

Abstract
Thyroid cancer (TC) is the mainly frequent endocrine cancer by different incidence rate in worldwide. However, early prediction of this cancer is still challenging due to the unclear pathogenicity. In this study with the aid of systems biology approach, performed a holistic study on GSE65144 dataset containing anaplastic thyroid carcinoma tissues. Co-expression network analysis by WGCNA suggested that highly preserved turquoise module with 1,480 genes was significantly correlated to TC. Most of the top 54 hub-genes of this module are functionality correlated to thyroid hormone generation (GO:0006590). Of these 54 hub-genes, FOXE1 has been reported previously to contain mutation asosiated to TC and chosen for experimental validation step. To this end, we conducted a case-control study including 81 TC patients and 165 controls individuals to evaluate the effects of FOXE1 functional polymorphisms (rs1867277) on the development of TC in Sistan and Balouchestan province of Iran. The polymorphisms of FOXE1 gene (rs1867277) assessed by tetra-ARMS PCR technique. Homozygous (GG) and (AA) variant of rs1867277 polymorphism were detected in 26 (32.1%) and 15 (18.5 %) of TC patients, and 66 (40.0%), and 15 (9.1%) in controls, respectively (p-value= 0.03, OR= 2.53). The A allele frequency was 70 (43.2%) in TC patients and 114 (34.5%) in controls (p-value= 0.06, OR= 1.44). Overall, our results suggested that FOXE1 gene could be used as a prognostic marker in TC and also provides information related to FOXE1 functional polymorphisms (rs1867277) in Sistan and Balouchestan province of Iran.
Key Words: Systems biology, WGCNA, thyroid cancer, FOXE1, polymorphism, genotype
Introduction
Thyroid cancer (TC) is the most widespread endocrine neoplasm by accounts 1% of entire human malignancies (Kondo et al., 2006). TC has a ninth place in the ranking of malignancies responsible for more than 500,000 cases worldwide. The global TC prevalence is 11 per 100,000 in women, and it is 3 times more than men population. In addition, TC represents 1 case in 20 cancer patients in 2018 and has low mortality rates (Bray et al., 2018; Horner et al., 2009). According to differentiation of cancer cells, TC classified into three groups, including papillary TC (PTC) as the most common type by 80 to 85% incidence rate, follicular TC (FTC) by 5 to 10% prevalence and Hurthle cell cancer as a rare type of TC (Davies and Welch, 2006). It seems that TC is the result of low to moderate penetration of multiple genes interactions with transcriptional regulators and collaboration of environmental risk factors that modulate TC susceptibility (Goldgar et al., 1994). Lack of iodine intake, exposure to ionizing radiation and certain pollutants particularly during childhood defined as environmental factors associated with TC risk (He et al., 2015). However, these explain just a small part of the TC etiology, thus diverse molecular signaling pathways and cellular conformations involved in TC development (Sierra et al., 2016).
Even though TC harbors several highly universal genetic alterations, some of which are unique to this cancer (Xing, 2005). It is still very challenging to predict this cancer due to the complex disease progression process and complicated molecular interactions involved in it. Network medicine is a new approach that focuses on the application of systems biology to holistically study the molecular complexity of a particular disease (Fiscon et al., 2018; Lee and Loscalzo, 2019). On the basis of a theory that genes with a similar pattern of expression can have similar functions or take part in specific pathways, weighted gene co-expression network analysis (WGCNA) is used to explain gene correlation patterns across microarray and RNA-seq data in order to obtain co-expressed gene networks related to various diseases (Chen et al., 2019; Guo et al., 2018; Huayan and Runhong, 2019; Wang et al., 2019). WGCNA constructs a gene co-expression network that captures transcript relationships as defined in the pattern of gene expression in which genes in the same module may have similar functionality or may be regulated by regulatory factors (Langfelder and Horvath, 2007).
In this analysis, WGCNA was used to classify the significant modules of the co-expressed gene networks associated with TC. Finally, a case-control study on TC tissue samples was conducted to approve WGCNA results as well as finding new prognosis markers for TC.
