

Abstract
The choice of repair pathways of DNA double-strand breaks (DSBs) is dependent upon the cell cycle phases. While homologous recombination repair (HRR) is active between the S and G2 phases, its involvement in mitotic DSB repair has not been examined in detail. In the present study, we developed a new reporter assay system to detect homology-directed repair (HDR), a major pathway used for HRR, in combination with an inducible DSB-generation system. As expected, the maximal HDR activity was observed in the late S phase, along with minimal activity in the G1 phase and at the G1/S boundary. Surprisingly, significant HDR activity was observed in M phase, and the repair efficiency was similar to that observed in late S phase. HDR was also confirmed in metaphase cells collected with continuous colcemid exposure. ChIP assays revealed the recruitment of RAD51 to the vicinity of DSBs in M phase. In addition, the ChIP assay for gamma-H2AX and phosphorylated DNA-PKcs indicated that a part of M-phase cells with DSBs could proceed into the next G1 phase. These results provide evidence showing that a portion of mitotic cell DSBs are undoubtedly repaired through action of the HDR repair pathway.
INTRODUCTION
A DNA double-strand break (DSB) is one of the most serious types of damage that can be experienced by cells and can often be induced by physiological mechanisms involved in DNA metabolism, or by DNA damaging agents such as ionizing radiation. To protect themselves from DSBs, higher eukaryote cells possess several DSB repair pathways, and among these pathways canonical non-homologous end joining (c-NHEJ) and homologous recombination repair (HRR) are considered to be the two major repair pathways [1, 2].
The radiation sensitivity of mammalian cells depends on the cell cycle position when damage occurs. S-Phase cells are the most resistant to damage, G1-cells are marginally resistant, cells at the G1/S boundary are slightly sensitive, and M-phase cells are the most sensitive to radiation [3, 4]. This difference in sensitivity to damage is thought to be due to the cell cycle position when damage occurs, because this determines the choice of which DSB repair pathway will be utilized. Which one of the two major DSB repair pathways will be utilized is determined by the expression of repair factors [5–8] or by the existence of repair templates during the cell cycle [9–12].
The c-NHEJ pathway can be functional throughout the cell cycle phases including G1, S and G2, whereas its involvement in DSB repair in M phase still remains to be determined [13–17]. Previously, inhibition of the c-NHEJ pathway in mitosis was demonstrated by several studies [16, 17]. In fact, Cdk1 as well as Plk1 inhibitory phosphorylation was shown to suppress c-NHEJ in mitotic cells [16]. There is also no recruitment of RNF8, RNF168 and 53BP1 to DSB sites in M phase [8, 13, 14, 16, 17]. However, Godinez et al. reported that some of the DSBs in M phase can be repaired by c-NHEJ [18], while the majority of DSBs are maintained until the next G1 phase, and are then repaired by the c-NHEJ pathway [14–16].
In contrast to the c-NHEJ pathway, the HRR pathway functions only in the S and G2 phases because the HRR pathway requires sister chromatids to be available for use as repair templates [9, 19]. The HRR pathway has never been examined to see if there was any involvement in mitotic DSB repair in somatic cells, although there have been reports indicating the occurrence of defective RAD51 filament formation from CDK1 activity [18, 20]. In germinal cells, of course, DSBs are generated by endonuclease activity between two homologous chromosomes, and recombination repair can re-join the DSBs to generate genetic diversity in offspring during meiosis [21–26], indicating that DSBs in chromosomes can be repaired through the HRR pathway.
Thus, accumulating evidence has suggested the existence of mitotic DSB repair, and this is further supported by recent results showing the existence of mitotic DNA repair synthesis [18, 20, 27, 28]. Sister chromatid non-disjunction, which results in ultrafine bridges (UFBs) in anaphase, should be repaired in mitosis, and resolution of UFBs requires DNA polymerase delta subunit 3 (POLD3)-dependent unscheduled DNA synthesis, so this could provide evidence for mitotic DSB repair in somatic cells [20]. In the present study, we introduced a newly developed reporter assay system to detect homology-directed repair (HDR), and assessed HDR in synchronized mitotic cells. The results clearly show, for the first time, that mitotic DSB repair occurs through the HDR pathway.
MATERIALS AND METHODS
Plasmids and constructs
A modified SCneo vector was prepared as previously described [29]. Two modified mutated estrogen receptor peptides (Mer) [30] were fused to both the N- and C-terminals of the I-Sce I endonuclease sequence in the pCMV-I-Sce I vector [31] to form the Mer-I-Sce I-Mer fusion protein expression vector.
