

Abstract
There is a growing appreciation that insect distribution and abundance are associated with the limits of thermal tolerance, but the physiology underlying thermal tolerance remains poorly understood. Many insects, like the migratory locust (Locusta migratoria), suffer a loss of ion and water balance leading to hyperkalaemia (high extracellular [K+]) in the cold that indirectly causes cell death. Cells can die in several ways under stress, and how they die is of critical importance to identifying and understanding the nature of thermal adaptation. Whether apoptotic or necrotic cell death pathways are responsible for low-temperature injury is unclear. Here, we use a caspase-3 specific assay to indirectly quantify apoptotic cell death in three locust tissues (muscle, nerves and midgut) following prolonged chilling and recovery from an injury-inducing cold exposure. Furthermore, we obtain matching measurements of injury, extracellular [K+] and muscle caspase-3 activity in individual locusts to gain further insight into the mechanistic nature of chilling injury. We found a significant increase in muscle caspase-3 activity, but no such increase was observed in either nervous or gut tissue from the same animals, suggesting that chill injury primarily relates to muscle cell death. Levels of chilling injury measured at the whole animal level, however, were strongly correlated with the degree of haemolymph hyperkalaemia, and not apoptosis. These results support the notion that cold-induced ion balance disruption triggers cell death but also that apoptosis is not the main form of cell damage driving low-temperature injury.
Keywords: cold tolerance, thermal performance, thermal limits, neuromuscular system, programmed cell death, ionoregulatory collapse
1. Introduction
An understanding of the physiology underlying thermal limits to performance is essential to interpreting and identifying target genes or the evolutionary lability of these traits. The majority of insects are chill-susceptible, meaning they lack physiological mechanisms capable of protecting them from low-temperature injury [1]. These insects enter a state of paralysis called chill coma [2] that can be reversed following rewarming. The temperature of this paralysis event and the time required to recover the ability to stand following a cold stress (chill coma recovery time; CCRT) are non-lethal and widely used measures of insect chill tolerance [3–6]. If a cold exposure is severe enough (defined depending on the species/population under study and its prior thermal history), however, chill-susceptible insects suffer from cold-induced injuries—termed chilling injury—that can be sublethal or lethal [1].
Chilling injury typically manifests as defects in an insect's ability to fly, walk or stand following chilling, while mortality is often quantified as a complete inability to move, or to undergo a critical phase of development, like adult emergence [7–9]. Although the term chill injury is used to describe multiple organismal outcomes, it most often refers to an insect's dexterity following cold stress. As such, cell death in the nerves and/or muscles is likely to directly underlie several common cold tolerance metrics.
Cell death is a common consequence of cold exposure in chill-susceptible insects, and has been associated with a systemic loss of ion and water homeostasis that occurs during chronic chilling [1]. Low temperatures suppress active ion transport [8,10] and damage paracellular barriers [11,12]. During prolonged chilling, a net leak of ions down their concentration gradients across cell membranes and epithelia is commonly observed [7,13,14], and a consequence of this mismatch is a systemic rise in extracellular [K+] [7,14]. The combined effects of slowed active ion transport and elevated extracellular [K+] depolarize excitable cells [15–17], triggering excessive Ca2+ influx that is proposed to directly initiate cell death, and both apoptotic and/or necrotic cell death have been blamed for insect chilling injury [18–20].
Understanding when, where and how cell death occurs in insects during or following chilling is essential to determining the primary causes of organismal chilling injury, but is also critical to understanding how insects modulate cold tolerance within the lifetime of an individual (e.g. acclimation) or over evolutionary time. Changes to cold tolerance within an insect appear to arise from adjustments that attenuate the physiological cascade of failure [1]. For example, cold-acclimated and adapted insects rely less on Na+ as an extracellular osmolyte [21,22], better maintain paracellular barrier function in the cold [11,12], have renal systems more efficient at clearing excess K+ from the haemolymph [12,23–25], and defend against muscle depolarization [17,26,27]. All of these adjustments serve to protect against injury by targeting upstream causes of failure, but chill tolerance may also be intimately tied to the ability to prevent cell death in the face of homeostatic collapse [20,27], or even the ability to clear damaged tissue following rewarming [28].
Cellular damage has been observed in insect muscles, fat body and gut epithelia following cold stress, and this damage appears to correlate with chilling injury phenotypes measured at the organismal level [15,17,18]. These observations of tissue damage, however, have been derived using one of two approaches. In some cases, they have been quantified from live/dead cell viability assays that (1) do not distinguish among necrotic (uncontrolled) and apoptotic (caspase-mediated/regulated) cell death, and (2) cannot penetrate the blood–brain barrier and thus have not been used to assess nervous damage following chilling [29]. With an alternative approach, Yi et al. used a TUNEL assay to quantify DNA fragmentation and interpreted their findings as cell death in the flight muscles of Drosophila following chilling occurring primarily via apoptosis [30]. Brief pre-exposure to chilling in a manner that improves chill tolerance (a rapid cold-hardening treatment) could inhibit this effect in tissues of flesh flies [20]. Like live/dead assays, however, TUNEL assays cannot distinguish among multiple forms of cell death [31], as DNA fragmentation is a common consequence of cell death. It is therefore likely that caspase-mediated apoptosis is not acting alone to cause chilling injury. Furthermore, since the nervous system has not been explored in the context of apoptotic or necrotic cell death, whether muscle or nerve damage (or both) cause organismal chilling injury in insect phenotypes remains entirely unclear.
Caspases serve multiple functions in insects [32,33], but their primary role is in programmed cell death cascades where they are produced in advance of cell death and maintained in an inactive precursor form (pro-caspase). Regulated cell death pathways are generally well-conserved among animals, and the roles of individual caspases are increasingly well-understood [34]. In Drosophila, Drice (a caspase-3 orthologue) is the major executioner caspase that is essential for programmed cell death during development and in response to tissue/cell damage [35–37]. This central role of caspase-3 and its orthologues as important drivers of cell destruction is conserved among insects and mammals. Because caspase-3 and its orthologues are mainly associated with apoptotic cell death and not necrosis [38], they can be a useful tool for understanding the ultimate causes of chilling injury.
