

Capsule Summary
A patient with a novel homozygous mutation in ITK presented with autoimmune lymphoproliferative syndrome, and had impaired TCR-driven Fas ligand upregulation, providing a mechanism for the T cell lymphoproliferation.
Keywords: ITK, autoimmune lymphoproliferative syndrome, primary immunodeficiencies, FAS ligand
To the Editor:
Activated T cells employ apoptosis as a regulatory mechanism to prevent an exaggerated immune response. Repetitive or sustained stimulation of the T cell receptor (TCR) induces surface expression of Fas and its ligand (FasL). Binding of FasL to its receptor initiates a signaling pathway resulting in the death of Fas-expressing T cells. Defects in Fas-mediated apoptosis cause autoimmune lymphoproliferative syndrome (ALPS), characterized by lymphoproliferation, autoimmune cytopenia, susceptibility to lymphoma, and accumulation of TCRαβ+CD4−CD8− (double negative) T cells1. The majority of ALPS patients have germline mutations in FAS, FASLG, CASP8 or CASP10, but as many as twenty percent lack a known genetic etiology1. Interleukin-2-inducible T cell kinase (ITK) is a member of the TEC family of non-receptor protein tyrosine kinases expressed in T lymphocytes, NK cells, invariant NKT cells (iNKT), and mast cells2.To date, all reported 20 patients with ITK deficiency have presented with Epstein Barr virus (EBV) related complications, including EBV driven B-cell lymphoproliferative disease and EBV-associated malignancies, because of inability to clear the virus and persistent EBV viremia3–6.Here, we report a patient with a novel homozygous ITK variant who presented with ALPS.
The patient is the daughter of consanguineous Iraqi parents (Fig 1A). She suffered from recurrent pulmonary infections starting at one year of age. She subsequently developed a Coombs positive chronic autoimmune hemolytic anemia, thrombocytopenia, vitiligo, diffuse lymphadenopathy, and splenomegaly. On presentation to us at 13 years of age, immunologic evaluation revealed decreased CD4+ T cells with normal numbers of CD8+ T cells, B cells and NK cells (Table 1). The percentages of CD45RA−CCR7+ central memory CD8+ cells and CD45RA−CCR7− effector memory CD8+ cells were reduced. Serum IgM was elevated; serum IgG and IgA were normal (Table 1). Notably, in addition to a Coombs positive anemia and thrombocytopenia, she had an elevated percentage of CD3+TCR αβ+CD4−CD8− T cells and an elevated vitamin B12 level, thus fulfilling the criteria for a probable diagnosis of ALPS (Table 1). Quantitative PCR tests on blood for EBV and cytomegalovirus were negative. IgG antibody to EBV viral capsid antigen (VCA) was 120 RU/ml (positive: >22 RU/ml), IgM antibody to VCA was undetectable. A computed tomography scan revealed numerous enlarged mediastinal and abdominal lymph nodes, splenomegaly, and bronchiolar and interstitial lung lesions. Histologic examination of an abdominal lymph node revealed abundant non-necrotizing granulomas, poor germinal center formation and paracortical T cell depletion (Fig. 1B). There was no expansion of the T cell zones or follicular hyperplasia typical of ALPS7.CD30, a marker expressed by Hodgkin/Reed-Sternberg cells, and subsets of plasmablasts and immunoblasts was undetected. Culture and staining of bronchoalveolar lavage fluid and lymph node tissue for bacteria, mycobacteria, fungi, and Pneumocystis jirovecii was negative. The patient was treated with oral prednisone, followed by mycophenolic acid as a steroid-sparing agent, resulting in reduction of her lymphadenopathy and improvement of her cytopenia. No HLA-matched donor was available for a hematopoietic stem cell transplant.
Figure 1.
