

Abstract
The last 25 years have witnessed tremendous progress in identifying and characterizing proteins that regulate the uptake, intracellular trafficking and export of copper. Although dietary copper is required in trace amounts, sufficient quantities of this metal are needed to sustain growth and development in humans and other mammals. However, copper is also a rate-limiting nutrient for the growth and proliferation of cancer cells. Oral copper chelators taken with food have been shown to confer anti-neoplastic and anti-metastatic benefits in animals and humans. Recent studies have begun to identify specific roles for copper in pathways of oncogenic signaling and resistance to anti-neoplastic drugs. Here, we review the general mechanisms of cellular copper homeostasis and discuss roles of copper in cancer progression, highlighting metabolic vulnerabilities that may be targetable in the development of anticancer therapies.
Keywords: Copper homeostasis, copper chelation, cancer, nutrition, metallochaperone, cisplatin chemotherapy
Introduction
Copper (Cu) is an indispensable micronutrient for the development and replication of all eukaryotes [1]. Because copper cannot be created or destroyed by metabolic processes, it must be acquired from external sources. The incorporation of copper into living systems is thought to have coincided with the release of molecular oxygen into earth’s early atmosphere [2, 3]. By harnessing the ability of copper to cycle between two oxidation states, Cu (I) and Cu (II), cuproenzymes participate in a wide spectrum of metabolic processes including aerobic respiration, pigmentation, peptide amidation, catecholamine biosynthesis, iron transport, superoxide dismutation and biosynthesis of the extracellular matrix (Table 1). Not surprisingly, sufficient quantities of copper are necessary to sustain the growth and development of fungi, plants and animals, however, copper is also potentially toxic when present at elevated concentrations. Under these conditions, the formation of free ionic Cu(I) within the cell can inhibit the function of enzymes by oxidizing cysteines within iron-sulfur cluster proteins, or react with hydrogen peroxide to produce highly-reactive hydroxyl radicals [4]. Accordingly, organisms have evolved a sophisticated cadre of transporters and copper-binding proteins to control the uptake, intracellular movement, storage and efflux of copper, while simultaneously preventing formation of the free ion within the cytoplasm (Figure 1). In mammals, copper is acquired from the diet and is absorbed across the intestinal epithelium for delivery to the liver via portal circulation. The liver is the main repository of copper in the body and serves to distribute copper to peripheral organs via the bloodstream, or to excrete copper from the body via the bile [5]. Once growth and development are completed, whole-body copper homeostasis is maintained by balancing the amounts of copper absorbed and excreted [6]. However, pregnancy and neonatal development are stages of the mammalian life cycle that require significantly higher intakes of dietary copper to meet the metabolic demands of growth and development [7]. Cancer cells also have a higher demand for copper compared to non-dividing cells, which is an Achilles’ heel that can be exploited through the use of oral copper chelation to suppress tumor growth and metastasis in animal cancer models and human patients [8]. Biological processes in which copper has been linked to cancer include mitochondrial respiration, collagen cross-linking, immune system modulation, antioxidant defense, mitogenic signaling and autophagy. Here, we review the current understanding of copper homeostasis in mammalian cells, with a focus on newly discovered roles for copper in cancer progression, potential roles of copper in transition metal signaling and the prospect of targeting copper homeostasis pathways in cancer therapy.
Table 1.
Mammalian copper trafficking proteins and cuproenzymes
| Copper trafficking |
Function |
|---|---|
| CTR1 | Copper uptake |
| CTR2 | Regulation of CTR1 |
| ATOX1 | Copper delivery to ATP7A/B |
| ATP7A | Copper transport to the secretory pathway and copper export |
| ATP7B | Copper transport to the secretory pathway and copper export |
| CCS | Copper delivery to SOD1 |
| MT | Copper sequestration |
| SCO1 | Copper delivery to COX |
| SCO2 | Copper delivery to COX |
| COX17 | Copper delivery to COX |
| Cuproenzymes | Function |
| COX | Aerobic respiration |
| LOX, LOXL1-4 | Extracellular matrix formation |
| TYR | Melanin production |
| PAM | Synthesis of amidated peptides |
| DβH | Synthesis of catecholamines |
| CP | Iron homeostasis |
| HEPH | Iron homeostasis |
| SOD1 | Superoxide detoxification |
| SOD3 | Superoxide detoxification |
| MEK1 | Mitogenic signaling |
| ULK1/2 | Autophagy signaling |
| MEMO1 | Regulation of cell motility and ROS production |
Figure 1. Model of cellular copper homeostasis in mammals.

The transport of Cu(I) (black circles) into the cytoplasm occurs via the CTR1 copper importer, which is located at the plasma membrane and within endosomal compartments. The uptake of Cu(I) via CTR1 is thought to depend on the reduction of Cu(II) to Cu(I) by members of the STEAP family of metalloreductases. The oligomerization of CTR1 and another SLC31 protein, CTR2, facilitates the cleavage of the CTR1 ectodomain by cathepsin L to enhance Cu(I) transport from endosomal compartments into the cytoplasm. Cytoplasmic Cu(I) is bound to glutathione (GSH) and can be transferred to metallochaperones for targeted trafficking to proteins. The CCS metallochaperone is required for insertion of Cu(I) into apo-SOD1 in both the cytoplasm and the mitochondrial intermembrane space. The metalation of cuproenzymes within the secretory pathway occurs via ATOX1-mediated Cu(I) delivery to the P-type ATPases, ATP7A or ATP7B, which are located in the trans-Golgi network (TGN) and facilitate Cu(I) loading into this organelle. Cu(I) transport into the mitochondrial matrix is facilitated by SLC25A3 located within the inner membrane. The metalation of COX occurs within the intermembrane space of the mitochondria and requires copper to be exported from the matrix via an unknown transporter. COX metalation and maturation requires the metallochaperones COX17, SCO1 and COX11, and other assembly / redox control factors. Protective mechanisms of Cu tolerance include sequestration by metallothioneins (MTs) as well as Cu(I)-stimulated trafficking of copper transporters, including the endocytosis and degradation of CTR1 which reduces Cu(I) import and exocytic trafficking of ATP7A and ATP7B to post-Golgi vesicles and the plasma membrane to facilitate copper export from the cell.
Copper acquisition in mammals
Copper uptake in mammalian cells is mediated by the CTR1 (SLC31A1) plasma membrane high affinity copper importer, which belongs to the SLC31 family of copper permeases found in all eukaryotes [1, 9]. Consistent with the essentiality of copper for normal growth and development, global deletion of the Ctr1 gene in mice results in embryonic lethality [10]. CTR1 is also essential for dietary Cu absorption since enterocyte-specific deletion of the Ctr1 gene in mice causes systemic Cu deficiency and neonatal lethality [11]. An alternative pathway for Cu uptake via the divalent metal transporter 1 (DMT 1; SLC11A2) is suggested by studies in certain cultured cell lines [12]. However, the finding that enterocyte-specific deletion of Dmt1 does not alter intestinal Cu absorption suggests that it is not essential for dietary Cu transport [13].
The CTR1 protein exhibits a three-domain topology that is conserved in all members of the SLC31 family which includes an amino terminal ectodomain containing Cu(I)-binding histidine- and methionine-rich sequences, three transmembrane domains, and a cytoplasmic C-terminal tail. Biochemical studies demonstrate that SLC31 family members form functional trimers that comprise nine-membrane spanning domains that selectively permit the inward flow of Cu(I) down a concentration gradient [14, 15]. Recently, the crystal structure of a CTR1 orthologue from Atlantic salmon (sCtr1) was solved, revealing a goblet-like architecture in which two rings of methionine residues were found to line the entrance to the ion conduction pore, and are postulated to function as selectivity filters for Cu(I) (Figure 2) [16]. These methionine rings are created by an MXXXM motif that is conserved in the second transmembrane domain of all SLC31 family members and is essential for copper transport activity of human CTR1 [17]. Another interesting feature revealed by the crystal structure of sCtr1 is the presence of an intramembranous Zn atom [16]. While this could be an experimental artifact, it suggests an intriguing possibility that Zn2+ may be required to modulate the function of CTR1.
Figure 2. Structure models of the human CTR1 copper importer.

A) Homology-based model of the human CTR1 homotrimer derived from the crystal structure of sCtr1 from Atlantic salmon. Each hCTR1 monomer is depicted with a different color for each monomer. The copper-binding MXXM motif within the second transmembrane domain of each monomer forms two methionine rings that are thought to function as a Cu(I) selective filter at the extracellular entrance. B) A top view highlighting copper co-ordination within the methionine rings.
Approximately 95% of copper in the plasma is found within ceruloplasmin, a multicopper oxidase that facilitates cellular iron export by oxidizing Fe2+ as it exits the iron exporter, ferroportin [18]. Although there is some evidence from cell culture experiments that ceruloplasmin-bound copper may be used as a source of copper by CTR1 [19], the fact that aceruloplasminemic mice and humans do not exhibit copper deficiency indicates that ceruloplasmin is not a major contributor to copper uptake in vivo [20]. Rather, minor fractions of copper bound to amino acids and other small molecular weight carriers are thought to be the major sources of plasma copper for cellular uptake [21, 22]. Since copper uptake via CTR1 requires prior reduction to Cu(I) [14], this implicates the requirement of a metalloreductase. In the baker’s yeast, Saccharomyces cerevisiae, the FRE1 protein is required for Cu2+ reduction prior to CTR1-mediated Cu1+ uptake [23]. In mammals, the STEAP family of ferric reductases (STEAP 1 – 4) appears to serve this role based on the finding that forced expression of STEAP 2, 3 or 4 in cultured HEK293 cells confers cupric reductase activity and stimulates copper uptake [24] (Figure 1). A recent study demonstrated that STEAP4 expression in colon cancer cells was induced by the inflammatory cytokine IL-17 leading to increased copper uptake and the promotion of tumorigenesis [25].
Studies in yeast, flies and worms indicate that CTR1-dependent copper uptake is regulated at the level of metal-responsive transcription [1]. In cultured cell lines, one lab has reported that the human CTR1 gene is transcriptionally induced in response to low Cu concentrations by the zinc-finger transcription factor Specificity Protein 1 (Sp1) [26, 27]. However, the in vivo relevance of these findings is unclear since studies have shown that copper deficiency in rats does not alter Ctr1 mRNA levels in the brain, intestine or liver compared to copper adequate controls [28]. There is considerably more support for post-translational regulation of mammalian orthologues of CTR1. Elevated copper concentrations stimulate CTR1 internalization into endosomes as a regulatory mechanism to reduce copper uptake across the plasma membrane [29, 30]. A recent study suggests that this process is regulated by retromer [31], a heterotrimeric protein complex of VPS35, VPS29 and VPS26 that plays a central role in the sorting of proteins within endosomal compartments [32]. Copper-stimulated internalized CTR1 protein within endosomes colocalizes with the retromer complex, which is required to recycle CTR1 back to the plasma membrane in response to a reduction in copper concentrations [31].
Studies have also identified a second mechanism of CTR1 regulation that involves the proteolytic removal of its high-affinity copper-binding ectodomain [33, 34]. Compared to the full-length protein, truncated CTR1 has reduced copper uptake activity at the plasma membrane, but increased copper export from endosomal compartments into the cytoplasm. The cleavage of CTR1 occurs via cathepsin proteases within endosomal compartments and requires interactions with CTR2, which is the only other SLC31 protein in mammals. Although CTR2 was initially proposed as a low-affinity copper transporter, a recent study suggests that during the evolution of metazoans, prototypic CTR2 lost the ability to transport copper as it acquired the ability to regulate CTR1 cleavage [35]. It is currently unknown whether the cleavage of CTR1 requires formation of a CTR1/CTR2 heterotrimer and whether CTR2 recruits cathepsin proteases to CTR1. Interestingly, the MXXXM-containing Cu(I) selectivity filter in CTR1 is also present in CTR2, and although mutational analyses indicates this motif in CTR2 is not required for CTR1 cleavage, it nonetheless may act as a copper sensor within endosomal compartments [34]. Consistent with a role for CTR2 as a positive regulator of CTR1-mediated copper export from endosomes, deletion of CTR2 in mice causes an age-dependent hyperaccumulation of copper within various organs, with highest levels in the brain [34]. Continued efforts to understand CTR2-dependent cleavage of CTR1 will be important to address how copper uptake is regulated in different tissues and under different physiological conditions or disease states.
Intracellular copper buffering and trafficking
Because copper is potentially toxic when present as the free ion, cytoplasmic concentrations of free copper are maintained at exceedingly low levels – estimated between 10−15 M and 10−21 M, or less than one free copper ion per cell [36]. Such an extraordinary limitation on free copper is achieved in part through the metal binding properties of metallothioneins and glutathione (Figure 1). Metallothioneins are small cysteine-rich proteins that are transcriptionally induced by excess copper (and other metals) and protect against copper toxicity by irreversible sequestration within metal-thiolate clusters [37]. In contrast, copper bound to reduced glutathione (GSH) can undergo facile exchange with higher affinity ligands such as metallochaperones (discussed below). Cytoplasmic concentrations of GSH are estimated to be in the millimolar range, which vastly exceed copper concentrations under steady state conditions [38]. This allows GSH to function as a cytosolic copper buffer that not only prevents the formation of free copper ions, but also maintains a negative concentration gradient across the plasma membrane that drives CTR1-mediated copper uptake [39]. Once in the cytoplasm, copper must be ferried to the active sites of cuproenzymes during their biosynthesis or shortly thereafter. This raises the question of how this highly reactive metal is delivered to the appropriate enzyme while avoiding adventitious interactions along the way. Nature has solved this problem with the evolution of a class of small copper-binding proteins known as metallochaperones that deliver copper to specific proteins. This process is facilitated by direct protein-protein interactions which induce metal transfer down an increasing affinity gradient [40-43].
Copper trafficking to mitochondrial cytochrome c oxidase
Mitochondria are one of the most important destinations of copper in the cell for the metalation of cytochrome c oxidase (COX), a critical enzyme that functions in aerobic respiration (Figure 1). COX is a large multimeric complex at the terminus of the electron transport chain that transfers electrons from cytochrome c to molecular oxygen and contributes protons to the electrochemical gradient used to power ATP production. The two copper-binding subunits of cytochrome c oxidase, COX1 and COX2, are metallated during COX assembly within the intermembrane space (IMS) of the mitochondria [44], however, it is currently unknown how cytoplasmic copper enters this organelle. The source of copper for COX metalation is derived from an unknown non-proteinaceous anionic complex within the mitochondrial matrix [45]. Recent studies in yeast have shown that a member of the mitochondrial carrier family, PIC2, and an iron transporter MRS3, can transport copper from the IMS into the matrix and are necessary for COX biogenesis [46, 47]. The PIC2 orthologue, SLC25A3, performs this function in mammalian cells [48], however, the mechanism by which copper is exported from the matrix into the IMS for COX biogenesis is currently unknown. The metalation and assembly of COX is a complex modular process that requires the metallochaperone COX17 and several other metallochaperones and assembly proteins (Figure 1). Copper insertion into COX2 requires the transfer of copper from COX17 to SCO1, a membrane-spanning metallochaperone whose copper-binding site faces the IMS. This process is facilitated by several accessory proteins including SCO2, COA6 and COX20. The insertion of copper into COX1 also requires COX17, the metallochaperone COX11, and other ancillary proteins that are yet to be identified [49].