Here, we use the migratory locust (Locusta migratoria) to test the hypothesis that ionoregulatory collapse drives caspase-mediated cell death in both the nerves and muscles and is responsible for insect chilling injury. We exposed locusts to up to 48 h at −2°C to determine a duration of exposure that caused significant and variable sublethal chilling injury and used this treatment to examine activation of caspase-3-like proteins (executioner caspases associated with apoptosis) in a thoracic muscle, the metathoracic ganglion and the midgut (as a negative control as midgut cells use autophagy, not caspase activation for programmed cell death [39]). Since caspase-3 activation occurred specifically in the muscles in the cold, we obtained matching measurements of survival, haemolymph [K+], and muscle executioner caspase activity from individual locusts during cold exposure. This allowed us to investigate the links between these parameters and generate the first data relating individual variation among these measures in any insect. With this approach, we provide evidence that injury to the muscles, and not the central ganglia, is probably responsible for motor defects following cold exposure, and that while cold stress activates muscle caspase, the degree of hyperkalaemia is a far better quantitative predictor for organismal chilling injury than muscle executioner caspase activity. Thus, other cell death pathways and/or other neuromuscular impairments (e.g. synaptic failure) are probably more prominent factors responsible for chilling injury.
2. Material and methods
(a). Animal husbandry
Our colony of Locusta migratoria is maintained at Carleton University, Ottawa, ON. This colony is continuously breeding under crowded conditions. Locusts are held at 30°C, with a 16 : 8 day/night cycle, fed on wheatgrass and an oat mixture (65% oats, 10% wheat germ, 10% wheat bran, 5% skim milk powder). For all experiments, locusts were taken from a crowded cage at three to four weeks post-final ecdysis and were used in an approximately 1 : 1 sex ratio for all experiments.
(b). Chill coma recovery time and survival following exposure to −2°C
CCRT and chilling injury were assessed following exposure to −2°C following previously described methods [40]. Each locust was placed in a 50 ml ventilated polypropylene centrifuge tube before being placed in a mixture of ethylene glycol and water (with holes in the tube lid in contact with the air) inside a refrigerated circulator (28 l with advanced programmable controller, VWR International, Radnor, USA). Temperature was set to hold locusts at 20°C for 15 min and then decrease to −2°C at a rate of −0.1°C min–1 and held there for up to 48 h. Groups (n = 10 per group) of locusts were removed from the bath at four-time points (2, 6, 24 and 48 h), and a control group was held in tubes at room temperature (approx. 22°C) for 24 h. The control group was not fed nor allowed to drink for the entire 24 h to best match the experimental groups. Cooling bath temperature was confirmed to keep locusts at −2°C (±0.5°C) using three type-K thermocouples (connected to a TC-08 data logger, Pico Technology Inc., St Neots, Cambridgeshire, UK) in three different tubes containing locusts.
Once removed from the cooling bath, locusts were placed at room temperature (22 ± 0.5°C) and gently stimulated every 5 min until they were observed to stand, or until 60 min had passed. Locusts were then returned to their respective 50 ml tube, with access to food and water, until survival score was assessed 24 h later. Survival score was rated on a scale of 0–5 in a manner similar to that used previously [16] by removing each locust from the tube and gently coaxing them to move. Survival was scored as follows: 0 = motionless/dead; 1 = twitching without coordinated movement; 2 = able to move but unable to stand; 3 = able to stand; 4 = able to walk, jump and initiate flight, but with slow reaction time; 5 = able to walk, jump and initiate flight with no observable defects or delays in reaction time. Movement was assessed in each locust independently, with a focus on wing movement (including free flight), leg movement (including jumping ability) and overall reaction time.
(c). Caspase-3 activity following cold exposure
Caspase-3-like activity was measured in three tissues dissected from locusts from three treatment groups (n = 6 per treatment): (1) Controls held at 28°C for 24 h, (2) cold exposed and dissected immediately after 24 h at −2°C and (3) cold exposed and dissected after a 2 h recovery period to test for delayed activation of caspase-3. The cooling bath followed an identical ramping regime used to assess chill coma recovery and chilling injury.
To isolate tissues, locusts were quickly decapitated, and all appendages were removed before a single incision was made in the anterior–posterior axis of the dorsal cuticle. The body cavity was pinned open, submerged in standard locust saline (in mmol l–1: 140 NaCl, 8 KCl, 3 CaCl2, 2 MgCl2, 90 sucrose, 5 glucose, 5 trehalose, 1 proline, 10 HEPES; pH 7.2), and a sample of the posterior midgut (excluding the caeca) was taken and cleaned with an aliquot of clean saline. Then the posterior metathoracic tergocoxal muscle (M90 following [41], a flight muscle) and the metathoracic ganglion were dissected out. All tissues were snap-frozen in liquid nitrogen after dissection and stored at −80°C until use.
Caspase-3-like activity was quantified using the EnzChek Caspase-3 Assay kit #1 (Molecular Probes, Eugene, OR, USA). Tissue samples were thawed on ice for 5 min, before being suspended in 100 µl mg−1 lysis buffer (10 mmol l−1 TRIS; pH 7.5, 0.1 mmol l−1 NaCl, 1 mmol l−1 EDTA, 0.01% Triton X-100, in dH2O). Each sample was sonicated for rounds of 5 s (with 15 s breaks on ice between rounds to prevent overheating) until fully homogenized. Samples were then centrifuged for 5 min at 2000 × g at 5°C. A 50 µl aliquot of sample supernatant was transferred to a black, clear bottomed, 96-well microplate.
Along with blank samples (containing only 100 µl lysis buffer), two additional controls were run in each assay plate. First, a subset of samples containing 1 µl of (1 mmol l−1 in DMSO) Ac-DEVD-CHO (a specific inhibitor of caspase-3-like proteases) were included in a subset of duplicate wells to confirm that the fluorescence observed was specifically caused by the activity of caspase-3-like proteases (confirmed). Secondly, samples with 1 µl of the DMSO solution were measured to control for the effect of the DMSO itself (there was none).
A 2× working solution was prepared by adding 2% V : V Z-DEVD-AMC substrate (10 mmol l−1 in DMSO) to the 2× reaction buffer (2.5 mmol l−1 PIPES, 0.5 mmol l−1 EDTA, 0.025% CHAPS, diluted in dH2O, pH 7.4 and 1% V : V DTT (in 1 mmol l−1 in DMSO)). Fifty millilitres of the working solution was added to each sample and control (combined volume of 100 µl). The samples and controls were left to incubate for 30 min at room temperature. To quantify caspase-3 activity through the DEVD-AMC substrate, serial dilutions of AMC ranging from 0 to 100 µM (from a stock solution also containing 10 mmol l−1 DMSO) were added to single wells (100 µl each). Fluorescence of the samples (excitation: 324 nm, emission: 441 nm) was measured with a CYTATION5 fluorescence spectrophotometer (BioTek Instruments, Winooski, VT, USA).
