

Abstract
The contribution of type II alveolar epithelial stem cells (AEC II) to radiation-induced lung fibrosis (RILF) is largely unknown. Cell differentiation phenotypes are determined by the balance between Lin28 and lethal-7 microRNA (let-7 miRNA). Lin28 is activated by β-catenin. The aim of this study was to track AEC II phenotypes at different phases of injury following thoracic irradiation and examine the expression of β-catenin, Lin28 and let-7 to identify their role in AEC II differentiation. Results showed that coexpression of prosurfactant protein C (proSP-C, an AEC II biomarker) and HOPX (homeobox only protein X, an AEC I biomarker) or vimentin (a differentiation marker) was detected in AEC II post-irradiation. The protein expression levels of HOPX and proSP-C were significantly downregulated, but vimentin was significantly upregulated following irradiation. The expression of E-cadherin, which prevents β-catenin from translocating to the nucleus, was downregulated, and the expression of β-catenin and Lin28 was upregulated after irradiation (P < 0.05 to P < 0.001). Four let-7 miRNA members (a, b, c and d) were upregulated in irradiated lungs (P < 0.05 to P < 0.001), but let-7d was significantly downregulated at 5 and 6 months (P < 0.001). The ratios of Lin28 to four let-7 members were low during the early phase of injury and were slightly higher after 2 months. Intriguingly, the Lin28/let-7d ratio was strikingly increased after 4 months. We concluded that β-catenin contributed to RILF by promoting Lin28 expression, which increased the number of AEC II and the transcription of profibrotic molecules. In this study, the downregulation of let-7d miRNA by Lin28 resulted in the inability of AEC II to differentiate into type I alveolar epithelial cells (AEC I).
Keywords: type II alveolar stem cells, dynamic differentiation phenotypes, radiation-induced lung fibrosis, transdifferentiation, β-catenin/Lin28/let-7 network
INTRODUCTION
Thoracic radiotherapy is an important therapeutic modality for treating lung cancer, breast cancer and various lymphomas. Pulmonary fibrosis is one of the major radiation-induced complications following thoracic irradiation, and its symptoms range from mild dyspnea to chronic pulmonary insufficiency [1]. The main histopathological features of radiation-induced lung fibrosis (RILF) are the loss of alveolar epithelium and deposition of excess collagen and extracellular matrix (ECM) in lung tissues [2, 3]. All the current therapies for this life-threatening complication are palliative at best because the mechanisms of RILF are largely unknown.
Type II alveolar epithelial stem cells (AEC II), which are intrinsic to the lungs, are quiescent cells, and this characteristic confers some degree of radioresistance. AEC II produce surfactants and are responsible for alveolar injury repair [4]. During the normal repair process, quiescent AEC II are activated and acquire the potential to proliferate and differentiate into type I alveolar epithelial cells (AEC I). Recently, a primitive and putative adult stem cell (ASC) with both epithelial and mesenchymal differentiation potential was identified in lung tissues from an experimental murine model [5]. This discovery led us to hypothesize that resident lung AEC II may be induced to undergo transdifferentiation into mesenchymal lineage cells (myofibroblasts) after thoracic irradiation.
Human miRNA profiles showed that the lethal-7 microRNA (let-7 miRNA) family is significantly downregulated in lung epithelial cells after irradiation [6, 7], which suggests that let-7 miRNA is vulnerable to radiation. Transforming growth factor-β1 (TGF-β1) has long been considered a typical profibrotic cytokine that participates in the pathogenesis of pulmonary fibrosis [8]. Recent studies confirmed that the downregulation of let-7 miRNA by TGF-β1 induced AEC II to express mesenchymal markers in mice, leading to thicker alveolar septa and increased collagen, subsequently leading to fibrosis [9].
It is well known that Lin28 can inhibit let-7 expression and that let-7 can repress Lin28 expression. Therefore, Lin28 and let-7 control cell differentiation via a mechanism that involves their mutual negative regulation [10]. Lin28 is activated by the Wnt/β-catenin pathway [11, 12]. Lin28, which is a stem cell marker, is vital for the initiation or maintenance of stem cell phenotypes [13]. Thus, β-catenin, Lin28 and let-7 may compose a network that regulates cell differentiation. To date, no previous studies have investigated the role of crosstalk among β-catenin, Lin28 and let-7 in determining AEC II differentiation states. In this study, we tracked AEC II phenotypes at different phases of injury and explored the correlation between the differentiation phenotypes of AEC II and the expression levels of β-catenin, Lin28 and four let-7 members following irradiation, in order to illustrate the effect of the β-catenin/Lin28/let-7 network on the differentiation fate of AEC II.
MATERIALS AND METHODS
Animals and treatments
The mouse studies were reviewed and approved by the Institutional Review Board and performed in accordance with the guidelines of the Institute of Laboratory Animal Resources, National Research Council, China (approved No. SHSY-IEC-KY-4.0/17–135/01). Female C57BL/6 J mice, 6–7 weeks old, (Vital River Laboratory Animal Technology Co. Ltd., Beijing, China) were maintained under standard vivarium conditions and were allowed to acclimate for 1 week prior to thoracic irradiation. Then, the mice were randomly divided into two groups: the control group (n = 40) and the irradiated group (n = 50). The irradiated mice received thoracic irradiation with a single dose of 20 Gy (average dose rate of 6 Gy/min and energy of 6 MV) to induce lung injury using an X-ray irradiator (Elekta Synergy). The mice in the irradiated group were anesthetized by intraperitoneal injection of 50 mg/kg 1% sodium pentobarbital, and then, the mice were positioned side by side on a plexiglass pedestal on their backs with their bodies fully stretched prior to thoracic irradiation. The irradiated field size (2 × 3 cm per mouse) was set to provide adequate coverage of the whole lung. A lead board (12-mm thick) with a 2 x 20 cm long strip window was used to shield the heads and abdomens of 7 mice during each irradiation. After irradiation, the mice were then divided into 8 subgroups, and they were euthanized with 100% CO2 at various time points ranging from 24 h to 6 months post-irradiation. The mice in the corresponding sham-irradiated control subgroups (n = 5/time point) were sacrificed at the same time points as those in the irradiated subgroups. The left lungs were fixed in 10% neutral formalin for morphological analyses, and the right lungs were first placed in liquid nitrogen and then stored at −80 °C for miRNA or protein analysis.
Histopathological analysis
Lung sections (5 μm) were stained either with hematoxylin/eosin (H&E) to assess histological changes or with Picro-Sirius Red to determine the sites and amounts of collagen deposition. All the analyses were performed on labeled slides by a blinded pathologist.
Immunopathological analysis
Lung sections (3 μm) mounted on slides were deparaffinized and hydrated in xylene, 95% ethanol, 80% ethanol, 75% ethanol and 1× phosphate-buffered saline (PBS at pH 7.4). Following antigen retrieval with citrate buffer, the tissue sections were preincubated with 3% H2O2 for 20 min and 3% bovine serum albumin (BSA) for 30 min at room temperature to prevent non-specific staining. Then, the slides were incubated with an anti-E-cadherin antibody (1:200, 3195, Cell Signaling Technology, Danvers, MA, USA), anti-Lin28 antibody (1:300, ab63740, Abcam) or anti-β-catenin antibody (1:300, ab32572, Abcam) at 4°C overnight. The control slides were incubated with the appropriate IgG instead of the target antibodies. The next day, the slides were washed in 1× PBS and incubated with Cy™3-conjugated donkey anti-rabbit IgG (1:500, 711–165-152, Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA) for 2 h at 37°C prior to counterstaining the cell nucleus with 4′,6-diamidino-2-phenylindole (DAPI, 1:100, 10236276001, Roche, Basel, Switzerland) for 1 min. The slides were mounted using Vectashield mounting medium (P0128M, Beyotime Biotechnology, Shanghai, China). Images were captured using a fluorescence microscope (Leica, Model DM2500, German Leica Microsystems) using a fluorescence image processing system (Image-Pro Plus 6 software, Media Cybernetics, USA).