CCS and copper trafficking to superoxide dismutase
Cu/Zn-superoxide dismutase (SOD1) requires both copper and zinc, and plays a protective role against oxidative damage by catalyzing the disproportionation of superoxide to molecular oxygen and hydrogen peroxide. In humans, SOD1 maturation requires the insertion of copper into its active site by the CCS metallochaperone [50, 51] (Figure 1). This process is facilitated by an intermolecular disulfide between CCS and monomeric SOD1 that is subsequently converted to an intramolecular disulfide between SOD1 dimers [52, 53]. Although the majority of SOD1 resides within the cytoplasm, a small fraction (<5%) is also found in the mitochondrial IMS [54]. The metalation of SOD1 in the IMS is also dependent on CCS, and, as with COX metalation, the source of this copper is derived from the matrix [55]. Interestingly, CCS also regulates the distribution of SOD1 between the IMS and the cytoplasm in an oxygen-dependent manner [56]. Under oxygen replete conditions, CCS interacts with SOD1 in the cytosol to facilitate the metalation and dimerization of SOD1 which prevents entry to the mitochondria. However, under low oxygen concentrations, the MIA40-ERV1 disulfide relay catalyzes the formation of an intramolecular disulfide in CCS, trapping it within the IMS [56, 57]. As apo-SOD1 enters the mitochondria as a disulfide-reduced monomer, it too is trapped within the IMS by conversion to the mature oxidized dimeric holoenzyme by CCS. The hypoxia-dependent retention of SOD1 in the IMS is thought to protect the mitochondria from elevated levels of reactive oxygen species that are produced as a byproduct of the electron transport chain under low oxygen conditions [54].
Metalation of cuproenzymes within the secretory pathway
The majority of known eukaryote cuproenzymes are either secreted across the plasma membrane or reside within vesicular compartments, and because their active sites are lumenally oriented, copper must be inserted within the secretory pathway [58]. In mammals, this process is facilitated by either ATP7A or ATP7B, copper pumps that are embedded in the trans-Golgi network (TGN) (Figure 1) [59, 60]. Both ATP7A and ATP7B (ATP7A/B) belong to the P1B subgroup of heavy-metal transporting P-type ATPases, which harness the energy released from ATP hydrolysis to translocate metal ions from the cytoplasm across lipid bilayers. Mammalian ATP7A/B are specific for Cu(I) ions and share approximately 60% amino acid identity across their entire length, although the homology is much higher within signature motifs involved in catalysis. ATP7A/B also share a common molecular architecture that includes eight transmembrane helices, a long cytoplasmic amino terminal region containing six metal-binding MXCXXC sites, an intramembranous copper-binding CPC motif, two large cytoplasmic loops containing catalytic domains for ATP binding, phosphorylation and phosphatase activities, and a cytoplasmic C-terminal region containing endocytosis signals (Figure 3A) [61].
Figure 3. Schematic model of ATP7A/B structure and predicted Cu entry site.

A) A schematic illustration of ATP7A/B highlighting domains that are important for transport and trafficking function. The copper-binding MXCXXC motifs in the N-terminal domain are required for copper-induced trafficking from the TGN and to receive copper from the ATOX1 metallochaperone. Canonical sequences conserved in all P-type ATPases include the GDGIND required for ATP binding, DKTGT which contains the aspartic acid that is phosphorylated during catalysis, and TGE within the phosphatase domain required for removing the aspartyl phosphate. A conserved Met-Asp-Glu triad at the channel entrance and an intramembranous Cys-Pr-Cys motif are thought to coordinate copper during transport. The C-terminal tail harbors a di-leucine (ATP7A) or trileucine (ATP7B) required for endocytosis. A C-terminal DTAL motif is required for basolateral targeting of ATP7A in polarized epithelial cells, whereas an N-terminal FAFDNVGYE motif is required for apical targeting of ATP7B. B and C) Homology-derived structure of human ATP7A based on the crystal structure of LCopA from Legionella pneumophila. A bend in TM2 domain (helical ribbon) is predicted to form a platform at the cytosolic-membrane interface adjacent to the Met-Glu-Asp triad that may serve as a metallochaperone docking site for copper delivery.
The current model of the Cu-ATPase catalytic cycle follows the classic E1/E2 Post-Albers reaction that was originally described for the Na+/K+-ATPase [62], with additional information from biochemical assays and the crystal structure of the LCopA Cu-ATPase from Legionella pneumophila [63]. Copper transport is predicted to be initiated by the binding of Cu(I) to a conserved triad comprised of Met-Asp-Glu residues located at the entrance to the ion conduction pore (Figure 3B), which stimulates ATP binding to the large cytosolic nucleotide binding domain. The hydrolysis of ATP and transfer of the released γ-phosphate to an aspartic acid within a conserved DKTGT motif generates a high-energy phosphorylated intermediate (E1P), which is thought to energetically favor copper translocation to intramembrane ligands, including the CPC motif (Figure 3A). An intrinsic phosphatase activity requiring a TGE motif within the actuator domain is coupled with the release of copper into the lumenal/extracellular space to complete the reaction cycle (Figure 3A). One of the most notable features of LCopA is a bend in the second transmembrane domain as it emerges into the cytosol, which creates a “platform” adjacent to the Met-Asp-Glu triad at the pore opening (Figure 3B and 3C) [63]. Positively charged residues that line the cytoplasmic face of the platform may provide a docking site for the amino terminal copper-binding domain or ATOX1 to facilitate copper transfer to the triad (Figure 3C). Since LCopA has been solved only in the copper-free E2 conformation [63], additional configurations will be needed to decipher how copper moves from the triad to the intramembrane CPC and other sites within the transport pathway. As discussed below, inhibitors of ATP7A/B have potential therapeutic applications in cancer, thus the structures of Cu-ATPases in different catalytic conformations will be important to design the development of small molecule inhibitors of these pumps.
Regulation of copper export
Cu-ATPases are found in a wide range of organisms including archaea, bacteria and eukaryotes, indicating that the requirement to transport copper was an early and constant evolutionary pressure. In addition to metalating cuproenzymes within the TGN, both ATP7A and ATP7B also export copper from cells. This process involves copper-stimulated trafficking of ATP7A and ATP7B from the TGN into post-Golgi vesicles, which are loaded with copper and released into the extracellular milieu upon fusion with the cell surface (Figure 1) [59, 60]. Approximately 10 - 20% of ATP7A also accumulates at the cell surface in elevated copper conditions [64]. Upon the restoration of normal copper levels, both ATP7A and ATP7B are trafficked through endosomal compartments back to the TGN via the activities of several protein complexes including adaptor complex AP-1, retromer, Arp2/3, WASH, BLOC-1 and COMMD/CCDC22/CCDC93 complexes [65]. In addition to providing a homeostatic mechanism that protects cells against rising copper levels, the exocytic trafficking of ATP7A and ATP7B in polarized cells also plays an important role in controlling systemic copper homeostasis. In intestinal epithelial cells, ATP7A traffics toward the basolateral membrane to facilitate the absorption of dietary copper [66]. Whereas in polarized hepatocytes, the copper-stimulated trafficking of ATP7B controls the movement of copper into lysosomes and its subsequent release across the canalicular (apical) membrane into the bile for excretion from the body [67, 68].
Site-directed mutagenesis has identified sequences within ATP7A/B that regulate both localization within the TGN and copper-stimulated trafficking. The C-terminal domains of both ATP7A and ATP7B contain di-leucine and tri-leucine sequences [69, 70], respectively, that are required for endocytic retrieval via interactions with clathrin adaptor complexes (Figure 3A) [71]. In addition, the third transmembrane domain of ATP7A has been shown to function as an autonomous Golgi-retention signal [72]. A DTAL motif at the C-terminus of ATP7A is required for basolateral targeting in polarized kidney cells [73] whereas apical targeting of ATP7B in polarized hepatoma cells requires a unique 9-amino acid sequence at the N-terminus that is absent in ATP7A [74]. Copper-stimulated exocytic trafficking of ATP7A from the TGN requires at least one of the six amino terminal CXXC motifs, although one study suggest that the 5th or 6th motif closest to the membrane channel are necessary [75], whereas another study suggests any one of the six motifs will suffice [76]. Mutations in different regions of ATP7A/B are known to inhibit copper-stimulated trafficking, suggesting that trafficking is coupled to the catalytic cycling. Additional studies have narrowed this requirement to the formation of the aspartyl-phosphate catalytic intermediate in both ATP7A and ATP7B [77, 78], suggesting that this conformation is recognized by sorting machinery in the TGN. Interestingly, catalysis can be uncoupled from trafficking because mutations have been identified that block copper-stimulated trafficking of ATP7A but retain transport activity [79, 80]. Future studies elucidating the phospho-intermediate structures of such mutants will aid in addressing how different catalytic conformations of ATP7A/B are recognized by sorting machinery in the TGN.
Regulation of ATP7A and ATP7B by ATOX1 and the glutathione redox system
The delivery of cytoplasmic copper to ATP7A/B is mediated by interactions with ATOX1, a small metallochaperone that binds Cu(I) via a single MXCXXC motif and subsequently transfers this metal to one of six MXCXXC motifs located within the N-terminal domain of both transporters (Figure 3). This domain is largely unstructured and may provide a mechanism of copper delivery to the transmembrane entrance. However, it is worth noting that neither the amino terminal domain of ATP7A/B, nor ATOX1, are absolute requirements for transport activity, which implicates additional mechanisms by which copper is delivered to the channel opening. Studies in cultured cells indicate that both copper transport and trafficking functions of ATP7A and ATP7B are regulated by post-translational modifications. The N- and C- terminal domains of ATP7A and ATP7B contain sites for kinase-mediated phosphorylation, which are thought to modulate catalytic activity or interactions with trafficking machinery [81, 82]. S-glutathionylation of redox-sensitive cysteines (i.e., Cys-SSG) is another post-translational modification that regulates the activity and trafficking of ATP7A/B. Copper stimulates the interaction of glutaredoxin-1 (GRX1) with the amino terminal domains of ATP7A and ATP7B resulting in deglutathionylation of the MXCXXC motifs [83]. This process is thought to increase copper binding to these cysteines, thus providing a regulatory mechanism for stimulating trafficking and transport activity. Recent studies suggest that the oxidation state of ATOX1 may also play a pivotal role in this process. In proliferating cells, a high ratio of reduced to oxidized glutathione (GSH/GSSG) maintains the copper-binding cysteines of ATOX1 in a mostly reduced state. However, under oxidizing conditions, GSSG oxidizes the copper-coordinating cysteines of ATOX1 to create an intramolecular disulfide that is incapable of binding copper, thus reducing ATP7B-mediated copper transport into the secretory pathway [84]. In contrast, the reduction of ATOX1 by GRX1 was found to enhance copper delivery to ATP7B and stimulate metalation of ceruloplasmin [84]. Consistent with these findings, silencing of GRX1 in MEF cells results in copper retention and reduced copper tolerance, whereas forced expression of GRX1 lowers intracellular copper levels [85]. Intriguingly, recent in vitro studies have implicated a novel function for GRX1 as a metallochaperone by showing that the CXXC motif within its active site possesses high-affinity Cu(I)-binding activity [86], and can deliver Cu(I) to ATOX1 and the 5th and 6th MXCXXC motifs of ATP7B [87]. Taken together, these results suggest that a reducing cytosolic milieu promotes copper transport to the secretory pathway via GRX1-mediated reduction of copper-binding cysteines in ATOX1 and ATP7A/B. It will be important in future studies to test if GRX1 regulates copper homeostasis in vivo, and whether copper binding to GRX1 confers general changes in protein glutathionylation status. The regulation of ATP7A/B by glutathionylation raises the question of whether these proteins might be regulated by physiological conditions that alter cellular redox status. Recently, an elegant study by Lutsenko and colleagues suggests that copper transport into the secretory pathway via ATOX1/ATP7A is regulated by the ratio of GSH to GSSG [88]. During the differentiation of motor neurons, an increased GSH:GSSG ratio was found to shift the redox status of the CXXC copper-binding site of ATOX1 from partially oxidized to fully reduced, thus increasing its copper binding capacity and copper delivery to the secretory pathway via ATP7A [88]. Of relevance to cancer, transformation of mouse embryonic fibroblasts with oncogenic kinases, BRAFV600E and KRASG12D, was recently shown to lower labile pools of copper and decrease the GSH:GSSG ratio, which was attributable to reduced expression of glutathione reductase [89]. It will be interesting to determine the extent to which redox-mediated changes in copper homeostasis regulate normal physiological and pathophysiological processes.
Disorders of copper metabolism
Inherited mutations that disrupt copper transport or intracellular trafficking are responsible for several human diseases. Certain COX deficiency disorders are caused by mutations in mitochondrial metallochaperones such as SCO1 (OMIM 603644) [90], SCO2 (OMIM 604272) [91], COA6 (OMIM 614772) [92] and COX20 (OMIM 614698) [93]. Other diseases of copper metabolism arise from mutations that impact copper absorption and its distribution in the body. For example, loss of function mutations in ATP7A give rise to Menkes disease, a pediatric disorder of copper deficiency that primarily affects males due to its X-linked inheritance. Patients exhibit a wide range of defects attributable to a loss of cuproenzyme activity, including neurological degeneration, seizure, hypotonia, hypothermia, hypopigmentation and connective tissue defects [94]. The deficiency of copper in affected individuals is attributable to reduced ATP7A-dependent copper export from intestinal enterocytes into the plasma, which is exacerbated by reduced copper export into the central nervous system [95, 96]. Menkes patients are treated with injections of copper salts (typically copper histidinate), however this therapy rarely improves the overall prognosis and most patients die in early childhood [97]. Other treatments being actively pursued include ATP7A gene therapy using adeno-associated viral vectors, which have shown therapeutic efficacy in the Brindled mouse model of Menkes disease [98]. The copper ionophore elesclomol is another promising treatment based on its ability to alleviate Menkes pathology and rescue mortality in the Brindled mice [99]. Importantly, elesclomol has an excellent safety profile having been previously developed as an anticancer drug [100]. Thus, repurposing elesclomol as a treatment for Menkes disease is an exciting prospect.
While complete loss of ATP7A function causes Menkes disease, less severe mutations in ATP7A give rise to occipital horn syndrome (OMIM 304150), a disease in which neurological symptoms are relatively mild and connective tissue and skeletal defects predominate [101]. SMAX3 (spinal muscular atrophy X-linked type 3; OMIM 300489) is another disorder caused by two hypomorphic missense mutations in ATP7A, resulting in a distal motor neuropathy, progressive gait deterioration, and age-dependent muscle atrophy in the lower extremities [102]. Studies in our lab have shown that motor neuron-specific deletion of Atp7a in mice phenocopies many of the features of SMAX3 patients, indicating that ATP7A is functionally important in this neuronal cell type [103]. Two hypomorphic missense mutations in ATP7A are known to cause SMAX3 without generating a detectable copper deficiency in patients, and in vitro studies suggest that these mutations may impair the function of ATP7A only within motor neurons as a result of unique protein interactions in this cell type [104]. It is also possible that motor neurons are more sensitive than other cell types to subtle changes in copper homeostasis caused by hypomorphic mutations in copper trafficking pathways.
