


Abstract
Transfer RNA-derived fragments (tRFs) are a new class of small non-coding RNAs and play important roles in biological and physiological processes. Prediction of tRF target genes and binding sites is crucial in understanding the biological functions of tRFs in the molecular mechanisms of human diseases. We developed a publicly accessible web-based database, tRFtarget (http://trftarget.net), for tRF target prediction. It contains the computationally predicted interactions between tRFs and mRNA transcripts using the two state-of-the-art prediction tools RNAhybrid and IntaRNA, including location of the binding sites on the target, the binding region, and free energy of the binding stability with graphic illustration. tRFtarget covers 936 tRFs and 135 thousand predicted targets in eight species. It allows researchers to search either target genes by tRF IDs or tRFs by gene symbols/transcript names. We also integrated the manually curated experimental evidence of the predicted interactions into the database. Furthermore, we provided a convenient link to the DAVID® web server to perform downstream functional pathway analysis and gene ontology annotation on the predicted target genes. This database provides useful information for the scientific community to experimentally validate tRF target genes and facilitate the investigation of the molecular functions and mechanisms of tRFs.
INTRODUCTION
Transfer RNA-derived fragments (tRFs) are a new class of small non-coding RNA (ncRNA) in the length of 13–48 nucleotides (nts) (1). tRFs are the products of non-random cleavage of either the precursor or mature transfer RNAs (tRNAs). Based on the cleavage sites of tRNA, tRFs are classified into five categories: tRF-1s (also termed as tRNA-derived small RNAs [tsRNAs]), tRF-3s, tRF-5s, tRF-2s and stress-induced tRNAs (tiRs) (Figure 1) (2). tRF-1s are generated from the 3′ end of the precursor tRNAs, whereas the other four types of tRFs are derived from different parts of the mature tRNAs with the 3′ end for tRF-3s and the 5′ end for tRF-5s, respectively (2). tRF-3s can be further divided into two subcategories with different lengths: short as tRF-3a and long as tRF-3b. Similarly, based on the length from short to long, tRF-5s can be divided into three subcategories: tRF-5a, tRF-5b and tRF-5c (3). tiRs have two subcategories: a 5tiR starts from the 5′ end of a mature tRNA to the end of the anticodon loop, and a 3tiR from the 3′ end to the end of the anticodon loop. tRF-2s, a new type of tRFs, are derived from the anticodon loop of the mature tRNAs (2).
Figure 1.

Illustration of the five categories of tRFs. tRF-1 or tsRNA is generated from the 3′ end of the precursor tRNA, whereas tRF-3, tRF-5, tiR and tRF-2 are derived from different parts of the mature tRNA. tRF, transfer RNA-derived fragment. tsRNA, tRNA-derived small RNA. tiR, stress-induced tRNA. ANG, angiogenin.
tRFs have been found to be conserved in diverse organisms from bacteria to humans (4). They are involved in many biological and physiological processes such as regulation of gene expression, RNA processing, tumor suppression, and cell proliferation (2). tRF dysregulation may play important roles in human diseases including cancer (5–7). Recently, several studies revisited datasets from The Cancer Genome Atlas (TCGA) and the NCI-60 human tumor cell lines screen, and reported tRFs as potential biomarkers in human cancer (8,9). Experimental results suggested that tRFs function as microRNAs (miRNAs) in post-transcriptional regulation of gene expression by partially complementary to target messenger RNAs (mRNAs), leading to the degradation or translational repression of target mRNAs (10,11). The results of photoactivatable-ribonucleoside-enhanced crosslinking and immunoprecipitation (PAR-CLIP) in human HEK293 cells showed the silencing complex formation of tRF-5s and tRF-3s in combination with Argonautes 1, 3 and 4 (4,12). RNA sequencing analysis further demonstrated the tRF–mRNA chimeric formation in the cross-linking, ligation and sequencing of hybrids (CLASH) data (4,13,14).