To track the AEC II phenotypes at different phases of injury in the lungs post-irradiation, double-color immunofluorescence analysis was performed to detect the coexpression of prosurfactant protein C (proSP-C, a unique AEC II marker) with phenotypic markers of different cell types in the lung tissues. Epithelial cellular adhesion molecule (EpCAM), homeobox only protein X homeobox only protein X (HOPX) and vimentin were used in this study to represent markers of epithelial stem cells, alveolar epithelial cells and mesenchymal cells, respectively. Briefly, after deparaffinization, antigen retrieval, and incubation with 3% H2O2 and 3% BSA as described above, the sections were incubated with anti-proSP-C antibodies (1:300, ab40897, Abcam) at 4°C overnight, followed by incubation with Fluor488 (red)-conjugated anti-rabbit IgG (H + L) (1:300, 711–516-152, Jackson ImmunoResearch Laboratories, Inc., West Grove, PA, USA). After proSP-C staining was performed, the slides were incubated with an anti-EpCAM antibody (1:100, ab71916, Abcam), anti-HOPX antibody (1:100, ab195974, Abcam) or anti-vimentin antibody (1:200, ab92547, Abcam) again for 5 h at 37°C. Subsequently, Cy™3 (green)-conjugated anti-rabbit IgG (1:500, 711–165-152, Jackson ImmunoResearch Laboratories, Inc.) was added to the sections and incubated, and the next steps were repeated as described above.
Quantitative real-time polymerase chain reaction assays
Total RNA was extracted from lung tissue using TRIzol reagent (Invitrogen, USA) according to the manufacturer’s recommendations. Briefly, after lysis, the RNA was extracted with chloroform, precipitated with isopropanol and washed with 75% ethanol. The RNA was dissolved in RNase-free water and quantified using a NanoDrop™ One/One c spectrophotometer (Thermo Scientific).
To evaluate the changes in the mRNA expression of the components of the regulatory network following ionizing radiation (IR), first-strand cDNA was synthesized using the HiScript III 1st Strand cDNA Synthesis Kit with cDNA wiper (Vazyme Biotech Co., Ltd.), followed by quantitative real-time polymerase chain reaction (qRT-PCR) assays using the ChamQ Universal SYBR qPCR Master Mix (Vazyme Biotech Co., Ltd.) in a QuantStudio® 5 Real-time PCR machine (Applied Biosystems Inc., Foster City, CA). The relative expression levels were calculated using the 2-△△CT method and β-actin was used as the internal control. All the samples were analyzed in triplicate.
To analyze the four let-7 miRNAs, total RNA was extracted from right lung tissue of both the control and irradiated groups using RISO™ reagent (BR0009, Biomics Biotechnologies Co., Ltd., Nantong, China). The RNA quality was confirmed by 1% formaldehyde agarose gel electrophoresis. Subsequently, the quantitative PCR system was prepared according to the EzOmics™ One-Step qPCR Kit (BK2100, Biomics Biotechnologies Co., Ltd., China), and the reaction mixtures were incubated on an ABI QuantStudio 5 Real-time PCR System. All the samples were analyzed in triplicate. The expression levels of let-7 miRNA family members a, b, c and d were normalized to the expression level of U6 and calculated using the 2-ΔΔCt method. The qRT-PCR assays to analyze all the mRNAs and miRNAs were performed by Biomics Biotechnologies Co., Ltd., China. The forward and reverse primers for all the genes analyzed by PCR are listed in Table 1.
Table 1.
Primers used for real-time quantitative PCR analysis
| Gene | Sequence (5′ to 3′) | |
|---|---|---|
| let-7a | Forward | GCGCGTGAGGTAGTAGGTTGT |
| Reverse | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAACTAT | |
| let-7b | Forward | GCGCGTGAGGTAGTAGGTTGT |
| Reverse | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAACCAC | |
| let-7c | Forward | GCGCGTGAGGTAGTAGGTTGT |
| Reverse | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAACCAT | |
| let-7d | Forward | GCGCGAGAGGTAGTAGGTTGC |
| Reverse | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAACTAT | |
| U6 | Forward | CTCGCTTGGCACA |
| Reverse | AACGCTTCACGAATTTGCGT | |
| β-Catenin | Forward | CAGATCCCATCCACGCAGTT |
| Reverse | TACCACCTGGTCCTCATCGT | |
| β-Actin | Forward | AGGTGACAGCATTGCTTCTG |
| Reverse | GCTGCCTCAACACCTCAAC | |
| E-Cadherin | Forward | CACAGGCGGTGAGACCTATC |
| Reverse | TGGGTTTAAATCGGCCAGCA | |
| HOPX | Forward | GATGTGGGAACCGTCTTCGT |
| Reverse | GTTCCCCTGCCTGTTCTGTT | |
| Lin28 | Forward | GTTGGTGATTGCTAGGTGGC |
| Reverse | TTTGAGAAGAGGACCGCACA | |
| proSP-c | Forward | AACGCCTTCTCATCGTGGTT |
| Reverse | GGGCTAGGCGTTTCTGAGTT | |
| TGF-β | Forward | TGCTAATGGTGGACCGCAA |
| Reverse | CACTGCTTCCCGAATGTCTGA | |
| Vimentin | Forward | GCTGCAGGCCCAGATTCA |
| Reverse | TTCATACTGCTGGCGCACAT |
Western blot assays
Frozen right lung tissue (50 mg) was ground in a mortar with liquid nitrogen, homogenized in 1 mL of radioimmunoprecipitation assay (RIPA) buffer with protease inhibitors (M250, Amresco, OH, USA) and stored in aliquots at −80°C until assayed. The protein concentration was determined using a standard BSA protein assay kit (A53226, Thermo Fisher Scientific, Waltham, MA, USA). Protein, 30 μg per well, was loaded and separated by 12% SDS-PAGE prior to being transferred to nitrocellulose membranes. After incubation with 5% non-fat milk, the nitrocellulose membranes were immunoblotted with the following primary antibodies: anti-E-cadherin antibody (1:1000, 3195, Cell Signaling Technology), anti-Lin28 antibody (1:800, ab63740, Abcam), anti-β-catenin antibody (1:1000, ab32572, Abcam), anti-proSP-C antibody (1:5000, ab40897, Abcam), anti-HOPX antibody (1:1000, ab195974, Abcam), anti-vimentin antibody (1:2000, ab92547, Abcam) and anti-β-actin antibody (1:3000, 4970, Cell Signaling Technology). Then, the membranes were rinsed and incubated with peroxidase-conjugated goat anti-rabbit IgG (1:10000, AP132P, Merck Millipore, Burlington, MA, USA) for 1 h at 37°C. The signals of the target proteins were detected by enhanced chemiluminescence (ECL Kit, 34075, Thermo Fisher Scientific). For densitometric analysis of all the protein bands on the membranes, the densitometric signal for a target protein (e.g. E-cadherin) was normalized to that of β-actin, and then the ratios obtained for each of the irradiated groups were compared to the ratios of the corresponding control groups. The differences in the ratios between the two groups were statistically analyzed.
Analysis of relative Lin28/let-7 ratios
The Lin28 protein binds to and represses let-7 miRNAs. Lin28 and let-7 control cell differentiation via a mechanism that involves their mutual negative regulation [14]. Therefore, the balance between Lin28 protein and let-7 miRNA expression is very important for determining cell differentiation fate. To determine the relative Lin28/let-7 ratios, we first separately calculated the mean expression of Lin28 and 4 let-7 members. Then, the mean expression of Lin28 in the IR group at each phase of injury was divided by the mean expression of each of the four let-7 miRNA family members at the corresponding phase of injury.