Materials and Methods
Data collection and preprocessing
The microarray data were taken from NCBI Gene Expression Omnibus (GEO) database with accession number GSE65144. This dataset is based on the GPL570 platforms and contain a total of 25 samples, including 13 anaplastic thyroid carcinoma tissue and 12 normal thyroid tissue. The raw data were corrected and quantile-normalized with the affy package of R 3.4.1 in Bioconductor. The annotation file published by Affymetrix (Affymetrix Human Genome U133 Plus 2.0 Array) was applied to assign probes for related gene IDs and symbols. Data of non-convertible probe IDs were excluded. The average statistics of identification expression for each sample were then conducted. Because of their varied, gene symbols were filtered through all samples, only the genes in the top 4,000 were selected for ensuing analysis.
Co-expression modules Construction of TC
Co-expression network for patient and control group gene expression data maintained through two matrixes using WGCNA package protocols. The first matrix was developed according to the Pearson test by transforming the gene expression profile into a matrix of similarity between pair genes, however, the second matrix named adjacency matrix evolved from similarity matrix conversion. The WGCNA package developed scale-free gene co-expression networks. When the value was set to 6, the adjacency matrix met the scale-free topology criteria. The topological overlap is a metric of gene-biological similarity based on the pairwise gene-co-expression correlation. Topological overlaps matrix (TOM) and TOM dissimilarity (diss TOM) were obtained through TOM similarity and adjacency matrix dissimilarity. Finally, a minimum module size of 30 genes per module and a cut height of 0.1 were described as clusters of extremely interconnected genes.
Module-trait relationships
To identify modules closely related to the clinical trait, the expression profiles of each module were identified using the module’s eigenegene (ME) and the association between the module and the trait was analyzed. The role of human genes in TC was evaluated using the gene significance (GS) value. The Module Membership (MM) has been established as the ME correlation and gene expression profile for each module. Therefore, they can be used to set up the network and determine the hub genes. Specifically, genes with both GS ≥0. 74 and MM ≥0. 86 are considered to be hub genes.
Functional enrichment of hub-genes
Functional enrichment analysis of hub-genes was performed using Gene Ontology (http://geneontology.org/) and Kyoto Encyclopedia of Genes and Genomes (KEGG) (https://www.genome.jp/kegg/) databases. Enriched ontological terms and pathways with the threshold of Benjamin-adjusted p-value<0.05 were selected and visualized using ClueGO in Cytoscape ver 3.7.2.
Hub-gene prioritization by genetic variant analysis
In order to investigate the impact of genetic variation, we extracted TC evidence for single-nucleotide polymorphisms (SNPs) from the GWAS catalog (Welter et al., 2013) resulting in 24 SNPs. Further on, these TC associated SNPs were filtered based on the similarity of mapped gene list and the obtained hub-genes from interest module. The similar genes were considered for next experimental validation.
Sample collection
In this case-control study, 81 TC patients were referred to the Imam Ali and khatam hospitals of Zahedan, City in the southeast of Iran during 2016-2018 and 165 healthy individuals without any history of neoplastic conditions were enrolled as volunteers. All of TC patients were histologically confirmed by pathologists and referred to Mehrazin nuclear medical center. The case and healthy participants were gender, age and ethnic matched. Informed consents were obtained from all participants before the sample collection. This study was signed informed consent code from the ethics committee of Zahedan University of Medical Science (IR. ZAUMS. REC.1397.289).
Genotyping assay
Genomic DNA was extracted from peripheral blood leukocytes of case and control group by routine extraction procure. FOXE1 gene single nucleotide polymorphisms (SNPs) were genotyped using tetra-ARMS PCR. The primer sequences and product size have listed in Table 2. FOXE1 amplification program included an initial denaturation step (95°C for 5 min) followed by 28 cycles, 95°C, 1min; 58°C, 30 s; 72°C, 30 s with a final extension 5 min at 72°C. The PCR products were detected on safe stained (Cinna Gen, Iran) 2 % agarose gel following UV light visualization.
Table 2.