(d). Heat shock controls
We were surprised to observe differences in caspase-3-like activity between the nerve and muscle tissues following chilling, so we examined whether this was a general pattern following thermal stress that causes organismal injury or was specific to our chilling protocol. We thus purposefully induced apoptosis in a separate group of locusts (n = 9) by exposing them to a lethal heat shock (60 ± 1°C for approx. 10 min). After resting at 28°C for 30 min, the locusts were dissected. While not all locusts were completely motionless directly after the heat shock, all of the locusts were scored as a 0 (dead/motionless) after the 30 min recovery period. The dissection and caspase detection protocol described above was then repeated for all three tissues collected from these locusts.
(e). Matching measurements of injury, haemolymph K+ concentration and muscle caspase-3 activity
In a separate set of experiments, locusts were exposed to −2°C for 0, 24 and 48 h (following the same procedure as above; the 0 h group was never exposed) before being moved to room temperature. After removal, a small haemolymph sample was collected between the head and the thorax with a glass capillary tube (approx. 1 µl). The haemolymph was then transferred to a small dish and kept under hydrated mineral oil. After 2 h of recovery, the locusts were scored for survival (0–5 as described above) and an additional wing-specific score was estimated (also 0–5) to rank motor function defects and injury to the wing muscles by observing range of motion and reaction time during predator escape simulations (hovering a hand over the locust and trying to pick them up), by gently prodding them, during free flight attempts, and by manually moving the wings while the locusts were handled after the flight attempts. The score was rated as follows: 0 = wing motionless; 1 = unresponsive but twitching; 2 = barely reactive; 3 = definite reaction to external stimulus but limited range of motion (e.g. wings not fully opening or closing); 4 = full range of motion, but uncoordinated wing beats (wings used during jumps to attempt flight, unsuccessfully), or with delayed reaction (still unable maintain flight); 5 = fully functional (i.e. maintained flight over several metres). After scoring locusts, a second haemolymph sample was taken, and the M90 flight muscle was dissected out under standard saline, quickly blotted dry, transferred to a pre-weighed Eppendorf tube and weighed, snap-frozen in liquid N2 and stored at −80°C until measurement of caspase-3-like activity (as described above).
Haemolymph [K+] was measured using ion-selective glass microelectrodes as described previously [25]. Briefly, glass capillaries (TW-150-4, World Precision Instruments (WPI), Sarasota, FL, USA) were pulled to a fine tip and silanized in an atmosphere of n,n-dimethyltrimethylsilylamine (Sigma Aldrich, St Louis, MO, USA). Silanized glass microelectrodes were then back-filled with 100 mmol l−1 KCl and front-filled with K+ ionophore (K+ ionophore I, cocktail B, Sigma Aldrich, St Louis, MO, USA). A thinly pulled glass electrode (IB200F-4, WPI) back-filled with 500 mmol l−1 KCl was used as a reference. Before every measurement, electrodes were calibrated in 10 and 100 mmol l−1 KCl solutions (LiCl was used to balance osmolality) to obtain the Nernstian slope (approx. 58.2 mV per 10-fold change in concentration at 25°C), and only electrodes with a slope between 50 and 62 mV were used (mean ± s.d. of 21 electrodes: 54.3 ± 2.4 mV). For this experiment, 6 locusts were used as controls and 24 locusts were exposed for both the 24 h and 48 h. It was not possible to obtain a second haemolymph sample from five locusts (three and two from the 24 h and 48 h exposure group, respectively), so the sample sizes here were n = 6, 21 and 22, and four muscle samples were lost during transfer out of the liquid N2 (three and one from the 24 h and 48 h exposure group, respectively), lowering the sample size for muscle caspase-3-like activity to n = 6, 21 and 23.
(f). Data analysis
All data analysis was completed in R v. 3.5.3 [42]. All datasets were tested for normality using boxplots and Shapiro-Wilk tests (shapiro.test() function), and non-parametric approaches were used when appropriate. All starting models included sex as a factor, but this factor was eliminated in all but one case where it interacted with exposure time: CCRTs following exposure to −2°C were analysed using a generalized linear model with exposure time as a continuous variable and sex as a factor. Survival scores were compared among exposure times using Kruskall–Wallis tests followed by Dunn's multiple comparisons tests using the kruskal.test() and dunnTest() (FSA package) functions, respectively. The effect of cold exposure on caspase-3 activity was analysed using separate one-way ANOVAs for each tissue, followed by Tukey HSD post hoc tests. Heat-activated caspase-3 activity (i.e. the positive control) in each tissue was compared to controls using Student's t-tests. For the dataset on individual variation, the effect of cold exposure on the survival score and caspase-3 activity were analysed using Kruskal–Wallis tests followed by Dunn's multiple comparison tests, while those of haemolymph K+ concentration was analysed using a one-way ANOVA followed by Tukey's HSD post hoc test. Correlations between survival scores and caspase-3 or haemolymph K+ concentration were tested using linear regression and nonlinear regression to a sigmoidal model (using the nls() function; model parameters specified in figure text), respectively, and the best fitting model (based on R2 values and AIC scores), if statistically significant, is displayed. All values listed are means ± s.e.m. unless otherwise stated, and the critical level for statistical significance was 0.05 in all analyses.
3. Results
(a). Chill coma recovery time and survival following exposure to −2°C
The cold tolerance of locusts was examined by measuring CCRT at specific time points during exposure to −2°C and was followed by a survival assessment (scale of 0–5) 24 h after the end of the cold exposure (figure 1). Exposure to −2°C gradually increased CCRT for both sexes (t2,21 = 13.8, p < 0.001 for exposure time; t1,21 = 0.8, p = 0.446 for sex), however, females became increasingly slower at recovering as exposure time increased (interaction: t2,21 = −2.8, p = 0.010) such that recovery took 9.2 ± 0.3 min and 9.0 ± 0.4 min for females and males, respectively, after 2 h of exposure and increased to 49.9 ± 3.9 min and 36.6 ± 9.2 min after 24 h. After 48 h no locusts recovered within the 60 min time limit (figure 1a). A similar decrease in post-exposure performance was found for the survival scores (no effect of sex); survival scores decreased from 4.9 ± 0.1 after 2 h of cold exposure to 1.0 ± 0.3 after 48 h (H = 25.5, p < 0.001; figure 1b).
Figure 1.