Statistical analysis
All the statistical analyses were performed using SPSS 17.0 software (Chicago, IL). All the values are expressed as the means ± SDs. Statistical analyses between groups were performed using Student’s t-test, and P < 0.05 was considered statistically significant. The data shown in the figures are the relative fold changes.
RESULTS
Histopathological changes in mouse lungs at different phases of injury after thoracic irradiation
Female C57BL/6 J mice received thoracic irradiation with a single dose of 20 Gy. After irradiation, mice in the irradiated and control groups were euthanized at different phases of injury ranging from acute injury to end-stage fibrosis. Lung sections (5 μm) were stained with H&E (Fig. 1A) or Picro-Sirius Red (Fig. 1B). The radiation-induced injuries progressed with time. No significant changes in lung architecture were observed at 24 h and 1 week post-irradiation, except for the presence of limited numbers of inflammatory cells. However, dilated and congestive capillaries were observed in the interstitium of the lungs at 1 and 2 months following irradiation (Fig. 1A, red and green arrows), and a few thickened alveolar septa were observed (Fig. 1A, black arrows). Compared to the lungs of the mice in the 1- and 2-month IR groups, the lungs of the mice in the 3- and 4-month IR groups exhibited more thickened alveolar septa and collapsed alveolar spaces (Fig. 1A). In addition, collagen accumulated in the alveoli and around bronchioles (Fig. 1A, blue arrows). After 5 months, the structure of the lung tissue was altered and many alveolar spaces disappeared. Fibrous components and a large number of lymphocytes were also clearly seen in the lung tissue (Fig. 1A, yellow arrows). Plasma cells, macrophages, mononucleolar cells and foam cells infiltrated into alveolar spaces (Fig. 1A, blue arrowheads). Picro-Sirius Red staining showed that collagen accumulation appeared in the lungs of the treated mice beginning in the first month after irradiation. This accumulation was time-dependent. The longer the interval between irradiation and analysis was, the more collagen was deposited in the lungs (Fig. 1A).
Fig. 1.
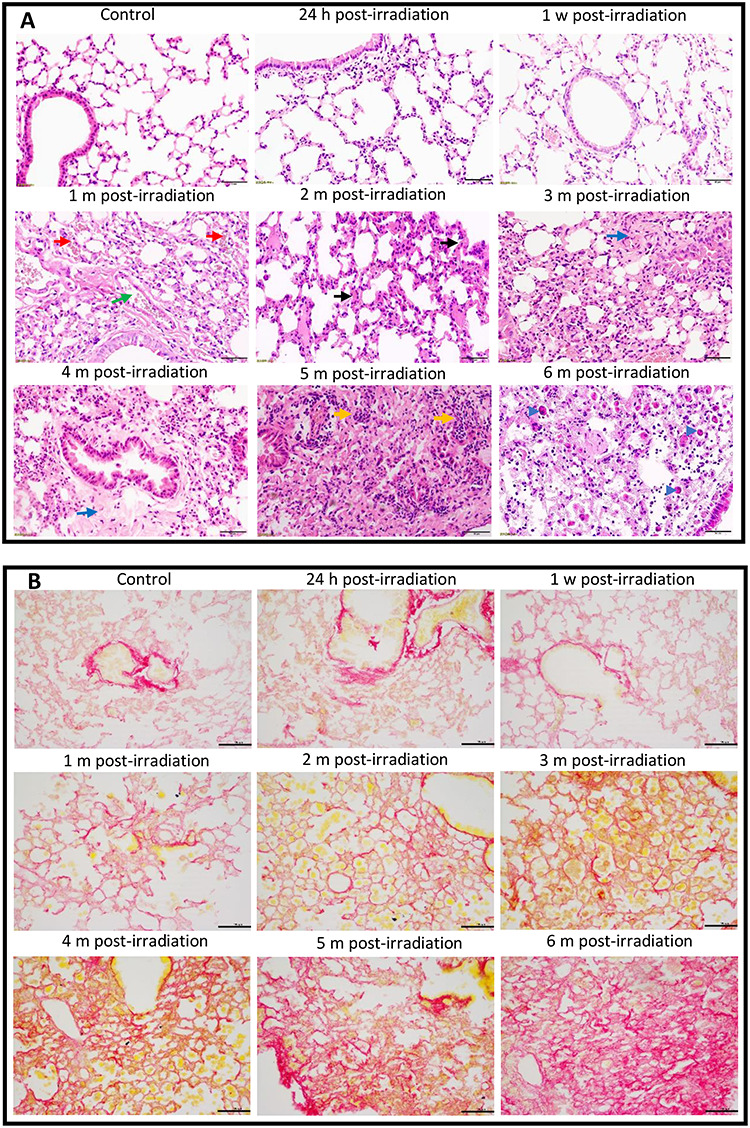
Histopathological changes in the lungs of mice after thoracic irradiation. Female C57BL/6 J mice received 20 Gy of X-rays in a single dose. The mice were euthanized at 24 h, 1 week, 1, 2, 3, 4, 5 or 6 months post-irradiation. The mice in the corresponding control groups were sacrificed at the same time points. (A) Photomicrographs of lung tissues stained with H&E. Green arrow, a dilated vessel; red arrows, congestive capillaries; black arrows, thickened septa; blue arrows, fibrotic tissue; yellow arrows, inflammatory cells; blue arrowheads, macrophages, mononucleolar cells or foam cells. (B) Images of lung tissues stained with Picro-Sirius Red. Collagen stains red. Scale bar = 50 μm. H, hour; w, week; m, month.
Among the 50 mice treated with thoracic irradiation, a total of 10 mice died within 6 months after irradiation (5 mice died by 8 weeks, 2 died at 9 weeks, and 1 died at 10, 21 and 24 weeks, respectively). These mice were excluded from this study.
Dynamic phenotypes of AEC II at different phases of injury after thoracic irradiation
An immunostaining method combining two or more antibodies against unique target cell biomarkers has been used for decades to determine cellular phenotypes [14, 15]. In this study, EpCAM, a putative epithelial stem cell biomarker, was used to further confirm the stemness trait of AEC II. As shown in the merged images of Fig. 2A, every AEC II in the lungs of both sham-irradiated and irradiated mice exhibited yellow or orange fluorescence due to the overlap of red (proSP-C) and green (EpCAM) fluorescence. Therefore, coexpression of EpCAM with proSP-C helped to confirm the stemness trait of AEC II since every proSP-C-positive cell (red fluorescence) also exhibited green fluorescence (EpCAM) (Fig. 2A).
Fig. 2.