Primers Sequencing and Amplicons Size were Used for FOXE1 Genotyping
| SNP | Primers sequence | Amplicon size (bp) |
|---|---|---|
| F-outer: 5′-TAA ACT AGC GGG CAC CAC AGA CC-3′ | Outers: 309 | |
| rs1867277 | R-outer: 5′-AGA GCT CAG GGG ATC GTC GC-3′ | Inner A:144 |
| F-inner: 5′-AGA GTC CAG TCC CGG GCG-3′ | Inner G: 201 | |
| R-inner: 5′-CAG CGG CGG TGG CCT AGT-3′ |
Statistical analysis
All statistical analysis was done by SPSS 19.0 software package (SPSS Inc., Chicago, IL). Descriptive statistics were carried out for determining the distribution of diverse genotypes within the case and healthy subjects. The p-value < 0.05 was considered significant.
Results
Identification of WGCNA modules
Preprocessing of data, including quantile normalization was performed to reduce the effects of technical noises. The plot of quantiles of expression levels across arrays is shown in Figure. S1. A total of 4,000 genes were included in WGCNA based on a variance of expression values. Three outliers were found by sample clustering in 25 samples and 22 samples were therefore included in the analysis (Figure S2). Subsequently, β=6 was identified as a soft-threshold power for construction of weighted co-expression network (Figure S3). As a result, the hierarchical clustering dendrogram described six modules represented in different color branches of the dendrogram (Figure S4). The number of genes for each module ranged from 144 (green) to 1480 (turquoise) (Table 1).
Table 1.
Module Colors Characterization. The co-expression modules identified by WGCNA
| Module color | #Genes | Correlation | p-value |
|---|---|---|---|
| Turquoise | 1480 | 0.87 | 7.00E-08 |
| Blue | 812 | 0.8 | 5.00E-06 |
| Green | 144 | 0.1 | 0.6 |
| Grey | 549 | 0.017 | 0.9 |
| Yellow | 210 | -0.13 | 0.6 |
| Brown | 805 | -0.17 | 0.4 |
Module-trait association analysis
Specific genes are determined for each module to evaluate the association of disease-presence modules in samples and module-module correlations the turquoise module as described was positively correlated with TC (r = 0.87, p-value = 7/00 E-08) (Figure 1). The most considerable pathways related to the turquoise module were visualized using the ClueGO tool. Hormone metabolic process and fertilization were the most important biological functions of the turquoise module (Figure 2A).
Figure 1.

Module-Trait Relationship. Each row corresponds to a module eigengene and column corresponds to TC status. Numbers in each cell represent the corresponding correlation and p-value
Figure 2.

Processes and Pathways Identified Within the (a) turquoise module and hub-genes (b). Node size corresponds to the number of associated genes, and node color reflects the statistical significance. The darker the pathway node, the more statistically significant it is, with a gradient from red (p-value 0.05-0.005) to black (p-value < 0.0005)
Identification of most significantly hub genes
The correlation between MM and GS of the turquoise module has contributed to the discovery of hub-genes that are closely correlated with TC (Figure S5). The top 54 hub-genes with GS ≥0.74 and MM ≥0.86 are listed in Figure S6. In our study, we examined the Enrichr analysis of our hub genes and found that thyroid hormone generation (GO: 0006590) including FOXE1, IYD, and DUOX2 is the most important biological process. In addition, KEGG 2019 Human analysis found that thyroid hormone synthesis is the most important pathway, including TPO, IYD, DUOXA2, LRP2, DUOX2, and TSHR (Figure 2B).
Furthermore, to find that which hub-gene SNPs were associated with TC pathogenicity, similarity analysis was conducted using Venny online program (https://bioinfogp.cnb.csic.es/tools/venny/) within obtained hub-genes and GWAS catalog containing 24 different SNPs for TC. The result indicated that only FOXE1 was common in both list. So, this gene was considered for next experimental validation (Figure 3).
Figure 3.