In contrast to ATP7A, mutations in ATP7B give rise to Wilson disease in which defective ATP7B-dependent biliary copper excretion causes hepatic copper overload, and if untreated, liver failure. Wilson patients may also exhibit neurological or psychiatric symptoms due to elevated copper in the brain [105]. Diagnostic features of Wilson disease include reduced ceruloplasmin activity and elevated copper concentrations in the serum, increased urinary copper and hyperpigmented copper deposits within the cornea (Kayser-Fleischer rings) [106]. The copper chelators penicillamine or trientine are often used as first-line treatments for symptomatic patients, whereas oral zinc therapy which is thought to compete for dietary copper absorption is indicated for asymptomatic patients [106]. As discussed below, the development of therapeutic copper chelators for Wilson disease have paved the way for copper depletion as a novel therapy in cancer. Disturbances in copper metabolism can also arise indirectly from mutations that affect general vesicular trafficking pathways. For example, in patients with MEDNIK syndrome (mental retardation, enteropathy, deafness, neuropathy, ichthyosis, keratodermia), mutations in the AP1 clathrin adapter complex give rise to hypocupremia and hypoceruloplasminemia as a consequence of defective trafficking of ATP7A and ATP7B [107]. Similarly, the hypopigmentation phenotype in patients with Hermansky-Pudlak syndrome is caused by mutations affecting the BLOC-1 trafficking complex, which is required for ATP7A trafficking to melanosomes for the metalation of tyrosinase [108].
Nutrient depletion as a cancer therapy – copper takes center stage
For almost a century it has been known that tumor cells adaptively modify pathways of nutrient metabolism to survive and proliferate within a myriad of different tissues and selective pressures. The concept that these adaptations might be vulnerable to targeted interventions is often attributed to the pediatric pathologist Sidney Farber, who in the 1940s observed that a diet rich in folic acid accelerated the progression of leukemia in children. Although the requirement for folic acid in DNA synthesis and thus cellular proliferation was unknown at the time, Farber showed that an inactive structural analogue of folate called aminopterin produced temporary remission in children with leukemia [109]. This breakthrough heralded the modern era of drug development, leading to more effective anti-folate drugs (e.g., methotrexate) and other “anti-metabolites”, which now constitute a major class of anti-neoplastic drugs.
In recent years, it has been increasingly recognized that the demand for copper is elevated in tumor cells relative to most other tissues, and represents a metabolic vulnerability that can be exploited by limiting copper availability [110]. As an element, copper cannot be created by metabolic processes, and its unique chemistry allows it to be selectively removed from dietary sources using highly specific copper chelators. The notion that tumor cells might have a higher copper requirement than normal tissues gained traction when copper concentrations within resected tumors of cancer patients were repeatedly found to be elevated compared to normal tissue, and that serum copper concentrations followed a similar trend [111-123] (Tables 2 and 3). The opportunity to directly test the effects of copper depletion on tumor growth was made possible with the advent of copper chelators such as penicillamine and trientine, which were developed for the treatment of copper overload in Wilson disease. By adding these chelators to the diets of animals, a state of copper deficiency could be achieved that reduced growth in various tumor models [8]. By the 1990s, a less toxic copper chelator called tetrathiomolybdate (TTM) was developed and has become the main drug used in copper depletion experiments in cancer models. There is now a substantial body of literature documenting the inhibitory effects of TTM on tumor growth and metastasis in various animal models of cancer (Table 4). This has led to a general hypothesis on the role of copper in cancer, which posits that 1) tumor cells have a higher demand for copper than normal cells; and 2) there is a window of copper depletion that can selectively block oncogenic processes without compromising essential metabolic functions. Several mechanisms have been ascribed to the therapeutic benefits of TTM including inhibition of cuproenzyme activities (e.g., LOX, SOD1 or COX), suppression of angiogenesis, and more recently, an attenuation of kinases that function in mitogenic signaling and autophagy [124-130]. One of the most striking demonstrations of the long-term therapeutic potential of TTM involved a study of spontaneous mammary tumor formation in the MMTV-neu mouse model [131]. In this study, the median time to develop tumors in untreated mice was 234 days compared to 460 days for the TTM-treated mice. However, the most intriguing finding of this study was that tumors appeared within weeks of stopping TTM therapy, suggesting that long-term copper depletion does not suppress neoplastic transformation, but rather maintains neoplastic cells in a continuous state of dormancy [132]. These findings provided important clues that copper depletion might function in preventing cancer malignancy.
Table 2.
Clinical studies of copper levels in tumors and normal tissue
| Type of Cancer |
Copper (μg/g) (Mean ± SD) |
p value | Reference | |
|---|---|---|---|---|
| Normal tissue |
Tumor | |||
| Breast | 9.3 ± 2.3* | 21.0 ± 10.7* | p < 0.0001 | PMID:6488192 |
| 6.13 ± 4.32 | 11.31 ± 7.83 | p < 0.01 | PMID:12413046 | |
| Gynecologic organs (cervix, uterus, ovary) | 1.26 ± 0.45 | 2.16 ± 0.6 | p < 0.01 | PMID:6871828 |
| Ovarian | 0.3 ± 0.1 | 0.7 ± 0.3 | p < 0.01 | PMID:17291257 |
| Lung | 5.08 ± 1.09 | 8.23 ± 4.88 | p < 0.01 | PMID:2740065 |
| Stomach | 1.44 ± 0.38* | 2.09 ± 0.52* | p < 0.02 | PMID:6871828 |
| 1.1 ± 0.4 | 1.7 ± 0.1 | p < 0.01 | PMID:17278230 | |
| Gallbladder | 16.30 ± 7.28 | 21.01 ± 6.67 | p = 0.01 | PMID:23942528 |
Measurements were obtained from tumor and normal tissue of the same patient. All other measurements were from different individuals.
Table 3:
Serum copper levels in cancer patients and healthy controls
| Type of Cancer | Serum copper (μg/dL) (Mean ± SD) |
p value | Reference | |
|---|---|---|---|---|
| Normal values |
Cancer patients |
|||
| Breast | 96 ± 11.52 | 152.96 ± 21.76 | p < 0.05 | PMID:11697759 |
| 101.62 ± 10.0 | 115.9 ± 14.4 | p < 0.01 | PMID:21968912 | |
| 109.56 ± 30.71 | 202.21 ± 89.18 | p < 0.001 | PMID:25737978 | |
| 96.5 ± 7.3 | 125.2 ± 15.0 | p < 0.01 | PMID:12413046 | |
| 65.2 ± 15.0 | 95.3 ± 4.9 | p < 0.01 | PMID:26837992 | |
| 113.2 ± 13.6 | 137.2 ± 36.2 | p < 0.001 | PMID:25520207 | |
| Leukemia | 86.7 ± 25.3 | 139.9 ± 51.2 | p < 0.001 | PMID:17205986 |
| 104.7 ± 0.16 | 122.6 ± 0.18 | p = 0.003 | PMID:22471499 | |
| 124.22 ± 10.36 | 146.25 ± 10.47 | p < 0.01 | PMID:26225654 | |
| 114 ± 29 | 328 ± 74 | p < 0.01 | PMID:3455680 | |
| 126.8 ± 21.2 | 256.8 ± 73.8 | p < 0.001 | PMID:4514972 | |
| Lung | 101.1 ± 7.96 | 128.6 ± 6.85 | p < 0.05 | PMID:7981002 |
| 128.5 ± 5.23 | 162.4 ± 8.18 | p < 0.01 | PMID:20638923 | |
| 112.5 ± 17 | 150.3 ± 33.3 | p < 0.001 | PMID:2914279 | |
| Oral | 114.20 ± 38.69 | 209.85 ± 160.28 | p < 0.001 | PMID:25179706 |
| 105.5 ± 18.81 | 141.99 ± 21.44 | p < 0.001 | PMID:27737355 | |
| Gastrointestinal | 96 ± 11.52 | 128.64 ± 20.48 | p < 0.05 | PMID:11697759 |
| Thyroid | 75.5 ± 8.1 | 85.4 ± 8.7 | p < 0.05 | PMID:27263537 |
| Gall bladder | 125.97 ± 38.93 | 159.19 ± 24.04 | p < 0.0002 | PMID:23942528 |
| Prostate | 97 ± 22 | 169.0 ± 31 | p < 0.005 | PMID:32398996 |
| Colorectal | 135.8 ± 30.5 | 138.6 ± 30.8 | p < 0.01 | PMID:28575311 |
Table 4.
Studies of TTM therapy in animal models of cancer
| Type of cancer | Predicted mechanism |
Reference |
|---|---|---|
| Breast | Angiogenesis inhibition | PMID:12208730 |
| NF-kappaB suppression and enhancement of doxorubicin induced apoptosis | PMID:12883034 | |
| Angiogenesis inhibition and hypoplastic remodeling of the mammary gland | PMID:19934283 | |
| Reduced LOX levels | PMID:27769988 | |
| Head and neck | Angiogenesis inhibition | PMID:15781759 |
| Angiogenesis inhibition | PMID:17363496 | |
| Angiogenesis inhibition | PMID:11359142 | |
| Reduced LOX activity | PMID:20682068 | |
| Lung | Angiogenesis inhibition | PMID:11896571 |
| Prostate | Angiogenesis inhibition | PMID:12192595 |
| Mesothelioma | Angiogenesis inhibition | PMID:24013775 |
| Papillary thyroid cancer | Inhibition of MAPK signaling | PMID:30065097 |
| Melanoma | Inhibition of MAPK signaling | PMID:28986383 |
| BRAF-driven MEF tumors | Inhibition of MAPK signaling | PMID:24717435 |
| Pancreatic | Inhibition of mitochondrial respiration | PMID:24218578 |
Clinical trials of therapeutic copper deficiency in cancer patients
To date, there have been seven clinical trials of oral TTM in cancer patients, each using a very similar protocol in which the copper chelator was taken up to 4 times daily with meals [8]. TTM binds to dietary copper in the small intestine and blocks its transport into the body, however, because excessive copper deficiency can be harmful, the dose of TTM is carefully titrated to reduce serum ceruloplasmin levels to ~20% of baseline (~5-15 mg/dL) [8, 133, 134]. Ceruloplasmin is rapidly degraded in the metal-free form, thus its abundance or activity in serum provides a reasonably accurate readout of whole-body copper status [135, 136]. This target level of ceruloplasmin corresponds to a state of moderate copper deficiency since it is reportedly well-tolerated by patients for multiple years with low rates of neutropenia or other indicators of severe copper deficiency [126, 133, 134, 137-140]. Unfortunately, the early clinical trials of TTM found no benefit in patients with metastatic prostate and kidney cancer [133, 134, 137]. However, because therapeutic levels of copper deficiency may take months to achieve [8], it was proposed that TTM may be more efficacious when used as an adjuvant to primary therapies, or when used at early stages of disease. In support of this concept, when administered as an adjuvant following surgery, TTM doubled the time to disease progression in patients with Stage I/II mesothelioma by two-fold (from 10 to 20 months), however there was no benefit in Stage III patients [139]. A more recent Phase II clinical trial tested the therapeutic efficacy of adjuvant TTM following chemotherapy in breast cancer patients, who showed no evidence of disease, but were at high risk for recurrence [126]. A total of 75 patients comprising Stage II/III (60%) and Stage IV (40%) disease were enrolled in this trial, of whom 48% were diagnosed with treat triple-negative breast cancer (TNBC), which is the most lethal and difficult to treat type of breast cancer. At a median follow-up time of 6.3 years, the overall survival and the event free survival rates for the entire cohort was 84% and 72%, respectively. Importantly, for TNBC patients, the event free survival was 90% (Stage II/III) and 50% (Stage IV). These results are striking considering that the average survival rate for the overall distant metastatic TNBC population is currently 11% at 5 years [141]. Because copper plays widespread roles in metabolism, a key question for the field is which oncogenic targets are susceptible to copper depletion? Below, we review recent studies that have begun to address this question.
Roles of copper in oncogenic kinases
It has been known for many decades that copper is essential for embryogenesis and cell proliferation, however, it was only recently that studies have identified specific functions of copper in mitogenic signaling pathways controlling both normal development and oncogenesis. Using genetic approaches in flies and later confirmed in mice, Thiele and colleagues showed that copper uptake via CTR1 stimulates the mitogen-activated protein kinase (MAPK) pathway (also known as the MAPK-ERK or RAS-RAF-MEK-ERK pathway) [142]. The MAPK pathway controls a wide range of normal biological processes such as proliferation, differentiation and motility, which are regulated by the interaction of growth factors with receptor tyrosine kinases at the plasma membrane. Biochemical studies demonstrated that the penultimate kinase in this pathway, MEK1, contains a high-affinity copper-binding site comprising two histidines and a methionine residue, which, upon copper binding, stimulate MEK1-dependent phosphorylation of ERK1/2 [129, 143] (Figure 4). Importantly, copper chelators were found to significantly reduce MEK1 signaling activity suggesting that MAPK-driven cancers may be a target of therapeutic copper depletion. In a series of elegant studies, Brady and colleagues demonstrated that orally administered TTM in mice could suppress the growth of melanoma xenografts containing an oncogenic mutation in the BRAF kinase, which lies upstream of MEK1 [129]. BRAF mutations are a common driver of metastatic melanoma, however resistance to BRAF inhibitors can frequently arise via activating mutations in downstream kinases such as MEK1. Therefore, it is especially notable that TTM showed efficacy against tumors expressing an activating MEK1 mutation that confers tolerance to BRAF inhibitors [129]. These studies suggest that oral copper chelators such as TTM may confer clinical benefit in BRAF-driven melanoma, particularly in the setting of acquired resistance to BRAF inhibitors.
Figure 4. Copper-dependent mechanisms of oncogenic signaling.
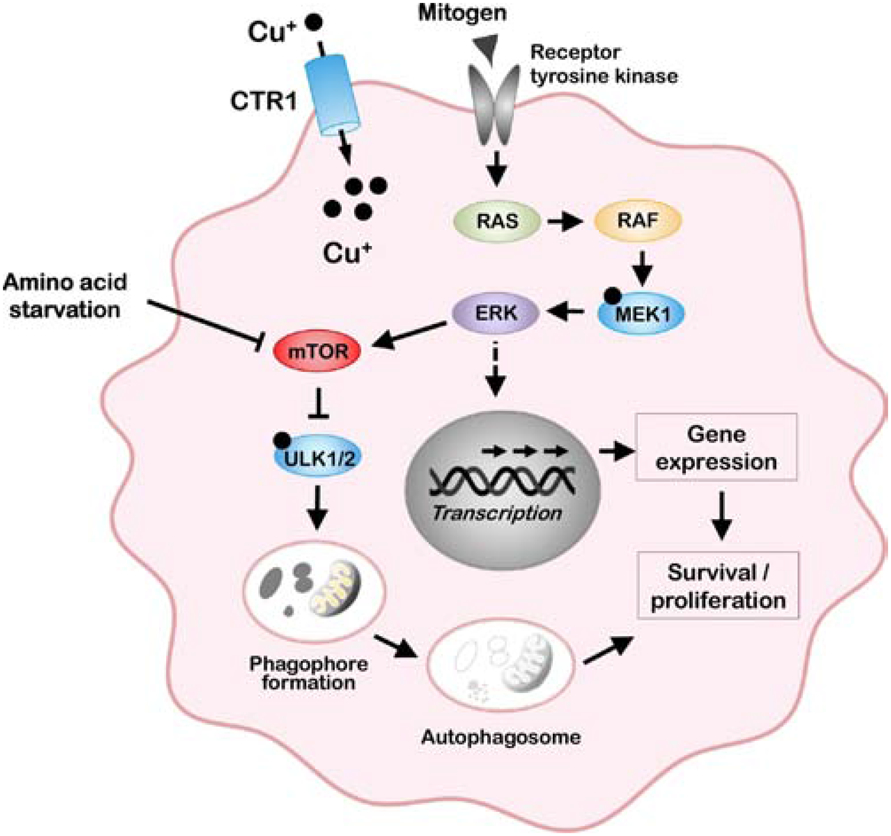
Copper uptake via CTR1 allosterically activates MEK1 to potentiate oncogenic signaling via the MAP kinase pathway. Copper is also an allosteric activator of the ULK1/2 kinases, which, under amino acid starvation conditions, stimulate formation of the autophagosome for the degradative recycling of biomolecules.