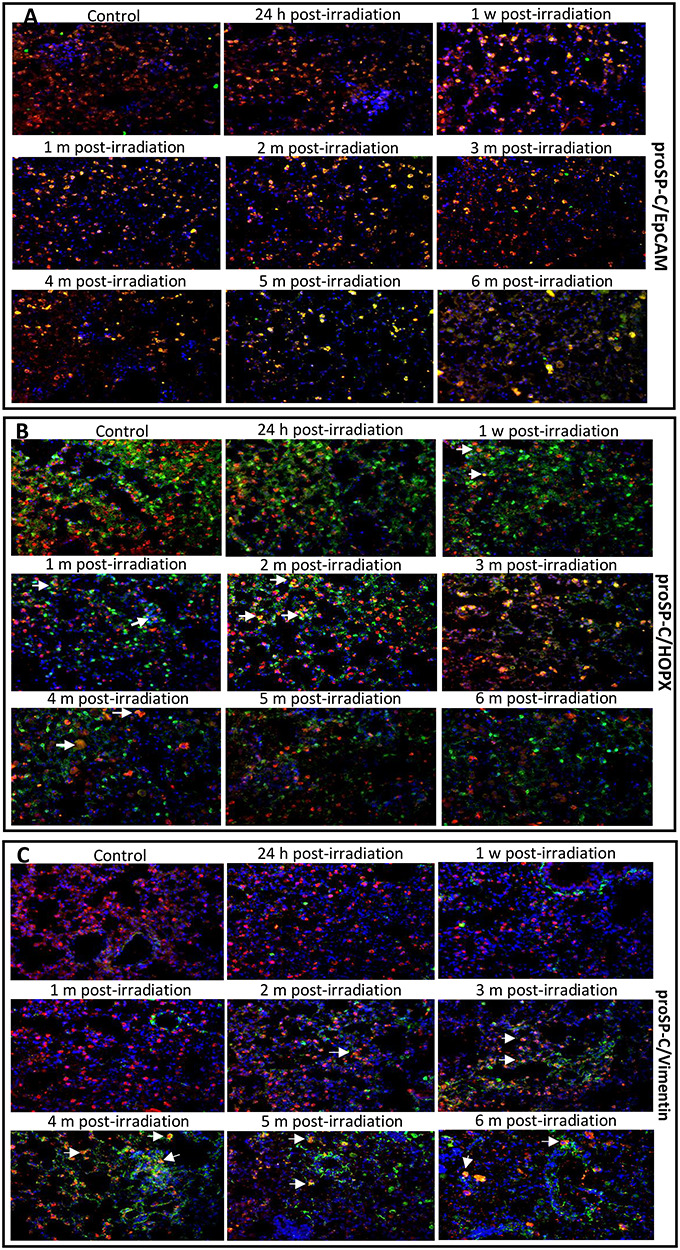
The merged images of the coexpression of proSP-C (AEC II cell marker) with EpCAM (epithelial stem cell marker) (A), HOPX (AEC I cell marker) (B), or vimentin (mesenchymal cell marker) (C) in the lungs of mice treated with IR and control-treated mice. Double immunofluorescence staining was used to detect protein expression. In the representative photomicrographs, the color of the pro-SP-C protein is red, the EpCAM, HOPX and vimentin proteins are green, and the nuclei are blue. The yellow fluorescence in AEC II is due to the overlap of red and green (arrows). Coexpression of EpCAM/proSP-C, HOPX/proSP-C or vimentin/proSP-C in the lung tissues was used to track AEC II dynamic phenotypes at different phases of injury after irradiation. Magnification: 400x. h, hour; w, week; m, month.
AEC II cells are essential for the repair and regeneration of injured alveolar epithelium. To track AEC II differentiation states at different phases of lung injury post-irradiation, we used a monoclonal antibody against proSP-C protein, a unique AEC II surface biomarker [16], to co-stain lung tissue either with an antibody against the HOPX protein, which was reported to be expressed only in AEC I in the lungs [17], or with an antibody against vimentin, which has been used as a biomarker of mesenchymal cells (indicative of myofibroblast differentiation) [18]. Our double-color immunofluorescence staining results showed that the AEC II differentiation status varied with different phases of injury following irradiation, and these results were consistent with the histopathological changes. In the merged immunostaining images, very few cells exhibiting yellow fluorescence (Fig. 2B, arrows) due to coexpression of HOPX (green) with proSP-C (red) were observed at 24 h and 1 month (Fig. 2B). In the 2- and 3-month groups, many cells coexpressed proSP-C with HOPX. This result suggests that most quiescent AEC II were activated to undergo differentiation into AEC I, leading to a transitional differentiation state (HOPX+/proSP-C+ phenotype). Few cells exhibited pure green fluorescence at 3 months, suggesting that the AEC II were damaged by IR. Then, more cells exhibiting green fluorescence and fewer cells exhibiting co-staining were observed at 1 month than at 3 months. This finding may be a result of the AEC II being driven to differentiate into AEC I. However, no yellow or orange cells were observed at 5 and 6 months after irradiation, and the numbers of cells exhibiting green or red fluorescence markedly were decreased compared to those at the other timepoints, except 2 and 3 months (Fig. 2A). This decrease in the numbers of AEC I (green cells) may be related to the long-term effect of IR.
As described above, irradiation induced regional pneumonitis during the early phase of injury, and this pneumonitis later progressed to fibrosis. In this study, we also examined the expression of mesenchymal cell markers by AEC II cells at different phases of injury following irradiation to test our hypothesis that AEC II could be induced to undergo transdifferentiation into non-epithelial cells. We found that coexpression of the vimentin protein (green fluorescence) with proSP-C was detected in AEC II by 2 months after irradiation (Fig. 2C, arrows), but this coexpression was more pronounced by 3 months, which indicates that AEC II acquired a profibrotic phenotype. In addition, the merged images showed that the numbers of cells exhibiting only green fluorescence were remarkably increased beginning at 3 months and continuing as time elapsed after irradiation, but the numbers of cells exhibiting red fluorescence were markedly decreased from 4 to 6 months (Fig. 2C) after irradiation, indicating that the AEC II had been driven to transdifferentiate into vimentin+ cells. A decrease in the numbers of HOPX+ cells and an increase in the numbers of vimentin+ cells suggested that AEC II preferentially differentiated into mesenchymal cells instead of AEC I after 3 months post-irradiation.
The expression of proSP-C, HOPX and vimentin at different phases of injury after thoracic irradiation
In this study, in addition to the detection of cell phenotype marker expression by immunostaining, the relative levels of these markers in the lungs were also quantified by qRT-PCR and western blotting. The qRT-PCR results showed that compared to that in the controls, proSP-C mRNA was slightly decreased at 1 week, 1 month and 4–6 months following irradiation, and mildly increased at 24 h (Fig. 3). Compared to that in the controls, the expression of hopx was increased in the IR-treated groups at 24 h, 1 week, 1 month and 3 months. By 4 months after irradiation, hopx expression was downregulated (Fig. 3). The expression of vimentin, a mesenchymal marker, was elevated in all the IR groups compared with the control groups.
Fig. 3.
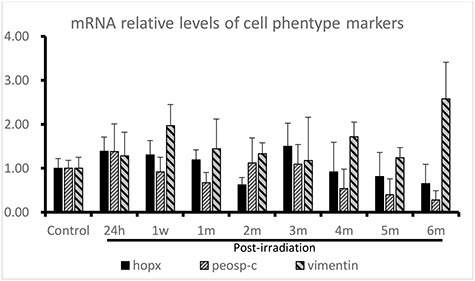
Gene expression of cell phenotype biomarkers detected by qRT-PCR in thoracically irradiated lungs at different phases of injury post-irradiation. The relative expression levels were calculated using the 2-△△CT method, and β-actin was used as the internal control. All the samples were analyzed in triplicate.
Western blotting was used to quantify the protein expression of three biomarkers of AEC I, AEC II or mesenchymal cells at different phases of injury post-irradiation to further confirm the effect of IR on the lungs. Compared with that in the corresponding control groups, the expression level of proSP-C was significantly decreased in 6 of the 8 irradiated groups (P < 0.05 to P < 0.001) except at 24 h and 1 week (Fig. 4A and B). The level reached its nadir at 5 months and rebounded at 6 months. A reduced protein expression level of HOPX was observed in the lung tissues of all the irradiated mice except at 3 months, but significant differences were observed only in the 1- (P< 0.001), 5- (P < 0.05) and 6-month (P < 0.001) groups (Fig. 4A and C) compared with the corresponding control groups. In contrast, except for the 24-h and 1-week groups, the expression level of vimentin was obviously increased in the lung tissues of the mice in the IR-treated groups (P < 0.001). The graphs in Fig. 4 show the relative levels of these three phenotype markers after exposure to IR (Fig. 4B, C and D).