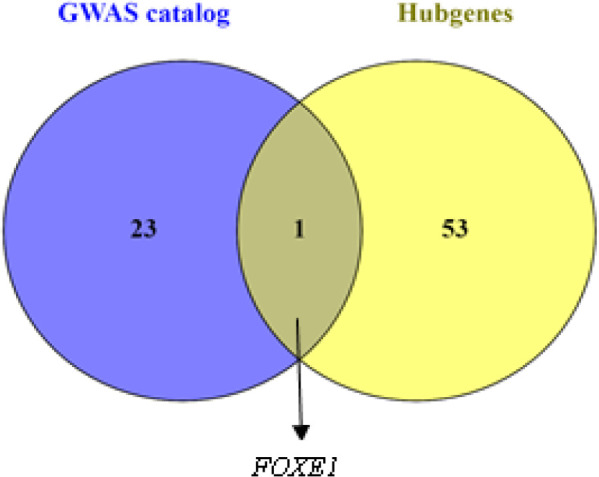
Similarity Analysis within Obtained Hub-Genes and GWAS Catalog. The result indicated that only FOXE1 was common to both list
Tissue validation for FOXE1 polymorphism (rs1867277)
In the current study, rs1867277 polymorphism of FOXE1 gene was assessed in 81 TC patients and 165 healthy individuals of southeast of Iran. From overall 246 individuals, 90 (36.5%) and 156 (63.4%) were males and females respectively. There was not any considerable difference in male and female distribution in healthy individuals 58 (35.2%) and 107 (64.8%) and TC patients 32 (39.5%) and 49 (60.5%), respectively. The mean age of the patients and healthy individuals were 46 ± 11.4 and 42±7.2, respectively. The results indicated that in rs1867277, the frequency of homozygous AA was 18.5% and 9.1% of patients and the control, respectively (p-value = 0.03, OR= 2.53). Also A allele frequency was 43.2 % of patients and 34.5% in the healthy group (p-value = 0.06, OR= 1.44) (Table 3).
Table 3.
FOXE1 Genotypic and Allelic Frequency in TC Patients (n=81) and Healthy (n=165) Objects
| Gene | Accession number | Chromosome location | Genotype | Case N (%) N = 81 | Control N (%) N = 165 | p-value | OR |
|---|---|---|---|---|---|---|---|
| GG | 26 (32.1) | 66 (40.0) | Ref:1 | ||||
| AG | 40 (49.4) | 84 (50.9) | 0.52 | 1.2 | |||
| FOXE1 | rs1867277 | 9q22.33 | AA | 15 (18.5) | 15 (9.1) | 0.03* | 2.53 |
| AG+AA | 55 (67.9) | 99 (60.0) | 0.23 | 1.41 | |||
| Allele G | 92 (56.8) | 216 (65.5) | Ref:1 | ||||
| Allele A | 70 (43.2) | 114 (34.5) | 0.06 | 1.44 |
*, Significant values in bold. FOXE1 genotypes (GG, wild type; AG, heterozygote; AA, mutant type).