Cold stress that causes injury also causes activation of caspase-3-like activity in the muscles of locusts. Prolonged exposure to −2°C gradually (a) increased the time needed for locusts to assume a standing position in both females (squares) and males (triangles) and (b) reduced the survival outcome after recovery at a permissive temperature. (c) During this cold exposure, caspase-3-like was increased in muscle tissue (orange, solid line) but remained the same in midgut (brown, dashed line) and nervous tissue (blue, dotted line). (d) Lethal heat exposure was used as a positive control and resulted in caspase-3-like activation in muscle and nervous tissue, while caspase-3-like activity decreased in the midgut. Individual data points are represented by small, empty symbols. Error bars not visible (for c,d) are occluded by the symbols. (Online version in colour.)
(b). Caspase-3 activity induced by exposure to thermal extremes
To test whether the observed reduction in survival was related to an increase in apoptotic activity, we measured caspase-3-like activity in muscle (flight muscle M90, after Snodgrass [41]), nervous tissue (metathoracic ganglion), and midgut (negative control) both after an intermediate cold exposure and after a brief recovery period, and followed each up with a positive heat exposure control (figure 1). Exposure to −2°C for 24 h increased caspase-3-like activity in muscle tissue from 0.8 ± 0.2 pmol AMC cleaved min−1 mg−1 in control locusts to 2.4 ± 0.4 pmol AMC cleaved min−1 mg−1, which was similar to the 2.8 ± 0.5 pmol AMC cleaved min−1 mg−1 measured after 2 h of recovery (F2,26 = 6.4, p = 0.006; figure 1c). Caspase-3-like activity remained unchanged in both midgut tissue and nervous tissue (F2,24 = 0.3, p = 0.755 and F2,23 = 1.6, p = 0.228, respectively) with activities ranging from ∼0.3 to 0.9 pmol AMC cleaved min−1 mg−1 (figure 1c). Brief exposure to 60°C was used a positive control for caspase activation (figure 1d), and increased caspase-3-like activity in flight muscle from 0.4 ± 0.1 pmol AMC cleaved min−1 mg−1 to 3.6 ± 0.8 pmol AMC cleaved min−1 mg−1 (t16 = −3.9, p = 0.001). Unlike the cold, lethal heat stress also increased caspase-3-like activity in nervous tissue from −0.4 ± 0.2 pmol AMC cleaved min−1 mg−1 to 0.9 ± 0.2 pmol AMC cleaved min−1 mg−1 (t15 = −4.9, p < 0.001), while it decreased in midgut tissue from 0.5 ± 0.1 pmol AMC cleaved min−1 mg−1 to 0.0 ± 0.2 pmol AMC cleaved min−1 mg−1 (t14 = 0.036).
(c). Individual variation in survival, haemolymph K+ concentration and caspase-3 activity
To gain further insight into the relationship between survival, ion balance disruption and caspase-3-like activity, we took advantage of the wide inter-individual variation noted in these variables in the first set of experiments. Here, we scored survival and measured haemolymph K+ concentration and flight muscle caspase-3-like activity in the same individuals, using unexposed locusts and locusts exposed to 24 and 48 h of exposure to −2°C (and 2 h of recovery; figure 2). As previously demonstrated, survival score decreased with longer cold exposures (H = 36.6, p < 0.001; figure 2a). In the same locusts, haemolymph K+ concentration increased during exposure and recovered over the 2 h of recovery before dissection of the muscle tissue (F5,97 = 51.1, p < 0.001; figure 2b). Specifically, haemolymph [K+] increased from 9.7 ± 0.6 mmol l−1 in controls to 23.8 ± 0.8 mmol l−1 after 24 h and was restored to 16.0 ± 0.8 mmol l−1 after recovery. In the group exposed for 48 h, it increased to 37.0 ± 1.4 mmol l−1 and returned to 24.6 ± 1.6 mmol l−1 after the recovery period. Correlating survival score and haemolymph [K+] for each locust revealed a tight, sigmoidal-like relationship with an IC50 (‘Injury Concentration 50'; haemolymph [K+] that correlates to a 50% reduction in survival score) of 34.8 ± 0.7 mmol l−1 (figure 2c). In the same animals, muscle caspase-3-like activity increased during cold exposure (samples taken after the 2 h recovery period) from −0.9 ± 0.2 pmol AMC cleaved min−1 mg−1 to 7.3 ± 2.2 and 5.3 ± 2.1 pmol AMC cleaved min−1 mg−1 after 24 and 48 h, respectively (H = 16.8, p < 0.001; figure 2d). Correlating survival scores and muscle caspase-3-like activities revealed no relationship between these parameters (linear regression: t1,48 = −0.7, p = 0.473; figure 2e). One would expect that flight muscle caspase-3 activity would correlate better with the wing-specific score, and although the correlation was stronger, the relationship did not reach statistical significance (t1,48 = −1.7, p = 0.089; see electronic supplementary material, figure S1). Furthermore, there was no relationship between caspase-3-like activity and haemolymph K+ concentration (linear regression: t1,48 = 1.0, p = 0.326, correlation not shown). The poor predictive power of muscle caspase-3-like activity is likely partially caused by the large variation in activity; a minority of muscle samples from cold exposed locusts have very high caspase-3-like activity (greater than 10 pmol AMC cleaved min−1 mg−1). When these samples are removed (using Grubb's test for outliers), all correlations became statistically significant using linear regression (apoptosis versus survival score: t1,39 = −2,9, p = 0.005, R5 = 0.164; apoptosis versus wing score: t1,39 = −4.4, p < 0.001, R2 = 0.316, apoptosis versus haemolymph [K+]: t1,39 = 3.1, p = 0.003, R2 = 0.178; see electronic supplementary material, figure S2). Taking this approach, however, (1) reduces our sample size to a degree we find uncomfortable (nine outliers out of 50 data points removed), and (2) yields relationships between muscle caspase activity and survival scores that, while significant, still do not come close to reaching the explanatory power of haemolymph [K+]. We therefore opted to retain the entire dataset in figure 2.
Figure 2.

Variation in cold-induced hyperkalaemia predicts individual survival outcomes while caspase-3-like activity in the muscles does not. (a) Exposure to stressful cold reduces survival and (b) increases haemolymph [K+] (hyperkalaemia) with (c) a strong sigmoidal correlation between the two . (d) Caspase-3-like-mediated apoptosis was activated during the same exposure, but (E) did not correlate with the survival score. (Online version in colour.)