Fig. 4.
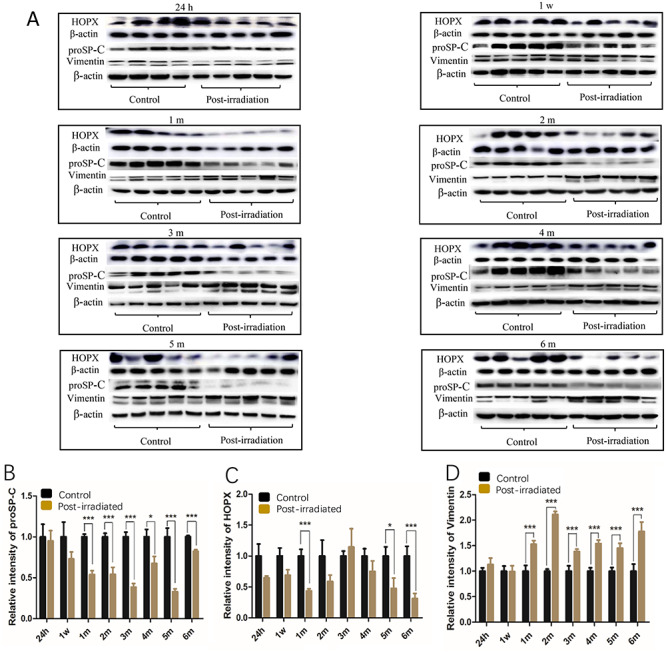
The expression of the pro-SP-C, HOPX and vimentin proteins in the mouse lungs at different phases of injury following irradiation. The relative levels of the proSP-C, HOPX and vimentin proteins were examined by western blot (A). Graphs (B), (C) and (D) show the relative levels of the three phenotype markers after exposure to IR. The relative amounts of proSP-C, HOPX and vimentin in the graphs were obtained by the ratios of the target protein pixels to the corresponding β-actin pixels. The data presented are shown as the mean ± SD. Statistical analyses between groups were performed using Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001. h, hour; w, week; m, month. Note: The proSP-C and vimentin proteins were detected on the same blot membranes, and the western blots for HOPX were performed separately.
The expression of E-cadherin, β-catenin and Lin28 in different phases of injury after thoracic irradiation
E-Cadherin, an epithelial cell marker, is capable of preventing β-catenin from translocating to the nucleus by fixing it to the cell membrane [19, 20]; as a result, E-cadherin prevents Wnt/β-catenin pathway activation. β-Catenin has the ability to upregulate the transcription of the Lin28 gene. Immunofluorescence staining showed that these three proteins were expressed by both alveolar epithelial cells and bronchiolar cells. Compared with the control, irradiation downregulated the expression of E-cadherin in both alveolar cells and bronchiolar epithelial cells in a time-dependent pattern (Fig. 5A), but this effect was more notable in alveolar cells than in bronchiolar epithelial cells. In contrast, compared to the control, irradiation of the lungs markedly increased the numbers of alveolar cells and bronchiolar cells exhibiting green fluorescence signals for β-catenin (Fig. 5B) in a time-dependent manner, particularly in alveolar cells after 1 month, and these numbers reached their highest levels at 6 months. The protein expression of Lin28 was also detected in both alveolar epithelial cells and bronchiolar cells (Fig. 5C). Many more Lin28-positive cells were observed in the IR groups at 3 and 4 months than in the other groups, but the number of cells exhibiting red florescence markedly decreased at 5 and 6 months.
Fig. 5.
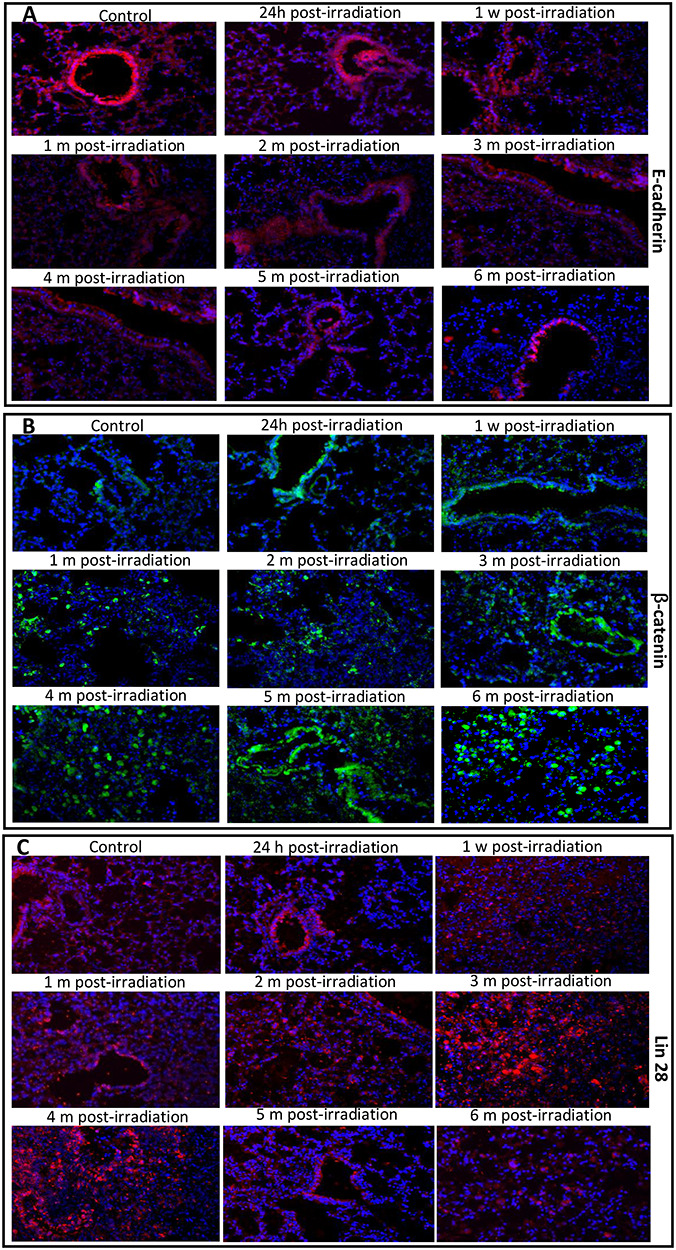
The images of immunofluorescence staining of the E-cadherin, β-catenin and Lin 28 proteins in the mouse lungs at different phases of injury after irradiation. In the representative photomicrographs, the color for the E-cadherin (A) and Lin28 (C) proteins is red, the β-catenin protein (B) is green, and the DAPI-stained nuclei are blue. Magnification: 400x. h, hour; w, week; m, month.
A time-course analysis of E-cadherin mRNA expression following IR showed decreased levels in 6 of the 8 IR groups, except in the 1-week and 3-month groups (Fig. 6). The expression of β-catenin was upregulated in all the IR groups, and its level peaked at 3 months following irradiation (Fig. 6). As a repressor of let-7 miRNA, Lin28 was downregulated at the early stages of injury (24 h and 1 week) and upregulated by 1 month following irradiation compared to the control (Fig. 6). The expression also reached its peak level at 3 months.
Fig. 6.