There are more than 30 web-based miRNA target prediction databases which have served as powerful tools for experimental validation of miRNA targets in multiple species (15,16), and accelerated the investigation of the biological functions of miRNAs by providing the binding sites on mRNAs. However, to our knowledge, there is no such target database available for tRFs. A previous study inferred the targeting modes of tRFs based on the limited experimental CLASH datasets that cover 26 human Argonaute-loaded tRFs (14). However, the bindings may be biased in the context of tissues and cells. Another approach to infer potential tRF targets is via co-expression network analysis or chromatin immunoprecipitation sequencing (ChIP-seq), which do not directly consider the complementary pairing between sequences (8,9). Site-directed mutations using reporter genes is a fundamental approach to seek the binding sites in the target genes, however, it requires the known regions where the binding site(s) is located.
In this study, we established a publicly accessible web-based transcriptome-wide tRF target prediction database, tRFtarget (http://trftarget.net), for eight species including human, mouse, Drosophila, Caenorhabditis elegans, Schizosaccharomyces pombe, Rhodobacter sphaeroides, Xenopus tropicalis and Zebrafish. It was hosted by the Extreme Science and Engineering Discovery Environment (XSEDE) (17). tRF target genes were computationally predicted based on the interactions between tRFs and mRNA transcripts using the two state-of-the-art algorithms RNAhybrid (18,19) and IntaRNA (20). The contents of the database include maximum complementary length (MCL) of the paired tRF and target transcript, binding sites on the transcript (from 5′ end) and binding regions of 5′ untranslated region (UTR), coding sequence (CDS), or 3′ UTR of the transcript, free energy which is a measure of the stability of the binding between a tRF and a candidate target transcript, and graphic illustration of the pairing sequences. tRFtarget allows researchers to search tRF–target interactions by either tRF ID, or transcript or gene symbol, and demonstrates the pairing regions between a tRF and target transcripts as well as their locations. It facilitates researchers to use such information to perform various experiments such as amplifying the interested regions, making mutations in the regions for clone and plasmid construction, and constructing reporter assays for validation of target genes. The database also provides experimental evidence of the predicted tRF–mRNA interactions and functional studies on tRFs based on manually curated publications. In addition, the predicted target genes can be further used for functional pathway analysis and gene ontology annotation, providing the potential biological functions of tRFs.
MATERIALS AND METHODS
Data source
A workflow of the tRFtarget database construction is shown in Figure 2. The tRF sequences were retrieved from tRFdb (http://genome.bioch.virginia.edu/trfdb/) (3) and a tsRNA study (21). The transcript sequences were currently restricted to protein-coding transcripts, and downloaded from GENCODE (version GRCh38.p13 for human and version GRCm38.p6 for mouse, https://www.gencodegenes.org/) (22). Human gene symbols and the corresponding Ensembl gene IDs were downloaded from HGNC BioMart (https://biomart.genenames.org/) (23), and mouse gene symbols and Ensembl gene IDs were downloaded from MGI (http://www.informatics.jax.org/) (24). The transcript names and the corresponding Ensembl transcript IDs were downloaded from Ensembl BioMart (version GRCh38.p13 for human and version GRCm38.p6 for mouse, http://www.ensembl.org/biomart/martview/) (25). The genes and protein-coding transcripts of other species were downloaded from Ensembl BioMart (version BDGP6.28 for Drosophila, version WBcel235 for C. elegans, version Xenopus_tropicalis_v9.1 for Xenopus tropicalis, and version GRCz11 for Zebrafish), EnsemblFungi BioMart (version ASM294v2 for S. pombe, http://fungi.ensembl.org/biomart/martview/) and EnsemblBacteria (version ASM1640v1 for R. sphaeroides, https://bacteria.ensembl.org/Rhodobacter_sphaeroides_atcc_17025/Info/Index) (25).
Figure 2.
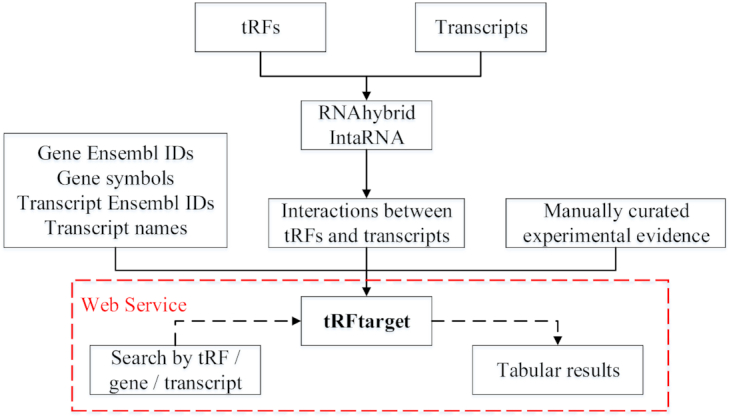
Flow diagram of constructing tRFtarget database. tRFtarget can be retrieved through web service. tRF, transfer RNA-derived fragment.
