

Abstract
The present study was carried out on 62 genome sequences of members of the family Anelloviridae, as there have been no reports of genome analysis of these DNA viruses using a bioinformatics approach. The genes were found to be rich in AC content with low codon usage bias (CUB). Relative synonymous codon usage (RSCU) values identified the preferred codons for each amino acid in the family. The codon AGA was overrepresented, while the codons TCG, TTG, CGG, CGT, ACG, GCG and GAT were underrepresented in all of the genomes. A significant correlation was found between the effective number of codons (ENC) and base constraints, indicating that compositional properties might have influenced the CUB. A highly significant correlation was observed between the overall base content and the base content at the third codon position, indicating that mutations might have affected the CUB. A highly significant positive correlation was observed between GC12 and GC3 (r = 0.904, p < 0.01), which indicated that directional mutation pressure influenced all three codon positions. A neutrality plot revealed that the contribution of mutation and natural selection in determining the CUB was 58.6% and 41.4%, respectively.
Electronic supplementary material
The online version of this article (10.1007/s00705-020-04890-2) contains supplementary material, which is available to authorized users.
Introduction
Amino acids play a crucial role in cellular metabolic activities of an organism. Amino acids are joined step by step to form proteins. In the standard genetic code, a set of 59 codons encode 18 standard amino acids. Here, methionine and tryptophan are the only two amino acids that are coded with a single codon while all other amino acids are encoded by more than one codons, thus making some codons seemingly redundant in transcript. A bias in synonymous substitution of codons resulting in preferential usage of a specific codon within a codon family is termed codon usage bias (CUB), and it is different for genes, genomes, transcriptomes, and species [6, 33, 51]. CUB is frequently observed in highly expressed genes, while genes that are expressed at a low level usually have less CUB [25]. The pattern of synonymous codon usage allows the identification of relevant isoacceptor tRNAs for efficient translation of a particular gene. Thus, genomes with highly expressed genes will have more bias in codon preference, leading to the formation of proteins with lower susceptibility to misfolding [2]. Studies have shown that variation in synonymous substitutions occurs between and within genes [23, 44]. Various researchers have pointed out that the study of CUB provides important information about the evolution of related organisms [55, 58]. Because viruses are replicated and their genes are translated in living host cells, investigation of codon usage patterns of viral genomes can potentially provide information about the interaction and co-evolution of viruses with their hosts [54].
Several theories have been propounded for the origin of CUB, two of which are the selection‐mutation‐drift theory and the neutral theory. In the selection‐mutation‐drift theory, the major determinants of CUB are mutational pressure, natural selection, and genetic drift [10, 34]. In the neutral theory, mutations in the third position of a codon should be neutral, resulting in a random choice of codons [16]. In addition to mutation, selection pressure and genetic drift, several other factors influence CUB, including base constraints [8], base skewness [14], gene length [17], gene stability, translational selection [53], replication [27, 40], protein structure [72], and protein properties [16]. Analysis of CUB is useful for understanding the expression of viral genes [67]. It is also relevant for designing vaccines [20].
Anelloviridae is a family of ssDNA viruses with icosahedral symmetry [52] and a diameter of around 18–30 nm [43]. Members of this family have a single capsid protein and a genomic size of 2000–4000 nucleotides [50], and they have high degree of genetic variability [13]. The virus replicates inside the host cell, where a double-stranded DNA intermediate is formed by DNA polymerase in the S (synthesis) phase of the cell cycle [43]. The natural hosts of anellovirids include chimpanzees, tupaias, African monkeys, chickens, cattle, pigs, sheep, cats, dogs, and humans [68]. These viruses are extremely prevalent, with a comparatively static distribution worldwide and high degree of genetic heterogeneity [63]. Major diseases associated with anellovirids include lupus, hepatitis, hematologic disorders, pulmonary diseases, and myopathy [49]. They can be transmitted by sexual contact or blood transfusion and possibly by the fecal-oral route [1, 7]. No vaccines or drugs have been developed against these viruses, and further research in this area is still needed.
It has been reported that CUB is a major driving force the evolution of small DNA viruses and astroviruses [54, 74]. Preliminary analysis of flaviviruses revealed that base constraints and codon usage were related to those of their hosts [9]. Karlin et al. showed that the pattern of codon usage of Epstein-Barr virus plays a major role in influencing latent infection, leading to productive infection [36]. A codon usage pattern analysis of 31 Newcastle disease virus isolates showed that codon usage was associated with gene functions and geographic location but not with host specificity [76]. The GC constraint has been found to be the major determinant of codon usage variation in members of the family Parvoviridae [61]. Xu, et al. showed that the codon usage pattern in New World begomoviruses is apparently different from that in Old World begomoviruses, supporting the notion that the New World bipartite begomoviruses might have developed from the Old World begomoviruses [80].
In the current study, we report base constraints, codon usage pattern, protein properties, and influencing factors in the genomes of members of the family Anelloviridae in order to identify their genetic characteristics and discern the role of mutation and natural selection in CUB in genes. We also report the preferred codon for each amino acid and the overrepresented and underrepresented codons for the family as a whole to facilitate genetic engineering for the design of effective vaccines and other therapeutics. Analysis of codon usage patterns in viral genomes might elucidate adaptive traits, the role of evolutionary forces, viral interaction strategies, and host adaptation.
Methodology
Data access
The coding sequences (cds) of 62 genomes of members of the family Anelloviridae with their accession numbers were accessed from National Centre for Biotechnology Information nucleotide database (www.ncbi.nlm.nih.gov). In our analysis, only base sequences with an exact multiple of three nucleobases that had proper start and stop codons were used. A list of accession numbers and genome names of Anelloviridae family members is shown in Supplementary Table S1.
Base constraint analysis
The base content for the entire family Anelloviridae was analysed using a Perl programme written by the corresponding author (SC) to identify constraints on base composition across the family. The compositional properties of coding sequences considered in our analysis were as follows: (a) overall base content (A, C, G, T%), (b) base content at the third position of the codon (A3, C3, G3, T3%) and (c) GC content (GC, GC1, GC2, GC3, GC12%). In addition, nucleotide skew values i.e., AT skew (A-T/A+T) and GC skew (G-C/G+C) values, were computed. A positive GC or AT skew indicates higher usage of G over C or A over T. The imbalanced usage of two bases can be seen from the skew values across the transcript [3]. Similarly, purine, pyrimidine, purine-pyrimidine, keto, and amino skew values were also determined to evaluate their impact on CUB.
Relative synonymous codon usage (RSCU)
RSCU is a CUB index evaluated as the ratio of the observed frequency of a codon to its expected frequency out of all synonymous codons for a particular amino acid, multiplied by the degeneracy level. An RSCU value of 1 indicates equal usage of all synonymous codons, while a value greater than 1 indicates that a particular codon is favored. A codon with an RSCU value greater than 1.6 is considered an overrepresented codon, and one with an RSCU value less than 0.6 is considered an underrepresented codon.
The RSCU value for each codon was computed using the following formula [56]:
where Xij indicates the frequency of the jth codon for ith amino acid and ni is the number of codons for the ith amino acid (ith codon family).