4. Discussion
(a). Stressful cold causes injury and activates programmed cell death in muscle tissue
Cold-induced cell death in insect muscle is thought to be the consequence of a debilitating cascade, at the centre of which is a loss of ionoregulatory capacity that drives haemolymph hyperkalaemia. This hyperkalaemia, in turn, depolarizes muscle tissue and induces an excessive Ca2+ influx, increasing the intracellular [Ca2+], and this is thought to activate apoptotic/necrotic pathways and thereby drive injury phenotypes [15,18–20]. In our experiments, we found that exposure to both prolonged cold and lethal heat (positive control) induced a marked increase in caspase-3-like activity in muscle tissue (figures 1c,d and 2d). Caspase-3 is one of the main executioner caspases responsible for programmed cell death, and while effector caspases can be activated by several up-stream initiator caspases, caspase-3, in particular, appears to be mainly associated with apoptotic rather than necrotic cell death [38], thus we demonstrate that muscle cell death caused by stressful temperatures is at least partially caused by caspase-3-mediated apoptosis. This is supported by the findings of Yi and Lee who demonstrated that cold-induced cell death in D. melanogaster was associated with DNA fragmentation [30], a common marker for cell death. The exposure used to induce cell death in the present study causes haemolymph hyperkalaemia and this hyperkalaemia is already known to cause muscle membrane depolarization (figure 2d) [16], thus our findings support a link between cold-induced ionoregulatory collapse and cell death [18,19]. Interestingly, the locust gut is also injured by haemolymph hyperkalaemia [18], however, we found no increase in caspase-3-like activity in the midgut in response to cold exposure (figure 1c). We noted a small but statistically significant decrease (rather than the expected increase) in caspase-3-like activity in the midgut after severe heat exposure. What, if anything, drove this small effect is unclear, but heat denaturation is a likely candidate (i.e. this effect is masked by the large activation of the enzyme in muscle and nervous tissues). Together, these results from our cold and heat-stress experiments suggest that unlike muscles and nervous tissue, cell death does not occur through activation of caspase-3 orthologues in the midgut of locusts, which is similar to what has been established for Drosophila [39].
(b). A lack of cold-induced apoptotic cell death in the central nervous system
Loss of coordinated movements after cold exposure can, as mentioned above, be caused by cold-induced injury to the integrating centres in the nervous system. To estimate injury to the central nervous system (CNS), we measured caspase-3-like activity in the metathoracic ganglion, and found increased activity only after exposure to lethal heat (figure 2c,d). This differs from the muscle tissue where both heat and cold initiated caspase-3-mediated cell death. One possible explanation for this lies in the differential distribution and abundance of Ca2+ channels in insect nerve and muscle tissue: Insect muscles use Ca2+ ions for action potential generation and have a high and relatively even distribution of voltage-gated Ca2+ channels resulting in the high Ca2+ currents necessary muscle excitation, whereas insect nerves use Na+ channels for action potential generation and have a highly localized Ca2+ channel distribution resulting in lower whole-cell currents [43–45]. Thus, if the onset of chilling injury is based purely on depolarization-mediated Ca2+ entry, tissue injury could in principle be driven entirely by the presence or absence of voltage-gated Ca2+ channels. This is supported by the finding that blockade of Ca2+ channels can prevent the onset of chilling injury [18].
The CNS not only distinguishes itself from muscle on the basis of Ca2+ channel distribution but also differs in its physiological response to stressful conditions: During exposure to thermal extremes, the CNS undergoes a phenomenon known as a spreading depolarization (SD) [46]. SD events are characterized by a rapid surge in interstitial [K+] that completely silences the CNS at a temperature closely associated with the loss of coordinated movements at the CTmin and CTmax [3,4,47]. However, while haemolymph hyperkalaemia appears to be detrimental for muscle viability, the SD event has been hypothesized to serve a neuroprotective function in insects [6,47]. Indeed, it has been proposed that the large shifts in interstitial ion concentrations that occur during SD could induce channel and/or spike arrest in the CNS such that the SD serves to lower metabolic demand during exposure to extreme conditions [4,48]. Furthermore, it was recently suggested that SD events themselves are benign unless occurring in metabolically compromised tissues [49]. Exposure to extreme heat severely challenges aerobic metabolism in insects while energy balance is generally maintained during cold exposure [50], and our finding that heat, and not cold, increases caspase-3-mediated cell death in the locust CNS therefore at least partially supports an adaptive nature of SD events.
(c). Individual variation in haemolymph [K+] predicts survival outcomes during cold exposure
The capacity to prevent hyperkalaemia during cold exposure is thought to underlie chill tolerance [1,51]. Until now, however, no study has quantified the degree of chilling injury and ion balance disruption in the same individual of any insect species. We took advantage of the variation in survival outcome in cold-exposed locusts to investigate the role of individual variation in ionoregulatory capacity in facilitating cold tolerance by measuring survival outcome, haemolymph [K+] and caspase-3-like activity in the muscles of individual locusts (figure 2). Poor survival outcomes were again associated with hyperkalaemia, but we also found a strong, negative sigmoidal relationship between the degree of chilling injury and degree of hyperkalaemia (figure 2a–c). We thus provide strong support for a link between ionoregulatory capacity and cold tolerance at the level of individual insects. Despite finding that caspase-3-like activity was also increased in cold exposed locusts, we found no relationship between caspase-3-like activity and survival score (figure 2e). The same was true for caspase-3-like activity and wing score, and caspase-3-like activity and haemolymph [K+] (electronic supplementary material, figure S1).
The current model for chilling injury implicates Ca2+ as a key signalling molecule in activating muscle cell apoptosis [18], but increased cytosolic [Ca2+] also activates other cell death pathways such as autophagy and necrosis [52]. It is therefore likely that not all cell death in locust muscle is driven by apoptosis. We thus argue that other cell death pathways play more critical roles in the cold-induced cell death that has been observed in insect muscle using other assays [15,17,18]. Indeed, damage to the cell membrane (used by live/dead assays to estimate viability) is a phenomenon commonly associated with necrosis caused by cell swelling [53].
Our inability to correlate caspase-3-like activity with survival outcomes could alternatively be explained by the use of a single flight muscle as a sample to predict injury at the organismal level. Some support for this can be found in the slightly stronger (but still not statistically significant) association between the wing-specific survival score and muscle caspase-3-like activity (see electronic supplementary material, figure S1). Lastly, the possibility remains that the mechanism underlying cold-induced behavioural deficits is not associated with muscle cell death, but via other detrimental effects of cold and/or hyperkalaemia on the neuromuscular system. For example, cold exposure has been shown to affect invertebrate synaptic function [54]. Additionally, the integrating neural centres (i.e. ganglia, where no apoptosis-activation was observed in the cold; figure 2c) are separated from the haemolymph by an effective blood–brain barrier [55], while peripheral nerves and the neuromuscular junctions are in direct contact with the haemolymph [56,57] and are therefore likely to be impaired by the combined effect of cold and hyperkalaemia (e.g. cell death). Thus, disruptions of synaptic function and/or cell death in peripheral neurons may also underlie behavioural impairments observed following rewarming.