RNAhybrid prediction
RNAhybrid (version 2.1.2, https://bibiserv.cebitec.uni-bielefeld.de/rnahybrid) is a computational tool for predicting interactions between a short RNA and a long target RNA based on free energy (18,19). We set an energy threshold of –15 kcal/mol and provided five best binding sites per RNA pair given by RNAhybrid. As this algorithm only allows an input of a target sequence of no more than 50 kilobases (kb) in length, all transcripts longer than 50 kb are excluded for prediction and archived in the database. Other parameters were set at the default values of the algorithm. We ran RNAhybrid for all pairs of tRFs and transcripts, parsed the unstructured results, extracted important features, and then stored them in the database. MCL is the length of the longest successively complementary sequences for a specific interaction. Entries with a MCL less than 6 nts were excluded because such a short complementary sequence is unlikely to result in a functional interaction (26).
IntaRNA prediction
IntaRNA (version 3.1.3, http://rna.informatik.uni-freiburg.de/IntaRNA/Input.jsp) is another computational tool to predict interactions between two RNA molecules based on a combination of free energy and accessibility (20,27). We used the exact mode in IntaRNA to get more accurate target gene prediction. We set a seed length threshold of 6 nts and provided five best binding sites per RNA pair. Other parameters were set at the default values of the algorithm. We ran IntaRNA for all pairs of tRFs and transcripts, parsed the results, and then stored them in the database. Interactions between the same tRF and transcript with similar pairing sequences and almost identical binding locations were considered as duplicated entries, and only the entry with a minimal free energy was kept (see manual webpage (http://trftarget.net/manual) for an example of duplicated entries).
Consensus between predictions with RNAhybrid and IntaRNA
We evaluated the concordance of binding sites predicted from RNAhybrid and IntaRNA. A consensus pair of predictions was defined as similar pairing sequences and almost identical binding locations on a transcript (allowing 2 nts offset in the start and/or end of the pairing sequences, see manual webpage (http://trftarget.net/manual) for an example of consensus predictions). Users have an option to search for consensus predictions in the database.
Functional pathway analysis
In the database, we provided users a list of the predicted target genes to perform downstream functional pathway analysis and gene ontology annotation. The gene list was obtained based on the free energy and MCL of the interactions with a tRF. The interactions between a tRF and all target transcripts were ranked by an ascending order of free energy, and the interactions with the same free energy were further ranked by a descending order of MCL. When ranking genes from consensus interactions, free energy generated by IntaRNA was used. We then selected the top interactions to create a list of gene Ensemble IDs. The default number of the top interactions is 2000. Users can also specify the number of genes to be included in the top gene list. The gene list is copied to clipboard automatically and can be pasted into the input box on the DAVID® web server (https://david.ncifcrf.gov/) for functional pathway analysis (28,29).
RESULTS
Database overview
tRFtarget includes 936 tRFs, 135 000 target genes and 294 million interactions across eight species. The database can be queried in three ways:
search by tRF ID for all predicted target transcripts;
search by transcript Ensembl ID or name for all potential interacting tRFs;
search by gene Ensembl ID or symbol for all tRFs which potentially interact with any transcripts of a given gene.
In each query method, additional filter criteria on the binding regions, free energy and MCL can be specified by users to narrow the prediction results. Results are displayed in a tabular form and can be downloaded as a CSV file. Sorting or filtering by table column is also supported. There are two columns in the table that demonstrate the experimental evidence at the gene and site levels for each predicted interaction, respectively. Furthermore, hyperlinks to the relevant information from other databases were provided in the search results for users to browse detailed information of tRFs, genes and transcripts. A click button allows users to perform downstream functional pathway analysis on tRF target genes. Table 1 summarizes the total number of tRFs, genes and transcripts, and the prediction results by RNAhybrid and IntaRNA across eight species. A detailed description of this information can be found on the statistics webpage of the database (http://trftarget.net/statistics).