The discovery that copper functions as an allosteric regulator of MEK1 raises the question of whether other oncogenic kinases might be similarly regulated by copper. Recent studies have identified MEK1-like copper-binding histidine and methionine sequences in the ULK1 and ULK2 kinases. Biochemical and genetic analyses show that copper binding to these sequences stimulates ULK1/2-kinase activity which was inhibited by TTM [130]. The major function of ULK1/2 kinases is to initiate autophagy, which is an adaptive response to starvation and other stresses whereby damaged or redundant proteins or mitochondria are engulfed and degraded within membrane-bound autolysosomes (Figure 4). A growing body of evidence points to important roles for autophagy in the maintenance of certain tumors, including those driven by mutations in the MAP kinase, KRAS [144]. Genetic ablation of CTR1-dependent copper uptake in several KRAS mutant cell lines decreased copper-stimulated ULK1/2 activity and autophagic flux, and diminished tumor growth in a mouse model of KRAS-driven lung cancer [130]. Using a fluorescent copper probe, the induction of autophagy by amino acid starvation resulted in a significant rise in labile pools of intracellular copper within approximately 20 minutes [130]. Taken together, these studies reveal a novel function of copper in the growth of KRAS-driven tumors via ULK1/2-dependent autophagy and suggest copper may act as a second messenger in this signaling cascade. A precedent exists for labile copper pools regulating signaling within neurons and adipocytes [145], however, the source of these labile pools of copper is unknown. The role of copper in both MEK1/2 and ULK1/2 kinases underscores the potential benefits of copper chelation as a way to target separate signaling pathways within a single cancer (Figure 4), and, more broadly, prompts the question of whether copper chelation might be efficacious across the landscape of kinase-mediated oncogenic signaling pathways. The answers to such questions will be aided by continued efforts to uncover additional kinases regulated by copper.
Copper-dependent metabolic changes in cancer cells
The role of copper-trafficking to the mitochondria plays an important role in the Warburg effect, an early metabolic switch by which most cancer cells rely on glycolysis to drive energy production instead of mitochondrial oxidative phosphorylation. This process is controlled, in part, by the loss of the p53 tumor suppressor, a transcription factor that is mutated in many different types of cancer [146]. Oxygen consumption is diminished in p53-deficient cancer cells due to the loss of p53-dependent transcription of the SCO2 gene, which is required for COX assembly in the IMS of the mitochondria (Figure 1). Normal oxygen consumption is restored to p53-deficient cells by forced expression of a SCO2 transgene, suggesting that most, if not all, of the reduced oxygen consumption is attributable to a loss of SCO2 activity [146]. The reduced expression of SCO2 in p53-null cells is thought to protect against mitochondrially-produced reactive oxygen species, which are inherently elevated in cancer cells. It is also possible that the diminished expression of SCO2 in p53-null cells provides a selective advantage by increasing copper availability for oncogenic kinases such MEK1/2 and ULK1/2. In this context, it would be interesting to test the extent to which copper chelators protect against p53−/− tumors.
Recently, copper was shown to regulate expression of the programmed death ligand 1 (PD-L1), a transmembrane protein upregulated on the surface of certain cancer cells that allows evasion of the immune system [147]. The binding of PD-L1 to its receptor, PD-1, on the surface of lymphocytes is known to suppress the killing of cancer cells by cytotoxic T-cells [148]. In neuroblastoma cells, elevated copper accumulation resulting from increased CTR1 expression was found to upregulate STAT and EGFR signaling pathways leading to increased PD-L1 abundance [147]. The same study found that the copper chelator, TEPA, when given to mice reduced PD-L1 expression in neuroblastoma xenografts, resulting in higher numbers of tumor-infiltrating T cells and increased survival. Since checkpoint inhibitors targeting PD-L1/PD-1 are used clinically to treat melanoma and lung cancer [149], it would be interesting to test whether such therapies can be augmented by copper chelation.
Mechanisms of copper-dependent metastasis
The primary cause of morbidity and mortality in cancer is the metastasis of neoplastic cells from primary tumor sites to distant organs. The family of copper-dependent lysyl oxidase enzymes (LOX and LOXL1-4) appear to play key roles in this process. LOX enzymes catalyze the oxidation of lysine residues within collagen and/or elastin, stabilizing crosslinks between these proteins during the development or remodeling of the extracellular matrix [150] (Figure 5). Although LOX proteins exhibit poor sequence conservation at their N-termini, they share a conserved catalytic Cu-binding domain in their C-terminal regions. To date, oncogenic roles for LOX or LOXL enzymes have been documented in breast, colorectal, prostate, gastric, hepatic, pancreatic and head and neck cancers, as well as cancers of the skin, including melanoma [151-154]. Regions of increased breast density detected via mammography are attributable to increased collagen crosslinking by LOX enzymes, and are the second-most important risk factor in breast cancer after ageing [155]. Several oncogenic mechanisms have been ascribed to LOX proteins including tumor cell migration via the activation of focal adhesion kinase (FAK1) [156, 157], promoting the epithelial-to-mesenchymal transition [158], and the recruitment of myeloid cells and endothelial precursor cells to pre-metastatic sites to promote tumor seeding [153, 154, 159].
Figure 5. Copper-dependent mechanisms of cancer cell migration and metastasis.

The copper-binding protein MEMO1 is required for motility and metastasis of cancer cells in response to receptor tyrosine kinase signaling. MEMO1 is postulated to produce ROS at the leading edge of cancer cells which and activates pathways that influence cancer cell migration and invasion. Copper uptake via CTR1 facilitates ATOX1-mediated copper delivery to the ATP7A copper pump, which transports copper to the lysyl oxidase family of enzymes (LOX) within the secretory pathway. The LOX family of enzymes mediate the cross linking of collagen fibers generating H2O2 which facilitates activation of integrin-associated focal adhesion kinase (FAK1) and proto-oncogene tyrosine-protein kinase (SRC) which promote subsequent migration and metastasis. LOX enzymes also facilitate the formation of pre-metastatic niches at distant metastatic sites.
Recent studies from our group have demonstrated that the ATP7A copper transporter is required to deliver copper to multiple members of the LOX family (Figure 5). Deletion of ATP7A inhibited LOX activity in breast and lung cancer cell lines, resulting in a significant loss of tumor growth and metastatic potential of these cells in mice [160]. This was associated with reduced oncogenic activities known to be LOX-dependent such as myeloid cell recruitment to the lungs, FAK1 phosphorylation, EGFR-mediated signaling, and tumor-associated collagen fibril assembly. Consistent with a pro-oncogenic role for ATP7A, elevated tumor expression of ATP7A mRNA is correlated with higher patient mortality in breast cancer [160]. In another recent study, ATP7A was found to exert oncogenic activities in colorectal cancer cells expressing oncogenic mutant KRAS protein [161]. Compared to control cells, mutant KRAS-expressing cells exhibited elevated total and surface levels of ATP7A which was essential to protect against high levels of intracellular copper accumulated via enhanced rates of macropinocytosis. Silencing of ATP7A was found to significantly reduce tumor growth in a mouse xenograft model of KRAS-expressing IEC6 intestinal cells [161]. Taken together, these studies suggest that a small molecule inhibitor of ATP7A may have therapeutic benefits in treating multiple types of cancer.
Other studies have shown that silencing of ATOX1, which delivers copper to ATP7A, reduces both LOX activity and cell migration in breast cancer cells [162]. An inhibitor with dual specificity against ATOX1 and domain I of CCS was shown to attenuate the proliferation of several cancer cells line in vitro, and block tumor growth in mouse xenograft models [163]. As with ATP7A, high tumor expression of ATOX1 is significantly correlated with poor survival in breast cancer patients [164]. Genetic deletion or pharmacological inhibition of ATOX1 was found to attenuate MAPK signaling and significantly reduce the proliferation of BRAF-driven melanoma cell lines, thus implicating a role for ATOX1 in copper delivery to MEK1 [165]. Other notable functions of ATOX1 include a reported ability to stimulate cell proliferation as a transcriptional activator of cyclin D1 (Figure 5), although this function has not been directly assessed in a cancer model [166]. Another copper-dependent enzyme with roles in tumor metastasis is MEMO1 (MEdiator of cell MOtility 1), which accumulates at the leading edge of cancer cells and promotes cell migration in response to several receptor tyrosinase kinases [167]. In a recent study, the expression level of MEMO1 in human breast tumors was found to be prognostic of poor outcome, and MEMO1 silencing in human breast cancer cell lines significantly reduced secondary lung metastases in a mouse xenograft model [168]. While the role of MEMO1 in cancer is not well understood, it is postulated to regulate the redox status of several proteins involved in cancer cell migration by producing localized ROS at focal adhesion sites at the leading edge (Figure 5). Additional studies are warranted to understand the pathways by which MEMO1 is metallated, and the extent to which copper chelation therapy interferes with the oncogenic activities of this novel cuproenzyme.
The intersection of copper metabolism and chemoresistance
Cisplatin is a platinum-based DNA crosslinking drug used to treat 40% of all cancer patients undergoing chemotherapy [169, 170], however, acquired resistance by tumor cells often limits its efficacy [171, 172]. Several different mechanisms of cisplatin resistance have been described including enhanced repair of DNA adducts, increased sequestration by endogenous nucleophiles (e.g., MT, GSH) or reduced accumulation of the drug [173]. Numerous genetic and biochemical studies suggest that copper transporters contribute to cisplatin resistance by controlling its uptake and export from tumor cells. For many years it was thought that cisplatin entered cells via passive diffusion across the plasma membrane until a genetic screen in yeast, and later confirmed in mammalian cells, demonstrated that CTR1 functions in the cellular uptake of cisplatin. Silencing of CTR1 was shown to reduce cellular levels of cisplatin compared to control cells resulting in a two- to three-fold increase in cisplatin tolerance [174, 175]. Consistent with its contribution to cisplatin sensitivity, CTR1 expression is reduced in certain cisplatin-resistant ovarian carcinoma cell lines [176]. However, a direct role of CTR1 in cisplatin accumulation remains controversial with some studies finding no evidence of altered cisplatin uptake or changes in sensitivity upon CTR1 silencing or overexpression in cultured cell lines [177-179]. Moreover, the recently reported crystal structure of CTR1 does not appear to readily accommodate a relatively large molecule like cisplatin or its structural analogues such as carboplatin [16]. It is possible that CTR1-driven mechanisms of cisplatin resistance may be variable in different cancer types and/or occur indirectly as a consequence of altered copper uptake. Indeed, depletion of antioxidant reserves, which are known to respond to changes in copper homeostasis, increase cisplatin sensitivity whereas elevated synthesis or conjugation to GSH is known to confer cisplatin chemoresistance [173]. Other studies suggest that CTR1-dependent accumulation of copper and cisplatin occur via different mechanisms based on findings that certain mutations block the uptake of one substrate but not the other [180]. We and others speculate that CTR1 mediates the uptake of cisplatin via an endocytic mechanism, rather than direct transport across the lipid bilayer [33, 181]. Consistent with this concept, cisplatin has been shown to induce the endocytosis and degradation of CTR1 in several cell types [182]. Once internalized, the ability of cisplatin to access DNA targets in the nucleus may occur via an unknown endosomal exporter or by direct endosomal fusion with the nuclear envelope [183].
Numerous studies have found compelling evidence of a role for either ATP7A or ATP7B in cisplatin resistance. In ovarian carcinoma cell lines, cisplatin resistance results in elevated expression of ATP7A or ATP7B and cross-resistance to copper [184, 185]. Cultured cell lines in which ATP7A is deleted or overexpressed demonstrate that ATP7A is necessary and sufficient to confer chemoresistance to cisplatin or other platinum-based analogues carboplatin and oxaliplatin [186]. Recent studies from our laboratory demonstrated that deletion of ATP7A increases cisplatin sensitivity and limits tumor growth in mice [187]. In cancer patients, ATP7A mRNA levels were found to be elevated in surgically resected cisplatin-resistant specimens of non-small cell lung cancer (NSCLC) relative to cisplatin-sensitive controls [188]. In breast carcinoma cell lines, a recent study demonstrated that ATP7A expression and cisplatin chemoresistance is negatively regulated by the miR-148a-3p microRNA [189], which has documented roles in metastasis of triple negative breast cancer [190]. Elevated levels of ATP7B are significantly correlated with poor clinical response to cisplatin chemotherapy in patients with multiple cancer types [191-194]. The mechanism by which ATP7A/B confer cisplatin resistance is poorly understood. Like copper, cisplatin stimulates the exocytic trafficking of both transporters from the TGN into cytoplasmic vesicles [195]. Moreover, ATP7A/B-dependent chemoresistance is associated with a reduction in cisplatin accumulation and DNA adduct formation [184]. Thus, it is thought that both transporters actively transport cisplatin into cytoplasmic vesicles, thus limiting access to the nucleus. However, such a model has been criticized based on the fact that both ATP7A and ATP7B are highly specific for Cu(I) and that copper and cisplatin exhibit considerable differences in size and electrochemical properties. Nonetheless, in vitro studies suggest that, like copper, cisplatin stimulates acyl-phosphorylation of ATP7A and ATP7B in microsomal preparations [196, 197]. Moreover, the sixth amino terminal MXCXXC domain of ATP7B required for copper-stimulated trafficking and catalysis also appears to be crucial for cisplatin-induced trafficking and chemoresistance [198]. Consistent with the movement of cisplatin along copper-transport pathways, studies also suggest a role for the ATOX1 metallochaperone in cisplatin resistance. Cisplatin binds to the CXXC copper-binding site of ATOX1 and can be transferred to the metal binding domain of ATP7B, suggesting that ATOX1 may be important for ATP7A/B-mediated cisplatin detoxification [199, 200]. Elevated expression of ATOX1 has been associated with increased cisplatin tolerance within tumor cell lines [201]. In contrast, however, other studies have found that deletion of ATOX1 either increases [202] or has no effect [179] on cisplatin resistance. Since ATOX1 is reported to enter the nucleus in the metal-bound state [166], it is possible that in addition to delivering cisplatin to ATP7A/B, ATOX1 may also carry cisplatin into the nucleus to promote DNA damage. Thus, whether ATOX1 enhances or reduces cisplatin sensitivity may be dependent on the net balance of these two activities.