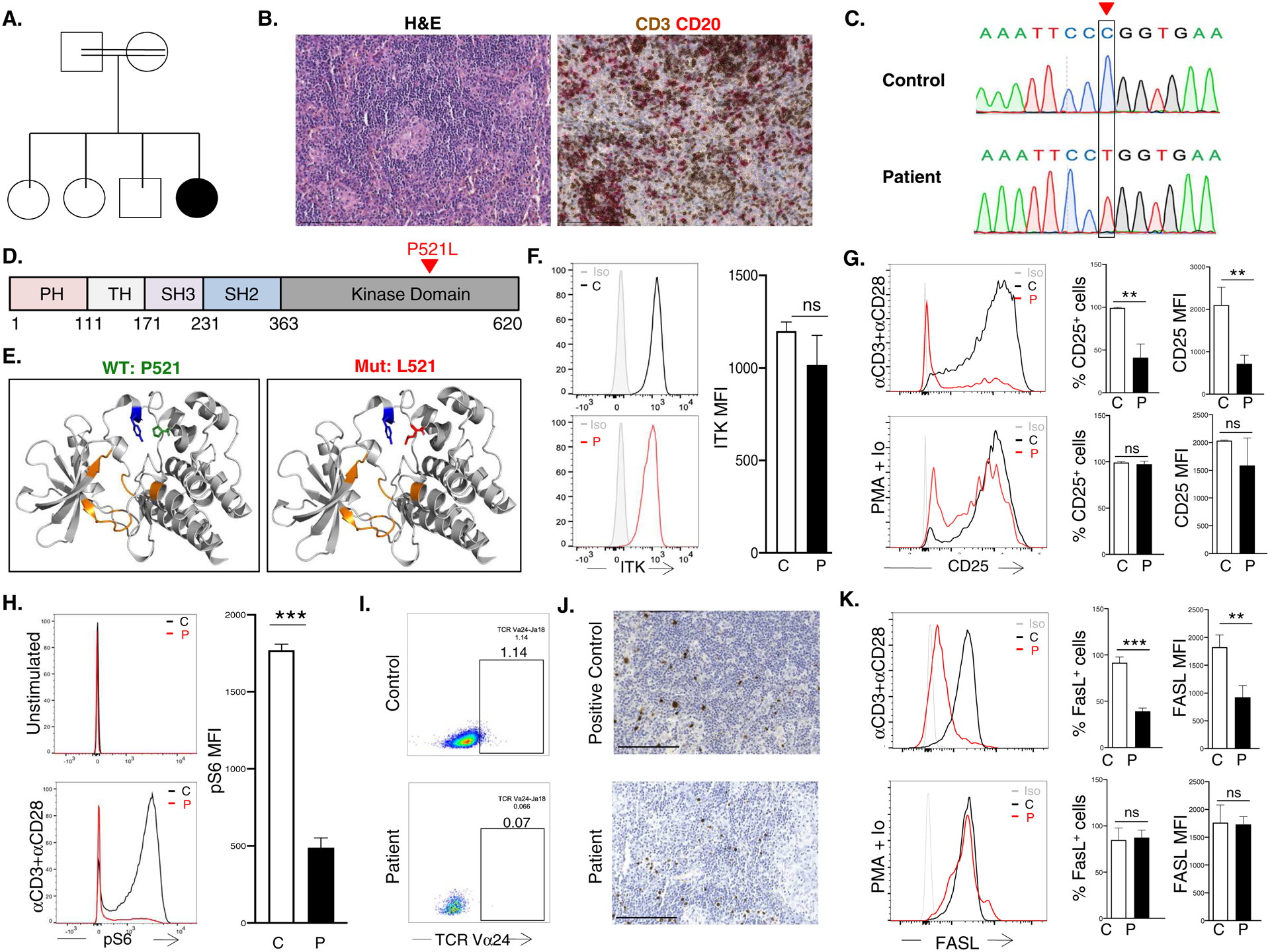
A. Pedigree. B. H&E (left) and IHC (right) of abdominal LN. Scale bar 150μm. C. Sanger sequencing of the ITK c.1562C->T mutation. D. Linear map of ITK with domains shown in different colors.. E. Ribbon diagram of the ITK kinase domain (gray): Y512 (blue), ATP binding domain (orange), P521 on the left (green) and mutant L521 (red) ion the right. F. Representative FACS (left) and quantitative analysis of mean fluorecence intensity (MFI) (right) of ITK in T cells G. Representative (left), and quantitative analysis of the percent CD25+ cells (middle) and CD25 MFI (right) in T cells stimulated with αCD3+αCD28 or PMA+Ionomycin. H. Representative (left) and quantitative analysis (right) of S6 phosphorylation in αCD3+αCD28 simulated T cells. I. FACS analysis of blood TCR Vα24+ iNKT cells among CD4+ cells. J. EBV-encoded RNA in situ hybridization in LN from positive control and patient. Scale bar 150μm. K. Representative FACS (left) and quantitative analysis of the percent FASL+ cells (middle) and FasL MFI (right) in T cells stimulated with αCD3+αCD28 or PMA + Ionomycin. Columns and bars are mean±SD of 3 independent determinations in the patient and 3 controls. ns, not significant; *p<0.05; **p<0.01, ***p<0.001; using one-way ANOVA for multiple comparisons or Student’s t test for two groups.