Table 1.
Summary statistics of tRFtarget database
| Species | tRFs | Target transcripts | Target genes | Target interactions |
|---|---|---|---|---|
| Human | 270 | 100 291 | 20 355 | 153 284 308 |
| Mouse | 164 | 67 125 | 22 385 | 60 074 771 |
| Drosophila | 86 | 30 588 | 13 947 | 14 822 386 |
| C. elegans | 47 | 33 552 | 20 191 | 7 685 677 |
| S. pombe | 104 | 5146 | 5145 | 3 105 075 |
| R. sphaeroides | 87 | 3111 | 3111 | 1 433 321 |
| Xenopus tropicalis | 51 | 54 848 | 19 983 | 16 449 592 |
| Zebrafish | 127 | 51 259 | 30 149 | 37 389 780 |
Validation of prediction
A previous study investigated interactions between small ncRNAs and mRNAs in the HEK293 cells using the CLASH experiments (13) and identified many tRF–mRNA chimeras, especially tRF-3–mRNA chimeras (4,14). We used the top 10 most abundant tRF-3–mRNA experimental interactions (4) for validation of our prediction results. Among the top 10 interactions in the CLASH data, four tRFs, tRF-3034a, 3035a, 3036a and 3037a, were not present in human tRFdb (3) and as a result were not included in the database. For tRF-3014a, the MCL of the reported interaction is 5 nts which is less than the threshold value of 6 nts, so this interaction structure was not indexed in the database. We then considered the experimental interactions from the remaining 5 tRFs to validate the prediction results. Figure 3 shows the interactions with mRNAs of the 5 tRFs illustrated by the predicted interactions of the CLASH tRF-3–mRNA chimeras using mfold, a computational tool for the prediction of the RNA secondary structure (30), and the predicted interactions in the tRFtarget database whose structures are most similar to the mfold predictions. All five CLASH chimeras have matched entries in the database. Moreover, our database provided more interactions with target mRNAs. For example, we found another interaction between tRF-3001a and the target mRNA RABEPK-203, besides the interactions of tRF-3001a with the two mRNAs DCTPP1-201 and DCTPP1-203 in the CLASH data. All three interactions have the same pairing sequences.
Figure 3.

Illustration of the interaction structures of tRF-3–mRNA chimeras from CLASH and the corresponding binding sites in tRFtarget. The interaction structures of tRF-3–mRNA chimeras were inferred by mfold RNA Folding Form using default settings, and the reads of the chimeras and the delta G values (dG) given by mfold are shown at the right. The binding sites in tRFtarget are shown below the mfold prediction. The transcript names are shown at the left, and the prediction algorithms and free energy (FE) are shown at the right. Black represents non-interaction sequences; blue represents interaction sequences of tRFs, green and red represent paired and non-paired bases of mRNA, respectively. CLASH, cross-linking, ligation and sequencing of hybrids.
Comparison with gene correlation analysis
In our previous study of tRFs in breast cancer, five tRFs, tRF-5024a, ts-34, ts-49, ts-58 and 5P_tRNA-Leu-CAA-4-1, were significantly associated with breast cancer patient survival. Among the five tRFs, tRF-5024a had the largest number of correlations with mRNA transcripts in which there were 404 positively and 2,292 negatively correlated genes (31). Here, we compared the predicted targets of tRF-5024a with the genes identified in correlation analysis. Among the 2292 genes that were negatively correlated with tRF-5024a, 625 genes are non-coding genes and were not indexed in the database. In the remaining 1667 genes, 1506 genes (90.3%) had at least one consensus interaction with tRF-5024a. Among the 404 genes that were positively correlated with tRF-5024a, 50 genes are non-coding genes and were not indexed in the database. In the remaining 354 genes, 296 genes (83.6%) had at least one consensus interaction with tRF-5024a. The target gene information and binding sites for the 1667 positively and 354 negatively correlated genes with tRF-5024a were shown in Supplementary Table S1.