The hijacking of copper trafficking pathways by cisplatin presents potential therapeutic strategies to attenuate cisplatin resistance by manipulating copper homeostasis in cancer patients. The copper chelator, TTM, was found to augment the inhibitory effects of cisplatin on tumor growth in mice and was associated with a reduction in the expression of ATP7A or ATP7B in tumor cells [203, 204]. Additionally, TTM has been shown to form intramolecular crosslinks between individual amino terminal metal binding domains of ATP7B, which would be expected to block cisplatin binding to these sites [205]. In addition, TTM may sensitize cells to cisplatin by forming a stable complex with ATOX1 [206]. More broadly, these studies raise the question of whether other molecules that attenuate ATP7A/B activity might have therapeutic value in overcoming cisplatin resistance. Recently, a high-throughput screen identified three clinically approved drugs (Tranilast, Telmisartan, and Amphotericin B) which reduced ATP7B abundance or trafficking and enhanced cisplatin-mediated toxicity in cisplatin-resistant ovarian carcinoma cells [207]. These effects were specific to cisplatin-resistant cells since the same compounds failed to augment cisplatin toxicity or attenuate ATP7B trafficking in HepG2 cells. Taken together, these studies provide proof-of-concept that cisplatin resistance might be overcome by targeted inhibition of ATP7A/B in tumor cells.
Conclusions
In 1993, an exceptionally rare case of a female child with Menkes disease provided the key to discovering the first copper transporter, ATP7A [208-210]. Over the ensuing decades, the discovery of additional copper transporters, metallochaperones and cuproenzymes is a testament to the extraordinary progress in the field. Nonetheless, many questions remain regarding the mechanisms by which copper is trafficked and utilized within the cell. The need to address these outstanding questions is amplified by a growing chorus of experimental and clinical data that brings into focus the importance of copper in oncogenic pathways, and the therapeutic potential of manipulating copper metabolism to prevent cancer progression. Over the next several years it will be important to identify the molecular underpinnings of altered copper homeostasis in different types of cancer, and to categorize the types of oncogenic mutations and chemotherapy regimens that are responsive to copper chelation therapy. The continued investigation of copper transporters, metallochaperones and cuproenzymes in oncogenic pathways will provide a rich menu of targets for developing innovative therapies with the potential to expand the existing landscape of cancer treatments.
Highlights.
Regulatory mechanisms of copper homeostasis in mammals
Disorders of copper metabolism
Roles of copper in tumor growth and metastasis
Copper depletion as an anticancer strategy
Acknowledgements
We acknowledge the financial support of the following grants from the National Institutes of Health: CA190265 (NCI) and DK116859 (NIDDK).
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Nevitt T, Ohrvik H, Thiele DJ, Charting the travels of copper in eukaryotes from yeast to mammals, Biochim Biophys Acta, 1823 (2012) 1580–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ochiai EI, Copper and the biological evolution, Biosystems, 16 (1983) 81–86. [DOI] [PubMed] [Google Scholar]
- [3].Chi Fru E, Rodriguez NP, Partin CA, Lalonde SV, Andersson P, Weiss DJ, El Albani A, Rodushkin I, Konhauser KO, Cu isotopes in marine black shales record the Great Oxidation Event, Proc Natl Acad Sci U S A, 113 (2016) 4941–4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Macomber L, Imlay JA, The iron-sulfur clusters of dehydratases are primary intracellular targets of copper toxicity, Proc Natl Acad Sci U S A, 106 (2009) 8344–8349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Pierson H, Yang H, Lutsenko S, Copper Transport and Disease: What Can We Learn from Organoids?, Annu Rev Nutr, 39 (2019) 75–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Turnlund JR, Keyes WR, Anderson HL, Acord LL, Copper absorption and retention in young men at three levels of dietary copper by use of the stable isotope 65Cu, Am J Clin Nutr, 49 (1989) 870–878. [DOI] [PubMed] [Google Scholar]
- [7].Prohaska JR, Impact of copper deficiency in humans, Ann N Y Acad Sci, 1314 (2014) 1–5. [DOI] [PubMed] [Google Scholar]
- [8].Lopez J, Ramchandani D, Vahdat L, Copper Depletion as a Therapeutic Strategy in Cancer, Met Ions Life Sci, 19 (2019). [DOI] [PubMed] [Google Scholar]
- [9].Zhou B, Gitschier J, hCTR1: a human gene for copper uptake identified by complementation in yeast, Proc Natl Acad Sci U S A, 94 (1997) 7481–7486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kuo YM, Zhou B, Cosco D, Gitschier J, The copper transporter CTR1 provides an essential function in mammalian embryonic development, Proc Natl Acad Sci U S A, 98 (2001) 6836–6841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Nose Y, Kim BE, Thiele DJ, Ctr1 drives intestinal copper absorption and is essential for growth, iron metabolism, and neonatal cardiac function, Cell Metab, 4 (2006) 235–244. [DOI] [PubMed] [Google Scholar]
- [12].Lin C, Zhang Z, Wang T, Chen C, James Kang Y, Copper uptake by DMT1: a compensatory mechanism for CTR1 deficiency in human umbilical vein endothelial cells, Metallomics, 7 (2015) 1285–1289. [DOI] [PubMed] [Google Scholar]
- [13].Shawki A, Anthony SR, Nose Y, Engevik MA, Niespodzany EJ, Barrientos T, Ohrvik H, Worrell RT, Thiele DJ, Mackenzie B, Intestinal DMT1 is critical for iron absorption in the mouse but is not required for the absorption of copper or manganese, Am J Physiol Gastrointest Liver Physiol, 309 (2015) G635–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lee J, Pena MM, Nose Y, Thiele DJ, Biochemical characterization of the human copper transporter Ctr1, J Biol Chem, 277 (2002) 4380–4387. [DOI] [PubMed] [Google Scholar]
- [15].Aller SG, Unger VM, Projection structure of the human copper transporter CTR1 at 6-A resolution reveals a compact trimer with a novel channel-like architecture, Proc Natl Acad Sci U S A, 103 (2006) 3627–3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ren F, Logeman BL, Zhang X, Liu Y, Thiele DJ, Yuan P, X-ray structures of the high-affinity copper transporter Ctr1, Nat Commun, 10 (2019) 1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Puig S, Lee J, Lau M, Thiele DJ, Biochemical and genetic analyses of yeast and human high affinity copper transporters suggest a conserved mechanism for copper uptake, J Biol Chem, 277 (2002) 26021–26030. [DOI] [PubMed] [Google Scholar]
- [18].Drakesmith H, Nemeth E, Ganz T, Ironing out Ferroportin, Cell Metab, 22 (2015) 777–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ramos D, Mar D, Ishida M, Vargas R, Gaite M, Montgomery A, Linder MC, Mechanism of Copper Uptake from Blood Plasma Ceruloplasmin by Mammalian Cells, PLoS One, 11 (2016) e0149516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Meyer LA, Durley AP, Prohaska JR, Harris ZL, Copper transport and metabolism are normal in aceruloplasminemic mice, J Biol Chem, 276 (2001) 36857–36861. [DOI] [PubMed] [Google Scholar]
- [21].Linder MC, Ceruloplasmin and other copper binding components of blood plasma and their functions: an update, Metallomics, 8 (2016) 887–905. [DOI] [PubMed] [Google Scholar]
- [22].Gray LW, Peng F, Molloy SA, Pendyala VS, Muchenditsi A, Muzik O, Lee J, Kaplan JH, Lutsenko S, Urinary copper elevation in a mouse model of Wilson's disease is a regulated process to specifically decrease the hepatic copper load, PLoS One, 7 (2012) e38327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hassett R, Kosman DJ, Evidence for Cu(II) reduction as a component of copper uptake by Saccharomyces cerevisiae, J Biol Chem, 270 (1995) 128–134. [DOI] [PubMed] [Google Scholar]
- [24].Ohgami RS, Campagna DR, McDonald A, Fleming MD, The Steap proteins are metalloreductases, Blood, 108 (2006) 1388–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liao Y, Zhao J, Bulek K, Tang F, Chen X, Cai G, Jia S, Fox PL, Huang E, Pizarro TT, Kalady MF, Jackson MW, Bao S, Sen GC, Stark GR, Chang CJ, Li X, Inflammation mobilizes copper metabolism to promote colon tumorigenesis via an IL-17-STEAP4-XIAP axis, Nat Commun, 11 (2020) 900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Liang ZD, Tsai WB, Lee MY, Savaraj N, Kuo MT, Specificity protein 1 (sp1) oscillation is involved in copper homeostasis maintenance by regulating human high-affinity copper transporter 1 expression, Mol Pharmacol, 81 (2012) 455–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Song IS, Chen HH, Aiba I, Hossain A, Liang ZD, Klomp LW, Kuo MT, Transcription factor Sp1 plays an important role in the regulation of copper homeostasis in mammalian cells, Mol Pharmacol, 74 (2008) 705–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lee J, Prohaska JR, Dagenais SL, Glover TW, Thiele DJ, Isolation of a murine copper transporter gene, tissue specific expression and functional complementation of a yeast copper transport mutant, Gene, 254 (2000) 87–96. [DOI] [PubMed] [Google Scholar]
- [29].Petris MJ, Smith K, Lee J, Thiele DJ, Copper-stimulated endocytosis and degradation of the human copper transporter, hCtr1, J Biol Chem, 278 (2003) 9639–9646. [DOI] [PubMed] [Google Scholar]
- [30].Maryon EB, Molloy SA, Ivy K, Yu H, Kaplan JH, Rate and regulation of copper transport by human copper transporter 1 (hCTR1), J Biol Chem, 288 (2013) 18035–18046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Curnock R, Cullen PJ, Mammalian copper homeostasis requires retromer-dependent recycling of the high-affinity copper transporter 1, J Cell Sci, 133 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang J, Fedoseienko A, Chen B, Burstein E, Jia D, Billadeau DD, Endosomal receptor trafficking: Retromer and beyond, Traffic, 19 (2018) 578–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ohrvik H, Logeman B, Turk B, Reinheckel T, Thiele DJ, Cathepsin Protease Controls Copper and Cisplatin Accumulation via Cleavage of the Ctr1 Metal-binding Ectodomain, J Biol Chem, 291 (2016) 13905–13916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ohrvik H, Nose Y, Wood LK, Kim BE, Gleber SC, Ralle M, Thiele DJ, Ctr2 regulates biogenesis of a cleaved form of mammalian Ctr1 metal transporter lacking the copper- and cisplatin-binding ecto-domain, Proc Natl Acad Sci U S A, 110 (2013) E4279–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Logeman BL, Wood LK, Lee J, Thiele DJ, Gene duplication and neo-functionalization in the evolutionary and functional divergence of the metazoan copper transporters Ctr1 and Ctr2, J Biol Chem, 292 (2017) 11531–11546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Culotta V, Scott RA, Metals in Cells, Wiley, Place Published, 2016. [Google Scholar]
- [37].Gudekar N, Shanbhag V, Wang Y, Ralle M, Weisman GA, Petris MJ, Metallothioneins regulate ATP7A trafficking and control cell viability during copper deficiency and excess, Sci Rep, 10 (2020) 7856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Jiang X, Chen J, Bajic A, Zhang C, Song X, Carroll SL, Cai ZL, Tang M, Xue M, Cheng N, Schaaf CP, Li F, MacKenzie KR, Ferreon ACM, Xia F, Wang MC, Maletic-Savatic M, Wang J, Quantitative real-time imaging of glutathione, Nat Commun, 8 (2017) 16087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Maryon EB, Molloy SA, Kaplan JH, Cellular glutathione plays a key role in copper uptake mediated by human copper transporter 1, Am J Physiol Cell Physiol, 304 (2013) C768–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Meister A, Anderson ME, Glutathione, Annu Rev Biochem, 52 (1983) 711–760. [DOI] [PubMed] [Google Scholar]
- [41].DeLeve LD, Kaplowitz N, Importance and regulation of hepatic glutathione, Semin Liver Dis, 10 (1990) 251–266. [DOI] [PubMed] [Google Scholar]
- [42].Suthanthiran M, Anderson ME, Sharma VK, Meister A, Glutathione regulates activation-dependent DNA synthesis in highly purified normal human T lymphocytes stimulated via the CD2 and CD3 antigens, Proc Natl Acad Sci U S A, 87 (1990) 3343–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Forman HJ, Zhang H, Rinna A, Glutathione: overview of its protective roles, measurement, and biosynthesis, Mol Aspects Med, 30 (2009) 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Timon-Gomez A, Nyvltova E, Abriata LA, Vila AJ, Hosler J, Barrientos A, Mitochondrial cytochrome c oxidase biogenesis: Recent developments, Semin Cell Dev Biol, 76 (2018) 163–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Cobine PA, Ojeda LD, Rigby KM, Winge DR, Yeast contain a non-proteinaceous pool of copper in the mitochondrial matrix, J Biol Chem, 279 (2004) 14447–14455. [DOI] [PubMed] [Google Scholar]
- [46].Vest KE, Leary SC, Winge DR, Cobine PA, Copper import into the mitochondrial matrix in Saccharomyces cerevisiae is mediated by Pic2, a mitochondrial carrier family protein, J Biol Chem, 288 (2013) 23884–23892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Vest KE, Wang J, Gammon MG, Maynard MK, White OL, Cobine JA, Mahone WK, Cobine PA, Overlap of copper and iron uptake systems in mitochondria in Saccharomyces cerevisiae, Open Biol, 6 (2016) 150223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Boulet A, Vest KE, Maynard MK, Gammon MG, Russell AC, Mathews AT, Cole SE, Zhu X, Phillips CB, Kwong JQ, Dodani SC, Leary SC, Cobine PA, The mammalian phosphate carrier SLC25A3 is a mitochondrial copper transporter required for cytochrome c oxidase biogenesis, J Biol Chem, 293 (2018) 1887–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Winge DR, Filling the mitochondrial copper pool, J Biol Chem, 293 (2018) 1897–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Culotta VC, Klomp LW, Strain J, Casareno RL, Krems B, Gitlin JD, The copper chaperone for superoxide dismutase, J Biol Chem, 272 (1997) 23469–23472. [DOI] [PubMed] [Google Scholar]