Effective number of codons
The effective number of codons (ENC) measures the extent of bias in codon usage in a particular gene (cds), independent of the gene length and the protein encoded by the gene. The ENC value of a cds varies from 20 to 61 in the standard genetic code. If only one codon is used for each individual amino acid, the ENC value of that cds is 20, whereas equal usage of all synonymous codons would lead to higher ENC value. The bias in codon usage is inversely related to the ENC value. An ENC value greater than 35 indicates lower CUB, while a value less than 35 indicates higher CUB [79]. The ENC value of a cds was computed using the formula [79]:
where Fa (a = 2, 3, 4 or 6) is the average of the Fa values of the amino acids with a-fold degeneracy.
Multivariate statistical analysis
Correspondence analysis (COA) acts as a multivariate statistical platform for visualization of the major trends in CUB studies of nucleotide sequences. A COA plot is implemented to understand the variation in codon usage, with RSCU values of 59 synonymous codons across two axes F1 (axis 1) and F2 (axis 2) [62]. We used “Past” software to plot the COA graph.
Parity plot (PR2) analysis
A PR2 plot is implemented to understand the role of evolutionary forces affecting the CUB. A PR2 plot was made for 2-fold, 4-fold and 6-fold degenerate codon families with [G3/(G3+C3) along the x-axis and [A/(A+T) along the y-axis for the third position of codons. The midpoint of the plot is 0.5, where no bias exists between complementary nucleotide sequences, where G = C and A = T [66].
Neutrality plot analysis
A neutrality plot is used to quantify the magnitude of evolutionary forces, i.e., the role of mutation and natural selection in the determination of CUB (Zhang et al. [82]. It is framed with GC12 along the y-axis and GC3 along the x-axis. Each dot in the plot represents an independent genome. If the dots are diagonally distributed with a regression coefficient value approaching 1, this suggests a more important role of mutation [64], whereas a scattered distribution of dots suggests a significant role of natural selection, with the regression coefficient approaching zero.
Protein properties
Biochemical properties of proteins, i.e., aromaticity and hydropathicity, have been reported to be associated with CUB [16]. The general average hydropathicity score (GRAVY) is the average of the hydropathicity scores of the amino acids in the coding sequence of a gene, and its value ranges from -2 to +2, with a negative value indicating that the protein is hydrophilic in character and vice versa [31]. The aromaticity of a protein indicates the distribution of aromatic amino acids (tryptophan, tyrosine, and phenylalanine) in the protein [42].
Mutation responsive index (MRI)
The MRI value is used to estimate the rate of mutational drift in a cds. A positive MRI value suggests the impact of directional mutation, while the opposite indicates the influence of translational selection in a gene [21, 22].
Translational selection (P2)
The P2 value estimates the rate of codon-anticodon interactions and suggests the impact of translational efficiency in a gene. A P2 value greater than 0.5 indicates a bias favouring translational selection [22]. The P2 value of a gene is calculated using the following formula [22]:
where W = T or A, S = G or C, and Y = T or C
Statistical analysis
The correlation coefficients for various parameters, namely, effective number of codons, base content, skew values, protein properties, mRNA free energy, and mRNA stability index, were computed using the statistical software SPSS 16.0 for Windows. A heat map was constructed for comparing GC3 values and codon usage values, using the XLSTAT software [38].
Results
Codon usage bias
To determine the impact of bias on codon usage in the genomes of members of the family Anelloviridae, the effective number of codons (ENC) was computed for each genome Table 1. The ENC values ranged from 36.65 to 57.40, with a mean ENC value of 51.93 indicating a low CUB [11]. However, a high degree of variation in codon usage was found among the different genomes. Analysis of RSCU values of 59 sense codons revealed that 27 of them were used frequently in the cds. Thus, more than one codon was preferred for each amino acid, supporting our finding of a low CUB in the genes.
Table 1.
Average ENC value of the genes of members of the family Anelloviridae
| Virus | ENC value |
|---|---|
| Avian gyrovirus 2 | 57.40 |
| California sea lion anellovirus | 50.67 |
| Chicken anemia virus | 53.83 |
| Giant panda anellovirus strain gpan20688 | 50.40 |
| Gyrovirus 4 | 54.20 |
| Gyrovirus GyV3 | 52.17 |
| Gyrovirus GyV7_SF | 51.30 |
| Gyrovirus GyV8 | 52.57 |
| Gyrovirus Tu243 | 54.83 |
| Gyrovirus Tu789 | 51.87 |
| Rodent torque teno virus 2 isolate RN_2_Se15 | 48.67 |
| Seal anellovirus 2 | 56.00 |
| Seal anellovirus 3 | 55.33 |
| Seal anellovirus TFFN_USA_2006 | 56.53 |
| Small anellovirus 1 | 51.93 |
| Small anellovirus 2 | 46.02 |
| Simian torque teno virus 31 isolate VGA001233 | 52.75 |
| Simian torque teno virus 32 isolate VGA001542 | 55.48 |
| Simian torque teno virus 33 isolate VWP0052211 | 49.93 |
| Simian torque teno virus 34 isolate VGA001201 | 53.65 |
| Torque teno virus 1 | 53.37 |
| Torque teno virus 2 | 54.65 |
| Torque teno virus 3 | 51.27 |
| Torque teno virus 4 | 54.95 |
| Torque teno virus 6 | 53.73 |
| Torque teno virus 7 | 53.90 |
| Torque teno virus 8 | 54.73 |
| Torque teno virus 10 | 55.83 |
| Torque teno virus 12 | 55.48 |
| Torque teno virus 14 | 55.55 |
| Torque teno virus 15 | 52.85 |
| Torque teno virus 16 | 51.40 |
| Torque teno virus 19 | 56.25 |
| Torque teno virus 25 | 48.77 |
| Torque teno virus 26 | 50.50 |
| Torque teno virus 27 | 54.43 |
| Torque teno virus 28 | 52.18 |
| Torque teno mini virus 1 | 51.53 |
| Torque teno mini virus 2 | 53.83 |
| Torque teno mini virus 3 | 52.43 |
| Torque teno mini virus 4 | 50.15 |
| Torque teno mini virus 5 | 51.87 |
| Torque teno mini virus 6 | 53.70 |
| Torque teno mini virus 7 | 46.60 |
| Torque teno mini virus 8 | 41.65 |
| Torque teno mini virus 9 | 42.80 |
| Torque teno mini virus ALH8 | 36.65 |
| Torque teno mini virus ALA22 | 51.43 |
| Torque teno mini virus 18 isolate 222 | 45.53 |
| Torque teno midi virus 1 | 49.13 |
| Torque teno midi virus 2 | 53.08 |
| Torque teno indri virus 1 | 49.67 |
| TTV_like mini virus isolate TTMV_LY1 | 48.97 |
| Torque teno canis virus | 55.27 |
| Torque teno felis virus | 54.07 |
| Torque teno Leptonychotes weddellii virus_2 isolate TTLwV_2_gt3_wsp24 | 55.03 |
| Torque teno douroucouli virus | 50.33 |
| Torque teno sus virus 1a | 56.00 |
| Torque teno sus virus 1b isolate 1p | 52.80 |
| Torque teno sus virus k2a isolate 2p | 53.40 |
| Torque teno Tadarida brasiliensis virus | 50.37 |
| Torque teno tamarin virus | 47.88 |
Base composition
The nucleotide base content can affect the CUB of a genome [34]. Thus, we computed the overall base composition of the genomes of various members of the family Anelloviridae. The mean percentage of A (31.78) was found to be the highest, followed by C (27.11) and G (22.06), while T (19.05) had the lowest frequency Fig. 1. This indicated that the nucleobases A and C occurred more frequently than the bases G and T in the coding sequence. At the third codon position, the percentage of A was the highest (32.87), followed by C (29.39), T (21.54), and G (16.19), suggesting that A- and C-ended codons might have been preferred to G- and T-ended codons. The mean GC content of the genomes was 49.21%, with an almost equal GC and AT content across the genomes. The overall GC content is an important factor contributing to bias in codon usage across genomes [75]. Here, analysis of the GC content at the first, second, and third codon positions revealed the frequency of that GC1 (54.67%) was higher than that of GC2 (47.28%) and GC3 (45.67%) Fig. 1. Correlation analysis of ENC value and base content showed a significant correlation of ENC with A%, G%, A3%, G3%, GC%, GC1%, GC2%, GC3% and GC12%, at p < 0.01 Table 2, suggesting that these bases might be responsible for the CUB of genes in members of the family Anelloviridae.