5. Conclusion
Overall, our findings suggest that cold stress activates apoptotic signalling cascades in the muscles, but not nervous tissues of a chill-susceptible insect. Hyperkalaemia has been repeatedly observed as a consequence of chilling in insects, and we found for the first time that it is a strong predictor of individual neuromuscular defects following rewarming. Although cold activates apoptosis in the muscles of locusts, and this apoptosis likely contributes to chilling injury phenotypes, caspase activity does not correlate with individual organismal injury phenotypes. We argue that haemolymph K+ is a better predictor of chilling injury primarily because (1) K+ imbalance is central to determining whether or not an insect is injured, and (2) other cell death pathways (most likely necrosis) and neuromuscular impairments are at play. To integrate these new findings into our current understanding of chilling injury, we present a revised model of the mechanisms driving organismal chilling injury in chill-susceptible insects (figure 3), which highlights the critical importance of distinguishing among apoptosis and other forms of cell death in furthering our understanding of insect cold tolerance, while also emphasizing the importance of other adverse effects on the neuromuscular system. Only by doing so can we understand how cold-adapted species and populations can avoid and repair cellular damage during and following cold stress.
Figure 3.
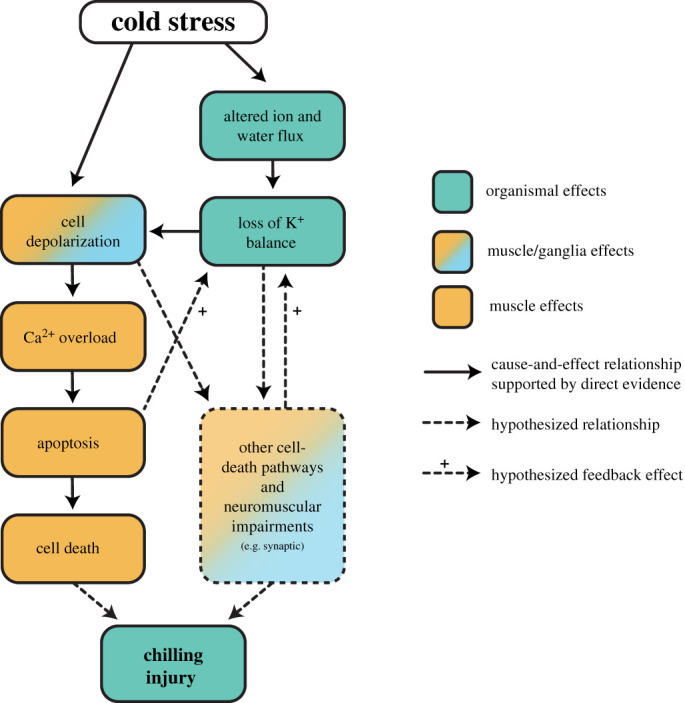
A revised model of cause-and-effect relationships between cold exposure and chilling injury phenotypes in insects. Exposure to stressful cold directly depolarizes cell membranes, and this effect is exacerbated by both a systemic (haemolymph; impacting muscles) and local (spreading depolarization; impacting the central nervous system) loss of K+ balance. This causes cell membrane depolarization that drives a catastrophic increase in cytosolic [Ca+] in muscle cells which activates executioner caspases and the resulting apoptotic cell death likely contributes to injury at the organismal level. Based on the findings of the present study, however, it is likely that other cell-death pathways (e.g. necrosis) or deleterious (and potentially Ca2+-overload-independent) mechanisms in the neuromuscular system are activated by membrane depolarization and cause the majority of chilling injury. (Online version in colour.)
Supplementary Material
Supplementary Material
Acknowledgements
The authors wish to thank Marshall Ritchie for taking care of the locust colony during the time this research was being conducted.
Data accessibility
All data are provided as electronic supplementary material file.
Competing interests
The authors declare no competing interests.
Funding
This work was supported by a Natural Sciences and Engineering Research Council (NSERC) Discovery Grant to H.A.M. (RGPIN-2018-05322), a Postdoctoral Fellowship grant to M.K.A. from the Carlsberg Foundation (CF18-0940), and infrastructure funding to H.A.M. from the Canadian Foundation for Innovation and Ontario Research Fund Small Infrastructure Fund.
References
- 1.Overgaard J, MacMillan HA. 2017. The integrative physiology of insect chill tolerance. Annu. Rev. Physiol. 79, 187–208. ( 10.1146/annurev-physiol-022516-034142) [DOI] [PubMed] [Google Scholar]
- 2.Ransberry VE, MacMillan HA, Sinclair BJ. 2011. The relationship between chill-coma onset and recovery at the extremes of the thermal window of Drosophila melanogaster. Physiol. Biochem. Zool. 84, 553–559. ( 10.1086/662642) [DOI] [PubMed] [Google Scholar]
- 3.Andersen MK, Jensen NJS, Meldrum Robertson R, Overgaard J. 2018. Central nervous system shutdown underlies acute cold tolerance in tropical and temperate Drosophila species. J. Exp. Biol. 221, jeb.179598 ( 10.1242/jeb.179598) [DOI] [PubMed] [Google Scholar]
- 4.Robertson RM, Spong KE, Srithiphaphirom P. 2017. Chill coma in the locust, Locusta migratoria, is initiated by spreading depolarization in the central nervous system. Sci. Rep. 7, 10297 ( 10.1038/s41598-017-10586-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.David RJ, Gibert P, Pla E, Petavy G, Karan D, Moreteau B. 1998. Cold stress tolerance in Drosophila: analysis of chill coma recovery in D. melanogaster. J. Therm. Biol. 23, 291–299. ( 10.1016/S0306-4565(98)00020-5) [DOI] [Google Scholar]
- 6.Robertson RM, Dawson-Scully KD, David Andrew R. 2020. Neural shutdown under stress: an evolutionary perspective on spreading depolarization. J. Neurophysiol. 123, 885–895. ( 10.1152/JN.00724.2019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koštál V, Yanagimoto M, Bastl J. 2006. Chilling-injury and disturbance of ion homeostasis in the coxal muscle of the tropical cockroach (Nauphoeta cinerea). Comp. Biochem. Physiol. Part B, Biochem. Mol. Biol. 143, 171–179. ( 10.1016/j.cbpb.2005.11.005) [DOI] [PubMed] [Google Scholar]
- 8.MacMillan HA, Sinclair BJ. 2011. Mechanisms underlying insect chill-coma. J. Insect Physiol. 57, 12–20. ( 10.1016/j.jinsphys.2010.10.004) [DOI] [PubMed] [Google Scholar]
- 9.Rojas RR, Leopold RA. 1996. Chilling injury in the housefly: evidence for the role of oxidative stress between pupariation and emergence. Cryobiology 33, 447–458. ( 10.1006/cryo.1996.0045) [DOI] [Google Scholar]
- 10.Zachariassen KE, Kristiansen E, Pedersen SA. 2004. Inorganic ions in cold-hardiness. Cryobiology 48, 126–133. ( 10.1016/j.cryobiol.2004.01.004) [DOI] [PubMed] [Google Scholar]