Establishment of the SCneo-Mer-I-Sce I cell line
A modified SCneo vector [29] was transfected into MRC5/SV cells (obtained from the Riken Bank). A cell line containing one copy of the SCneo construct was selected using Southern blotting analysis as previously described [29]. The Mer-I-Sce I-Mer vector was then co-transfected along with a puromycin-resistant vector pApuro [32], and stable clones were selected by culture in medium containing puromycin (0.1 μg/ml). Expression of the Mer-I-Sce I-Mer fusion protein in the puromycin-selected clones was checked with western blotting using an anti-estrogen receptor antibody (Merck-Millipore, #06–935). The HDR frequency in the Mer-I-Sce I-Mer-expressing cells was tested, and a cell line showing the highest stable HDR frequency with a low background frequency was obtained after additional selection. Using this established cell clone, a site-specific DSB can be induced in the S2neo fragment in the SCneo reporter construct by adding 4-hydroxytamoxifen (OH-TAM) to the medium. heat shock protein 90 (HSP90), which binds to the Mer subunit under normal conditions, is replaced by OH-TAM, and the Mer-I-Sce I-Mer nuclease can then be transported into the nucleus. This nuclear translocation allows the Mer-I-Sce I-Mer nuclease access to its recognition sequence on the S2neo fragment (Fig. 1A). All the experiments described in this work were performed using one specific clone.
Fig. 1.
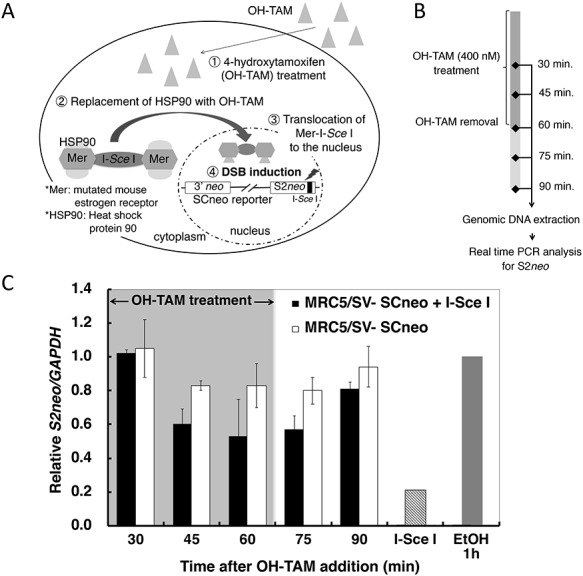
Kinetics of DSB induction by OH-TAM treatment and DSB rejoining after removal of OH-TAM. (A) Diagram showing the SCneo-Mer-I-Sce I cell system. A site-specific DSB is induced in the SCneo reporter gene by the Mer-I-Sce I-Mer fusion nuclease after the addition of OH-TAM. Access of the Mer-I-Sce I-Mer nuclease to its recognition sequence on S2neo occurs when HSP90 is replaced by OH-TAM, and the Mer-I-Sce I-Mer nuclease is then transported into the nucleus. (B) Schedule for DSB induction assay samples following OH-TAM treatment. (C) Asynchronous cells were treated with OH-TAM (400 nM) for 60 min. OH-TAM was then removed and the cells were further incubated. The amount of intact S2neo was monitored by real time PCR analysis. Values are expressed as the relative amount of S2neo compared to control (ethanol-treated) cells. Black vertical bars represent Mer-I-Sce I-Mer-expressing cells treated with OH-TAM and white vertical bars represent OH-TAM-treated cells without Mer-I-Sce I-Mer expression. The gray vertical bar represents EtOH-treated cells (0.1% ethanol). The gray hatched bar labeled I-Sce I represents I-Sce I-digested genomic DNA.
Analysis of DSB induction and rejoining with real-time PCR
Synchronized or asynchronous SCneo/Mer-I-Sce I cells were treated with OH-TAM (400 nM) for 1 h. At 30, 45, 60, 75 and 90 min after the addition of OH-TAM, the cells were lysed with Tris–HCl buffer (pH 7.5) supplemented with 2% sodium dodecylsulfate (SDS) and 0.1 mg/ml proteinase K (Wako), and genomic DNA was extracted. Real time PCR analysis was used to determine the ratio of intact S2neo fragments relative to total number of S2neo sequences, because DSB induction at the I-Sce I recognition site decreases the copy number of intact S2neo sequences. The assays were performed following the manufacturer’s instructions using TaqMan MGB master mix (Thermo-Fischer) and a Step-One Real Time PCR System (Thermo-Fischer). The sequence of the TaqMan probe and a primer set for S2neo were as follows: the TaqMan probe was 5′-AATATCATGGTGGAAAATG-3′; in the primer set, the forward primer was 5′-GCGAGGATCTCGTCGTGACT-3′ and the reverse primer was 5′-ACGGGTAGCCAACGCTATGT-3′. The TaqMan expression assay for glutaraldehyde 3-phosphate dehydrogenase (GAPDH) (Thermo-Fischer) was used as an internal control to quantitate genomic DNA. The relative value for delta-Ct for S2neo to delta-Ct for GAPDH (the relative value of intact S2neo) was calculated, and the relative number of DSBs induced was indicated by the decreasing value of intact S2neo relative to the value expressed in ethanol-treated cells. The DSB-positive control was the I-Sce I-digested genomic DNA extracted from untreated cells.
Analysis of HDR efficiency and repair products
Cells were cultured in serum-free medium containing OH-TAM (400 nM) for 1 h. The cells were washed twice with normal culture medium and were then cultured for an additional 24 h. The cells were then plated into medium containing G418 (Calbiochem, 400 μg/ml) to allow colony formation. The HDR (gene conversion) frequency was calculated from the number of G418-resistant colonies. Analysis of HDR products was performed as previously described [29, 33]. Briefly, the S2neo sequence in G418-resistant clones was amplified with PCR using a specific primer set (5′-CGTCGAGCAGTGTGGTTTTCA-3′ and 5′-AAAGCACGAGGAAGCGGTCAG-3′) and ExTaq DNA polymerase (TaKaRa). The amplified DNA was digested with NcoI or I-SceI in order to identify two types of HDR products: short-tract gene conversion (STGC) or long-tract gene conversion/sister chromatid exchange (LTGC/SCE).