Table 1.
Immunological profile of the patient.
| Patient | (normal range) | |
|---|---|---|
| Age at testing (years) | 13 | |
| Hemogram, 103 cells/ μL | ||
| White blood cells | 9.40 | (4.00 – 10.0) |
| Neutrophils | 5.92 | (2.00 – 7.50) |
| Lymphocytes | 2.35 | (1.00 – 4.00) |
| Monocytes | 0.54 | (0.20 – 1.00) |
| Lymphocyte subsets | ||
| CD3+, 103 cells/μL | 1.68 | (1.50 – 3.90) |
| CD3+CD4+, 103 cells/μL | 0.79 | (1.30 – 3.40) |
| CD45RA+CCR7+ naïve, %CD4+ | 47.5 | (31.3 – 70.0) |
| CD45RA−CCR7+central memory, % CD4+ | 23.5 | (21.0 – 41.3) |
| CD45RA−CCR7−effector memory, % CD4+ | 28.0 | (7.80 – 25.9) |
| CD45RA+CCR7−TEMRA, % CD4+ | 0.82 | (0.20 – 2.10) |
| CD3+CD8+, 103 cells/μL | 0.47 | (0.30 – 0.92) |
| CD45RA+CCR7+ naïve, % CD8+ | 67.0 | (31.1 – 73.2) |
| CD45RA−CCR7+central memory, % CD8+ | 0.76 | (2.60 – 8.70) |
| CD45RA−CCR7− effector memory, % CD8+ | 4.24 | (8.80 – 44.4) |
| CD45RA+CCR7−, TEMRA, % CD8+ | 28.0 | (8.70 – 38.0) |
| CD19+, 103 cells/μL | 0.24 | (0.21 – 0.57) |
| CD3−CD56+, 103 cells/μL | 0.31 | (0.07 – 0.48) |
| Immunoglobulins, mg/dL | ||
| IgG | 1097 | (639 – 1344) |
| IgM | 989 | (34 – 215) |
| IgA | 76 | (55 – 276) |
| Laboratory data supportive of ALPS | ||
| Hemoglobin | 9.3 | (11.0 – 13.5) |
| Platelets, 103 cells/μL | 108 | (150 – 450) |
| % CD8 CD4− cells among TCRαβ+ cells | 12.1 | (< 2.5) |
| Direct Coombs test | Positive | (negative) |
| Vitamin B12 (pg/mL) | 807 | (190 – 778) |
Bold values are outside the normal range.
Targeted next-generation DNA sequencing of 264 genes associated with primary immunodeficiency identified a novel homozygous missense variant in ITK (c.1562C>T; p.Pro521Leu) confirmed by Sanger sequencing (Fig 1C). No exonic mutations were detected in genes are associated with ALPS (FAS, FASLG, CASP8, CASP10). The mutation was predicted to be deleterious by PolyPhen-2, SIFT, and CADD, with a minor allelic frequency of 7.96 × 10−7 in the GnomAD database without homozygous instances. Both healthy parents were heterozygous for the ITK mutation. Samples from the three healthy siblings were not available for testing. The mutated residue P521 is in the kinase domain of ITK (Fig 1D), four angstroms away from residue Y512 in the activation site (Fig 1E). Disruption of this region may impair Y512 phosphorylation necessary for the protein’s catalytic activity2.ITK protein expression in the patient’s CD3+ cells was comparable to that of controls (Fig 1F).
