

Abstract
Glioblastoma (GBM) is an incurable brain tumor with inevitable recurrence. This is in part due to a highly malignant cancer stem cell (CSC) subpopulation of tumor cells that is particularly resistant to conventional treatments, including radiotherapy. Here we show that CBL0137, a small molecule anti-cancer agent, sensitizes GBM CSCs to radiotherapy. CBL0137 sequesters the FACT (facilitates chromatin transcription) complex to chromatin, resulting in cytotoxicity preferentially within tumor cells. We show that when combined with radiotherapy, CBL0137 inhibited GBM CSC growth and resulted in more DNA damage in the CSCs compared to irradiation or drug alone. Using an in vivo subcutaneous model, we showed that the frequency of GBM CSCs was reduced when tumors were pretreated with CBL0137 and then exposed to irradiation. Survival studies with orthotopic GBM models resulted in significantly extended survival for mice treated with combinatorial therapy. As GBM CSCs contribute to the inevitable recurrence in patients, targeting them is imperative. This work establishes a new treatment paradigm for GBM that sensitizes CSCs to irradiation and may ultimately reduce tumor recurrence.
Keywords: glioblastoma, cancer stem cells, FACT, CBL0137, DNA damage
1. Introduction
Glioblastoma (GBM) is a highly malignant brain tumor with a dismal prognosis. The current standard of care is maximal surgical resection, followed by radiotherapy and chemotherapy (1). Despite aggressive treatment, recurrence is nearly universal and the 5-year overall survival rate remains at approximately 5% (2). The extensive locally invasive nature of GBM creates challenges in complete surgical resection and tumor resistance to radiotherapy is common (3,4). Additionally, there is a subpopulation of cells within the tumor, called cancer stem cells (CSCs), that has been shown to be particularly unresponsive to the conventional treatment of radiotherapy and chemotherapy (3,5–7). In regards to CSC radioresistance, it has been shown that CSCs survive exposure to even high doses of irradiation and drive tumor growth after treatment (3). Therefore, it is important to inclusively target this subpopulation when considering new therapies and especially combination therapies aimed at radiosensitization of the tumor.
Previously, our lab has utilized an anti-cancer compound, CBL0137, to target GBM CSCs (8). CBL0137 indirectly inhibits facilitates chromatin transcription (FACT), a complex that has been shown to be elevated in GBM CSCs as well as other cells with an undifferentiated phenotype (8,9). FACT is a heterodimer, comprised of SSRP1 (structure specific recognition protein 1) and SPT16 (suppressor of Ty 16), and is involved in transcription elongation (10,11). It plays a role in building, maintaining, and overcoming the chromatin barrier as well as in depositing histones and forming nucleosomes (10). FACT has also been shown to play various roles in the DNA damage response, such as depositing histone variants at sites of DNA damage to stimulate signaling for both double and single stranded breaks (12,13). FACT is also upregulated in GBM tumors in comparison to normal brain tissue, rendering it an attractive, specific therapeutic target (9).
CBL0137 binds to the minor groove of DNA causing a conformational change, and thus indirectly sequesters FACT on the chromatin (14,15). This trapping of FACT leads to inhibition of a subset of NF-κB target genes and can also lead to the activation of p53 through phosphorylation at Ser392 by casein kinase 2 (CK2) (14). CBL0137 is currently in several clinical trials for both hematological cancers and solid tumors. Previously, we and others have shown that CBL0137 is able to increase survival in orthotopic models of GBM as a monotherapy as well as in combination with temozolomide (8,9). Although it has been reported that CBL0137 synergized with DNA damaging agents, like cisplatin and etoposide, it alone has not been shown to cause DNA damage (16,17). In regards to targeting CSCs, we demonstrated that CBL0137 has a biphasic impact whereby acute exposure reduced cancer stem cell gene expression, self-renewal, and tumor initiation whereas prolonged exposure led to cell death (8). The ability of CBL0137 to shift the CSC phenotype as well as the previous reports that the drug could synergize with genotoxic agents led us to hypothesize that pretreatment with CBL0137 could radiosensitize GBM CSCs.
Hence, our aim in this study was to test that hypothesis and determine if CBL0137 treatment could sensitize GBM to radiotherapy and if this treatment aided in the specific resistance seen in GBM associated CSCs. Both in vitro and in vivo models were utilized to evaluate the impact of combination therapy on the extent of DNA damage, cell growth, cancer stem cell frequency within tumors, and the ability to prolong survival in mice bearing orthotopic GBM. Radiotherapy after surgery is currently the most effective treatment for GBM. However, high rates of recurrence due to radioresistance causes prognoses to remain dismal. Therefore, identification of a therapeutic that radiosensitizes the CSC subpopulation while targeting the tumor is critical.