Cell cycle synchronization
Cells were synchronized by using a sequential treatment consisting of serum starvation, exposure to colcemid and mitotic shake-off. First, cells were cultured under serum-starved conditions for 31 h. Under these conditions, cells are partially synchronized in the G1 phase without any increase in the number of apoptotic cells. The medium was then removed and replaced with normal medium (7% fetal bovine serum) and the cells were cultured for 6 h to allow them to progress through the cell cycle. These partially synchronized cells were further incubated for 8 h in colcemid (25 ng/ml)-containing medium. Finally, mitotic cells were detached from the culture dish with mild vibrations and pipetting. Detached cells were collected with centrifugation (500 x g for 5 min) and were washed twice with normal medium. The collected mitotic cells were then incubated. The cell cycle stages in the synchronized population were monitored with flow cytometric analysis using a FACS machine (BD-Bioscience) or a Tali image-based cytometer (Thermo-Fischer).
Chromatin immunoprecipitation (ChIP) assay
Recruitment of DSB repair factors to the I-Sce I-induced DSB site was analyzed with a ChIP assay. Briefly, cells were treated with 1% (vol/vol) formaldehyde in phosphate buffered saline (PBS) for 10 min, and the cross-link reaction was terminated by adding 1.5 M glycine (for a final concentration of 77.5 mM) and additional incubation for 5 min. The cell nuclei were isolated by incubation in IP buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.5 mM EDTA pH 8.0, 1.0% Nonidet P-40) for 15 min on ice, and were then sonicated with a Bioruptor UCD-300 (Cosmo-Bio) to get 600–800 bp chromatin fragments. The fragments in solution were suspended in ChIP dilution buffer (50 mM Tris–HCl pH 8.0, 167 mM NaCl, 1.1% Triton X-100, 0.11% sodium deoxycholate, 1/200 protease inhibitor cocktail, 1/200 phosphatase inhibitor) and incubated with the desired antibody for 2 h at 4°C. Protein G sepharose (GE Healthcare) was added and incubated overnight. The antibodies used were anti-histone H2A.X-pS139 (ab81299, Abcam, [1:200]), anti-NBS1 (GTX70224, GeneTex, [1:200]), anti-RAD51 (Bio Academia, [1:200]), anti-53BP1 (A300-272A, Bethyl, [1:200]) and anti-DNA dependent protein kinase catalytic subunit (DNA-PKcs) phospho-S2056 (ab18192, Abcam, [1:200]). After the incubation, the protein G sepharose was washed with a series of 1 × RIPA buffer (50 mM Tris–HCl pH 8.0, 150 mM NaCl, 1 mM EDTA pH 8.0, 1% Triton X-100, 0.1% SDS, 0.1% sodium deoxycholate), 1 × RIPA buffer/500 mM NaCl (50 mM Tris–HCl pH 8.0, 500 mM NaCl, 1 mM EDTA pH 8.0, 1% Triton X-100, 0.1% SDS, 0.1% sodium deoxycholate), LiCl wash buffer (10 mM Tris–HCl pH 8.0, 0.25 M LiCl, 1 mM EDTA pH 8.0, 0.5% NP-40, 0.5% sodium deoxycholate) and 1 × TE buffer (10 mM Tris–HCl pH 8.0, 1 mM EDTA). To elute the chromatin fragments, the washed Protein G sepharose was incubated overnight with 200 μL of ChIP direct elution buffer (10 mM Tris–HCl pH 8.0, 300 mM NaCl, 5 mM EDTA pH 8.0, 0.5% SDS) at 65°C. Eluted chromatin fragments were incubated with 0.1 mg/ml Proteinase K for 1 h at 55°C and the genomic DNA fragments were then ethanol precipitated. Real time PCR analysis was performed as described. The PCR primers and TaqMan probes used were: S2neo#341 (594 nt from the DSB), 5′-CGACCCTGCAGCCAATATG, 5′-AGAACCTGCGTGCAATCCA, and the probe was 5′-ATCGGCCATTGAAC; S2neo#729 (206 nt from the DSB), 5′-CGGCTGCATACGCTTGATC, 5′-GATGCGATGTTTCGCTTGGT, and the probe was 5′-ACCTGCCCATTCGA.
Statistical analysis
All the data were obtained from at least three independent experiments. Statistical analysis was performed using the Student’s t-test. For HDR assays, pooled data obtained from at least four independent experiments were used for statistical analysis.