- [51].Casareno RL, Waggoner D, Gitlin JD, The copper chaperone CCS directly interacts with copper/zinc superoxide dismutase, J Biol Chem, 273 (1998) 23625–23628. [DOI] [PubMed] [Google Scholar]
- [52].Lamb AL, Torres AS, O'Halloran TV, Rosenzweig AC, Heterodimeric structure of superoxide dismutase in complex with its metallochaperone, Nat Struct Biol, 8 (2001) 751–755. [DOI] [PubMed] [Google Scholar]
- [53].Torres AS, Petri V, Rae TD, O'Halloran TV, Copper stabilizes a heterodimer of the yCCS metallochaperone and its target superoxide dismutase, J Biol Chem, 276 (2001) 38410–38416. [DOI] [PubMed] [Google Scholar]
- [54].Sturtz LA, Diekert K, Jensen LT, Lill R, Culotta VC, A fraction of yeast Cu,Zn-superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria. A physiological role for SOD1 in guarding against mitochondrial oxidative damage, J Biol Chem, 276 (2001) 38084–38089. [DOI] [PubMed] [Google Scholar]
- [55].Cobine PA, Pierrel F, Bestwick ML, Winge DR, Mitochondrial matrix copper complex used in metallation of cytochrome oxidase and superoxide dismutase, J Biol Chem, 281 (2006) 36552–36559. [DOI] [PubMed] [Google Scholar]
- [56].Kawamata H, Manfredi G, Different regulation of wild-type and mutant Cu,Zn superoxide dismutase localization in mammalian mitochondria, Hum Mol Genet, 17 (2008) 3303–3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kloppel C, Suzuki Y, Kojer K, Petrungaro C, Longen S, Fiedler S, Keller S, Riemer J, Mia40-dependent oxidation of cysteines in domain I of Ccs1 controls its distribution between mitochondria and the cytosol, Mol Biol Cell, 22 (2011) 3749–3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Polishchuk R, Lutsenko S, Golgi in copper homeostasis: a view from the membrane trafficking field, Histochemistry and cell biology, 140 (2013) 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Petris MJ, Mercer JF, Culvenor JG, Lockhart P, Gleeson PA, Camakaris J, Ligand-regulated transport of the Menkes copper P-type ATPase efflux pump from the Golgi apparatus to the plasma membrane: a novel mechanism of regulated trafficking, EMBO J, 15 (1996) 6084–6095. [PMC free article] [PubMed] [Google Scholar]
- [60].Hung IH, Suzuki M, Yamaguchi Y, Yuan DS, Klausner RD, Gitlin JD, Biochemical characterization of the Wilson disease protein and functional expression in the yeast Saccharomyces cerevisiae, J Biol Chem, 272 (1997) 21461–21466. [DOI] [PubMed] [Google Scholar]
- [61].Barry AN, Shinde U, Lutsenko S, Structural organization of human Cu-transporting ATPases: learning from building blocks, J Biol Inorg Chem, 15 (2010) 47–59. [DOI] [PubMed] [Google Scholar]
- [62].Dyla M, Kjaergaard M, Poulsen H, Nissen P, Structure and Mechanism of P-Type ATPase Ion Pumps, Annu Rev Biochem, 89 (2020) 583–603. [DOI] [PubMed] [Google Scholar]
- [63].Gourdon P, Liu XY, Skjorringe T, Morth JP, Moller LB, Pedersen BP, Nissen P, Crystal structure of a copper-transporting PIB-type ATPase, Nature, 475 (2011) 59–64. [DOI] [PubMed] [Google Scholar]
- [64].Pase L, Voskoboinik I, Greenough M, Camakaris J, Copper stimulates trafficking of a distinct pool of the Menkes copper ATPase (ATP7A) to the plasma membrane and diverts it into a rapid recycling pool, Biochem J, 378 (2004) 1031–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Hartwig C, Zlatic SA, Wallin M, Vrailas-Mortimer A, Fahrni CJ, Faundez V, Trafficking mechanisms of P-type ATPase copper transporters, Curr Opin Cell Biol, 59 (2019) 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Monty JF, Llanos RM, Mercer JF, Kramer DR, Copper exposure induces trafficking of the menkes protein in intestinal epithelium of ATP7A transgenic mice, J Nutr, 135 (2005) 2762–2766. [DOI] [PubMed] [Google Scholar]
- [67].Roelofsen H, Wolters H, Van Luyn MJ, Miura N, Kuipers F, Vonk RJ, Copper-induced apical trafficking of ATP7B in polarized hepatoma cells provides a mechanism for biliary copper excretion, Gastroenterology, 119 (2000) 782–793. [DOI] [PubMed] [Google Scholar]
- [68].Polishchuk EV, Concilli M, Iacobacci S, Chesi G, Pastore N, Piccolo P, Paladino S, Baldantoni D, van ISC, Chan J, Chang CJ, Amoresano A, Pane F, Pucci P, Tarallo A, Parenti G, Brunetti-Pierri N, Settembre C, Ballabio A, Polishchuk RS, Wilson disease protein ATP7B utilizes lysosomal exocytosis to maintain copper homeostasis, Dev Cell, 29 (2014) 686–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Zhu S, Shanbhag V, Hodgkinson VL, Petris MJ, Multiple di-leucines in the ATP7A copper transporter are required for retrograde trafficking to the trans-Golgi network, Metallomics, 8 (2016) 993–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Francis MJ, Jones EE, Levy ER, Martin RL, Ponnambalam S, Monaco AP, Identification of a di-leucine motif within the C terminus domain of the Menkes disease protein that mediates endocytosis from the plasma membrane, J Cell Sci, 112 (Pt 11) (1999) 1721–1732. [DOI] [PubMed] [Google Scholar]
- [71].Holloway ZG, Velayos-Baeza A, Howell GJ, Levecque C, Ponnambalam S, Sztul E, Monaco AP, Trafficking of the Menkes copper transporter ATP7A is regulated by clathrin-, AP-2-, AP-1-, and Rab22-dependent steps, Mol Biol Cell, 24 (2013) 1735–1748, S1731-1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Francis MJ, Jones EE, Levy ER, Ponnambalam S, Chelly J, Monaco AP, A Golgi localization signal identified in the Menkes recombinant protein, Hum Mol Genet, 7 (1998) 1245–1252. [DOI] [PubMed] [Google Scholar]
- [73].Greenough M, Pase L, Voskoboinik I, Petris MJ, O'Brien AW, Camakaris J, Signals regulating trafficking of Menkes (MNK; ATP7A) copper-translocating P-type ATPase in polarized MDCK cells, Am J Physiol Cell Physiol, 287 (2004) C1463–1471. [DOI] [PubMed] [Google Scholar]
- [74].Braiterman L, Nyasae L, Guo Y, Bustos R, Lutsenko S, Hubbard A, Apical targeting and Golgi retention signals reside within a 9-amino acid sequence in the copper-ATPase, ATP7B, Am J Physiol Gastrointest Liver Physiol, 296 (2009) G433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Strausak D, La Fontaine S, Hill J, Firth SD, Lockhart PJ, Mercer JF, The role of GMXCXXC metal binding sites in the copper-induced redistribution of the Menkes protein, J Biol Chem, 274 (1999) 11170–11177. [DOI] [PubMed] [Google Scholar]
- [76].Goodyer ID, Jones EE, Monaco AP, Francis MJ, Characterization of the Menkes protein copper-binding domains and their role in copper-induced protein relocalization, Hum Mol Genet, 8 (1999) 1473–1478. [DOI] [PubMed] [Google Scholar]
- [77].Petris MJ, Voskoboinik I, Cater M, Smith K, Kim BE, Llanos RM, Strausak D, Camakaris J, Mercer JF, Copper-regulated trafficking of the Menkes disease copper ATPase is associated with formation of a phosphorylated catalytic intermediate, J Biol Chem, 277 (2002) 46736–46742. [DOI] [PubMed] [Google Scholar]
- [78].Cater MA, La Fontaine S, Mercer JF, Copper binding to the N-terminal metal-binding sites or the CPC motif is not essential for copper-induced trafficking of the human Wilson protein (ATP7B), Biochem J, 401 (2007) 143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Skjorringe T, Amstrup Pedersen P, Salling Thorborg S, Nissen P, Gourdon P, Birk Moller L, Characterization of ATP7A missense mutants suggests a correlation between intracellular trafficking and severity of Menkes disease, Sci Rep, 7 (2017) 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Kim BE, Smith K, Petris MJ, A copper treatable Menkes disease mutation associated with defective trafficking of a functional Menkes copper ATPase, J Med Genet, 40 (2003) 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Braiterman LT, Gupta A, Chaerkady R, Cole RN, Hubbard AL, Communication between the N and C termini is required for copper-stimulated Ser/Thr phosphorylation of Cu(I)-ATPase (ATP7B), J Biol Chem, 290 (2015) 8803–8819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Veldhuis NA, Valova VA, Gaeth AP, Palstra N, Hannan KM, Michell BJ, Kelly LE, Jennings I, Kemp BE, Pearson RB, Robinson PJ, Camakaris J, Phosphorylation regulates copper-responsive trafficking of the Menkes copper transporting P-type ATPase, Int J Biochem Cell Biol, 41 (2009) 2403–2412. [DOI] [PubMed] [Google Scholar]
- [83].Singleton WC, McInnes KT, Cater MA, Winnall WR, McKirdy R, Yu Y, Taylor PE, Ke BX, Richardson DR, Mercer JF, La Fontaine S, Role of glutaredoxin1 and glutathione in regulating the activity of the copper-transporting P-type ATPases, ATP7A and ATP7B, J Biol Chem, 285 (2010) 27111–27121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Hatori Y, Clasen S, Hasan NM, Barry AN, Lutsenko S, Functional partnership of the copper export machinery and glutathione balance in human cells, J Biol Chem, 287 (2012) 26678–26687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Ho YS, Xiong Y, Ho DS, Gao J, Chua BH, Pai H, Mieyal JJ, Targeted disruption of the glutaredoxin 1 gene does not sensitize adult mice to tissue injury induced by ischemia/reperfusion and hyperoxia, Free Radic Biol Med, 43 (2007) 1299–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Brose J, La Fontaine S, Wedd AG, Xiao Z, Redox sulfur chemistry of the copper chaperone Atox1 is regulated by the enzyme glutaredoxin 1, the reduction potential of the glutathione couple GSSG/2GSH and the availability of Cu(I), Metallomics, 6 (2014) 793–808. [DOI] [PubMed] [Google Scholar]
- [87].Maghool S, Fontaine S, Roberts BR, Kwan AH, Maher MJ, Human glutaredoxin-1 can transfer copper to isolated metal binding domains of the P1B-type ATPase, ATP7B, Sci Rep, 10 (2020) 4157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Hatori Y, Yan Y, Schmidt K, Furukawa E, Hasan NM, Yang N, Liu CN, Sockanathan S, Lutsenko S, Neuronal differentiation is associated with a redox-regulated increase of copper flow to the secretory pathway, Nat Commun, 7 (2016) 10640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Chung CY, Posimo JM, Lee S, Tsang T, Davis JM, Brady DC, Chang CJ, Activity-based ratiometric FRET probe reveals oncogene-driven changes in labile copper pools induced by altered glutathione metabolism, Proc Natl Acad Sci U S A, 116 (2019) 18285–18294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Valnot I, Osmond S, Gigarel N, Mehaye B, Amiel J, Cormier-Daire V, Munnich A, Bonnefont JP, Rustin P, Rotig A, Mutations of the SCO1 gene in mitochondrial cytochrome c oxidase deficiency with neonatal-onset hepatic failure and encephalopathy, Am J Hum Genet, 67 (2000) 1104–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Papadopoulou LC, Sue CM, Davidson MM, Tanji K, Nishino I, Sadlock JE, Krishna S, Walker W, Selby J, Glerum DM, Coster RV, Lyon G, Scalais E, Lebel R, Kaplan P, Shanske S, De Vivo DC, Bonilla E, Hirano M, DiMauro S, Schon EA, Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene, Nat Genet, 23 (1999) 333–337. [DOI] [PubMed] [Google Scholar]
- [92].Ghosh A, Trivedi PP, Timbalia SA, Griffin AT, Rahn JJ, Chan SS, Gohil VM, Copper supplementation restores cytochrome c oxidase assembly defect in a mitochondrial disease model of COA6 deficiency, Hum Mol Genet, 23 (2014) 3596–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Szklarczyk R, Wanschers BF, Nijtmans LG, Rodenburg RJ, Zschocke J, Dikow N, van den Brand MA, Hendriks-Franssen MG, Gilissen C, Veltman JA, Nooteboom M, Koopman WJ, Willems PH, Smeitink JA, Huynen MA, van den Heuvel LP, A mutation in the FAM36A gene, the human ortholog of COX20, impairs cytochrome c oxidase assembly and is associated with ataxia and muscle hypotonia, Hum Mol Genet, 22 (2013) 656–667. [DOI] [PubMed] [Google Scholar]
- [94].Kaler SG, ATP7A-related copper transport diseases-emerging concepts and future trends, Nature reviews. Neurology, 7 (2011) 15–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Wang Y, Zhu S, Hodgkinson V, Prohaska JR, Weisman GA, Gitlin JD, Petris MJ, Maternofetal and neonatal copper requirements revealed by enterocyte-specific deletion of the Menkes disease protein, Am J Physiol Gastrointest Liver Physiol, 303 (2012) G1236–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Telianidis J, Hung YH, Materia S, Fontaine SL, Role of the P-Type ATPases, ATP7A and ATP7B in brain copper homeostasis, Front Aging Neurosci, 5 (2013) 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Kaler SG, Translational research investigations on ATP7A: an important human copper ATPase, Ann N Y Acad Sci, 1314 (2014) 64–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Haddad MR, Choi EY, Zerfas PM, Yi L, Martinelli D, Sullivan P, Goldstein DS, Centeno JA, Brinster LR, Ralle M, Kaler SG, Cerebrospinal Fluid-Directed rAAV9-rsATP7A Plus Subcutaneous Copper Histidinate Advance Survival and Outcomes in a Menkes Disease Mouse Model, Mol Ther Methods Clin Dev, 10 (2018) 165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Guthrie LM, Soma S, Yuan S, Silva A, Zulkifli M, Snavely TC, Greene HF, Nunez E, Lynch B, De Ville C, Shanbhag V, Lopez FR, Acharya A, Petris MJ, Kim BE, Gohil VM, Sacchettini JC, Elesclomol alleviates Menkes pathology and mortality by escorting Cu to cuproenzymes in mice, Science, 368 (2020) 620–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Hedley D, Shamas-Din A, Chow S, Sanfelice D, Schuh AC, Brandwein JM, Seftel MD, Gupta V, Yee KW, Schimmer AD, A phase I study of elesclomol sodium in patients with acute myeloid leukemia, Leuk Lymphoma, 57 (2016) 2437–2440. [DOI] [PubMed] [Google Scholar]
- [101].Kaler SG, Gallo LK, Proud VK, Percy AK, Mark Y, Segal NA, Goldstein DS, Holmes CS, Gahl WA, Occipital horn syndrome and a mild Menkes phenotype associated with splice site mutations at the MNK locus, Nat Genet, 8 (1994) 195–202. [DOI] [PubMed] [Google Scholar]