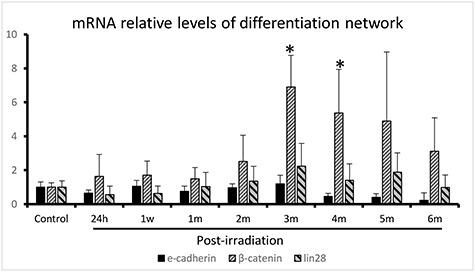
Gene expression of the E-cadherin/β-catenin/Lin28 network detected by qRT-PCR in thoracically irradiated lungs at different phases of injury post-irradiation. The relative expression levels were calculated using the 2-△△CT method, and β-actin was used as the internal control. All the samples were analyzed in triplicate. *P < 0.05.
Western blot analysis showed that the expression of E-cadherin was downregulated in all the irradiated groups compared with the corresponding control groups (P < 0.05 to P ~ 0.001) except at 24 h (Fig. 7A and B). In contrast, the expression levels of β-catenin (Fig. 7C and D) and Lin 28 (Fig. 7E and F) were significantly upregulated in the lungs of the mice in all the irradiated groups (P < 0.05 to P ~ 0.001), except Lin28 was not significantly upregulated at 1 week. The peak levels were seen at 4 and 3 months, respectively.
Fig. 7.
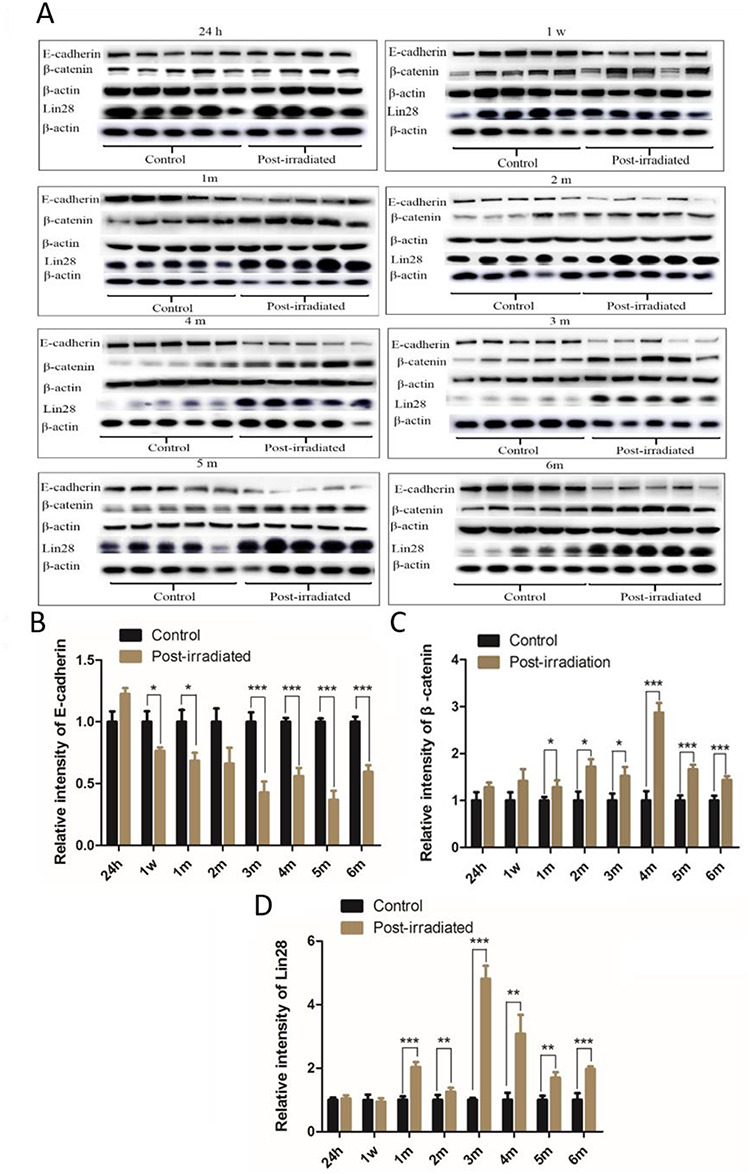
The expression of the E-cadherin, β-catenin and Lin 28 proteins detected by western blot (A). The relative expression levels of E-cadherin (B), β-catenin (C) and Lin28 (D) relative to that of β-actin are shown in the graphs. The data presented are shown as the mean ± SD. Statistical analyses between groups were performed using Student’s t-test. *P < 0.05, **P < 0 .01, ***P < 0.001. h, hour; w, week; m, month. Note: The E-catenin and β-catenin proteins were detected on the same blot membranes, and the western blots for Lin28 were performed separately.
The expression levels of let-7 miRNAs at different phases of injury after thoracic irradiation
Since let-7 miRNA participates in cell differentiation, we investigated the dynamic levels of let-7a, let-7b, let-7c and let-7d in the mouse lungs at different phases of injury. The results showed that compared to that in the corresponding control groups, let-7a expression was increased in all the IR groups, but significant differences were seen from 24 h to 2 months after irradiation (P < 0.01 to P < 0.001) (Fig. 8A). The expression of let-7b and let-7c was significantly elevated from 24 h to 3 months post-irradiation (P < 0.05 to P < 0.001) (Fig. 8B and C). Let-7d was significantly upregulated from 24 h to 1 month after irradiation (P < 0.001) but was significantly downregulated at 5 and 6 months (P < 0.001) (Fig. 8D). However, when analysis was performed among all the irradiated groups, the expression levels of all the let-7 miRNA members were strikingly increased at the early phases of injury (24 h and 1 week), but these levels abruptly decreased from 1 month to 6 months (Fig. 8).
Fig. 8.
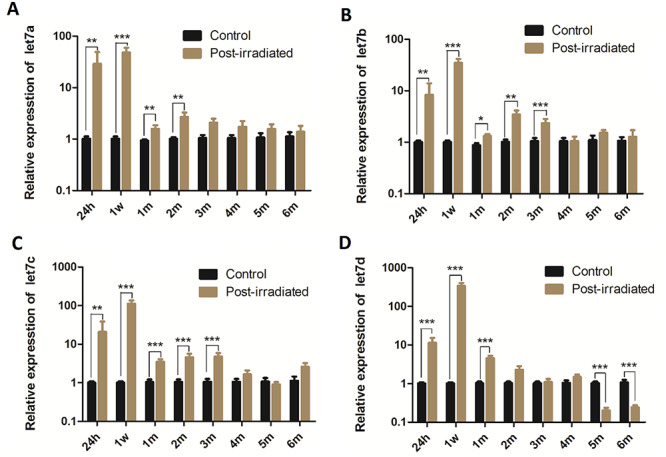
The relative expression levels of four let-7 miRNA family members (a, b, c and d) [(shown in (A), (B), (C) and (D) respectively] were detected by RT-qPCR in mouse lungs at different phases of injury following irradiation. The expression levels of the let-7 miRNA family members were normalized to that of U6 and calculated using the 2-ΔΔCt method. The data presented were obtained from three independent experiments per sample and are shown as the mean ± SD. Statistical analyses between groups were performed using Student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001. h, hour, w, week, m, month.
Relative Lin28/let-7 ratios
Since the Lin28 protein represses let-7 miRNAs by binding to them, we analyzed the ratios of Lin28 protein expression to let-7 miRNA expression in this study. Because Lin28 and let-7 regulate cell differentiation via a mechanism that involves their mutual negative regulation [10], separate analyses of their individual expression levels post-irradiation do not illustrate the true roles of Lin28 and let-7 in determining cell differentiation status. Therefore, the carefully regulated balance between Lin28 and let-7 is an important factor in determining cell differentiation state. When the Lin28/let-7 ratio is high (more Lin28 than let-7), cells will be in an undifferentiated state and will maintain stemness, and vice versa. In the current study, we calculated and analyzed the relative Lin28/let-7 ratios throughout the phases of injury. The curves in Fig. 9 show that during the early phase of injury, the four Lin28/let-7 ratios were low (Fig. 9). After 2 months, the ratios gradually trended upward as time elapsed following irradiation. Most notably, the Lin28/let-7d ratio markedly increased at 3 months. Then, the ratio experienced a transient slight decrease at 4 months and then strikingly increased at 5 and 6 months, reaching levels >7–10-fold higher than those observed at the early time points, respectively (Fig. 10).