Fig. 1.

(a) Overall base content and base content at the third codon position of genes of members of the family Anelloviridae, (b) Overall GC content and GC content at the first, second, third and first and second codon positions
Table 2.
Correlation between ENC and base content of genes of members of the family Anelloviridae
| A% | T% | G% | C% | GC% | A3% | T3% | G3% | C3% | GC1% | GC2% | GC3% | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ENC | -0.521** | -0.019 | 0.458** | 0.249 | 0.420** | -0.578** | 0.102 | 0.524** | 0.204 | 0.374** | 0.364** | 0.419** |
**Significant at p < 0.01
Codon usage pattern
To understand the pattern of codon usage in each synonymous codon family, we performed relative synonymous codon usage (RSCU) analysis of 59 sense codons, as shown in Fig. 2. The pattern of codon usage differed from genome to genome (Supplementary Table S2). Further, we obtained mean RSCU value of codons for 62 genomes of Anelloviridae members and categorized them into four groups: RSCV value >1.6, overrepresented codons,<0.6, underrepresented codons; >1, more frequently used codons; < 1, less frequently used codons. The codon AGA was overrepresented in all of the genomes, while the codons TCG, TTG, CGG, CGT, ACG, GCG and GAT were underrepresented in all of the genomes. The preferred codons for amino acids are reported in Supplementary Table S3. Of the 27 frequently used codons, 13 were A-ended, 10 were C-ended, three codons were T-ended, and one was G-ended. Thus, A- and C-ended codons were more likely to be abundant in coding sequences. Of the 32 less frequently used codons, 13 were T-ended, 11 were G-ended, six were C-ended, and one was A-ended. Our analysis of base compositional properties and codon usage patterns across the genomes suggested that compositional features under mutation pressure contributed to the observed codon usage pattern [4].
Fig. 2.

(a) Overall RSCU values of codons in genomes of members of the family Anelloviridae. Red indicates underrepresented codons, (b) more frequently used codons, and (c) less frequently used codons
We generated a heat map by correlating the codon usage value with GC3 and found that, except for the codons TTG and AAC, all of the G- and C-ended codons were positively correlated with GC3, while, except for CGA, AGT, GTT, GGT, GCT and CGT, all of the A- and T-ended codons were negatively correlated with GC3 Fig 3. These findings confirm that codon usage variation is subject to GC constraints, and this acquires importance for understanding the molecular architecture of the genomes of members of the family Anelloviridae [30].
Fig. 3.

Heat map with codon usage values for members of the family Anelloviridae. Green indicates negative correlation, red indicates positive correlation, and black indicates no correlation of GC3 with A-, T-, G- and C-ended codons
Relationship between codon usage patterns in the genomes of members of the family Anelloviridae and those of their hosts
Viruses, being obligate parasites, can be sustained only within living hosts [86]. Here, the RSCU values of different members of the family Anelloviridae were compared with those of a few of their host organisms (humans, African monkeys, chickens, sheep, pigs, and dogs). It was found that they had several of their most frequently used codons (RSCU value > 1) and less frequently used codons (RSCU value < 1) in common (Supplementary Table S4), suggesting that these viruses were adapted to their host. Similar associations between viruses and their hosts in their pattern of codon usage have also been reported for chikungunya virus [11], poliovirus [47], and coronaviruses [78].
Role of mutational pressure
Alterations in the base sequence of the genome are usually related to mutational pressure, which is an important factor in determining CUB. The correlation between the overall base content and the base content at the third codon position was investigated to determine whether mutational pressure alone was responsible for the observed CUB. The results of Pearson’s correlation analysis, shown in Table 3, indicate a highly significant positive correlation for A-A3%, T-A3%, T-T3%, G-G3%, C-G3%, GC-G3%, G-C3%, C-C3%, GC-C3%, G-GC3%, C-GC3% and GC-GC3% at p < 0.01, indicating that these bases were proportionally related to each other, while highly significant negative correlation was observed for G-A3%, C-A3%, GC-A3%, C-T3%, GC-T3%, A-G3%, T-G3%, A-C3%, T-C3%, A-GC3% and T-GC3%, at p < 0.01, i.e., these bases were inversely related to each other in their abundance. Our results suggest that mutational pressure and natural selection influenced the CUB of members of the family Anelloviridae, in agreement with previous observations [2].
Table 3.
Interrelationships of overall base composition with the base composition at third codon positions
| A3% | T3% | G3% | C3% | GC3% | |
|---|---|---|---|---|---|
| A% | 0.972** | 0.194 | -0.908** | -0.580** | -0.877** |
| T% | 0.407** | 0.903** | -0.377** | -0.851** | -0.714** |
| G% | -0.899** | -0.231 | 0.935** | 0.470** | 0.831** |
| C% | -0.672** | -0.655** | 0.507** | 0.917** | 0.828** |
| GC% | -0.917** | -0.483** | 0.856** | 0.769** | 0.954** |
**Significant at p < 0.01
Using regression analysis of A-A3%, G-G3%, T- T3%, and C- C3%, as shown in Fig. 4, we investigated the extent to which each base was affected by mutational pressure. G-G3% and A-A3% were found to be more strongly affected by mutation than C-C3% and T-T3%, in agreement with an earlier study [72].
Fig. 4.

Regression analysis comparing overall base content and base content at the third codon position. The regression coefficient value of the four plots indicates a greater contribution of mutation in base G, G3% and A, A3% over C, C3% and T, T3%
Trends of codon usage bias
The trends of variation in the codon usage pattern were analyzed using correspondence analysis (COA), a multivariate technique. COA was performed using the RSCU values of 59 sense codons. In COA analysis, all genomes were marked with blue colour and found scattered across the rectangular plot, G/C- and A/T-ended codons were represented as green and red colour dots, respectively, with a few overlapping ones along the x-axis Fig. 5. The graph revealed that G/C- and A/T-ended codons were separated along the axes, and the differences in codon distribution in the plot were mainly due to variation in the frequency of G/C- and A/T-ended codons. Here, some codons were found very close to the axes, suggesting that mutational pressure might have governed the CUB.