RESULTS
We established a cell line that carries one copy of the SCneo reporter gene and expresses the Mer-I-Sce I-Mer fusion nuclease (Fig. 1A). In this SCneo-based system, a site-specific DSB can be induced at an I-Sce I recognition site on the HDR reporter gene by treating the cells with OH-TAM. If the DSB was repaired with a HDR process, an active neo gene is reconstructed and the cells then become neomycin (G418)-resistant. Therefore, one can monitor the HDR frequency by examining the frequency of neo+ colonies after OH-TAM treatment. Using this cell line, we monitored DSB induction after OH-TAM (400 nM) exposure and DSB rejoining after the removal of OH-TAM (Fig. 1B). As shown in Fig. 1C, the relative number of DSBs in the S2neo sequence was maximal at 45–60 min after the addition of OH-TAM. Although the number of induced DSBs was stable for 15 min (75 min after addition of OH-TAM), >60% of the induced DSBs were rejoined within 30 min after the removal of OH-TAM (at 90 min after addition of OH-TAM). From these results we chose to use an OH-TAM treatment period of 1 h.
Figure 2A shows flow cytometric analysis of the synchronized cell population. More than 80% of the cells were synchronized at the G2/M stage, and microscopic analysis indicated that those cells were in the mitotic phase (Fig. 2B). Cells were in the G1 phase from 3 to 12 h, in S phase from 12 to 16 h and in G2 phase at 20 h after release from the colcemid block, respectively. Using a synchronized population, we induced DSBs with a 1 h OH-TAM treatment, and analyzed the frequency of HDR events. The DSB-induction/rejoining kinetics of the synchronized population were also monitored at each cell cycle stage. In a similar manner, DSB-induction/rejoining kinetics were observed in an asynchronous population and were observed at all stages of the cell cycle (Supplementary Fig. 1, see online supplementary material). Therefore, DSB induction by the Mer-I-Sce I-Mer fusion nuclease is functional throughout the cell cycle. It should be noted that the M-phase-synchronized cells treated with OH-TAM for 1 h show cell cycle progress into the subsequent G1 phase, whereas they stayed in M phase if colcemid was continuously present (Fig. 2C).
Fig. 2.
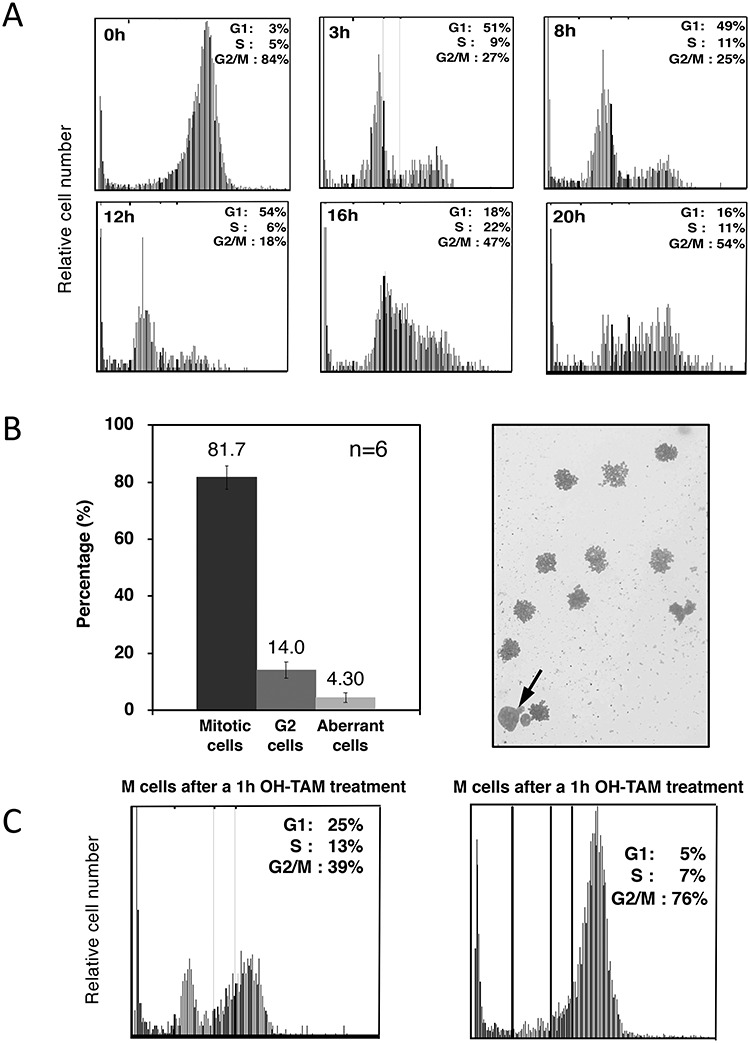
Cell cycle position at HDR analysis where cells were treated with OH-TAM. (A) The cell cycle distribution was measured with flow cytometry (image-based cytometer) at intervals after release from a colcemid block followed by a mitotic-shake off. G1 phase is from 3 to 12 h; S phase from 12 to 16 h; and G2 phase at 20 h. (B) Microscopic analysis of cells collected with a mitotic shake-off. The percentages of mitotic, interphase (G2) and aberrant cells are shown. A representative photograph is shown on the right side. An arrow indicates a G2 (interphase) cell while the others are mitotic cells. The number of independent experiments performed to obtain the values is shown (n = 6). (C) Cell cycle analysis for cells with or without continuous colcemid treatment during a 1 h OH-TAM treatment.