Fig. 9.
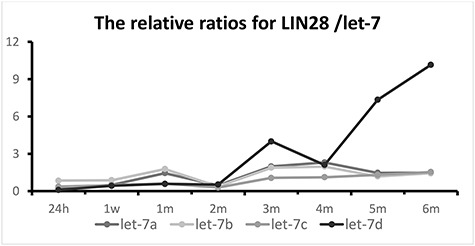
Trends of four relative Lin28/let-7 ratios in thoracically irradiated lungs at different phases of injury following irradiation. Since Lin28 and let-7 regulate cell differentiation via a mechanism that involves their mutual negative regulation, the balance between Lin28 and let-7 is important for determining cell differentiation fate. In this study, the four relative Lin28/let-7 ratios were analyzed to illustrate the role of the Lin28/let-7 axis in determining cell differentiation phenotypes.
Fig. 10.
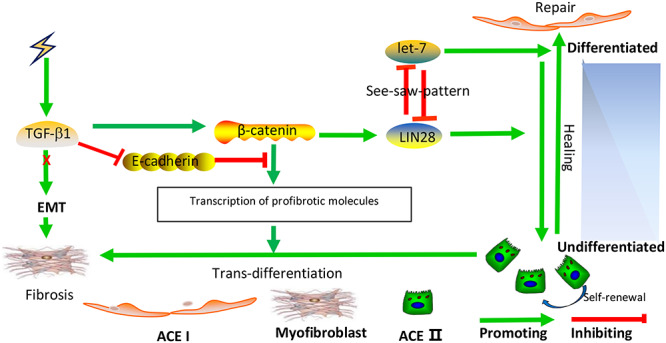
Diagram of a signaling regulatory network that determines the AEC II differentiation phenotype. TGF-β1 promotes RILF via the induction of β-catenin to trigger AEC II transdifferentiation. After irradiation, TGF-β1 expression is induced. Subsequently, the suppression of E-cadherin and activation of β-catenin occur; as a result, Lin28 transcription is induced, which in turn, maintains AEC II stemness in order to promote injury repair by repressing let-7 via a mechanism that involves the mutual negative regulation of these molecules. On the other hand, significantly increased β-catenin expression is capable of inducing AEC II transdifferentiation by promoting the transcription of several profibrotic cytokine and factor genes. This is a fibrotic network. High levels of let-7 miRNAs are required for AEC II to AEC I differentiation. This pathway may occur during normal injury repair.
DISCUSSION
Since lungs are constantly exposed to insults from the external environmental, they contain numerous resident adult stem cells that are responsible for injury repair [21]. Obviously, the repair and regeneration of injured lungs rely on these resident quiescent stem cells. EpCAM is a putative biomarker of epithelial stem cells [22]. In this study, the fact that every single proSP-C-positive cell in the lungs expressed the EpCAM protein further demonstrated the AEC II stemness trait. To the best of our knowledge, this is the first study to further demonstrate the AEC II stemness trait with a double-color immunofluorescent staining technique using these 2 antibodies (Fig. 2).
To date, no mechanisms of RILF can explain why injured alveolar epithelial cells are not repopulated by AEC II, which is responsible for alveolar repair. In this study, we tracked the dynamic phenotypes of AEC II at different phases of injury to test our hypothesis, i.e. AEC II were induced to transdifferentiate into non-epithelial cells by IR. The coexpression of proSP-C/vimentin by 3 months demonstrated that AEC II acquired a new cell differentiation status with a mesenchymal phenotype. These hybrid phenotype cells may continue to fully differentiate into cells with a vimentin+ phenotype, which is supported by increased numbers of new cells exhibiting pure green fluorescence (vimentin+ phenotype) and decreased numbers of cells exhibiting pure red fluorescence (proSP-C+ phenotype) after 3 months following irradiation, compared with the numbers observed at the early phases of injury (from 24 h to 2 months). The immunostaining findings suggested that the third month following irradiation seems to be a critical stage for determining AEC II differentiation fate, and these cells were most likely induced to transdifferentiate into mesenchymal cells after this stage of injury due to IR.
Our western blot analysis showed that the expression levels of the HOPX protein at different phases of injury were consistent with those observed by immunostaining. Decreased HOPX expression may contribute to failed regenerative processes in end-stage pulmonary fibrosis since Ota et al. found that the knockdown of hopx by siRNA resulted in the suppression of AEC II to AEC I differentiation in vitro [17].
In this study, the decreased expression of the proSP-C protein may occur due to either a lower number of AEC II compared to the reactive proliferation of various pulmonary cells (there are at least 40 cell types in lung tissue) or a lower number of AEC II cells after some AEC II were driven to transdifferentiate into other cell types following irradiation.
β-Catenin is a transcription factor and a mesenchymal marker [23, 24]. When β-catenin translocates to the nucleus, it activates the transcription of genes involved in myofibroblastic differentiation [23]. E-Cadherin is capable of preventing β-catenin from translocating to the nucleus by fixing it to the membrane [19,20]. In this study, both immunofluorescence staining and western blotting showed that E-cadherin expression was significantly downregulated by radiation. The decrease in E-cadherin expression resulted in the release of free β-catenin; as a result, β-catenin entered the nucleus. On the other hand, the reduced expression of E-cadherin suggested a loss of the epithelial phenotype due to epithelial damage by IR.
At the molecular level, stem cell differentiation is tightly regulated by a complex signaling network. Numerous investigations have demonstrated that various cytokines and profibrotic factors are released into the niche after exposure to radiation. In this study, after irradiation, signaling pathways downstream of TGF-β1, a profibrotic cytokine, were activated, which in turn promoted the expression of β-catenin. Increased β-catenin upregulated Lin28, which maintained AEC II stemness and augmented the AEC II population by repressing let-7 via a mechanism that involves the mutual negative regulation of these molecules. In addition to being an activator of Lin28, significantly increased β-catenin expression was capable of inducing AEC II transdifferentiation into mesenchymal cells by promoting the transcription of several profibrotic cytokine and factor genes (Fig. 10). This is a fibrotic network. In this network that regulates differentiation, the AEC II differentiation phenotype was directly determined by
the Lin28/let-7 axis. During the early phases of injury (from 24 hto 2 months), low Lin28/let-7 ratios facilitated alveolar repair since high levels of let-7 miRNAs may be required for resident AEC II to differentiate into AEC I. Beginning in the third month post-irradiation, higher ratios of Lin28/let-7 in the lungs allowed adult stem cells to maintain stemness and augmented the population. However, among the four let-7 miRNA members, the substantial downregulation of let-7d at the late phase of injury seemed to play a predominant role in RILF and led to the inability of AEC II to differentiate into alveolar epithelial cells. Pandit et al. corroborated our findings that downregulation of let-7d by TGF-β1 caused lung fibrosis [9].
Taken together, the balance between Lin28 and let-7 varied with injury stage. At the early phase of injury, a high level of let-7 was necessary for resident ASCs to repair normally, leading to low Lin28/let-7 ratios (Fig. 10). At the late phase of injury, Lin28 was required for the stemness and self-renewal of AEC II in order to continue to repair injured tissue due to the long-term effect of IR. Although TGF-β1 was activated in the irradiated lungs, type I alveolar epithelial–mesenchymal transition (EMT) was less likely since alveolar epithelial cells are radiosensitive. The doses of IR used in clinical treatment of cancer are capable of damaging alveolar epithelial cells and inducing AEC I apoptosis rather than transdifferentiation into mesenchymal cells.