Fig. 5.
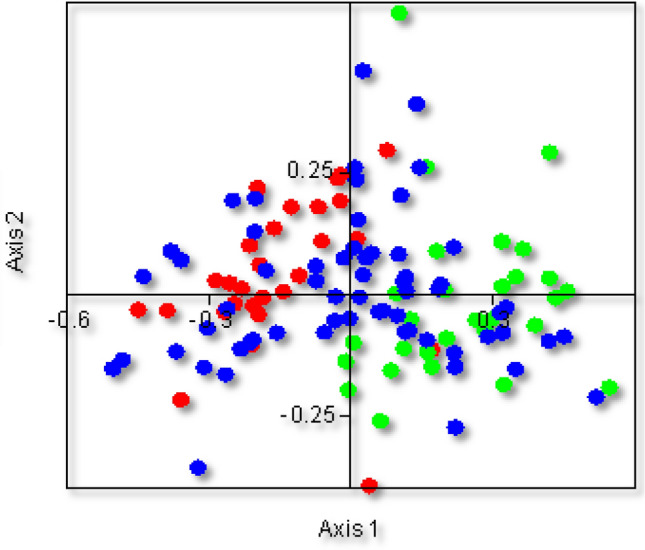
Correspondence analysis of genomes of members of the family Anelloviridae. Green indicates GC-ended codons, red indicates AT-ended codons, and blue indicates each genome. The trends of variations of the codons are presented in the plot
Further, we used the UPGMA algorithm to determine the Euclidean similarity index in Past 3 software by performing cluster analysis [28]. The results revealed two major clusters, as shown in Fig. 6. One cluster included seven genomes, and another one included 55 genomes, revealing intraspecific and interspecific relationships between them.
Fig. 6.

Cluster analysis of genomes of members of the family Anelloviridae
PR- 2 bias plot analysis
PR-2 bias plots were generated to assess the role of mutational pressure and natural selection in determining the CUB. If mutational pressure is the sole factor determining CUB, the GC and AT content will be proportional along the axes, while a deviation from proportional distribution might be due to the combined effect of mutational pressure and natural selection [65]. We constructed PR-2 bias plots for the 2-fold, 4-fold and 6-fold degenerate codon families with G3/G3+C3 and A3/A3+T3 on the x- and y-axis, respectively Fig. 7, and found unequal distribution of AT and GC bases, suggesting that both mutation and natural selection might have shaped the CUB of these genomes.
Fig. 7.

Parity rule 2 bias plot of genomes of members of the family Anelloviridae. A non-uniform distribution of bases suggests that both mutation pressure and natural selection might have influenced their CUB
Neutrality plot analysis
A neutrality plot comparing GC12 (y-axis) and GC3 (x-axis) was made to determine the effect of mutation pressure and natural selection on compositional bias Fig. 8. A highly significant correlation was observed between GC12 and GC3 (r = 0.904** at p < 0.01), suggesting that directional mutation pressure acted on all codon positions. The points were diagonally distributed with a wide range of GC3 distribution, indicating that mutational pressure influenced the CUB. Moreover, the slope of the regression line of GC12 vs. GC3 was 0.586. These results suggest a major effect of mutational pressure (58.6%) and a minor effect of natural selection (41.4%) in determining the CUB of the genes.
Fig. 8.
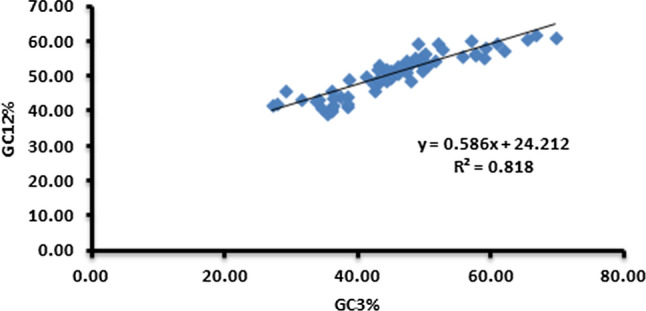
Neutrality plot of genomes of members of the family Anelloviridae. The regression coefficient value suggests that mutation contributed 58.6% and natural selection contributed 41.4% to the CUB
Role of protein properties
Several studies have revealed that aromatic and hydrophilic properties of proteins are related to the bias in codon usage [69]. We performed correlation analysis using ENC, GRAVY, hydrophilicity and aromaticity using Pearson’s correlation method and found a highly significant positive correlation between ENC and GRAVY (0.333**) at p < 0.01 Table 4, indicating proportional relationship between them. Highly significant negative correlation was observed between ENC and hydrophilicity (-0.349**) at p < 0.01, indicating an inverse relationship. These results suggest that the extent of CUB was associated with the properties of protein, namely, GRAVY and hydrophilicity.
Table 4.
Correlation between ENC and protein properties of genes of members of the family Anelloviridae
| GRAVY | Hydrophilicity | Aromaticity | |
|---|---|---|---|
| ENC | 0.333** | -0.349** | 0.236 |
**Significant at p < 0.01
Role of skewness
Previously, it was shown that skewness in the base composition of coding sequences is reflected in their transcriptional products [5]. Here, the overall GC skew value for all members of the family Anelloviridae was -0.11, indicating that the base C was more abundant than G, and the mean AT skew value was 0.24, indicating that the base A was more abundant than T. To further examine the role of skewness on CUB, correlation analysis was performed between the ENC value and the AT skew, GC skew, keto skew, amino skew, purine skew, pyrimidine skew and purine-pyrimidine skew Table 5. The results showed a highly significant positive correlation of the GC skew with CUB at p < 0.01, i.e., the GC skew had a proportional relationship to CUB, while the AT skew, keto skew, amino skew, purine skew, and purine-pyrimidine skew had a highly significant negative correlation with CUB at p < 0.01, indicating an inverse relationship. These results together suggest that skewness of bases influenced the CUB of the genes.
Table 5.
Correlation between ENC and skew properties of genes of members of the family Anelloviridae
| GC skew | AT skew | PU skew | PY skew | Keto skew | Amino skew | PU-PY skew | |
|---|---|---|---|---|---|---|---|
| ENC | 0.381** | -0.526** | -0.490** | -0.118 | -0.337** | -0.410** | -0.363** |
**Significant at p < 0.01
Mutational responsive index (MRI) and translational selection (P2)
The mutational responsive index is a specific criterion to enumerate the impact of directional mutational pressure and translational selection across the genome. Here, the mean MRI value for 62 members of the family Anelloviridae was 0.50. This positive MRI value suggests a role of directional mutation across the genomes [21]. The mean P2 value was 0.16, i.e., less than 0.5, indicating a low impact of translational selection, consistent with previous observations [14].
Discussion
The choice of a preferred codon out of each synonymous codon family for an amino acid results in a phenomenon called codon usage bias (CUB) [46]. CUB is important for exogenous gene expression, which might require optimization of codons [45]. The degree of CUB varies from species to species [51] and is associated with mutational pressure, genetic drift, and natural selection [29]. Other factors contributing to CUB include base content, base skewness, gene expression level, gene length, protein structure, and the aromaticity and hydrophilicity of the protein. In the present study, we investigated the extent and the variation in the codon usage patterns of 62 genomes of members of the family Anelloviridae. We also examined the dynamics of base content and identified major factors influencing CUB. This study provides insights into the genetics and evolutionary relationships of organisms, and identification of overrepresented and underrepresented codons may assist in efforts to alter gene expression levels through codon optimization.