DISCUSSION
tRFs are a novel class of regulatory ncRNAs and have been found to be dysregulated in cancer (1). Although previous studies have revealed the roles of tRFs in biological processes, there are still many features of tRFs, such as the biogenesis of tRF-3s and tRF-5s, and the functions of tRFs, yet to be fully characterized (2). To facilitate the research on tRFs, serval tRF-related databases have been developed based on omics data. For example, PtRFdb collects detailed information of plant tRFs (32); tRFdb includes a standardized nomenclature of tRFs in eight organisms (3); tRFexplorer provides expression profiles of identified tRFs in every cell line in NCI-60 and for each TCGA cancer type (9); MINTbase contains the abundance of tRFs identified in all TCGA projects (33); and tRF2Cancer not only provides the expression of identified tRFs in TCGA but supports searching for novel tRFs from user uploaded sequencing data (34). However, to our knowledge, there is no database available for a comprehensive prediction of tRF targets. tRFtarget developed in this study is the first tRF target prediction database to fill this gap. In tRFtarget, interactions between tRFs and mRNA transcripts were predicted by two algorithms, RNAhybrid (18,19) and IntaRNA (20). Both methods are free energy-based tools to predict RNA–RNA interaction. RNAhybrid is a popular tool to discover the targeting modes of small ncRNAs (14) and achieves more precise prediction than alignment-based methods (35,36). It utilizes the dynamic programming technique to efficiently calculate the optimal binding sites and can be easily run in parallel. RNAhybrid achieves high sensitivity but low precision in a comprehensive comparison study (36). IntaRNA, on the other hand, uses the ‘accessibility’ feature (20,27) to improve the accuracy of target prediction (35–37). ‘Accessibility’ is calculated as the free energy needed to unfold the binding sites in both RNAs to make the sites accessible for hybridization. It measures the likelihood that a short RNA is able to bind a specific site on the target mRNA and leads to a higher but more diverse free energy. As a result, IntaRNA achieves high sensitivity and high precision in target prediction (35,36).
We provided five best binding sites per RNA pair rather than the one with minimal free energy in tRFtarget for the following considerations. First, similar to miRNAs, each tRF may have multiple binding sites with a target transcript in the process of gene regulation (38). Second, the accuracy of target prediction commonly decreases as the length of target transcript increases when we use prediction based on minimal free energy. Providing more binding sites will be more likely to include the true interaction even when the target transcript is long (35,36). Third, the interaction with minimal free energy may not be biologically active. We also provided the suboptimal interactions with a bit higher free energy than the minimal one for users’ reference (36). When comparing the predicted targets in tRFtarget with the CLASH data (4), our database covered the most abundant tRF–mRNA interaction structures of the CLASH dataset, suggesting the reliability of target prediction. We also evaluated the overlap of the predicted targets and the correlated genes of tRF-5024a. More than 80% of the correlated genes were found to interact with tRF-5024a. Because correlation based on gene expression does not necessarily suggest interaction, there were some correlated genes that were not the predicted targets of tRF-5024a.
tRFtarget can be further improved in the following aspects. First, tRFs in the current version of the database were retrieved from tRFdb (3), and novel tRFs will continuously be identified by experiments. We will regularly update the database to include more tRFs and their targets. Second, tRFs interacted with a variety of RNAs including mRNAs, miRNAs and long intergenic ncRNAs (lincRNAs) in the CLASH data (14). We will expand the database by including ncRNAs as potential targets. Third, the motifs of tRFs based on interactions with different transcripts will be included in the database for query. Lastly, tRFtarget may include false positive interactions, but still provides useful information for further cost-effective experimental validation.
In conclusion, we developed tRFtarget, a transcriptome-wide tRF target prediction database for querying interactions between tRFs and transcripts in eight species. Manually curated experimental evidence was integrated into the database. This database provides useful information to guide biological experiments and target validation, as well as accelerates the understanding of the function and mechanism of tRFs.
DATA AVAILABILITY
All data in tRFtarget are freely accessible at http://trftarget.net.
Supplementary Material
ACKNOWLEDGEMENTS
This work used the Extreme Science and Engineering Discovery Environment (XSEDE) which is supported by National Science Foundation grant number ACI-1548562, and the XSEDE Jetstream resource at the IU/TACC through allocation TG-MCB200036. The authors want to thank Heling Zhou for website design and Zhong Wang for help with the XSEDE resource application and utilization.
Contributor Information
Ningshan Li, SJTU-Yale Joint Center for Biostatistics and Data Science, Department of Bioinformatics and Biostatistics, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China; Department of Biostatistics, Yale School of Public Health, New Haven, CT 06520, USA.