- 11.MacMillan HA, Yerushalmi GY, Jonusaite S, Kelly SP, Donini A. 2017. Thermal acclimation mitigates cold-induced paracellular leak from the Drosophila gut. Sci. Rep. 7, 8807 ( 10.1038/s41598-017-08926-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andersen MK, MacMillan HA, Donini A, Overgaard J. 2017. Cold tolerance of Drosophila species is tightly linked to epithelial K+ transport capacity of the Malpighian tubules and rectal pads. J. Exp. Biol. 220, 4261–4269. ( 10.1242/jeb.168518) [DOI] [PubMed] [Google Scholar]
- 13.Koštál V, Vambera J, Bastl J. 2004. On the nature of pre-freeze mortality in insects: water balance, ion homeostasis and energy charge in the adults of Pyrrhocoris apterus. J. Exp. Biol. 207, 1509–1521. ( 10.1242/jeb.00923) [DOI] [PubMed] [Google Scholar]
- 14.MacMillan HA, Sinclair BJ. 2011. The role of the gut in insect chilling injury: cold-induced disruption of osmoregulation in the fall field cricket, Gryllus pennsylvanicus. J. Exp. Biol. 214, 726–734. ( 10.1242/jeb.051540) [DOI] [PubMed] [Google Scholar]
- 15.MacMillan HA, Baatrup E, Overgaard J. 2015. Concurrent effects of cold and hyperkalaemia cause insect chilling injury. Proc. R. Soc. B 282, 20151483 ( 10.1098/rspb.2015.1483) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.MacMillan HA, Findsen A, Pedersen TH, Overgaard J. 2014. Cold-induced depolarization of insect muscle: differing roles of extracellular K+ during acute and chronic chilling. J. Exp. Biol. 217, 2930–2938. ( 10.1242/jeb.107516) [DOI] [PubMed] [Google Scholar]
- 17.Andersen MK, Folkersen R, MacMillan HA, Overgaard J. 2017. Cold-acclimation improves chill tolerance in the migratory locust through preservation of ion balance and membrane potential. J. Exp. Biol. 220, 487–496. ( 10.1242/jeb.150813) [DOI] [PubMed] [Google Scholar]
- 18.Bayley JS, Winther CB, Andersen MK, Grønkjær C, Nielsen OB, Pedersen TH, Overgaard J. 2018. Cold exposure causes cell death by depolarization-mediated Ca2+ overload in a chill-susceptible insect. Proc. Natl Acad. Sci. USA 115, 201813532 ( 10.1073/pnas.1813532115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boutilier RG. 2001. Mechanisms of cell survival in hypoxia and hypothermia. J. Exp. Biol. 204, 3171–3181. [DOI] [PubMed] [Google Scholar]
- 20.Yi S-X, Lee REJ. 2011. Rapid cold-hardening blocks cold-induced apoptosis by inhibiting the activation of pro-caspases in the flesh fly Sarcophaga crassipalpis. Apoptosis 16, 249–255. ( 10.1007/s10495-010-0570-0) [DOI] [PubMed] [Google Scholar]
- 21.Olsson T, MacMillan HA, Nyberg N, Stærk D, Malmendal A, Overgaard J. 2016. Hemolymph metabolites and osmolality are tightly linked to cold tolerance of Drosophila species: a comparative study. J. Exp. Biol. 219, 2504–2513. ( 10.1242/jeb.140152) [DOI] [PubMed] [Google Scholar]
- 22.MacMillan HA, Andersen JL, Loeschcke V, Overgaard J. 2015. Sodium distribution predicts the chill tolerance of Drosophila melanogaster raised in different thermal conditions. Am. J. Physiol. Regul. Integr. Comp. Physiol. 308, 823–831. ( 10.1152/ajpregu.00465.2014) [DOI] [PubMed] [Google Scholar]
- 23.Yerushalmi GY, Misyura L, MacMillan HA, Donini A. 2018. Functional plasticity of the gut and the Malpighian tubules underlies cold acclimation and mitigates cold-induced hyperkalaemia in Drosophila melanogaster. J. Exp. Biol. 221, jeb.174904 ( 10.1242/jeb.174904) [DOI] [PubMed] [Google Scholar]
- 24.Andersen MK, Overgaard J. 2020. Maintenance of hindgut reabsorption during cold exposure is a key adaptation for Drosophila cold tolerance. J. Exp. Biol. 223, jeb.213934 ( 10.1242/jeb.213934) [DOI] [PubMed] [Google Scholar]
- 25.MacMillan HA, Andersen JL, Davies SA, Overgaard J. 2015. The capacity to maintain ion and water homeostasis underlies interspecific variation in Drosophila cold tolerance. Sci. Rep. 5, 18607 ( 10.1038/srep18607) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andersen JL, MacMillan HA, Overgaard J. 2015. Muscle membrane potential and insect chill coma. J. Exp. Biol. 218, 2492–2495. ( 10.1242/jeb.123760) [DOI] [PubMed] [Google Scholar]
- 27.Bayley JS, Sørensen JG, Moos M, Koštál V, Overgaard J. 2020. Cold-acclimation increases depolarization resistance and tolerance in muscle fibers from a chill-susceptible insect, Locusta migratoria. Am. J. Physiol. Regul. Integr. Comp. Physiol. 308, R823–R831. ( 10.1152/ajpregu.00068.2020) [DOI] [PubMed] [Google Scholar]
- 28.Gerken AR, Eller OC, Hahn DA, Morgan TJ. 2015. Constraints, independence, and evolution of thermal plasticity: probing genetic architecture of long- and short-term thermal acclimation. Proc. Natl Acad. Sci. USA 112, 4399–4404. ( 10.1073/pnas.1503456112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yi S-X, Lee RE. 2003. Detecting freeze injury and seasonal cold-hardening of cells and tissues in the gall fly larvae, Eurosta solidaginis (Diptera: Tephritidae) using fluorescent vital dyes. J. Insect Physiol. 49, 999–1004. ( 10.1016/S0022-1910(03)00168-9) [DOI] [PubMed] [Google Scholar]
- 30.Yi S-XX, Moore CW, Lee REJ. 2007. Rapid cold-hardening protects Drosophila melanogaster from cold-induced apoptosis. Apoptosis 12, 1183–1193. ( 10.1007/s10495-006-0048-2) [DOI] [PubMed] [Google Scholar]
- 31.Grasl-Kraupp B, Ruttkay-Nedecky B, Koudelka H, Bukowska K, Bursch W, Schulte-Hermann R. 1995. In situ detection of fragmented DNA (tunel assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology 21, 1465–1468. ( 10.1016/0270-9139(95)90071-3) [DOI] [PubMed] [Google Scholar]