The effective number of codons reflects the extent of CUB in the coding sequence of a particular gene [73]. A higher CUB is associated with a lower ENC value and vice versa [14]. The ENC values of 62 genomes of members of the family Anelloviridae ranged from 36.65 to 57.40, with an average ENC value of 51.93, indicating low CUB. The lower CUB suggests efficient usage of multiple codons for protein production [34]. Zhou et al. reported that the mean ENC value for H5N1 influenza virus was 50.91, ranging from 43.11 to 55.21, indicating low CUB [86].
The base content of a gene significantly influences the pattern of codon usage across the gene [14]. Our analysis of compositional properties indicated that the relative usage of bases was A > C > G > T. At the third codon position, A was more frequent than C and T was more frequent than G, with a preferred usage of A- and C-ended codons. The overall AT and GC content was almost equal. A highly significant correlation was observed between ENC and base content values,hence, the extent of CUB was dependent on compositional properties of the genome. Torque teno sus virus 1 had 32.72% A, 25.70% G, 22.39% C, and 19.20 % U(T). At the third codon position it had 37.45% A, 32.63% C, 30.82% G, and 25.90% U(T), with the increased preference for A-ended codons [Zhang Zhicheng et al. 2013]. The base composition of 17 human cytomegalovirus strains was 21.46% A, 21.07% T, 28.99% G, and 28.49% C, with G and C almost equal and A and T almost equal [32]. Tsai et al. reported that the GC content of 12 iridovirus genomes ranged from 27 to 55%, with distantly related viruses showing different patterns of synonymous CUB [70].
The RSCU value indicates the pattern of codon usage across the genome. We therefore determined the RSCU value for each member of the family Anelloviridae and found that 27 out of 59 codons were more frequently used, with preferred usage of A- and C-ended codons. The AGA codon was overrepresented, and the TCG, TTG, CGG, CGT, ACG, GCG and GAT codons were underrepresented in all of the genomes. Zhang et al. reported 18 frequently used codons with preferential usage of A- and C-ended codons and nine underrepresented codons in torque teno sus virus 1 [82]. RSCU analysis of Newcastle disease virus revealed eight underrepresented codons (CGC, CGA, CGT, CGG, CCG, ACG, TCG, and GCG) that were markedly suppressed [76]. A heat map comparing codon usage values and GC3 content showed that the usage of C- and G-ended codons was positively correlated with GC constraints, while the usage of T- and A-ended codons was negatively correlated with GC constraints, with a few exceptions. Similarly, a positive correlation of G- and C-ended codons with GC3 and a negative correlation of A- and T-ended codons with GC3 content has also been found in yeast, bacteria, humans [48], birds, and mammals [71]. This suggests that GC constraints are positively related to the CUB of genes.
Correlation analysis between the overall base content and the base content at the third codon position showed a highly significant correlation (p < 0.01), suggesting that mutational pressure might have influenced the CUB together with other environmental determinants. The bias in favour of codons with a higher content of one base over the other three also revealed the predominant role of mutational pressure [35, 60, 83, 85]. It has been reported that mutational pressure leads to alterations in biochemical mechanisms, with recurrent changes in certain bases thus contributing to the CUB [19, 24]. In torque teno sus virus 1, a significant negative correlation of A3% with G% and GC% (p < 0.01), and G3% and C3% with G% (p < 0.01) has been reported, indicating that base constraints and mutational pressure together determined the CUB [82].
A COA plot indicated that mutational pressure and natural selection together contributed to the CUB, consistent with a previous report [3]. A COA plot of 30 strains of hepatitis A virus revealed a major role of base content in CUB determination [15]. Fu performed COA analysis of herpesviruses and reported that genes with lower GC content were distributed along the right side of the plot, while genes with higher GC content were at the left side of the plot, supporting the notion that base compositional properties highly influenced synonymous codon changes [20].
To investigate the evolutionary relatedness of anellovirid genomes, a cluster analysis was performed that revealed two major clusters, one with seven genomes and another with 55 genomes, suggesting intraspecific and interspecific relationships. Cluster analysis of 11 isolates of human bocavirus performed based on RSCU values showed that genes with similar functions, even if they were from different isolates, grouped in the same lineage, irrespective of geographical location [84]. Wong et al. performed cluster analysis of influenza A viruses and reported that three genes, NA, HA and PB1, of human H2N2 influenza viruses of avian origin were also found in a cluster of avian virus, while avian-origin PB1 and HA genes of human H3N2 influenza viruses were extended from the cluster and a few of the H1 viral genes of human (PA) were also represented in the human H3 cluster [77].
PR-2 bias plot analysis in this study showed a disproportional distribution of AT and GC content. This pattern of base arrangement across the graph suggested that natural selection and mutational pressure both contributed to the CUB [72]. Parity plot analysis of the PB2 gene of influenza A H7N9 virus revealed no constraints in mutation and natural selection between the two complementary strands of a DNA duplex [26].
Neutrality plot analysis revealed that mutation pressure was more important than selection pressure in members of the family Anelloviridae. The higher mutational bias might be due to chemical decay of nucleotides, non-uniform DNA repair, and non-random replication errors [37]. Mutations usually occur spontaneously without any external driving force [73]. Neutrality plot analysis showed a significant correlation between GC3 and GC12 (0.904) at p < 0.01, suggesting that directional mutation pressure acted at all codon positions [20]. It also suggested that mutational pressure played a more important role than natural selection. Neutrality analysis of the PB2 gene of influenza A H7N9 virus revealed a greater role of selection pressure than mutational bias [26].
A significant correlation was found between the ENC value and biochemical properties of the protein, with the extent of CUB being associated with the GRAVY and hydrophilicity scores of the proteins across the genomes. Zhao et al. reported that hydrophilicity scores were critical factors in the codon usage of 11 isolates of bocavirus [84]. Liu et al. reported a highly significant positive correlation of GRAVY with codon usage variation and a highly significant negative correlation of aromaticity with codon usage variation in porcine circovirus [41].
Analysis of base skewness revealed higher usage of C over G and A over T. Skew values of genomes also correlated significantly with ENC values. AT skew, GC skew, amino skew, keto skew, purine skew, pyrimidine skew, and purine-pyrimidine skew were found to affect CUB. Transcription has been reported to have a likely relationship to base skew properties [12]. A significant correlation of base skewness with CUB has also been found in Nipah virus genes [14].
The MRI index and P2 values suggested a directional role of mutation over translational selection in members of the family Anelloviridae in the present study. Zhang et al. reported that mutational pressure arising from compositional constraint along with translational selection was responsible for CUB in torque teno sus virus 1 [82]. Similarly, in various other organisms, mutation and translational selection were identified as important determinants of variations in codon usage [35, 39, 57, 59].