Cell cycle effect on HDR
Figure 3 shows the HDR frequency in cells in which DSBs were induced at each cell cycle stage. There were no differences between the DSB induction-rejoining kinetics throughout the cell cycle (see Supplementary Fig. 1). A maximal HDR frequency was observed at mid-late S phase (16 h) as expected, and it was minimal in G1 and at the G1/S boundary (3–12 h). Surprisingly, a significant level of HDR activity was observed in cells in which a DSB was induced in the mitotic phase. This significant level of HDR in the mitotic phase could not have resulted from higher numbers of induced DSBs by the OH-TAM treatment because there was no difference in DSB induction kinetics throughout the cell cycle (Fig. 4A and Supplementary Fig. 1).
Fig. 3.
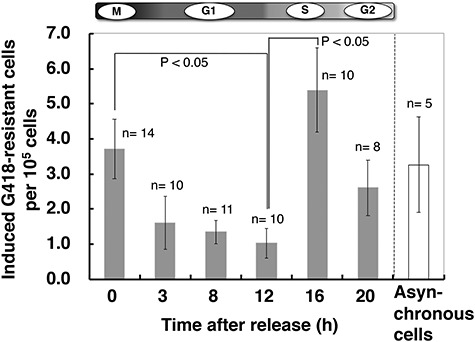
HDR frequency induced by the DSBs generated at each cell cycle stage. Synchronized cells were treated with OH-TAM (400 nM) for 60 min at the desired cell cycle stages. The HDR frequency was calculated as the number of G418-resistant clones per 105 OH-TAM-treated cells. The white bar at the right indicates results for asynchronous cells. The number n indicates the number of independent experiments performed to obtain each of the values in the graph.
Fig. 4.
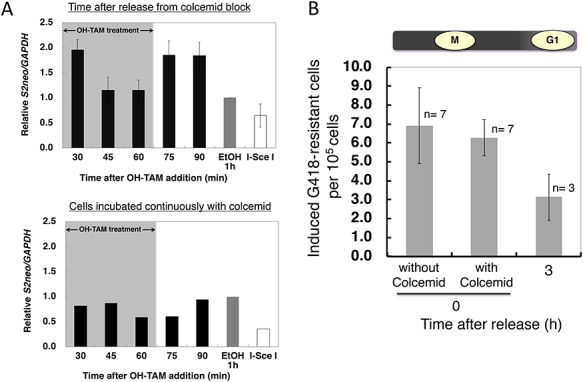
(A) Rejoining kinetics and (B) HDR frequency for Mer-I-Sce I Mer-induced DSBs in M-phase cells with or without a prolonged exposure to colcemid. (A) The relative S2neo/GAPDH quantity indicates the ratio of DSB induction-rejoining events (black bars) after release from colcemid block at zero time (upper panel). The gray bar indicates EtOH-treated controls, and the white bar indicates I-Sce I-digested genomic DNA. The black bars in the lower panel show the relative S2neo/GAPDH quantiity as a function of time after OH-TAM addition in cells continuously incubated with colcemid. The gray bar indicates EtOH treated controls and the white bar indicates I-Sce I digested genomic DNA. (B) HDR frequency in M-phase cells (immediately after release from colcemid) with or without prolonged colcemid exposure during a 1-h OH-TAM treatment. HDR frequency for G1 cells at 3 h after release from colcemid is also shown. The number n over each bar represents the number of independent experiments performed for that time point.
To further confirm the existence of DSB rejoining in M phase, we examined DSB induction and rejoining kinetics with or without continuous colcemid treatment. When cells were allowed to undergo DSB repair in the presence of colcemid, the efficiency of DSB rejoining was significantly decreased, although a significant number of DSBs were rejoined (Fig. 4A). In addition, a similar HDR efficiency was seen in the M phase populations, even with continuous colcemid treatment (Fig. 4B). These observations indicate that at least 20% of DSBs can be repaired during M phase, and some of the DSBs might be left unrepaired and transported into the next cell cycle stage.
Table 1 shows a summary of the HDR events. Non-crossover events (STGC) were dominant throughout the cell cycle, but a slight increase in crossover events (LTGC/SCE) was observed in the S and M phases.
Table 1.
Analysis of recombination products. STGC and LTGC/SCE repair products were scored after OH-TAM treatment and G418 selection. The total number of G418-resistant clones analyzed at each time point after cell cycle synchronization is shown. The total number of STGC and LTGC events at each time point is 100%. The great majority of events were STGC
| Time point (h) | Percentage of repair products | |||
|---|---|---|---|---|
| STGC | LTGC | |||
| No. of clones | % | No. of clones | % | |
| 0 | 25 | 86.2 | 4 | 13.8 |
| 3 | 42 | 95.5 | 2 | 4.6 |
| 8 | 31 | 96.9 | 1 | 3.1 |
| 12 | 27 | 81.8 | 6 | 18.2 |
| 16 | 38 | 90.5 | 4 | 9.5 |
| 20 | 36 | 94.7 | 2 | 5.3 |
Cells in M phase can repair DSBs via HDR
To obtain further evidence that HDR can occur in M phase, we performed a ChIP analysis on genomic DNA adjacent to the DSB. Two positions in the vicinity of the I-Sce I-cutting site (206 and 594 bases from the DSB end) were monitored for the presence of gamma-H2AX, RAD51, NBS1, 53BP1 and phospho-(S2056) DNA-PKcs (Fig. 5A). The relative values were calculated by comparing results seen in ethanol-treated control cells. The amount of gamma-H2AX and RAD51 accumulated at the ‘distal site’ (594 nt) was observed at 1 h after the removal of OH-TAM. A slight accumulation of NBS1 was also seen at the same time. At the ‘proximal DSB site’ (206 nt) from the DSB, a large amount of gamma-H2AX and RAD51 was seen at 1 h after the OH-TAM treatment, but these disappeared within 1 h (Fig. 5B). A significant accumulation of gamma-H2AX at the proximal DSB site was again seen at 2 h following the OH-TAM treatment.