Discussion
TC is the multifactorial disease by 1% prevalence of the entire human cancers and known as the most frequent endocrine malignancy (Liyanarachchi et al., 2013; Nguyen et al., 2015). Genetic predisposition, radiation exposure and hormonal factors are the possible causes of TC etiology, but the exact reason is still poorly understood (Schmidbauer et al., 2017). Despite the recent development of diagnostic and therapeutic approaches, the prognosis of TC remains poorly understood (Saini et al., 2018). Nowadays, the main public health goal is to develop biomarkers and targeted therapy approaches for early diagnosis and treatment of TC, respectively (Abdullah et al., 2019). To this end, our study was conducted to identify biomarkers associated with progression of TC using WGCNA. In the present study, we have analyzed GSE65144 dataset containing 25 samples including 13 anaplastic thyroid carcinoma tissue and 12 normal thyroid tissue. Co-expression network analysis by WGCNA suggested that highly preserved turquoise module with 1,480 genes was significantly correlated to TC. The top 54 hub genes of this module were listed in Fig. S6, most of them are functionally correlated with thyroid hormone generation (GO: 0006590). Similarity analysis between obtaining hub-genes and GWAS catalog revealed that, from this hub-genes list, only FOXE1 has been reported previously to contain a mutation associated with TC. FOXE1 is a kind of thyroid transcription factors that play tumor suppressor role in the control of cell proliferation and tumor invasion in thyroid cells (Parlato et al., 2004). The cellular transcription factor 1 belongs to the basic helix-loop-helix group (Takahashi et al., 2010). The rs1867277 of FOXE1 gene is an A/G single-nucleotide variation in the 5′ untranslated region on human chromosome 9 (Landa et al., 2009). It was connected to a gene modulation of the transcriptional regulators. Previously, it has been demonstrated significant association between rs1867277 variation and differentiated TC of the many different populations (Pereda et al., 2015). On this basis, FOXE1 gene was considered for next experimental validation. Evidence indicated that FOXE1 gene can enhance the susceptibility of sporadic TC in a Japanese and European population (Gudmundsson et al., 2009; Matsuse et al., 2011). To this end, we conducted a case-control study to evaluate the effects of FOXE1 functional polymorphisms (rs1867277) on the development of TC in Sistan and Balouchestan province of Iran.
The result of our study showed a AA genotype frequency of 18% and 9% in case and control (p-value = 0.03, OR = 2.53). In onother studies, for example, Somuncu et al., (2015) it is shown that AA genotype of rs1867277 variation is considerably related to numerous histopathological parameters. In addition, AA genotype was notably linked with the classical variant that belongs to PTC. Kula et al., (2017)confirmed the positive association between rs1867277 and PTC predisposition in Polish population (p-value = 1×10-6, OR = 1.59) . In other study, Nikitski et al., (2017) indicated remarkable relation between FOXE1 coding region and PTC of functional SNPs rs1867277 in the Japanese and Belarusian populations by univariate analysis). Also, Landa et al., (2009) indicated that A allele of rs1867277 was related to TC risk. Furthermore, in another study, A allele reported significantly related to TC and they showed that FOXE1 polymorphisms were associated with risk of PTC (Somuncu et al., 2015). Further, Kula et al., (2010) showed, A allele was probably associated with TC in the metastatic form. FOXE1 is necessary for proliferation and migration of thyroid cells during morphogenesis. Hence, clarified of FOXE1 risk variants and their functions can be fundamental in tumor transformation (Tomaz et al. 2012). On the other hand, FOXE1 as thyroid particular DNA binding could identify thyroperoxidase and thyroglobulin (Cuesta et al., 2007; Fernández et al., 2013). In this regard, Three cohort studies, confirmed the positive association in the rs1867277 and the FOXE1 poly Ala tract polymorphism with TC in Portuguese, Australian and Spanish population (Bullock et al., 2012; Kallel et al., 2011; Tomaz et al., 2012). FOXE1 is essential for thyroid and pituitary gland formation and play in the key role in the regulatory function of transcription factors that involved in thyroid differentiation in the embryonic phase.
In conclusion, via systems biology approach and co-expression network analysis, we identified a novel module involved in TC pathogenicity and discovered main hub-genes e.g. FOXE1. To confirm bioinformatic result, a case-control study was conducted on tissue samples to evaluate associations between FOXE1 (rs1867277) polymorphisms with TC. The results showed a significant correlation among rs1867277 and TC pathogenicity, suggest that FOXE1 (rs1867277) SNP can be a potential predictor biomarker for TC diagnosis. Moreover, functional studies are required to confirm its role as a therapeutic target. We recommend detailed assessing these polymorphisms in larger populations of various geographical regions.
Ethical Approval
The ethical code for this study is IR.ZAUMS.REC. 1397.289.
Conflict of Interests
There is no conflict of interest.