Conclusion
We determined the base composition and codon usage pattern of the genes of 62 members of the Anelloviridae family of DNA viruses. The overall bias in the pattern of codon usage was low, with a high level of variation in synonymous codon usage in the viral genes. The base A was used more than C and G, and T was the least frequent. A- and C-ended codons were used preferentially. One codon (AGA) was overrepresented, while seven codons (TCG, TTG, CGG, CGT, ACG, GCG and GAT) were underrepresented in all of the genomes. Mutational pressure had a greater role than natural selection in determining the codon usage patterns of members of this family.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgement
The authors are grateful to Assam University, Silchar, Assam, India, for providing necessary facilities to carry out the research work.
Funding
Unfunded
Compliance with ethical standards
Conflict of interest
The authors declare no conflicts of interest in this work.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Aramouni M (2013) Role of Torque teno sus viruses during co-infection with other swine pathogens, Universitat Autònoma de Barcelona
- 2.Barbhuiya PA, Uddin A, Chakraborty S. Genome-wide comparison of codon usage dynamics in mitochondrial genes across different species of amphibian genus Bombina. J Exp Zool Part B Mol Deve Evol. 2019;332(3–4):99–112. doi: 10.1002/jez.b.22852. [DOI] [PubMed] [Google Scholar]
- 3.Barbhuiya PA, Uddin A, Chakraborty S. Compositional properties and codon usage of TP73 gene family. Gene. 2019;683:159–168. doi: 10.1016/j.gene.2018.10.030. [DOI] [PubMed] [Google Scholar]
- 4.Behura SK, Severson DW. Comparative analysis of codon usage bias and codon context patterns between dipteran and hymenopteran sequenced genomes. PLoS ONE. 2012;7:e43111. doi: 10.1371/journal.pone.0043111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beletskii A, Bhagwat AS. Transcription-induced cytosine-to-thymine mutations are not dependent on sequence context of the target cytosine. J Bacteriol. 2001;183:6491–6493. doi: 10.1128/JB.183.21.6491-6493.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bennetzen JL, Hall BD. Codon selection in yeast. J Biol Chem. 1982;257:3026–3031. doi: 10.1016/S0021-9258(19)81068-2. [DOI] [PubMed] [Google Scholar]
- 7.Bernardin F, Operskalski E, Busch M, Delwart E. Transfusion transmission of highly prevalent commensal human viruses. Transfusion. 2010;50:2474–2483. doi: 10.1111/j.1537-2995.2010.02699.x. [DOI] [PubMed] [Google Scholar]
- 8.Bibb M, Findlay P, Johnson M. The relationship between base composition and codon usage in bacterial genes and its use for the simple and reliable identification of protein-coding sequences. Gene. 1984;30:157–166. doi: 10.1016/0378-1119(84)90116-1. [DOI] [PubMed] [Google Scholar]
- 9.Blitvich B, Firth A. Insect-specific flaviviruses: a systematic review of their discovery, host range, mode of transmission, superinfection exclusion potential and genomic organization. Viruses. 2015;7:1927–1959. doi: 10.3390/v7041927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bulmer M. The selection-mutation-drift theory of synonymous codon usage. Genetics. 1991;129:897–907. doi: 10.1093/genetics/129.3.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Butt AM, Nasrullah I, Tong Y. Genome-wide analysis of codon usage and influencing factors in chikungunya viruses. PLoS ONE. 2014;9:e90905. doi: 10.1371/journal.pone.0090905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Butt AM, Nasrullah I, Qamar R, Tong Y. Evolution of codon usage in Zika virus genomes is host and vector specific. Emerg Microbes Infect. 2016;5:1–14. doi: 10.1038/emi.2016.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cadar D, Kiss T, Ádám D, Cságola A, Novosel D, Tuboly T. Phylogeny, spatio-temporal phylodynamics and evolutionary scenario of Torque teno sus virus 1 (TTSuV1) and 2 (TTSuV2) in wild boars: Fast dispersal and high genetic diversity. Vet Microbiol. 2013;166:200–213. doi: 10.1016/j.vetmic.2013.06.010. [DOI] [PubMed] [Google Scholar]
- 14.Chakraborty S, Deb B, Barbhuiya PA, Uddin A. Analysis of codon usage patterns and influencing factors in Nipah virus. Virus Res. 2019;263:129–138. doi: 10.1016/j.virusres.2019.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.D’Andrea L, Pintó RM, Bosch A, Musto H, Cristina J. A detailed comparative analysis on the overall codon usage patterns in hepatitis A virus. Virus Res. 2011;157:19–24. doi: 10.1016/j.virusres.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deb B, Uddin A, Mazumder GA, Chakraborty S. Analysis of codon usage pattern of mitochondrial protein-coding genes in different hookworms. Mol Biochem Parasitol. 2018;219:24–32. doi: 10.1016/j.molbiopara.2017.11.005. [DOI] [PubMed] [Google Scholar]
- 17.Eyre-Walker A. Synonymous codon bias is related to gene length in Escherichia coli: selection for translational accuracy? Mol Biol Evol. 1996;13:864–872. doi: 10.1093/oxfordjournals.molbev.a025646. [DOI] [PubMed] [Google Scholar]
- 18.Fickett JW. Recognition of protein coding regions in DNA sequences. Nucleic Acids Res. 1982;10:5303–5318. doi: 10.1093/nar/10.17.5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Francino MP, Ochman H. Deamination as the basis of strand-asymmetric evolution in transcribed Escherichia coli sequences. Mol Biol Evol. 2001;18:1147–1150. doi: 10.1093/oxfordjournals.molbev.a003888. [DOI] [PubMed] [Google Scholar]
- 20.Fu M. Codon usage bias in herpesvirus. Adv Virol. 2010;155:391–396. doi: 10.1007/s00705-010-0597-0. [DOI] [PubMed] [Google Scholar]
- 21.Gatherer D, McEwan NR. Small regions of preferential codon usage and their effect on overall codon bias-The case of the plp gene. IUBMB Life. 1997;43:107–114. doi: 10.1080/15216549700203871. [DOI] [PubMed] [Google Scholar]
- 22.Gouy M, Gautier C. Codon usage in bacteria: correlation with gene expressivity. Nucleic Acids Res. 1982;10:7055–7074. doi: 10.1093/nar/10.22.7055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grantham R, Gautier C, Gouy M, Mercier R, Pave A. Codon catalog usage and the genome hypothesis. Nucleic Acids Res. 1980;8:197–197. doi: 10.1093/nar/8.1.197-c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Green P, Ewing B, Miller W, Thomas PJ, Green ED, Program NCS. Transcription-associated mutational asymmetry in mammalian evolution. Nat Genet. 2003;33:514. doi: 10.1038/ng1103. [DOI] [PubMed] [Google Scholar]
- 25.Gu W, Zhou T, Ma J, Sun X, Lu Z. Analysis of synonymous codon usage in SARS Coronavirus and other viruses in the Nidovirales. Virus Res. 2004;101:155–161. doi: 10.1016/j.virusres.2004.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gun L, Haixian P, Yumiao R, Han T, Jingqi L, Liguang Z. Codon usage characteristics of PB2 gene in influenza A H7N9 virus from different host species. Infect Genet Evol. 2018;65:430–435. doi: 10.1016/j.meegid.2018.08.028. [DOI] [PubMed] [Google Scholar]