Due to the limited availability of the patient’s blood, flow cytometry was the only feasible approach for examining Y512 phosphorylation in the patient’s cells. However, commercially available pY512 ITK antibodies failed to detect phosphorylation of this residue in normal T cells activated by anti-CD3 mAb. ITK is an important transducer of TCR signaling that results in CD25 expression and activation of the PI3K/mTOR pathway2. Consistent with impaired ITK activation and TCR signaling, the patient CD3+ cells showed decreased expression of CD25 following stimulation with αCD3+αCD28, but not with phorbol myristate acetate (PMA) and ionomycin which bypasses the TCR (Fig 1G). In parallel, S6 phosphorylation, an event downstream of mTOR1 activation, was significantly decreased in the patient’s activated T cells compared to controls (Fig 1H). These results strongly suggest that ITKPro521Leu impairs TCR/CD3 signaling. Diminished T cell and T cell-dependent B cell function may underlie the patient’s recurrent infections. The patient exhibited other features associated with ITK deficiency: reduced naive CD4+ T cells and iNKT cells (Fig 1I). Although the patient had no detectable EBV viremia, in situ hybridization on the patient’s lymph nodes after the ITK variant was identified, revealed the presence of EBV-encoded small nuclear RNA (EBER) (Fig 1J). Collectively, these results strongly suggest that the patient had deficient ITK function.
Studies in mice implicate ITK in the upregulation of FasL expression and in Fas/FasL mediated activation-induced cell death (AICD)8.FasL upregulation was significantly reduced in the patient’s T cells compared to control following anti-CD3+anti-CD28 activation, but not PMA/IO stimulation (Fig 1K). Although we were unable to obtain blood from the patient to directly examine AICD, failure to upregulate TCR-driven FasL expression likely contributed to the development ALPS in our patient. Of note, FasL upregulation and AICD are defective in T cells from patients with SLAM-associated protein (SAP) deficiency, who also have a predisposition to EBV-driven lymphoproliferation and T cell expansion in lymph nodes3,9.
Our study expands the clinical phenotype of patients with ITK deficiency and suggests that failure to upregulate FasL provides an additional mechanism for T cell lymphoproliferation in ITK-deficient patients.
Supported by:
1K08AI116979-01 (J.C.), 1R01AI139633-01(R.S.G.) the Samara Turkel Foundation and the Perkin Fund (R.S.G.).
Subjects and Methods
Human subjects
All study participants provided written informed consent approved by the respective institutional review boards.
Genetic and Sanger sequencing
Genomic DNA was isolated from peripheral blood samples using the Gentra Puregene blood kit (Qiagen, Germantown, Md). Targeted next-generation DNA sequencing of 264 genes associated with primary immunodeficiency was performed. Sanger sequencing of the mutation in ITK was performed with the following primers: forward, 5′-GACTCCTTAACTACTGATGA-3′; reverse, 5′-TAGGGTTGTCTGCATTTGATA-3′.
Protein model:
The patient mutation was modeled using Pymol and the crystal structure 4M0Y from the Protein Data Bank.
Cell Culture:
PBMCs were isolated from heparinized blood using Ficoll-Hypaque (GE Healthcare). T cells were purified by negative selection (Miltenyi Biotec 130-096-535). Cells were suspended in RPMI-1640 containing 10% heat-inactivated FCS (HyClone), 2 mM L-glutamine, 50 μg/mL streptomycin and 100 U/mL penicillin (medium). T cells were stimulated with Gibco Dynabeads Human T-Activator αCD3/CD28 (Thermo Fisher: 25 μl of beads per 1M cells) or with phorbol myristate acetate (20 ng/mL) and ionomycin (0.5 ug/mL) for 3 days (CD25), overnight (pS6) or 3 days (FASL).
