and SNP markers. BMC Plant Biol. 2015; 15:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Filippi C.V., Merino G.A., Montecchia J.F., Aguirre N.C., Rivarola M., Naamati G., Fass M.I., Álvarez D., Di Rienzo J., Heinz R.A. et al.. Genetic diversity, population structure and linkage disequilibrium assessment among international sunflower breeding collections. Genes. 2020; 11:283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Maccaferri M., Harris N.S., Twardziok S.O., Pasam R.K., Gundlach H., Spannagl M., Ormanbekova D., Lux T., Prade V.M., Milner S.G. et al.. Durum wheat genome highlights past domestication signatures and future improvement targets. Nat. Genet. 2019; 51:885–895. [DOI] [PubMed] [Google Scholar]
- 30. Wilkinson P.A., Allen A.M., Tyrrell S., Wingen L.U., Bian X., Winfield M.O., Burridge A., Shaw D.S., Zaucha J., Griffiths S. et al.. CerealsDB-new tools for the analysis of the wheat genome: update 2020. Database. 2020; 2020:baaa060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Howe K.L., Contreras-Moreira B., De Silva N., Maslen G., Akanni W., Allen J., Alvarez-Jarreta J., Barba M., Bolser D.M., Cambell L. et al.. Ensembl Genomes 2020-enabling non-vertebrate genomic research. Nucleic Acids Res. 2020; 48:D689–D695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Grewal S., Hubbart-Edwards S., Yang C., Devi U., Baker L., Heath J., Ashling S., Scholefield D., Howells C., Yarde J. et al.. Rapid identification of homozygosity and site of wild relative introgressions in wheat through chromosome-specific KASP genotyping assays. Plant Biotechnol. J. 2020; 18:743–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. MacDonald J.R., Ziman R., Yuen R.K.C., Feuk L., Scherer S.W.. The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 2014; 42:D986–D992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R.S., Thormann A., Flicek P., Cunningham F.. The ensembl variant effect predictor. Genome Biol. 2016; 17:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Naithani S., Geniza M., Jaiswal P.. Variant effect prediction analysis using resources available at Gramene database. Methods Mol. Biol. 2017; 1533:279–297. [DOI] [PubMed] [Google Scholar]
- 36. Vilella A.J., Severin J., Ureta-Vidal A., Heng L., Durbin R., Birney E.. EnsemblCompara GeneTrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 2009; 19:327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Herrero J., Muffato M., Beal K., Fitzgerald S., Gordon L., Pignatelli M., Vilella A.J., Searle S.M.J., Amode R., Brent S. et al.. Ensembl comparative genomics resources. Database. 2016; 2016:bav096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mi H., Poudel S., Muruganujan A., Casagrande J.T., Thomas P.D.. PANTHER version 10: expanded protein families and functions, and analysis tools. Nucleic Acids Res. 2016; 44:D336–D342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Han M.V., Thomas G.W.C., Lugo-Martinez J., Hahn M.W.. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol. Biol. Evol. 2013; 30:1987–1997. [DOI] [PubMed] [Google Scholar]
- 40. De Bie T., Cristianini N., Demuth J.P., Hahn M.W.. CAFE: a computational tool for the study of gene family evolution. Bioinformatics. 2006; 22:1269–1271. [DOI] [PubMed] [Google Scholar]
- 41. Tello-Ruiz M.K., Marco C.F., Hsu F.-M., Khangura R.S., Qiao P., Sapkota S., Stitzer M.C., Wasikowski R., Wu H., Zhan J. et al.. Double triage to identify poorly annotated genes in maize: the missing link in community curation. PLoS One. 2019; 14:e0224086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Paten B., Herrero J., Fitzgerald S., Beal K., Flicek P., Holmes I., Birney E.. Genome-wide nucleotide-level mammalian ancestor reconstruction. Genome Res. 2008; 18:1829–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Paten B., Herrero J., Beal K., Fitzgerald S., Birney E.. Enredo and Pecan: genome-wide mammalian consistency-based multiple alignment with paralogs. Genome Res. 2008; 18:1814–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ryu K.H., Huang L., Kang H.M., Schiefelbein J.. Single-cell RNA sequencing resolves molecular relationships among individual plant cells. Plant Physiol. 2019; 179:1444–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jean-Baptiste K., McFaline-Figueroa J.L., Alexandre C.M., Dorrity M.W., Saunders L., Bubb K.L., Trapnell C., Fields S., Queitsch C., Cuperus J.T.. Dynamics of gene expression in single root cells of Arabidopsis thaliana. Plant Cell. 2019; 31:993–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shulse C.N., Cole B.J., Ciobanu D., Lin J., Yoshinaga Y., Gouran M., Turco G.M., Zhu Y., O’Malley R.C., Brady S.M. et al.. High-throughput single-cell transcriptome profiling of plant cell types. Cell Rep. 2019; 27:2241–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Turco G.M., Rodriguez-Medina J., Siebert S., Han D., Valderrama-Gómez M.Á., Vahldick H., Shulse C.N., Cole B.J., Juliano C.E., Dickel D.E. et al.. Molecular mechanisms driving switch behavior in xylem cell differentiation. Cell Rep. 2019; 28:342–351. [DOI] [PubMed] [Google Scholar]