- 27.Gupta S, Ghosh T. Gene expressivity is the main factor in dictating the codon usage variation among the genes in Pseudomonas aeruginosa. Gene. 2001;273:63–70. doi: 10.1016/S0378-1119(01)00576-5. [DOI] [PubMed] [Google Scholar]
- 28.Hammer Ø, Harper D, Ryan P. PAST-palaeontological statistics, ver. 1.89. Palaeontol. electron. 2001;4:1–9. [Google Scholar]
- 29.Harrison RJ, Charlesworth B. Biased gene conversion affects patterns of codon usage and amino acid usage in the Saccharomyces sensu stricto group of yeasts. Mol Biol Evol. 2010;28:117–129. doi: 10.1093/molbev/msq191. [DOI] [PubMed] [Google Scholar]
- 30.Hassan H, Mohamed M, Youssef AW, Hassan ER. Effect of using organic acids to substitute antibiotic growth promoters on performance and intestinal microflora of broilers. Asian-Australas J Anim Sci. 2010;23:1348–1353. doi: 10.5713/ajas.2010.10085. [DOI] [Google Scholar]
- 31.Hopp TP, Woods KR. Prediction of protein antigenic determinants from amino acid sequences. Proc Natl Acad Sci. 1981;78:3824–3828. doi: 10.1073/pnas.78.6.3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu C, Chen J, Ye L, Chen R, Zhang L, Xue X. Codon usage bias in human cytomegalovirus and its biological implication. Gene. 2014;545:5–14. doi: 10.1016/j.gene.2014.05.018. [DOI] [PubMed] [Google Scholar]
- 33.Ikemura T. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes: a proposal for a synonymous codon choice that is optimal for the E. coli translational system. J Mol Biol. 1981;151:389–409. doi: 10.1016/0022-2836(81)90003-6. [DOI] [PubMed] [Google Scholar]
- 34.Jenkins GM, Holmes EC. The extent of codon usage bias in human RNA viruses and its evolutionary origin. Virus Res. 2003;92:1–7. doi: 10.1016/S0168-1702(02)00309-X. [DOI] [PubMed] [Google Scholar]
- 35.Karlin S, Mrázek J. What drives codon choices in human genes? J Mol Biol. 1996;262:459–472. doi: 10.1006/jmbi.1996.0528. [DOI] [PubMed] [Google Scholar]
- 36.Karlin S, Blaisdell BE, Schachtel GA. Contrasts in codon usage of latent versus productive genes of Epstein-Barr virus: data and hypotheses. J Virol. 1990;64:4264–4273. doi: 10.1128/jvi.64.9.4264-4273.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaufmann WK, Paules RS. DNA damage and cell cycle checkpoints. FASEB J. 1996;10:238–247. doi: 10.1096/fasebj.10.2.8641557. [DOI] [PubMed] [Google Scholar]
- 38.Komurov K, White MA, Ram PT. Use of data-biased random walks on graphs for the retrieval of context-specific networks from genomic data. PLoS Comput Biol. 2010;6:e1000889. doi: 10.1371/journal.pcbi.1000889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lesnik T, Solomovici J, Deana A, Ehrlich R, Reiss C. Ribosome traffic in E. coli and regulation of gene expression. J Theor Biol. 2000;202:175–185. doi: 10.1006/jtbi.1999.1047. [DOI] [PubMed] [Google Scholar]
- 40.Liu Q. Analysis of codon usage pattern in the radioresistant bacterium Deinococcus radiodurans. Biosystems. 2006;85:99–106. doi: 10.1016/j.biosystems.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 41.Liu X-s, Zhang Y-g, Fang Y-z, Wang Y-l. Patterns and influencing factor of synonymous codon usage in porcine circovirus. Viro J. 2012;9:68. doi: 10.1186/1743-422X-9-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lobry J, Gautier C. Hydrophobicity, expressivity and aromaticity are the major trends of amino-acid usage in 999 Escherichia coli chromosome-encoded genes. Nucleic Acids Res. 1994;22:3174–3180. doi: 10.1093/nar/22.15.3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Louten J. Essential human virology. London: Academic Press; 2016. [Google Scholar]
- 44.Martin A, Bertranpetit J, Oliver J, Medina J. Variation in G+ C-content and codon choice: differences among synonymous codon groups in vertebrate genes. Nucleic Acids Res. 1989;17:6181–6189. doi: 10.1093/nar/17.15.6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mauro VP, Chappell SA. A critical analysis of codon optimization in human therapeutics. Trends Mol Med. 2014;20:604–613. doi: 10.1016/j.molmed.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mirsafian H, Mat Ripen A, Singh A, Teo PH, Merican AF, Mohamad SB (2014) A comparative analysis of synonymous codon usage bias pattern in human albumin superfamily. Sci World J 2014 [DOI] [PMC free article] [PubMed]
- 47.Mueller S, Papamichail D, Coleman JR, Skiena S, Wimmer E. Reduction of the rate of poliovirus protein synthesis through large-scale codon deoptimization causes attenuation of viral virulence by lowering specific infectivity. J Virol. 2006;80:9687–9696. doi: 10.1128/JVI.00738-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Palidwor GA, Perkins TJ, Xia X. A general model of codon bias due to GC mutational bias. PLoS ONE. 2010;5:e13431. doi: 10.1371/journal.pone.0013431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pavlovic M, Chatterjee S, Kats A. Parvovirus b19 and auto antibodies reactive with ssDNA in lupus disease: bioinformatics analysis and hypothesis, republic of Yemen. MOJ Immunol. 2018;6:281–286. [Google Scholar]
- 50.Payne S. Viruses: from understanding to investigation. Cambridge: Academic Press; 2017. [Google Scholar]
- 51.Plotkin JB, Robins H, Levine AJ. Tissue-specific codon usage and the expression of human genes. Proc Natl Acad Sci. 2004;101:12588–12591. doi: 10.1073/pnas.0404957101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Popgeorgiev N, Temmam S, Raoult D, Desnues C. Describing the silent human virome with an emphasis on giant viruses. Intervirology. 2013;56:395–412. doi: 10.1159/000354561. [DOI] [PubMed] [Google Scholar]
- 53.Reis Md, Savva R, Wernisch L. Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res. 2004;32:5036–5044. doi: 10.1093/nar/gkh834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sewatanon J, Srichatrapimuk S, Auewarakul P. Compositional bias and size of genomes of human DNA viruses. Intervirology. 2007;50:123–132. doi: 10.1159/000098238. [DOI] [PubMed] [Google Scholar]
- 55.Shackelton LA, Parrish CR, Holmes EC. Evolutionary basis of codon usage and nucleotide composition bias in vertebrate DNA viruses. J Mol Evol. 2006;62:551–563. doi: 10.1007/s00239-005-0221-1. [DOI] [PubMed] [Google Scholar]
- 56.Sharp PM, Li W-H. An evolutionary perspective on synonymous codon usage in unicellular organisms. J Mol Evol. 1986;24:28–38. doi: 10.1007/BF02099948. [DOI] [PubMed] [Google Scholar]