Fig. 5.
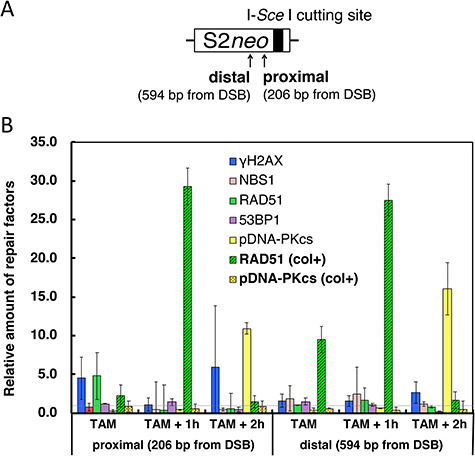
ChIP analysis of HDR factors in the vicinity of a DSB in the M phase. (A) Primer positions on the S2neo reporter. The I-Sce I site is indicated by the bold vertical line. (B) Relative quantity of HR factors (NBS1, RAD51) or c-NHEJ factors (53BP1, phospho-S2056 DNA-PKcs) detected at the two primer sites in the vicinity of the I-Sce I-induced DSB. The quantity of HDR (or c-NHEJ) factors was monitored just after a 1 h treatment with OH-TAM (TAM), 1 h after OH-TAM removal (TAM + 1 h) and 2 h after OH-TAM removal (TAM + 2 h). The relative amounts for each time point and position are expressed with the ethanol-treated control value being 1.0. The values were calculated from the values normalized against the amount of histone H3. Results for RAD51 and phospho-(S2056) DNA-PKcs for cells continuously exposed to colcemid during the OH-TAM treatment are also shown (col+).
The c-NHEJ factor, phospho-(S2056) DNA-PKcs had clearly accumulated in the vicinity of the DSB at both the proximal and distal sites at 2 h after OH-TAM removal. However, this accumulation was not seen when the cells were continuously treated with colcemid. This suggests that DNA-PK was able to accumulate when the cells went into G1 phase along with the ’second’ increase of gamma-H2AX at the proximal site. An extensive accumulation of RAD51 was detected at 1 h following OH-TAM removal at both the proximal and distal sites when the cells were continuously treated with colcemid during DSB induction and its repair period.
DISCUSSION
In the present study, we analyzed the cell cycle dependence of HDR at the site of a specifically induced DSB. A maximum HDR frequency was observed at mid- to late S phase (16 h after release from a colcemid block). Surprisingly, a significant elevation of the HDR frequency was also seen in the M phase (Fig. 3). This increase in HDR in M phase should not be due to the loss of the nuclear membrane because there are no differences in the DSB-induction efficiency throughout the cell cycle (Fig. 4A and Supplementary Fig. 1). To further confirm that DSBs were actually repaired during M phase, we induced DSBs in the continuous presence of colcemid. A significant HDR frequency was detected, although it was lower than that seen when DSBs were induced immediately after release from a colcemid block (Fig. 4B). In addition, the ChIP assay suggested there was recruitment of RAD51, a primary recombination factor involved in HDR, in the vicinity of the DSB, especially when the cells were continuously treated with colcemid (Fig. 5B). The c-NHEJ factor, phosphorylated DNA-PKcs, apparently accumulated in the vicinity of DSBs 2 h following OH-TAM removal, whereas it did not accumulate when the cells were continuously treated with colcemid. These observations strongly suggest that DSBs in M phase can be repaired before cell division through the activity of the HDR pathway.
Several reports have suggested the existence of mitotic-stage DSB repair, and this is further supported by recent results showing mitotic DNA repair synthesis [18, 20, 27, 28]. Sister chromatid non-disjunction, which results in UFBs in anaphase, should be repaired during mitosis, and resolution of UFBs requires POLD3-dependent unscheduled DNA synthesis, so this could provide additional evidence for mitotic DSB repair in somatic cells [20]. The present results provide the first direct evidence showing the existence of DSB repair through an HDR pathway in early to mid-phase mitotic phase cells, although a portion of the DSBs present in M-phase could progress and be present in the subsequent G1 phase.
Chromatin in M phase is highly condensed and it appears to be difficult to repair DSBs under these conditions. However, the fact that genomic DNA can be exchanged during meiosis [21, 22] and that somatic cells exhibit sister chromatid exchanges when they complete the cell cycle [34] supports our observation, because those phenomena indicate that DSB induction and repair can occur in the presence of condensed chromatin.