- 32.Accorsi A, Zibaee A, Malagoli D. 2015. The multifaceted activity of insect caspases. J. Insect Physiol. 76, 17–23. ( 10.1016/j.jinsphys.2015.03.007) [DOI] [PubMed] [Google Scholar]
- 33.Cooper DM, Granville DJ, Lowenberger C. 2009. The insect caspases. Apoptosis 14, 247–256. ( 10.1007/s10495-009-0322-1) [DOI] [PubMed] [Google Scholar]
- 34.Galluzzi L, López-Soto A, Kumar S, Kroemer G. 2016. Caspases connect cell-death signaling to organismal homeostasis. Immunity 44, 221–231. ( 10.1016/j.immuni.2016.01.020) [DOI] [PubMed] [Google Scholar]
- 35.Florentin A, Arama E. 2012. Caspase levels and execution efficiencies determine the apoptotic potential of the cell. J. Cell Biol. 196, 513–527. ( 10.1083/jcb.201107133) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kumar S. 2007. Caspase function in programmed cell death. Cell Death Differ. 14, 32–43. ( 10.1038/sj.cdd.4402060) [DOI] [PubMed] [Google Scholar]
- 37.Shalini S, Dorstyn L, Dawar S, Kumar S. 2015. Old, new and emerging functions of caspases. Cell Death Differ. 22, 526–539. ( 10.1038/cdd.2014.216) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yuan J, Najafov A, Py BF. 2016. Roles of caspases in necrotic cell death. Cell 167, 1693–1704. ( 10.1016/j.cell.2016.11.047) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Denton D, Shravage B, Simin R, Mills K, Berry DL, Baehrecke EH, Kumar S. 2009. Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr. Biol. 19, 1741–1746. ( 10.1016/j.cub.2009.08.042) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brzezinski K, MacMillan HA. 2020. Chilling induces unidirectional solute leak through the locust gut epithelia. J. Exp. Biol. 223, jeb215475 ( 10.1242/jeb.215475) [DOI] [PubMed] [Google Scholar]
- 41.Snodgrass RE. 1929. The thoracic mechanism of a grasshopper, and its antecedents. Smithson. Misc. Collect. 82, 1–111. [Google Scholar]
- 42.R Development Core Team. 2019. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; See http://www.r-project.org. [Google Scholar]
- 43.Pearson HA. 1993. Calcium channel currents in neurones from locust (Schistocerca gregaria) thoracic ganglia. J. Exp. Biol. 177, 201–221. [Google Scholar]
- 44.Bayley JS, Klepke MJ, Pedersen TH, Overgaard J. 2019. Cold acclimation modulates voltage gated Ca2+ channel currents and fiber excitability in skeletal muscles of Locusta migratoria. J. Insect Physiol. 114, 116–124. ( 10.1016/j.jinsphys.2019.03.003) [DOI] [PubMed] [Google Scholar]
- 45.Quintavalle A. 2013. Voltage-gated calcium channels in honey bees: physiological roles and potential targets for insecticides. MSc thesis, ENS de Lyon, France. [Google Scholar]
- 46.Rodgers CI, Armstrong GAB, Shoemaker KL, LaBrie JD, Moyes CD, Robertson RM. 2007. Stress preconditioning of spreading depression in the locust CNS. PLoS ONE 2, e1366 ( 10.1371/journal.pone.0001366) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jørgensen LB, Robertson RM, Overgaard J. 2020. Neural dysfunction correlates with heat coma and CTmax in Drosophila but does not set the boundaries for heat stress survival. J. Exp. Biol. 223, jeb218750 ( 10.1101/844316) [DOI] [PubMed] [Google Scholar]
- 48.Hochachka PW. 1986. Defense strategies against hypoxia and hypothermia. Science 231, 234–241. ( 10.1126/science.2417316) [DOI] [PubMed] [Google Scholar]
- 49.Shuttleworth CW, et al. 2020. Which spreading depolarizations are deleterious to brain tissue? Neurocrit. Care 32, 317–322. ( 10.1007/s12028-019-00776-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Verberk WCEP, Overgaard J, Ern R, Bayley M, Wang T, Boardman L, Terblanche JS. 2016. Does oxygen limit thermal tolerance in arthropods? A critical review of current evidence. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 192, 64–78. ( 10.1016/j.cbpa.2015.10.020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.MacMillan HA. 2019. Dissecting cause from consequence: a systematic approach to thermal limits. J. Exp. Biol. 222, jeb191593 ( 10.1242/jeb.191593) [DOI] [PubMed] [Google Scholar]
- 52.Zhivotovsky B, Orrenius S. 2011. Calcium and cell death mechanisms: a perspective from the cell death community. Cell Calcium 50, 211–221. ( 10.1016/j.ceca.2011.03.003) [DOI] [PubMed] [Google Scholar]
- 53.Fink SL, Cookson BT. 2005. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 73, 1907–1916. ( 10.1128/IAI.73.4.1907) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhu YC, Cooper RL. 2018. Cold exposure effects on cardiac function and synaptic transmission at the neuromuscular junction in invertebrates. Int. J. Zool. Res. 14, 49–60. ( 10.3923/ijzr.2018.49.60) [DOI] [Google Scholar]
- 55.Treherne J, Schofield P. 1981. Mechanisms of ionic homeostasis in the central nervous system of an insect. J. Exp. Biol. 95, 61–73. [DOI] [PubMed] [Google Scholar]
- 56.Lane NJ, Treherne JE. 1973. The ultrastructural organization of peripheral nerves in two insect species (Periplaneta americana and Schistocerca gregaria). Tissue Cell 5, 703–714. ( 10.1016/S0040-8166(73)80056-4) [DOI] [PubMed] [Google Scholar]
- 57.Lane NJ, Leslie RA, Swales LS. 1975. Insect peripheral nerves: accessibility of neurohaemal regions to lanthanum. J. Cell Sci. 18, 179–197. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are provided as electronic supplementary material file.