- 57.Sharp PM, Li W-H. Codon usage in regulatory genes in Escherichia coli does not reflect selection for ‘rare’codons. Nucleic Acids Res. 1986;14:7737–7749. doi: 10.1093/nar/14.19.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sharp PM, Matassi G. Codon usage and genome evolution. Curr Opin Genet Dev. 1994;4:851–860. doi: 10.1016/0959-437X(94)90070-1. [DOI] [PubMed] [Google Scholar]
- 59.Sharp PM, Tuohy TM, Mosurski KR. Codon usage in yeast: cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986;14:5125–5143. doi: 10.1093/nar/14.13.5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sharp PM, Stenico M, Peden JF, Lloyd AT. Codon usage: mutational bias, translational selection, or both? London: Portland Press Limited; 1993. [DOI] [PubMed] [Google Scholar]
- 61.Shi S-L, Jiang Y-R, Liu Y-Q, Xia R-X, Qin L. Selective pressure dominates the synonymous codon usage in parvoviridae. Virus Genes. 2013;46:10–19. doi: 10.1007/s11262-012-0818-6. [DOI] [PubMed] [Google Scholar]
- 62.Shields DC, Sharp PM. Synonymous codon usage in Bacillus subtilis reflects both translational selection and mutational biases. Nucleic Acids Res. 1987;15:8023–8040. doi: 10.1093/nar/15.19.8023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Spandole S, Cimponeriu D, Berca LM, Mihăescu G. Human anelloviruses: an update of molecular, epidemiological and clinical aspects. Adv Virol. 2015;160:893–908. doi: 10.1007/s00705-015-2363-9. [DOI] [PubMed] [Google Scholar]
- 64.Sueoka N. Directional mutation pressure and neutral molecular evolution. Proc Natl Acad Sci. 1988;85:2653–2657. doi: 10.1073/pnas.85.8.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sueoka N. Intrastrand parity rules of DNA base composition and usage biases of synonymous codons. J Mol Evol. 1995;40:318–325. doi: 10.1007/BF00163236. [DOI] [PubMed] [Google Scholar]
- 66.Sueoka N. Two aspects of DNA base composition: G+ C content and translation-coupled deviation from intra-strand rule of A= T and G= C. J Mol Evol. 1999;49:49–62. doi: 10.1007/PL00006534. [DOI] [PubMed] [Google Scholar]
- 67.Tao P, Dai L, Luo M, Tang F, Tien P, Pan Z. Analysis of synonymous codon usage in classical swine fever virus. Virus Genes. 2009;38:104–112. doi: 10.1007/s11262-008-0296-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tennant P, Fermin G, Foster JE. Viruses: Molecular Biology, Host Interactions, and Applications to Biotechnology. Cambridge: Academic Press; 2018. [Google Scholar]
- 69.Tillier ER, Collins RA. The contributions of replication orientation, gene direction, and signal sequences to base-composition asymmetries in bacterial genomes. J Mol Evol. 2000;50:249–257. doi: 10.1007/s002399910029. [DOI] [PubMed] [Google Scholar]
- 70.Tsai C-T, Lin C-H, Chang C-Y. Analysis of codon usage bias and base compositional constraints in iridovirus genomes. Virus Res. 2007;126:196–206. doi: 10.1016/j.virusres.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 71.Uddin A, Chakraborty S. Codon usage trend in mitochondrial CYB gene. Gene. 2016;586:105–114. doi: 10.1016/j.gene.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 72.Uddin A, Chakraborty S. Codon usage pattern of genes involved in central nervous system. Mol Neurobiol. 2019;56:1737–1748. doi: 10.1007/s12035-018-1173-y. [DOI] [PubMed] [Google Scholar]
- 73.Uddin A, Paul N, Chakraborty S. The codon usage pattern of genes involved in ovarian cancer. Ann N Y Acad Sci. 2019;1440:67–78. doi: 10.1111/nyas.14019. [DOI] [PubMed] [Google Scholar]
- 74.van Hemert FJ, Berkhout B, Lukashov VV. Host-related nucleotide composition and codon usage as driving forces in the recent evolution of the Astroviridae. Virology. 2007;361:447–454. doi: 10.1016/j.virol.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wan X-F, Xu D, Kleinhofs A, Zhou J. Quantitative relationship between synonymous codon usage bias and GC composition across unicellular genomes. BMC Evol Biol. 2004;4:19. doi: 10.1186/1471-2148-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang M, Liu Y-s, Zhou J-h, Chen H-t, Ma L-n, Ding Y-z, Liu W-q, Gu Y-x, Zhang J. Analysis of codon usage in Newcastle disease virus. Virus Genes. 2011;42:245–253. doi: 10.1007/s11262-011-0574-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wong EH, Smith DK, Rabadan R, Peiris M, Poon LL. Codon usage bias and the evolution of influenza A viruses Codon Usage Biases of Influenza Virus. BMC Evol Biol. 2010;10:253. doi: 10.1186/1471-2148-10-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Woo PC, Wong BH, Huang Y, Lau SK, Yuen K-Y. Cytosine deamination and selection of CpG suppressed clones are the two major independent biological forces that shape codon usage bias in coronaviruses. Virology. 2007;369:431–442. doi: 10.1016/j.virol.2007.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wright F. The ‘effective number of codons’ used in a gene. Gene. 1990;87:23–29. doi: 10.1016/0378-1119(90)90491-9. [DOI] [PubMed] [Google Scholar]
- 80.Xu X-z, Liu Q-p, Fan L-j, Cui X-f, Zhou X-p. Analysis of synonymous codon usage and evolution of begomoviruses. J Zhejiang Univ Sci B. 2008;9:667–674. doi: 10.1631/jzus.B0820005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang WJ, Zhou J, Li ZF, Wang L, Gu X, Zhong Y. Comparative analysis of codon usage patterns among mitochondrion, chloroplast and nuclear genes in Triticum aestivum L. J Integr Plant Biol. 2007;49:246–254. doi: 10.1111/j.1744-7909.2007.00404.x. [DOI] [Google Scholar]
- 82.Zhang Z, Dai W, Wang Y, Lu C, Fan H. Analysis of synonymous codon usage patterns in torque teno sus virus 1 (TTSuV1) Adv Virol. 2013;158:145–154. doi: 10.1007/s00705-012-1480-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhao S, Zhang Q, Chen Z, Zhao Y, Zhong J. The factors shaping synonymous codon usage in the genome of Burkholderia mallei. J Genet Genom. 2007;34:362–372. doi: 10.1016/S1673-8527(07)60039-3. [DOI] [PubMed] [Google Scholar]
- 84.Zhao S, Zhang Q, Liu X, Wang X, Zhang H, Wu Y, Jiang F. Analysis of synonymous codon usage in 11 Human Bocavirus isolates. Biosystems. 2008;92:207–214. doi: 10.1016/j.biosystems.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhong J, Li Y, Zhao S, Liu S, Zhang Z. Mutation pressure shapes codon usage in the GC-Rich genome of foot-and-mouth disease virus. Virus Genes. 2007;35:767–776. doi: 10.1007/s11262-007-0159-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhou H, Wang H, Huang L, Naylor M, Clifford P. Heterogeneity in codon usages of sobemovirus genes. Adv Virol. 2005;150:1591–1605. doi: 10.1007/s00705-005-0510-4. [DOI] [PubMed] [Google Scholar]
- 87.Zhou T, Gu W, Ma J, Sun X, Lu Z. Analysis of synonymous codon usage in H5N1 virus and other influenza A viruses. Biosystems. 2005;81:77–86. doi: 10.1016/j.biosystems.2005.03.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.