- 48. Cooper L., Meier A., Laporte M.-A., Elser J.L., Mungall C., Sinn B.T., Cavaliere D., Carbon S., Dunn N.A., Smith B. et al.. The Planteome database: an integrated resource for reference ontologies, plant genomics and phenomics. Nucleic Acids Res. 2018; 46:D1168–D1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jassal B., Matthews L., Viteri G., Gong C., Lorente P., Fabregat A., Sidiropoulos K., Cook J., Gillespie M., Haw R. et al.. The reactome pathway knowledgebase. Nucleic Acids Res. 2020; 48:D498–D503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Waese J., Provart N.J.. The bio-analytic resource for plant biology. Methods Mol. Biol. 2017; 1533:119–148. [DOI] [PubMed] [Google Scholar]
- 51. Orchard S., Ammari M., Aranda B., Breuza L., Briganti L., Broackes-Carter F., Campbell N.H., Chavali G., Chen C., del-Toro N. et al.. The MIntAct project–IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014; 42:D358–D363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. del-Toro N., Dumousseau M., Orchard S., Jimenez R.C., Galeota E., Launay G., Goll J., Breuer K., Ono K., Salwinski L. et al.. A new reference implementation of the PSICQUIC web service. Nucleic Acids Res. 2013; 41:W601–W606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. UniProt Consortium UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019; 47:D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Naithani S., Preece J., D’Eustachio P., Gupta P., Amarasinghe V., Dharmawardhana P.D., Wu G., Fabregat A., Elser J.L., Weiser J. et al.. Plant Reactome: a resource for plant pathways and comparative analysis. Nucleic Acids Res. 2017; 45:D1029–D1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kausch A.P., Nelson-Vasilchik K., Hague J., Mookkan M., Quemada H., Dellaporta S., Fragoso C., Zhang Z.J.. Edit at will: genotype independent plant transformation in the era of advanced genomics and genome editing. Plant Sci. 2019; 281:186–205. [DOI] [PubMed] [Google Scholar]
- 56. Hua K., Zhang J., Botella J.R., Ma C., Kong F., Liu B., Zhu J.-K.. Perspectives on the application of genome-editing technologies in crop breeding. Mol. Plant. 2019; 12:1047–1059. [DOI] [PubMed] [Google Scholar]
- 57. Doudna J.A., Charpentier E.. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014; 346:1258096. [DOI] [PubMed] [Google Scholar]
- 58. Frankish A., Diekhans M., Ferreira A.-M., Johnson R., Jungreis I., Loveland J., Mudge J.M., Sisu C., Wright J., Armstrong J. et al.. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019; 47:D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Dunn N.A., Unni D.R., Diesh C., Munoz-Torres M., Harris N.L., Yao E., Rasche H., Holmes I.H., Elsik C.G., Lewis S.E.. Apollo: democratizing genome annotation. PLoS Comput. Biol. 2019; 15:e1006790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Naithani S., Gupta P., Preece J., Garg P., Fraser V., Padgitt-Cobb L.K., Martin M., Vining K., Jaiswal P.. Involving community in genes and pathway curation. Database. 2019; 2019:bay146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Xu W., Gupta A., Jaiswal P., Taylor C., Lockhart P., Regala J.. Improving publication pipeline with automated biological entity detection and validation service. Data Inform. Manage. 2019; 3:3–17. [Google Scholar]
- 62. Gupta A., Xu W., Jaiswal P., Taylor C., Regala J.. Extracting Domain Information using Deep Learning. Proceedings of the Practice and Experience in Advanced Research Computing on Rise of the Machines (learning), PEARC ’19. 2019; NY: Association for Computing Machinery; 1–7. [Google Scholar]
- 63. Müller H.-M., Van Auken K.M., Li Y., Sternberg P.W.. Textpresso Central: a customizable platform for searching, text mining, viewing, and curating biomedical literature. BMC Bioinformatics. 2018; 19:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wei C.-H., Allot A., Leaman R., Lu Z.. PubTator central: automated concept annotation for biomedical full text articles. Nucleic Acids Res. 2019; 47:W587–W593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lee J., Yoon W., Kim S., Kim D., Kim S., So C.H., Kang J.. BioBERT: a pre-trained biomedical language representation model for biomedical text mining. Bioinformatics. 2020; 36:1234–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Devlin J., Chang M.-W., Lee K., Toutanova K.. BERT: pre-training of deep bidirectional transformers for language understanding. 2018; arXiv doi:24 May 2019, preprint: not peer reviewedhttps://arxiv.org/abs/1810.04805.
- 67. Füllgrabe A., George N., Green M., Nejad P., Aronow B., Clarke L., Fexova S.K., Fischer C., Freeberg M.A., Huerta L. et al.. Guidelines for reporting single-cell RNA-Seq experiments. 2019; arXiv doi:31 October 2019, preprint: not peer reviewedhttps://arxiv.org/abs/1910.14623. [DOI] [PMC free article] [PubMed]
- 68. Yates A.D., Achuthan P., Akanni W., Allen J., Allen J., Alvarez-Jarreta J., Amode M.R., Armean I.M., Azov A.G., Bennett R. et al.. Ensembl 2020. Nucleic Acids Res. 2020; 48:D682–D688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ware D.H., Jaiswal P., Ni J., Yap I.V., Pan X., Clark K.Y., Teytelman L., Schmidt S.C., Zhao W., Chang K. et al.. Gramene, a tool for grass genomics. Plant Physiol. 2002; 130:1606–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Alliance of Genome Resources Consortium Alliance of Genome Resources Portal: Unified Model Organism Research Platform. Nucleic Acids Res. 2020; 48:D650–D658. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.