Nayang Shan, Department of Biostatistics, Yale School of Public Health, New Haven, CT 06520, USA; Center for Statistical Science, Department of Industrial Engineering, Tsinghua University, Beijing 100084, China.
Lingeng Lu, Department of Chronic Disease Epidemiology, Yale School of Public Health, New Haven, CT 06520, USA.
Zuoheng Wang, SJTU-Yale Joint Center for Biostatistics and Data Science, Department of Bioinformatics and Biostatistics, School of Life Sciences and Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [K01AA023321]; National Science Foundation [DMS1916246]; N.L. was supported in part by China Scholarship Council; the Neil Shen's SJTU Medical Research Fund; and SJTU-Yale Collaborative Research Seed Fund. Funding for open access charge: National Science Foundation.
Conflict of interest statement. None declared.
REFERENCES
- 1. Lee Y.S., Shibata Y., Malhotra A., Dutta A.. A novel class of small RNAs: tRNA-derived RNA fragments (tRFs). Genes Dev. 2009; 23:2639–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kumar P., Kuscu C., Dutta A.. Biogenesis and function of transfer RNA-related fragments (tRFs). Trends Biochem. Sci. 2016; 41:679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kumar P., Mudunuri S.B., Anaya J., Dutta A.. tRFdb: a database for transfer RNA fragments. Nucleic Acids Res. 2014; 43:D141–D145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kumar P., Anaya J., Mudunuri S.B., Dutta A.. Meta-analysis of tRNA derived RNA fragments reveals that they are evolutionarily conserved and associate with AGO proteins to recognize specific RNA targets. BMC Biol. 2014; 12:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Balatti V., Pekarsky Y., Croce C.M.. Croce C.M., Fisher P.B.. Advances in Cancer Research. 2017; 135:Academic Press; 173–187. [DOI] [PubMed] [Google Scholar]
- 6. Sun C., Fu Z., Wang S., Li J., Li Y., Zhang Y., Yang F., Chu J., Wu H., Huang X. et al.. Roles of tRNA-derived fragments in human cancers. Cancer Lett. 2018; 414:16–25. [DOI] [PubMed] [Google Scholar]
- 7. Zhu L., Liu X., Pu W., Peng Y.. tRNA-derived small non-coding RNAs in human disease. Cancer Lett. 2018; 419:1–7. [DOI] [PubMed] [Google Scholar]
- 8. Telonis A.G., Loher P., Magee R., Pliatsika V., Londin E., Kirino Y., Rigoutsos I.. tRNA fragments show intertwining with mRNAs of specific repeat content and have links to disparities. Cancer Res. 2019; 79:3034–3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. La Ferlita A., Alaimo S., Veneziano D., Nigita G., Balatti V., Croce C.M., Ferro A., Pulvirenti A.. Identification of tRNA-derived ncRNAs in TCGA and NCI-60 panel cell lines and development of the public database tRFexplorer. Database. 2019; 2019:baz115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sobala A., Hutvagner G.. Small RNAs derived from the 5′ end of tRNA can inhibit protein translation in human cells. RNA Biology. 2013; 10:553–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kuscu C., Kumar P., Kiran M., Su Z., Malik A., Dutta A.. tRNA fragments (tRFs) guide Ago to regulate gene expression post-transcriptionally in a Dicer-independent manner. RNA. 2018; 24:1093–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hafner M., Landthaler M., Burger L., Khorshid M., Hausser J., Berninger P., Rothballer A., Ascano M. Jr., Jungkamp A.-C., Munschauer M. et al.. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010; 141:129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Helwak A., Kudla G., Dudnakova T., Tollervey D.. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell. 2013; 153:654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guan L., Karaiskos S., Grigoriev A.. Inferring targeting modes of Argonaute-loaded tRNA fragments. RNA Biology. 2020; 17:1070–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Singh N.K. miRNAs target databases: developmental methods and target identification techniques with functional annotations. Cell. Mol. Life Sci. 2017; 74:2239–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Monga I., Kumar M.. Lai X., Gupta S.K., Vera J.. Computational Biology of Non-Coding RNA: Methods and Protocols. 2019; NY: Springer; 215–250. [Google Scholar]
- 17. Towns J., Cockerill T., Dahan M., Foster I., Gaither K., Grimshaw A., Hazlewood V., Lathrop S., Lifka D., Peterson G.D. et al.. XSEDE: accelerating scientific discovery. Comput. Sci. Eng. 2014; 16:62–74. [Google Scholar]