Analysis of HDR products indicated that there was a dominance of short-tract HDR activity throughout the cell cycle (Table 1). This is reasonable for the maintenance of genomic stability because exchange events cause a change in the genome’s structure. A slight increase in LTGC/SCE-type events was seen when a DSB was induced at late S or in M phase. This may be due to the elevated probability of inter-chromatid recombination events occurring.
A decreased HDR frequency was observed in cells maintained in M phase throughout the DSB repair period. In addition, the ChIP assay for phosphorylated-H2AX or phosphorylated DNA-PKcs showed a significant accumulation of these at both the proximal and distal DSB sites 2 h after OH-TAM treatment (Fig. 5B). Because the cells were able to enter the subsequent G1 phase even during OH-TAM treatment (Fig. 2C), these observations suggest that some of the DSBs present in M-phase cells might be preserved and propagated into the subsequent G1 phase. This is consistent with a previous report by Terasawa et al. [16].
DSBs must be rejoined as soon as possible, therefore cells repair DSBs using pathways that are available at the stage of the cell cycle when such breaks occur. When DSBs are present in M phase when chromatin is highly condensed, only the HDR pathway is available, but it may not be efficient. As a result, a portion of the DSBs induced in M phase may persist into the subsequent G1 phase and finally be rejoined by NHEJ. This is the first report showing the existence of DSB repair through homologous recombination during the mitotic stage.
We reported previously that cells irradiated during the G2/M phase, when the majority of the cells are in M phase, are hypersensitive to mutation induction from high LET radiation, and that irradiation during the G2/M phase results in large deletion-type mutations [35, 36]. This observation suggests the existence of DSB repair during the M phase. Combining the previous observations of mutation induction during M phase and the present results supports the existence of appropriate or inappropriate recombination processes which utilize any homologous sequences, whether they are inter-chromatid or intra-chromatid sequences.
Supplementary Material
ACKNOWLEDGMENTS
The authors are grateful to Dr Leon N. Kapp for editing the manuscript, and Drs Akira Tachibana, Kenshi Komatsu and Asako J. Nakamura for valuable discussions. They are also grateful to Keita Satoh, Yuka Ohashi, Yui Tabei and Naoki Hasegawa, for their technical assistance in obtaining portions of the experimental data. The pCMV-I-Sce I vector is a gift from Dr Maria Jasin at Sloan Kettering Cancer Center. A part of this work was performed as a collaborative program with the Radiation Biology Center, Kyoto University, and was supported by the Program for Network-type Joint Usage/Research Center for Radiation Disaster Medical Science.
Contributor Information
Yuki Sakamoto, Department of Biological Sciences, Ibaraki University, Bunkyo 2-1-1, Mito, Ibaraki 310-8512, Japan.
Tetsuya Kokuta, Department of Biological Sciences, Ibaraki University, Bunkyo 2-1-1, Mito, Ibaraki 310-8512, Japan.
Ai Teshigahara, Department of Biological Sciences, Ibaraki University, Bunkyo 2-1-1, Mito, Ibaraki 310-8512, Japan.
Kenta Iijima, Department of Biological Sciences, Ibaraki University, Bunkyo 2-1-1, Mito, Ibaraki 310-8512, Japan; Department of Cancer Biology, Nagoya University Graduate School of Medicine, 65 Tsurumai-cho, Showa-ku, Nagoya 466-8550, Japan.
Hiroyuki Kitao, Department of Molecular Cancer Biology, Graduate School of Pharmaceutical Sciences, Kyushu University, Maidashi 3-1-1, Higashi-ku, Fukuoka 812-8582, Japan.
Minoru Takata, Radiation Biology Center, Kyoto University, Yoshida-Konoe Cho, Sakyo-ku, Kyoto 606-8501, Japan.
Hiroshi Tauchi, Department of Biological Sciences, Ibaraki University, Bunkyo 2-1-1, Mito, Ibaraki 310-8512, Japan.
FUNDING
This work was supported in part by a Grant-in-Aid for Scientific Research (KAKENHI) from the Japan Society for the Promotion of Science (HT, #22310033, #24310038, and #19 K12318).
CONFLICT OF INTEREST
None declared.