Flow cytometry.
Standard flow cytometric methods were used for the staining of cell-surface and intra-cellular proteins. Anti-human mAbs with the appropriate isotype-matched controls were used for staining. All flow cytometry data was collected with an LSR Fortessa (BD Biosciences) cell analyzer and analyzed with FlowJo software (Tree Star). The following reagents and monoclonal antibodies were used as described by the manufacturer: Fixable Viability Dye (ebioscience 65-0865-14), TK (Cell Signaling 77215), anti-Rabbit IgG-FITC (SouthernBiotech 4030–02), pS6 (S235/236) (Cell Signaling 2211), CD4 (Biolegend 317410), CD3 (Biolegend 300415), CD8 (Biolegend 300912), Rabbit IgG (Cell Signaling 3900), FASL (Biolegend 106605) and CD25 Biolegend 302620).
Immunohistochemistry (IHC):
IHC for CD3 and CD20, and in situ hybridization for EBER of lymph node sections were performed according to standard procedures in the pathology laboratories of the Brigham and Women Hospital and Boston Children’s Hospital respectively.
Statistics
Statistical analyses were performed using Prism 8 software (GraphPad, La Jolla, Calif). Significance was calculated by using 2-tailed t tests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: The authors declare no conflicts of interest.
References
- 1.Bleesing JJH, Nagaraj CB, Zhang K. Autoimmune Lymphoproliferative Syndrome. In editors. GeneReviews R. Seattle (WA); 1993. [PubMed] [Google Scholar]
- 2.Bunnell SC, Diehn M, Yaffe MB, Findell PR, Cantley LC, Berg LJ. Biochemical interactions integrating Itk with the T cell receptor-initiated signaling cascade. J Biol Chem 2000; 275:2219–30. [DOI] [PubMed] [Google Scholar]
- 3.Cagdas D, Erman B, Hanoglu D, Tavil B, Kuskonmaz B, Aydin B, et al. Course of IL-2-inducible T-cell kinase deficiency in a family: lymphomatoid granulomatosis, lymphoma and allogeneic bone marrow transplantation in one sibling; and death in the other. Bone Marrow Transplant 2017; 52:126–9. [DOI] [PubMed] [Google Scholar]
- 4.Eken A, Cansever M, Somekh I, Mizoguchi Y, Zietara N, Okus FZ, et al. Genetic Deficiency and Biochemical Inhibition of ITK Affect Human Th17, Treg, and Innate Lymphoid Cells. J Clin Immunol 2019; 39:391–400. [DOI] [PubMed] [Google Scholar]
- 5.Ghosh S, Drexler I, Bhatia S, Adler H, Gennery AR, Borkhardt A. Interleukin-2-Inducible T-Cell Kinase Deficiency-New Patients, New Insight? Front Immunol 2018; 9:979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Howe MK, Dowdell K, Roy A, Niemela JE, Wilson W, McElwee JJ, et al. Magnesium Restores Activity to Peripheral Blood Cells in a Patient With Functionally Impaired Interleukin-2-Inducible T Cell Kinase. Front Immunol 2019; 10:2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lim MS, Straus SE, Dale JK, Fleisher TA, Stetler-Stevenson M, Strober W, et al. Pathological findings in human autoimmune lymphoproliferative syndrome. Am J Pathol 1998; 153:1541–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller AT, Berg LJ. Defective Fas ligand expression and activation-induced cell death in the absence of IL-2-inducible T cell kinase. J Immunol 2002; 168:2163–72. [DOI] [PubMed] [Google Scholar]
- 9.Snow AL, Marsh RA, Krummey SM, Roehrs P, Young LR, Zhang K, et al. Restimulation-induced apoptosis of T cells is impaired in patients with X-linked lymphoproliferative disease caused by SAP deficiency. J Clin Invest 2009; 119:2976–89. [DOI] [PMC free article] [PubMed] [Google Scholar]