- 18. Rehmsmeier M., Steffen P., Höchsmann M., Giegerich R.. Fast and effective prediction of microRNA/target duplexes. RNA. 2004; 10:1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Krüger J., Rehmsmeier M.. RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006; 34:W451–W454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mann M., Wright P.R., Backofen R.. IntaRNA 2.0: enhanced and customizable prediction of RNA–RNA interactions. Nucleic Acids Res. 2017; 45:W435–W439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Balatti V., Nigita G., Veneziano D., Drusco A., Stein G.S., Messier T.L., Farina N.H., Lian J.B., Tomasello L., Liu C.-g. et al.. tsRNA signatures in cancer. Proc. Natl. Acad. Sci. 2017; 114:8071–8076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Frankish A., Diekhans M., Ferreira A.-M., Johnson R., Jungreis I., Loveland J., Mudge J.M., Sisu C., Wright J., Armstrong J. et al.. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2018; 47:D766–D773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Braschi B., Denny P., Gray K., Jones T., Seal R., Tweedie S., Yates B., Bruford E.. Genenames.org: the HGNC and VGNC resources in 2019. Nucleic Acids Res. 2018; 47:D786–D792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bult C.J., Blake J.A., Smith C.L., Kadin J.A., Richardson J.E., Group t.M.G.D.. Mouse Genome Database (MGD) 2019. Nucleic Acids Res. 2018; 47:D801–D806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zerbino D.R., Achuthan P., Akanni W., Amode M R., Barrell D., Bhai J., Billis K., Cummins C., Gall A., Girón C.G. et al.. Ensembl 2018. Nucleic Acids Res. 2017; 46:D754–D761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brennecke J., Stark A., Russell R.B., Cohen S.M.. Principles of microRNA–target recognition. PLoS Biol. 2005; 3:e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Miladi M., Montaseri S., Backofen R., Raden M.. Integration of accessibility data from structure probing into RNA–RNA interaction prediction. Bioinformatics. 2018; 35:2862–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huang D.W., Sherman B.T., Lempicki R.A.. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2008; 37:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang D.W., Sherman B.T., Lempicki R.A.. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009; 4:44–57. [DOI] [PubMed] [Google Scholar]
- 30. Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003; 31:3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shan N., Li N., Dai Q., Hou L., Yan X., Amei A., Lu L., Wang Z.. Interplay of tRNA-derived fragments and T cell activation in breast cancer patient survival. Cancers. 2020; 12:2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gupta N., Singh A., Zahra S., Kumar S.. PtRFdb: a database for plant transfer RNA-derived fragments. Database. 2018; 2018:bay063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pliatsika V., Loher P., Magee R., Telonis A.G., Londin E., Shigematsu M., Kirino Y., Rigoutsos I.. MINTbase v2.0: a comprehensive database for tRNA-derived fragments that includes nuclear and mitochondrial fragments from all The Cancer Genome Atlas projects. Nucleic Acids Res. 2017; 46:D152–D159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zheng L.-L., Xu W.-L., Liu S., Sun W.-J., Li J.-H., Wu J., Yang J.-H., Qu L.-H.. tRF2Cancer: a web server to detect tRNA-derived small RNA fragments (tRFs) and their expression in multiple cancers. Nucleic Acids Res. 2016; 44:W185–W193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lai D., Meyer I.M.. A comprehensive comparison of general RNA–RNA interaction prediction methods. Nucleic Acids Res. 2015; 44:e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Umu S.U., Gardner P.P.. A comprehensive benchmark of RNA–RNA interaction prediction tools for all domains of life. Bioinformatics. 2016; 33:988–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Richter A., Backofen R.. Accessibility and conservation: general features of bacterial small RNA–mRNA interactions. RNA Biology. 2012; 9:954–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Millar A.A., Waterhouse P.M.. Plant and animal microRNAs: similarities and differences. Funct. Integr. Genomics. 2005; 5:129–135. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data in tRFtarget are freely accessible at http://trftarget.net.