References
- Abdullah MI, Junit SM, Ng KL, et al. 2019) Papillary thyroid cancer: Genetic alterations and molecular biomarker investigations. Int J Med Sci. 16:450. doi: 10.7150/ijms.29935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray F, Ferlay J, Soerjomataram I, et al. 2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- Bullock M, Duncan EL, O’Neill C, et al. 2012) Association of FOXE1 polyalanine repeat region with papillary thyroid cancer. J Clin Endocrinol Metab. 97:E1814–19. doi: 10.1210/jc.2012-1456. [DOI] [PubMed] [Google Scholar]
- Chen W, Chen X, Wang Y, et al. Construction and Analysis of lncRNA-Mediated ceRNA Network in Cervical Squamous Cell Carcinoma by Weighted Gene Co-Expression Network Analysis. Med Sci Monit. 2019;25:2609–22. doi: 10.12659/MSM.913471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuesta I, Zaret KS, Santisteban P. The forkhead factor FOXE1 binds to the thyroperoxidase promoter during thyroid cell differentiation and modifies compacted chromatin structure. Mol Cell Biol. 2007;27:7302–14. doi: 10.1128/MCB.00758-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies L, Welch HG. Increasing incidence of thyroid cancer in the United States, 1973-2002. JAMA. 2006;295:2164–67. doi: 10.1001/jama.295.18.2164. [DOI] [PubMed] [Google Scholar]
- Fernández LP, López-Márquez A, Martínez ÁM, et al. New insights into FOXE1 functions: identification of direct FOXE1 targets in thyroid cells. PLoS One. 2013;8:e62849. doi: 10.1371/journal.pone.0062849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiscon G, Conte F, Farina L, et al. Network-based approaches to explore complex biological systems towards network medicine. Genes. 2018;9:437. doi: 10.3390/genes9090437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldgar DE, Easton DF, Cannon-Albright LA, et al. Systematic population-based assessment of cancer risk in first-degree relatives of cancer probands. J Natl Cancer Inst. 1994;86:1600–08. doi: 10.1093/jnci/86.21.1600. [DOI] [PubMed] [Google Scholar]
- Gudmundsson J, Sulem P, Gudbjartsson DF, et al. Common variants on 9q22 33 and 14q13 3 predispose to thyroid cancer in European populations. Nat genet. 2009;41:460–64. doi: 10.1038/ng.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo N, Zhang N, Yan L, et al. Weighted gene coexpression network analysis in identification of key genes and networks for ischemicreperfusion remodeling myocardium. Mol Med Rep. 2018;18:1955–62. doi: 10.3892/mmr.2018.9161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H, Li W, Liyanarachchi S, et al. Genetic predisposition to papillary thyroid carcinoma: involvement of FOXE1, TSHR, and a novel lincRNA gene, PTCSC2. J Clin Endocrinol Metab. 2015;100:E164–E72. doi: 10.1210/jc.2014-2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang R, Liu H. Identification of temporal characteristic networks of peripheral blood changes in Alzheimer’s disease based on weighted correlation network analysis. Front Aging Neurosci. 2019;2019:11–83. doi: 10.3389/fnagi.2019.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallel R, Belguith-Maalej S, Akdi A, et al. Genetic investigation of FOXE1 polyalanine tract in thyroid diseases: new insight on the role of FOXE1 in thyroid carcinoma. Cancer Biomark. 2011;8:43–51. doi: 10.3233/DMA-2011-0824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo T, Ezzat S, Asa SL. Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nat Rev Cancer. 2006;6:292–306. doi: 10.1038/nrc1836. [DOI] [PubMed] [Google Scholar]
- Kula D, Kalemba M, Jurecka-Lubieniecka B, et al. Genetic predisposition to papillary thyroid cancer. Endokrynol Pol. 2010;61:486–89. [PubMed] [Google Scholar]
- Kula D, Kalemba M, Puch Z, et al. Age at diagnosis and gender modify the risk of 9q22 and 14q13 polymorphisms for papillary thyroid carcinoma. Endokrynol Pol. 2017;68:283–89. doi: 10.5603/EP.2017.0021. [DOI] [PubMed] [Google Scholar]