CONCLUSION
Twenty grays of X-rays, as used in this study, have the capability to cause fibrotic changes in mouse lung tissue. This study is the first to confirm the AEC II stemness trait and to track dynamic phenotypes at different phases of injury using a double-color immunofluorescence staining method. This study is also the first to propose a new mechanism of RILF. In future studies, it would be important to drive AEC II differentiation toward AEC I by manipulating the β-catenin/Lin28/let-7 network; such studies could lead to novel treatments for lung fibrosis.
ACKNOWLEDGMENTS
The authors would like to thank Biomics Biotechnologies Co., Ltd., China, for designing, synthesizing, providing qRT-PCR primer sequences specific for genes and miRNAs and performing qRT-PCR assays.
Contributor Information
Xiaozhuan Liu, Center for Clinical Single-Cell Biomedicine, Henan Provincial People’s Hospital, People's Hospital of Zhengzhou University, School of Clinical Medicine, Henan University, Zhengzhou, Henan 450003, China.
Tingting Zhang, Center for Clinical Single-Cell Biomedicine, Henan Provincial People’s Hospital, People's Hospital of Zhengzhou University, School of Clinical Medicine, Henan University, Zhengzhou, Henan 450003, China.
Jianwei Zhou, Department of Oncology, Henan Provincial People’s Hospital, People's Hospital of Zhengzhou University, School of Clinical Medicine, Henan University, Zhengzhou, Henan 450003, China.
Ziting Xiao, Department of Oncology, Henan Provincial People’s Hospital, People's Hospital of Zhengzhou University, School of Clinical Medicine, Henan University, Zhengzhou, Henan 450003, China.
Yanjun Li, Center for Clinical Single-Cell Biomedicine, Henan Provincial People’s Hospital, People's Hospital of Zhengzhou University, School of Clinical Medicine, Henan University, Zhengzhou, Henan 450003, China.
Yuwei Zhang, Department of Science Research and Discipline Construction, Henan Key Laboratory of Stem Cell Differentiation and Modification, Henan Provincial People’s Hospital, Zhengzhou, Henan 450003, China.
Haodi Yue, Center for Clinical Single-Cell Biomedicine, Henan Provincial People’s Hospital, People's Hospital of Zhengzhou University, School of Clinical Medicine, Henan University, Zhengzhou, Henan 450003, China.
Zhitao Li, Department of Immunology, Medical College of Henan University of Science and Technology, Luoyang, Henan, China, 471023.
Jian Tian, Department of Oncology, Henan Provincial People’s Hospital, People's Hospital of Zhengzhou University, School of Clinical Medicine, Henan University, Zhengzhou, Henan 450003, China.
CONFLICT OF INTEREST
None declared.
FUNDING
This study was supported by the National Natural Science Foundation of China (Grant No. 81673097 and 81801547).
References
- 1. Graves PR, Siddiqui F, Anscher MS et al. Radiation pulmonary toxicity: From mechanisms to management. Semin Radiat Oncol 2010;20:201–7. [DOI] [PubMed] [Google Scholar]
- 2. Almeida C, Nagarajan D, Tian J et al. The role of alveolar epithelium in radiation-induced lung injury. PLoS One 2013;8:e53628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tian J, Zhao W, Tian S et al. Expression of genes involved in mouse lung cell differentiation/regulation after acute exposure to photons and protons with or without low-dose preirradiation. Radiat Res 2011;176:553–64. [DOI] [PubMed] [Google Scholar]
- 4. Rock JR, Barkauskas CE, Cronce MJ et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci U S A 2011;108:E1475–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu XM, Driskell RR, Engelhardt JF. Stem cells in the lung. Methods Enzymol 2006;419:285–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dickey JS, Zemp FJ, Martin OA et al. The role of miRNA in the direct and indirect effects of ionizing radiation. Radiat Environ Biophys 2011;50:491–9. [DOI] [PubMed] [Google Scholar]
- 7. Simone NL, Soule BP, Ly D et al. Ionizing radiation-induced oxidative stress alters miRNA expression. PLoS One 2009;4:e6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Martin M, Lefaix J, Delanian S. TGF-beta1 and radiation fibrosis. A master switch and a specific therapeutic target? Int J Radiat Oncol Biol Phys 2000;47:277–90. [DOI] [PubMed] [Google Scholar]
- 9. Pandit KV, Corcoran D, Yousef H et al. Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2010;182:220–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McDaniel K, Hall C, Sato K et al. Lin28 and let-7: Role and regulation in liver diseases. Am J Physiol Gastrointest Liver Physiol 2016;310(10):G757-G765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cai WY, Wei TZ, Luo QC et al. The Wnt-beta-catenin pathway represses let-7 microRNA expression through transactivation of Lin28 to augment breast cancer stem cell expansion. J Cell Sci 2013;126:2877–89. [DOI] [PubMed] [Google Scholar]
- 12. Henderson WR Jr, Chi EY, Ye X et al. Inhibition of Wnt/beta-catenin/CREB binding protein (CBP) signaling reverses pulmonary fibrosis. Proc Natl Acad Sci U S A 2010;107:14309–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tsialikas J, Romer-Seigert J. Lin28: Roles and regulation in development and beyond. Development 2015;142:2379–404. [DOI] [PubMed] [Google Scholar]
- 14. Janossy G, Bollum FJ, Bradstock et al. cellular phenotype of normal and leukemia hemopoietic cells determined by analysis with selected antibody combinations. Blood 1980;56(3):430–441. [PubMed] [Google Scholar]
- 15. Ramshaw AL, Parums DV. Combined immunohistochemical and immunofluorescence method to determine the phenotype of proliferating cell population. J Clin Pathol 1992;45(11):1015–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Beers M, Lomax CA, Russo S. Synthetic processing of surfactant protein C by alveolar epithelial cells. J Biol Chem 1998;273:15287–93. [DOI] [PubMed] [Google Scholar]
- 17. Ota C, Ng-Blichfeldt JP, Korfei M et al. Dynamic expression of HOPX in alveolar epithelial cells reflects injury and repair during the progression of pulmonary fibrosis. Sci Rep 2018;8(1) 12983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mendez M, Kojima SI, Goldman RD. Vimentin induces changes in cell shape, motility and adhesion during the epithelial-mesenchymal transition. FASEB J 2010; 24(6):1838–1851.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Matsuzaki S, Darcha C, Maleysson E et al. Impaired down-regulation of E-cadherin and beta-catenin protein expression in endometrial epithelial cells in the mid-secretory endometrium of infertile patients with endometriosis. J Clin Endocrinol Metab 2010;95:3437–45. [DOI] [PubMed] [Google Scholar]
- 20. Ozawa M, Baribault H, Kemler R. The cytoplasmic domain of the cell adhesion molecule uvomorulin associates with three independent proteins structurally related in different species. EMBO J 1989;8:1711–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Volckaert T, De Langhe S. Lung epithelial stem cells and their niches: Fgf10 takes center stage. Fibrogenesis Tissue Repair 2014;7:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hasegawa K, Sato A, Tanimura K et al. Fraction of MHCII and EpCAM expression characterizes distal lung epithelial cells for alveolar type 2 cell isolation. Respir Res 2017;18:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gottardi CJ, Gumbiner BM. Distinct molecular forms of beta-catenin are targeted to adhesive or transcriptional complexes. J Cell Biochem 2004;167:339–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nelson WJ, Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science 2004;303:1483–7. [DOI] [PMC free article] [PubMed] [Google Scholar]