- [102].Kennerson ML, Nicholson GA, Kaler SG, Kowalski B, Mercer JF, Tang J, Llanos RM, Chu S, Takata RI, Speck-Martins CE, Baets J, Almeida-Souza L, Fischer D, Timmerman V, Taylor PE, Scherer SS, Ferguson TA, Bird TD, De Jonghe P, Feely SM, Shy ME, Garbern JY, Missense mutations in the copper transporter gene ATP7A cause X-linked distal hereditary motor neuropathy, Am J Hum Genet, 86 (2010) 343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Hodgkinson VL, Dale JM, Garcia ML, Weisman GA, Lee J, Gitlin JD, Petris MJ, X-linked spinal muscular atrophy in mice caused by autonomous loss of ATP7A in the motor neuron, J Pathol, 236 (2015) 241–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Yi L, Donsante A, Kennerson ML, Mercer JF, Garbern JY, Kaler SG, Altered intracellular localization and valosin-containing protein (p97 VCP) interaction underlie ATP7A-related distal motor neuropathy, Hum Mol Genet, 21 (2012) 1794–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Czlonkowska A, Litwin T, Dusek P, Ferenci P, Lutsenko S, Medici V, Rybakowski JK, Weiss KH, Schilsky ML, Wilson disease, Nat Rev Dis Primers, 4 (2018) 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Saroli Palumbo C, Schilsky ML, Clinical practice guidelines in Wilson disease, Ann Transl Med, 7 (2019) S65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Martinelli D, Travaglini L, Drouin CA, Ceballos-Picot I, Rizza T, Bertini E, Carrozzo R, Petrini S, de Lonlay P, El Hachem M, Hubert L, Montpetit A, Torre G, Dionisi-Vici C, MEDNIK syndrome: a novel defect of copper metabolism treatable by zinc acetate therapy, Brain, 136 (2013) 872–881. [DOI] [PubMed] [Google Scholar]
- [108].Setty SR, Tenza D, Sviderskaya EV, Bennett DC, Raposo G, Marks MS, Cell-specific ATP7A transport sustains copper-dependent tyrosinase activity in melanosomes, Nature, 454 (2008) 1142–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Farber S, Diamond LK, Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid, N Engl J Med, 238 (1948) 787–793. [DOI] [PubMed] [Google Scholar]
- [110].Brewer GJ, The promise of copper lowering therapy with tetrathiomolybdate in the cure of cancer and in the treatment of inflammatory disease, J Trace Elem Med Biol, 28 (2014) 372–378. [DOI] [PubMed] [Google Scholar]
- [111].Yaman M, Kaya G, Simsek M, Comparison of trace element concentrations in cancerous and noncancerous human endometrial and ovary tissues, Int J Gynecol Cancer, 17 (2007) 220–228. [DOI] [PubMed] [Google Scholar]
- [112].Peng F, Lu X, Janisse J, Muzik O, Shields AF, PET of human prostate cancer xenografts in mice with increased uptake of 64CuCl2, J Nucl Med, 47 (2006) 1649–1652. [PubMed] [Google Scholar]
- [113].Shah I, Lewkow LM, Khilanani U, Correlation of hypercupremia with other acute phase reactants in malignant lymphoma, Cancer, 51 (1983) 851–854. [DOI] [PubMed] [Google Scholar]
- [114].Vaidya SM, Kamalakar PL, Copper and ceruloplasmin levels in serum of women with breast cancer, Indian J Med Sci, 52 (1998) 184–187. [PubMed] [Google Scholar]
- [115].Sharma K, Mittal DK, Kesarwani RC, Kamboj VP, Chowdhery, Diagnostic and prognostic significance of serum and tissue trace elements in breast malignancy, Indian J Med Sci, 48 (1994) 227–232. [PubMed] [Google Scholar]
- [116].Mao S, Huang S, Zinc and copper levels in bladder cancer: a systematic review and meta-analysis, Biol Trace Elem Res, 153 (2013) 5–10. [DOI] [PubMed] [Google Scholar]
- [117].Dragutinovic VV, Tatic SB, Nikolic-Mandic SD, Tripkovic TM, Dunderovic DM, Paunovic IR, Copper as ancillary diagnostic tool in preoperative evaluation of possible papillary thyroid carcinoma in patients with benign thyroid disease, Biol Trace Elem Res, 160 (2014) 311–315. [DOI] [PubMed] [Google Scholar]
- [118].Baharvand M, Manifar S, Akkafan R, Mortazavi H, Sabour S, Serum levels of ferritin, copper, and zinc in patients with oral cancer, Biomed J, 37 (2014) 331–336. [DOI] [PubMed] [Google Scholar]
- [119].Kaba M, Pirincci N, Yuksel MB, Gecit I, Gunes M, Ozveren H, Eren H, Demir H, Serum levels of trace elements in patients with prostate cancer, Asian Pac J Cancer Prev, 15 (2014) 2625–2629. [DOI] [PubMed] [Google Scholar]
- [120].Kaba M, Pirincci N, Yuksel MB, Gecit I, Gunes M, Demir M, Akkoyun H, Demir H, Serum Levels of Trace Elements in Patients with Testicular Cancers, Int Braz J Urol, 41 (2015) 1101–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Boz A, Evliyaoglu O, Yildirim M, Erkan N, Karaca B, The value of serum zinc, copper, ceruloplasmin levels in patients with gastrointestinal tract cancers, Turk J Gastroenterol, 16 (2005) 81–84. [PubMed] [Google Scholar]
- [122].Diez M, Arroyo M, Cerdan FJ, Munoz M, Martin MA, Balibrea JL, Serum and tissue trace metal levels in lung cancer, Oncology, 46 (1989) 230–234. [DOI] [PubMed] [Google Scholar]
- [123].Zowczak M, Iskra M, Torlinski L, Cofta S, Analysis of serum copper and zinc concentrations in cancer patients, Biol Trace Elem Res, 82 (2001) 1–8. [DOI] [PubMed] [Google Scholar]
- [124].Lee K, Briehl MM, Mazar AP, Batinic-Haberle I, Reboucas JS, Glinsmann-Gibson B, Rimsza LM, Tome ME, The copper chelator ATN-224 induces peroxynitrite-dependent cell death in hematological malignancies, Free Radic Biol Med, 60 (2013) 157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Sajesh BV, McManus KJ, Targeting SOD1 induces synthetic lethal killing in BLM- and CHEK2-deficient colorectal cancer cells, Oncotarget, 6 (2015) 27907–27922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Chan N, Willis A, Kornhauser N, Ward MM, Lee SB, Nackos E, Seo BR, Chuang E, Cigler T, Moore A, Donovan D, Vallee Cobham M, Fitzpatrick V, Schneider S, Wiener A, Guillaume-Abraham J, Aljom E, Zelkowitz R, Warren JD, Lane ME, Fischbach C, Mittal V, Vahdat L, Influencing the Tumor Microenvironment: A Phase II Study of Copper Depletion Using Tetrathiomolybdate in Patients with Breast Cancer at High Risk for Recurrence and in Preclinical Models of Lung Metastases, Clin Cancer Res, 23 (2017) 666–676. [DOI] [PubMed] [Google Scholar]
- [127].Glasauer A, Sena LA, Diebold LP, Mazar AP, Chandel NS, Targeting SOD1 reduces experimental non-small-cell lung cancer, J Clin Invest, 124 (2014) 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Juarez JC, Betancourt O Jr., Pirie-Shepherd SR, Guan X, Price ML, Shaw DE, Mazar AP, Donate F, Copper binding by tetrathiomolybdate attenuates angiogenesis and tumor cell proliferation through the inhibition of superoxide dismutase 1, Clin Cancer Res, 12 (2006) 4974–4982. [DOI] [PubMed] [Google Scholar]
- [129].Brady DC, Crowe MS, Turski ML, Hobbs GA, Yao X, Chaikuad A, Knapp S, Xiao K, Campbell SL, Thiele DJ, Counter CM, Copper is required for oncogenic BRAF signalling and tumorigenesis, Nature, 509 (2014) 492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Tsang T, Posimo JM, Gudiel AA, Cicchini M, Feldser DM, Brady DC, Copper is an essential regulator of the autophagic kinases ULK1/2 to drive lung adenocarcinoma, Nat Cell Biol, 22 (2020) 412–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Pan Q, Kleer CG, van Golen KL, Irani J, Bottema KM, Bias C, De Carvalho M, Mesri EA, Robins DM, Dick RD, Brewer GJ, Merajver SD, Copper deficiency induced by tetrathiomolybdate suppresses tumor growth and angiogenesis, Cancer research, 62 (2002) 4854–4859. [PubMed] [Google Scholar]
- [132].Pan Q, Rosenthal DT, Bao L, Kleer CG, Merajver SD, Antiangiogenic tetrathiomolybdate protects against Her2/neu-induced breast carcinoma by hypoplastic remodeling of the mammary gland, Clin Cancer Res, 15 (2009) 7441–7446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Henry NL, Dunn R, Merjaver S, Pan Q, Pienta KJ, Brewer G, Smith DC, Phase II trial of copper depletion with tetrathiomolybdate as an antiangiogenesis strategy in patients with hormone-refractory prostate cancer, Oncology, 71 (2006) 168–175. [DOI] [PubMed] [Google Scholar]
- [134].Lin J, Zahurak M, Beer TM, Ryan CJ, Wilding G, Mathew P, Morris M, Callahan JA, Gordon G, Reich SD, Carducci MA, Antonarakis ES, A non-comparative randomized phase II study of 2 doses of ATN-224, a copper/zinc superoxide dismutase inhibitor, in patients with biochemically recurrent hormone-naive prostate cancer, Urol Oncol, 31 (2013) 581–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Matsuda I, Pearson T, Holtzman NA, Determination of apoceruloplasmin by radioimmunoassay in nutritional copper deficiency, Menkes' kinky hair syndrome, Wilson's disease, and umbilical cord blood, Pediatr Res, 8 (1974) 821–824. [DOI] [PubMed] [Google Scholar]
- [136].Gitlin D, Janeway CA, Turnover of the copper and protein moieties of ceruloplasmin, Nature, 185 (1960) 693. [DOI] [PubMed] [Google Scholar]
- [137].Redman BG, Esper P, Pan Q, Dunn RL, Hussain HK, Chenevert T, Brewer GJ, Merajver SD, Phase II trial of tetrathiomolybdate in patients with advanced kidney cancer, Clin Cancer Res, 9 (2003) 1666–1672. [PubMed] [Google Scholar]
- [138].Schneider BJ, Lee JS, Hayman JA, Chang AC, Orringer MB, Pickens A, Pan CC, Merajver SD, Urba SG, Pre-operative chemoradiation followed by post-operative adjuvant therapy with tetrathiomolybdate, a novel copper chelator, for patients with resectable esophageal cancer, Invest New Drugs, 31 (2013) 435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Pass HI, Brewer GJ, Dick R, Carbone M, Merajver S, A phase II trial of tetrathiomolybdate after surgery for malignant mesothelioma: final results, Ann Thorac Surg, 86 (2008) 383–389; discussion 390. [DOI] [PubMed] [Google Scholar]
- [140].Jain S, Cohen J, Ward MM, Kornhauser N, Chuang E, Cigler T, Moore A, Donovan D, Lam C, Cobham MV, Schneider S, Hurtado Rua SM, Benkert S, Mathijsen Greenwood C, Zelkowitz R, Warren JD, Lane ME, Mittal V, Rafii S, Vahdat LT, Tetrathiomolybdate-associated copper depletion decreases circulating endothelial progenitor cells in women with breast cancer at high risk of relapse, Ann Oncol, 24 (2013) 1491–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141]. https://www.cancer.org/cancer/breast-cancer/understanding-a-breast-cancer-diagnosis/types-of-breast-cancer/triple-negative.html.
- [142].Turski ML, Brady DC, Kim HJ, Kim BE, Nose Y, Counter CM, Winge DR, Thiele DJ, A novel role for copper in Ras/mitogen-activated protein kinase signaling, Mol Cell Biol, 32 (2012) 1284–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Brady DC, Crowe MS, Greenberg DN, Counter CM, Copper Chelation Inhibits BRAF(V600E)-Driven Melanomagenesis and Counters Resistance to BRAF(V600E) and MEK1/2 Inhibitors, Cancer research, 77 (2017) 6240–6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Shin HR, Zoncu R, The Lysosome at the Intersection of Cellular Growth and Destruction, Dev Cell, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Ackerman CM, Chang CJ, Copper signaling in the brain and beyond, J Biol Chem, 293 (2018) 4628–4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM, p53 regulates mitochondrial respiration, Science, 312 (2006) 1650–1653. [DOI] [PubMed] [Google Scholar]
- [147].Voli F, Valli E, Lerra L, Kimpton K, Saletta F, Giorgi FM, Mercatelli D, Rouaen JRC, Shen S, Murray JE, Ahmed-Cox A, Cirillo G, Mayoh C, Beavis PA, Haber M, Trapani JA, Kavallaris M, Vittorio O, Intratumoral Copper Modulates PD-L1 Expression and Influences Tumor Immune Evasion, Cancer research, 80 (2020) 4129–4144. [DOI] [PubMed] [Google Scholar]
- [148].Xu J, Brosseau JP, Shi H, Targeted degradation of immune checkpoint proteins: emerging strategies for cancer immunotherapy, Oncogene, (2020). [DOI] [PubMed] [Google Scholar]
- [149].Ackermann CJ, Adderley H, Ortega-Franco A, Khan A, Reck M, Califano R, First-Line Immune Checkpoint Inhibition for Advanced Non-Small-Cell Lung Cancer: State of the Art and Future Directions, Drugs, (2020). [DOI] [PubMed] [Google Scholar]
- [150].Kagan HM, Li W, Lysyl oxidase: properties, specificity, and biological roles inside and outside of the cell, Journal of cellular biochemistry, 88 (2003) 660–672. [DOI] [PubMed] [Google Scholar]
- [151].Barker HE, Cox TR, Erler JT, The rationale for targeting the LOX family in cancer, Nature reviews. Cancer, 12 (2012) 540–552. [DOI] [PubMed] [Google Scholar]
- [152].Baker AM, Cox TR, Bird D, Lang G, Murray GI, Sun XF, Southall SM, Wilson JR, Erler JT, The role of lysyl oxidase in SRC-dependent proliferation and metastasis of colorectal cancer, Journal of the National Cancer Institute, 103 (2011) 407–424. [DOI] [PubMed] [Google Scholar]
- [153].Salvador F, Martin A, Lopez-Menendez C, Moreno-Bueno G, Santos V, Vazquez-Naharro A, Santamaria PG, Morales S, Dubus PR, Muinelo-Romay L, Lopez-Lopez R, Tung JC, Weaver VM, Portillo F, Cano A, Lysyl Oxidase-like Protein LOXL2 Promotes Lung Metastasis of Breast Cancer, Cancer research, 77 (2017) 5846–5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Wong CC, Tse AP, Huang YP, Zhu YT, Chiu DK, Lai RK, Au SL, Kai AK, Lee JM, Wei LL, Tsang FH, Lo RC, Shi J, Zheng YP, Wong CM, Ng IO, Lysyl oxidase-like 2 is critical to tumor microenvironment and metastatic niche formation in hepatocellular carcinoma, Hepatology, 60 (2014) 1645–1658. [DOI] [PubMed] [Google Scholar]
- [155].Sherratt MJ, McConnell JC, Streuli CH, Raised mammographic density: causative mechanisms and biological consequences, Breast Cancer Res, 18 (2016) 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Baker AM, Bird D, Lang G, Cox TR, Erler JT, Lysyl oxidase enzymatic function increases stiffness to drive colorectal cancer progression through FAK, Oncogene, 32 (2013) 1863–1868. [DOI] [PubMed] [Google Scholar]
- [157].Payne SL, Fogelgren B, Hess AR, Seftor EA, Wiley EL, Fong SF, Csiszar K, Hendrix MJ, Kirschmann DA, Lysyl oxidase regulates breast cancer cell migration and adhesion through a hydrogen peroxide-mediated mechanism, Cancer research, 65 (2005) 11429–11436. [DOI] [PubMed] [Google Scholar]