- Landa I, Ruiz-Llorente S, Montero-Conde C, et al. The variant rs1867277 in FOXE1 gene confers thyroid cancer susceptibility through the recruitment of USF1/USF2 transcription factors. PLoS Genet. 2009;5:e1000637. doi: 10.1371/journal.pgen.1000637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. Eigengene networks for studying the relationships between co-expression modules. BMC Syst Biol. 2007;2007:1–54. doi: 10.1186/1752-0509-1-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee LY-H, Loscalzo J. Network medicine in pathobiology. Am J Pathol. 2019;189:1311–26. doi: 10.1016/j.ajpath.2019.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liyanarachchi S, Wojcicka A, Li W, et al. Cumulative risk impact of five genetic variants associated with papillary thyroid carcinoma. Thyroid. 2013;23:1532–40. doi: 10.1089/thy.2013.0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuse M, Takahashi M, Mitsutake N, et al. The FOXE1 and NKX2-1 loci are associated with susceptibility to papillary thyroid carcinoma in the Japanese population. J Med Genet. 2011;48:645–48. doi: 10.1136/jmedgenet-2011-100063. [DOI] [PubMed] [Google Scholar]
- Nguyen QT, Lee EJ, Huang MG, et al. Diagnosis and treatment of patients with thyroid cancer. Am Health Drug Benefits. 2015;8:30–40. [PMC free article] [PubMed] [Google Scholar]
- Nikitski AV, Rogounovitch TI, Bychkov A, et al. Genotype analyses in the Japanese and Belarusian populations reveal independent effects of rs965513 and rs1867277 but do not support the role of FOXE1 polyalanine tract length in conferring risk for papillary thyroid carcinoma. Thyroid. 2017;27:224–35. doi: 10.1089/thy.2015.0541. [DOI] [PubMed] [Google Scholar]
- Parlato R, Rosica A, Rodriguez-Mallon A, et al. An integrated regulatory network controlling survival and migration in thyroid organogenesis. Dev Biol. 2004;276:464–75. doi: 10.1016/j.ydbio.2004.08.048. [DOI] [PubMed] [Google Scholar]
- Pereda CM, Lesueur F, Pertesi M, et al. Common variants at the 9q22 33 14q13 3 and ATM loci and risk of differentiated thyroid cancer in the Cuban population. BMC Genet. 2015;2015:16–22. doi: 10.1186/s12863-015-0180-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini S, Tulla K, Maker AV, et al. Therapeutic advances in anaplastic thyroid cancer: a current perspective. Mol Cancer. 2018;17:154. doi: 10.1186/s12943-018-0903-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidbauer B, Menhart K, Hellwig D, et al. Differentiated thyroid cancer-treatment: State of the Art. Int J Mol Sci. 2017;18:1292. doi: 10.3390/ijms18061292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somuncu E, Karatas A, Ferahman S, et al. The investigation of FOXE1 variations in papillary thyroid carcinoma. Int J Clin Exp Pathol. 2015;8:13458–64. [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Saenko VA, Rogounovitch TI, et al. 2010) The FOXE1 locus is a major genetic determinant for radiation-related thyroid carcinoma in Chernobyl. Hum Mol Genet. 19:2516–23. doi: 10.1093/hmg/ddq123. [DOI] [PubMed] [Google Scholar]
- Tomaz RA, Sousa I, Silva JG, et al. 2012) FOXE1 polymorphisms are associated with familial and sporadic nonmedullary thyroid cancer susceptibility. Clin Endocrinol (Oxf) 77:926–33. doi: 10.1111/j.1365-2265.2012.04505.x. [DOI] [PubMed] [Google Scholar]
- Wang X, Song P, Huang C, et al. Weighted gene coexpression network analysis for identifying hub genes in association with prognosis in Wilms tumor. Mol Med Rep. 2019;19:2041–50. doi: 10.3892/mmr.2019.9881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welter D, MacArthur J, Morales J, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2013;42:D1001–D1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing M. BRAF mutation in thyroid cancer Endocrine-related cancer. Endocr Relat Cancer. 2005;12:245–62. doi: 10.1677/erc.1.0978. [DOI] [PubMed] [Google Scholar]