References
- 1. Ciccia A, Elledge SJ. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010;40:179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Scully R, Panday A, Elango R et al. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019;20:698–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Terashima T, Tolmach LJ. X-ray sensitivity and DNA synthesis in synchronous populations of HeLa cells. Science 1963;140:490–2. [DOI] [PubMed] [Google Scholar]
- 4. Sinclair WK. Cyclic X-ray responses in mammalian cells in vitro. Radiat. Res. 1968;33:620–43. [PubMed] [Google Scholar]
- 5. Orthwein A, Noordermeer SM, Wilson MD et al. A mechanism for the suppression of homologous recombination in G1 cells. Nature 2015;528:422–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yamamoto A, Taki T, Yagi H et al. Cell cycle-dependent expression of the mouse Rad51 gene in proliferating cells. Mol. Gen. Genet. 1996;251:1–12. [DOI] [PubMed] [Google Scholar]
- 7. Vaughn JP, Cirisano FD, Huper G et al. Cell cycle control of BRCA2. Cancer Res. 1996;56:4590–4. [PubMed] [Google Scholar]
- 8. Giunta S, Belotserkovskaya R, Jackson SP. DNA damage signaling in response to double-strand breaks during mitosis. J. Cell Biol. 2010;190:197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ceccaldi R, Rondinelli B, D'Andrea AD. Repair pathway choices and consequences at the double-strand break. Trends cell Biol. 2016;26:52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shibata A, Conrad S, Birraux J et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J. 2011;30:1079–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shibata A, Moiani D, Arvai AS et al. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol. Cell 2014;53:7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Biehs R, Steinlage M, Barton OJ et al. DNA double-strand break resection occurs during non-homologous end joining in G1 but is distinct from resection during homologous recombination. Mol. Cell 2017;65:671–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Orthwein A, Fradet-Turcotte A, Noordermeer SM et al. Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science 2014;344:189–93. [DOI] [PubMed] [Google Scholar]
- 14. Lee DH, Acharya SS, Kwon M et al. Dephosphorylation enables the recruitment of 53BP1 to double-strand DNA breaks. Mol. Cell 2014;54:512–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leimbacher PA, Jones SE, Shorrocks AK et al. MDC1 interacts with TOPBP1 to maintain chromosomal stability during mitosis. Mol. Cell 2019;74:571–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Terasawa M, Shinohara A, Shinohara M. Canonical non-homologous end joining in mitosis induces genome instability and is suppressed by M-phase-specific phosphorylation of XRCC4. PLoS Genet. 2014;10:e1004563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Benada J, Burdová K, Lidak T et al. Polo-like kinase 1 inhibits DNA damage response during mitosis. Cell Cycle 2015;14:219–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Godinez VG, Kabbara S, Sherman A et al. DNA damage induced during mitosis undergoes DNA repair synthesis. PLoS One 2020;15:e0227849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Karanam K, Kafri R, Loewer A et al. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol. Cell 2012;47:320–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Minocherhomji S, Ying S, Bjerregaard VA et al. Replication stress activates DNA repair synthesis in mitosis. Nature 2015;528:286–90. [DOI] [PubMed] [Google Scholar]
- 21. Hunter N. Meiotic recombination: The essence of heredity. Cold Spring Harb. Perspect. Biol. 2015;7:a016618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gray S, Cohen PE. Control of meiotic crossovers: From double-strand break formation to designation. Annu. Rev. Genet. 2016;50:175–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lam I, Keeney S. Mechanism and regulation of meiotic recombination initiation. Cold Spring Harb. Perspect. Biol. 2014;7:a016634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim KP, Weiner BM, Zhang L et al. Sister cohesion and structural axis components mediate homolog bias of meiotic recombination. Cell 2010;143:924–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Keeney S, Lange J, Mohibullah N. Self-organization of meiotic recombination initiation: General principles and molecular pathways. Annu. Rev. Genet. 2014;48:187–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zickler D, Kleckner N. A few of our favorite things: Pairing, the bouquet, crossover interference and evolution of meiosis. Semin. Cell Dev. Biol. 2016;54:135–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ovejero S, Bueno A, Sacristán MP. Working on genomic stability: From the S-phase to mitosis. Genes 2020;11:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thompson R, Gatenby R, Sidi S. How cells handle DNA breaks during mitotic detection, signaling, repair, and fate choice. Cells 2019;8:1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tauchi H, Kobayashi J, Morishima K et al. Nbs1 is essential for DNA repair by homologous recombination in higher vertebrate cells. Nature 2002;420:93–8. [DOI] [PubMed] [Google Scholar]
- 30. Zhang Y, Riesterer C, Ayrall AM et al. Inducible site-directed recombination in mouse embryonic stem cells. Nucleic Acids Res. 1996;24:543–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Johnson RD, Liu N, Jasin M. Mammalian XRCC2 promotes the repair of DNA double-strand breaks by homologous recombination. Nature 1999;401:397–9. [DOI] [PubMed] [Google Scholar]
- 32. Takata M, Sabe H, Hata A et al. Tyrosine kinases Lyn and Syk regulate B cell receptor-coupled Ca2+ mobilization through distinct pathways. EMBO J. 1994;13:1341–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sakamoto S, Iijima K, Mochizuki D et al. Homologous recombination repair is regulated by domains at the N- and C-terminus of NBS1 and is dissociated with ATM functions. Oncogene 2007;26:6002–9. [DOI] [PubMed] [Google Scholar]
- 34. Conrad S, Künzel J, Löbrich M. Sister chromatid exchanges occur in G2-irradiated cells. Cell Cycle 2011;10:222–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tauchi H, Nakamura N, Sawada S. Cell cycle dependence for the induction of 6-thioguanine-resistant mutations: G2/M stage is distinctively sensitive to 252Cf neutrons but not to 60Co gamma-rays. Int. J. Radiat. Biol. 1993;63:475–81. [DOI] [PubMed] [Google Scholar]
- 36. Tauchi H, Nakamura N, Komatsu K et al. Accumulation of cells at G2/M stage by low dose-rate irradiation renders the cell population more susceptible to the subsequent induction of 6-thioguanine-resistant mutations by 252Cf fission neutrons. J. Radiat. Res. 1996;37:49–57. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.