- [158].El-Haibi CP, Bell GW, Zhang J, Collmann AY, Wood D, Scherber CM, Csizmadia E, Mariani O, Zhu C, Campagne A, Toner M, Bhatia SN, Irimia D, Vincent-Salomon A, Karnoub AE, Critical role for lysyl oxidase in mesenchymal stem cell-driven breast cancer malignancy, Proc Natl Acad Sci U S A, 109 (2012) 17460–17465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Erler JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A, Le QT, Giaccia AJ, Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche, Cancer Cell, 15 (2009) 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Shanbhag V, Jasmer-McDonald K, Zhu S, Martin AL, Gudekar N, Khan A, Ladomersky E, Singh K, Weisman GA, Petris MJ, ATP7A delivers copper to the lysyl oxidase family of enzymes and promotes tumorigenesis and metastasis, Proceedings of the National Academy of Sciences of the United States of America, 116 (2019) 6836–6841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Aubert L, Nandagopal N, Steinhart Z, Lavoie G, Nourreddine S, Berman J, Saba-El-Leil MK, Papadopoli D, Lin S, Hart T, Macleod G, Topisirovic I, Gaboury L, Fahrni CJ, Schramek D, Meloche S, Angers S, Roux PP, Copper bioavailability is a KRAS-specific vulnerability in colorectal cancer, Nat Commun, 11 (2020) 3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Blockhuys S, Zhang X, Wittung-Stafshede P, Single-cell tracking demonstrates copper chaperone Atox1 to be required for breast cancer cell migration, Proc Natl Acad Sci U S A, 117 (2020) 2014–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Wang J, Luo C, Shan C, You Q, Lu J, Elf S, Zhou Y, Wen Y, Vinkenborg JL, Fan J, Kang H, Lin R, Han D, Xie Y, Karpus J, Chen S, Ouyang S, Luan C, Zhang N, Ding H, Merkx M, Liu H, Chen J, Jiang H, He C, Inhibition of human copper trafficking by a small molecule significantly attenuates cancer cell proliferation, Nat Chem, 7 (2015) 968–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Blockhuys S, Brady DC, Wittung-Stafshede P, Evaluation of copper chaperone ATOX1 as prognostic biomarker in breast cancer, Breast Cancer, 27 (2020) 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Kim YJ, Bond GJ, Tsang T, Posimo JM, Busino L, Brady DC, Copper chaperone ATOX1 is required for MAPK signaling and growth in BRAF mutation-positive melanoma, Metallomics, 11 (2019) 1430–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Itoh S, Kim HW, Nakagawa O, Ozumi K, Lessner SM, Aoki H, Akram K, McKinney RD, Ushio-Fukai M, Fukai T, Novel role of antioxidant-1 (Atox1) as a copper-dependent transcription factor involved in cell proliferation, J Biol Chem, 283 (2008) 9157–9167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Sorokin AV, Chen J, MEMO1, a new IRS1-interacting protein, induces epithelial-mesenchymal transition in mammary epithelial cells, Oncogene, 32 (2013) 3130–3138. [DOI] [PubMed] [Google Scholar]
- [168].MacDonald G, Nalvarte I, Smirnova T, Vecchi M, Aceto N, Dolemeyer A, Frei A, Lienhard S, Wyckoff J, Hess D, Seebacher J, Keusch JJ, Gut H, Salaun D, Mazzarol G, Disalvatore D, Bentires-Alj M, Di Fiore PP, Badache A, Hynes NE, Memo is a copper-dependent redox protein with an essential role in migration and metastasis, Sci Signal, 7 (2014) ra56. [DOI] [PubMed] [Google Scholar]
- [169].Berners-Price SJ, Activating platinum anticancer complexes with visible light, Angew Chem Int Ed Engl, 50 (2011) 804–805. [DOI] [PubMed] [Google Scholar]
- [170].Hall MD, Okabe M, Shen DW, Liang XJ, Gottesman MM, The role of cellular accumulation in determining sensitivity to platinum-based chemotherapy, Annual review of pharmacology and toxicology, 48 (2008) 495–535. [DOI] [PubMed] [Google Scholar]
- [171].Rybak LP, Mukherjea D, Jajoo S, Ramkumar V, Cisplatin ototoxicity and protection: clinical and experimental studies, Tohoku J Exp Med, 219 (2009) 177–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Yao X, Panichpisal K, Kurtzman N, Nugent K, Cisplatin nephrotoxicity: a review, Am J Med Sci, 334 (2007) 115–124. [DOI] [PubMed] [Google Scholar]
- [173].Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M, Kroemer G, Molecular mechanisms of cisplatin resistance, Oncogene, 31 (2012) 1869–1883. [DOI] [PubMed] [Google Scholar]
- [174].Holzer AK, Samimi G, Katano K, Naerdemann W, Lin X, Safaei R, Howell SB, The copper influx transporter human copper transport protein 1 regulates the uptake of cisplatin in human ovarian carcinoma cells, Mol Pharmacol, 66 (2004) 817–823. [DOI] [PubMed] [Google Scholar]
- [175].Ishida S, Lee J, Thiele DJ, Herskowitz I, Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals, Proc Natl Acad Sci U S A, 99 (2002) 14298–14302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Lee YY, Choi CH, Do IG, Song SY, Lee W, Park HS, Song TJ, Kim MK, Kim TJ, Lee JW, Bae DS, Kim BG, Prognostic value of the copper transporters, CTR1 and CTR2, in patients with ovarian carcinoma receiving platinum-based chemotherapy, Gynecol Oncol, 122 (2011) 361–365. [DOI] [PubMed] [Google Scholar]
- [177].Ivy KD, Kaplan JH, A re-evaluation of the role of hCTR1, the human high-affinity copper transporter, in platinum-drug entry into human cells, Mol Pharmacol, 83 (2013) 1237–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Beretta GL, Gatti L, Tinelli S, Corna E, Colangelo D, Zunino F, Perego P, Cellular pharmacology of cisplatin in relation to the expression of human copper transporter CTR1 in different pairs of cisplatin-sensitive and -resistant cells, Biochem Pharmacol, 68 (2004) 283–291. [DOI] [PubMed] [Google Scholar]
- [179].Bompiani KM, Tsai CY, Achatz FP, Liebig JK, Howell SB, Copper transporters and chaperones CTR1, CTR2, ATOX1, and CCS as determinants of cisplatin sensitivity, Metallomics, 8 (2016) 951–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Sinani D, Adle DJ, Kim H, Lee J, Distinct mechanisms for Ctr1-mediated copper and cisplatin transport, J Biol Chem, 282 (2007) 26775–26785. [DOI] [PubMed] [Google Scholar]
- [181].Guo Y, Smith K, Petris MJ, Cisplatin stabilizes a multimeric complex of the human Ctr1 copper transporter: requirement for the extracellular methionine-rich clusters, J Biol Chem, 279 (2004) 46393–46399. [DOI] [PubMed] [Google Scholar]
- [182].Holzer AK, Howell SB, The internalization and degradation of human copper transporter 1 following cisplatin exposure, Cancer research, 66 (2006) 10944–10952. [DOI] [PubMed] [Google Scholar]
- [183].Chaumet A, Wright GD, Seet SH, Tham KM, Gounko NV, Bard F, Nuclear envelope-associated endosomes deliver surface proteins to the nucleus, Nat Commun, 6 (2015) 8218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Katano K, Kondo A, Safaei R, Holzer A, Samimi G, Mishima M, Kuo YM, Rochdi M, Howell SB, Acquisition of resistance to cisplatin is accompanied by changes in the cellular pharmacology of copper, Cancer research, 62 (2002) 6559–6565. [PubMed] [Google Scholar]
- [185].Nakayama K, Miyazaki K, Kanzaki A, Fukumoto M, Takebayashi Y, Expression and cisplatin sensitivity of copper-transporting P-type adenosine triphosphatase (ATP7B) in human solid carcinoma cell lines, Oncol Rep, 8 (2001) 1285–1287. [DOI] [PubMed] [Google Scholar]
- [186].Samimi G, Safaei R, Katano K, Holzer AK, Rochdi M, Tomioka M, Goodman M, Howell SB, Increased expression of the copper efflux transporter ATP7A mediates resistance to cisplatin, carboplatin, and oxaliplatin in ovarian cancer cells, Clin Cancer Res, 10 (2004) 4661–4669. [DOI] [PubMed] [Google Scholar]
- [187].Zhu S, Shanbhag V, Wang Y, Lee J, Petris M, A Role for The ATP7A Copper Transporter in Tumorigenesis and Cisplatin Resistance, J Cancer, 8 (2017) 1952–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Inoue Y, Matsumoto H, Yamada S, Kawai K, Suemizu H, Gika M, Takanami I, Iwazaki M, Nakamura M, Association of ATP7A expression and in vitro sensitivity to cisplatin in non-small cell lung cancer, Oncol Lett, 1 (2010) 837–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Yu Z, Cao W, Ren Y, Zhang Q, Liu J, ATPase copper transporter A, negatively regulated by miR-148a-3p, contributes to cisplatin resistance in breast cancer cells, Clin Transl Med, 10 (2020) 57–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Xu X, Zhang Y, Jasper J, Lykken E, Alexander PB, Markowitz GJ, McDonnell DP, Li QJ, Wang XF, MiR-148a functions to suppress metastasis and serves as a prognostic indicator in triple-negative breast cancer, Oncotarget, 7 (2016) 20381–20394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Nakayama K, Kanzaki A, Terada K, Mutoh M, Ogawa K, Sugiyama T, Takenoshita S, Itoh K, Yaegashi N, Miyazaki K, Neamati N, Takebayashi Y, Prognostic value of the Cu-transporting ATPase in ovarian carcinoma patients receiving cisplatin-based chemotherapy, Clin Cancer Res, 10 (2004) 2804–2811. [DOI] [PubMed] [Google Scholar]
- [192].Miyashita H, Nitta Y, Mori S, Kanzaki A, Nakayama K, Terada K, Sugiyama T, Kawamura H, Sato A, Morikawa H, Motegi K, Takebayashi Y, Expression of copper-transporting P-type adenosine triphosphatase (ATP7B) as a chemoresistance marker in human oral squamous cell carcinoma treated with cisplatin, Oral Oncol, 39 (2003) 157–162. [DOI] [PubMed] [Google Scholar]
- [193].Higashimoto M, Kanzaki A, Shimakawa T, Konno S, Naritaka Y, Nitta Y, Mori S, Shirata S, Yoshida A, Terada K, Sugiyama T, Ogawa K, Takebayashi Y, Expression of copper-transporting P-type adenosine triphosphatase in human esophageal carcinoma, Int J Mol Med, 11 (2003) 337–341. [PubMed] [Google Scholar]
- [194].Komatsu M, Sumizawa T, Mutoh M, Chen ZS, Terada K, Furukawa T, Yang XL, Gao H, Miura N, Sugiyama T, Akiyama S, Copper-transporting P-type adenosine triphosphatase (ATP7B) is associated with cisplatin resistance, Cancer research, 60 (2000) 1312–1316. [PubMed] [Google Scholar]
- [195].Petruzzelli R, Polishchuk RS, Activity and Trafficking of Copper-Transporting ATPases in Tumor Development and Defense against Platinum-Based Drugs, Cells, 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Tadini-Buoninsegni F, Bartolommei G, Moncelli MR, Inesi G, Galliani A, Sinisi M, Losacco M, Natile G, Arnesano F, Translocation of platinum anticancer drugs by human copper ATPases ATP7A and ATP7B, Angew Chem Int Ed Engl, 53 (2014) 1297–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Safaei R, Otani S, Larson BJ, Rasmussen ML, Howell SB, Transport of cisplatin by the copper efflux transporter ATP7B, Mol Pharmacol, 73 (2008) 461–468. [DOI] [PubMed] [Google Scholar]
- [198].Safaei R, Adams PL, Mathews RA, Manorek G, Howell SB, The role of metal binding and phosphorylation domains in the regulation of cisplatin-induced trafficking of ATP7B, Metallomics, 5 (2013) 964–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Boal AK, Rosenzweig AC, Crystal structures of cisplatin bound to a human copper chaperone, J Am Chem Soc, 131 (2009) 14196–14197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Dolgova NV, Nokhrin S, Yu CH, George GN, Dmitriev OY, Copper chaperone Atox1 interacts with the metal-binding domain of Wilson's disease protein in cisplatin detoxification, Biochem J, 454 (2013) 147–156. [DOI] [PubMed] [Google Scholar]
- [201].Palm-Espling ME, Lundin C, Bjorn E, Naredi P, Wittung-Stafshede P, Interaction between the anticancer drug Cisplatin and the copper chaperone Atox1 in human melanoma cells, Protein Pept Lett, 21 (2014) 63–68. [DOI] [PubMed] [Google Scholar]
- [202].Safaei R, Maktabi MH, Blair BG, Larson CA, Howell SB, Effects of the loss of Atox1 on the cellular pharmacology of cisplatin, J Inorg Biochem, 103 (2009) 333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Chisholm CL, Wang H, Wong AH, Vazquez-Ortiz G, Chen W, Xu X, Deng CX, Ammonium tetrathiomolybdate treatment targets the copper transporter ATP7A and enhances sensitivity of breast cancer to cisplatin, Oncotarget, 7 (2016) 84439–84452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Ryumon S, Okui T, Kunisada Y, Kishimoto K, Shimo T, Hasegawa K, Ibaragi S, Akiyama K, Thu Ha NT, Monsur Hassan NM, Sasaki A, Ammonium tetrathiomolybdate enhances the antitumor effect of cisplatin via the suppression of ATPase copper transporting beta in head and neck squamous cell carcinoma, Oncol Rep, 42 (2019) 2611–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Fang T, Chen W, Sheng Y, Yuan S, Tang Q, Li G, Huang G, Su J, Zhang X, Zang J, Liu Y, Tetrathiomolybdate induces dimerization of the metal-binding domain of ATPase and inhibits platination of the protein, Nat Commun, 10 (2019) 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Alvarez HM, Xue Y, Robinson CD, Canalizo-Hernandez MA, Marvin RG, Kelly RA, Mondragon A, Penner-Hahn JE, O'Halloran TV, Tetrathiomolybdate inhibits copper trafficking proteins through metal cluster formation, Science, 327 (2010) 331–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Mariniello M, Petruzzelli R, Wanderlingh LG, La Montagna R, Carissimo A, Pane F, Amoresano A, Ilyechova EY, Galagudza MM, Catalano F, Crispino R, Puchkova LV, Medina DL, Polishchuk RS, Synthetic Lethality Screening Identifies FDA-Approved Drugs that Overcome ATP7B-Mediated Tolerance of Tumor Cells to Cisplatin, Cancers (Basel), 12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Chelly J, Tumer Z, Tonnesen T, Petterson A, Ishikawa-Brush Y, Tommerup N, Horn N, Monaco AP, Isolation of a candidate gene for Menkes disease that encodes a potential heavy metal binding protein, Nat Genet, 3 (1993) 14–19. [DOI] [PubMed] [Google Scholar]
- [209].Vulpe C, Levinson B, Whitney S, Packman S, Gitschier J, Isolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPase, Nat Genet, 3 (1993) 7–13. [DOI] [PubMed] [Google Scholar]
- [210].Mercer JF, Livingston J, Hall B, Paynter JA, Begy C, Chandrasekharappa S, Lockhart P, Grimes A, Bhave M, Siemieniak D, et al. , Isolation of a partial candidate gene for Menkes disease by positional cloning, Nat Genet, 3 (1993) 20–25. [DOI] [PubMed] [Google Scholar]