2. Materials and Methods
2.1. Cells and culture conditions
All cells were obtained as de-identified specimens that were initially acquired as primary human brain tumor patient specimens in accordance with appropriate, approved Institutional Review Board (IRB) protocols. 3691 and 08–387 were a kind gift from Dr. Jeremy Rich (University of California San Diego; IRBs from Duke University and The Cleveland Clinic); 1016 was a kind gift from Dr. Anita Hjelmeland (University of Alabama); and NU757 was kindly obtained from Dr. Craig Horbinski and the Northwestern University Nervous System Tumor Bank. CD133-positive cells were enriched for the CD133/1 (AC133) epitope by magnetic-activated cell sorting as per the manufacturer’s recommendations (MACS; Miltenyi Biotec) from dissociated subcutaneous xenografts (Papain Dissociation System; Worthington Biochemical) grown in the flanks of athymic nude (Nu/Nu; Crl:NU-Foxn1nu) mice and used for experiments within ten passages post-sorting. All cells were cultured at 37°C at 5% CO2 in Neurobasal media (minus phenol red; Gibco) with added B27 (minus Vitamin A; Gibco), human fibroblast growth factor-2 (10 ng/mL; Miltenyi Biotec), human epidermal growth factor (10 ng/mL; Miltenyi Biotec), L-glutamine (2 mM; Gibco), sodium pyruvate (1 mM; Gibco), and penicillin/streptomycin (100 I.U./ml/100 ug/ml; Gibco). Cell culture studies were done with cells plated adherently on Geltrex LDEV-Free hESC-Qualified, Reduced Growth Factor Basement Membrane Matrix (Gibco) whereas in vivo studies were done with cells grown in suspension as tumorspheres before dissociation and cell counting prior to implantation. For cell counting before each experiment, a single-cell suspension was achieved using TrypLE Express Enzyme (no phenol red; Gibco). Mycoplasma testing was performed quarterly (LookOut Mycoplasma PCR Detection Kit; Sigma-Aldrich) and cell line verification was performed annually (microsatellite genotyping; Ohio State University Comprehensive Cancer Center Genomics Shared Resource).
2.2. Animals and in vivo studies
All animal studies described were approved by the Ohio State University Institutional Animal Care and Use Committee and conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Male and female athymic Nu/Nu mice were used for all studies and were obtained from the Ohio State University Comprehensive Cancer Center Target Validation Shared Resource or from Charles River (strain #553). For subcutaneous tumor studies, 1 × 105 cells were injected in a total volume of 100 μL Neurobasal media (no additives) into the left flank of 6–8 week old mice. Once tumors reached approximately 0.12 cm3 in volume, mice were randomized into one of four treatment groups for a 7 day acute treatment schedule; vehicle, CBL0137 (10 mg/kg daily, intraperitoneal; Selleck Chemicals), vehicle and irradiation (2.5 Gray (Gy) on days 1, 3, and 5), or CBL0137 and irradiation. Irradiation was given 6 hours after vehicle or CBL0137 injections using the Small Animal Radiation Research Platform (SARRP; Xstrahl Medical and Life Sciences) for targeted dose delivery. Tumors were monitored and measured daily using perpendicular diameter measurements for 7 days. Tumor volume was calculated using the ellipsoid formula π/6 × larger diameter × (smaller diameter). Twenty four hours after the final treatment on day 8, tumors were dissociated into single cells and sorted for limiting dilution assays. For orthotopic injections, 1 × 104 cells were injected intracranially in a total volume of 2 μl Neurobasal media (no additives), 2 mm right lateral of bregma and at a depth of 2.5 mm from the dura in mice 6–8 weeks old. 7 days later (for 3691) or 14 days later (for 1016), mice were randomized into one of four treatment groups; vehicle, CBL0137 (70 mg/kg, intravenous), vehicle + irradiation (2.5 Gy), or CBL0137 and irradiation. Initiation of treatment for 3691 at 7 days post-implantation and 1016 at 14 days post-implantation was based on previous studies whereby tumor burden was known to have been established at that time point using similar implantation conditions (8,18,19). Vehicle or CBL0137 were delivered via tail vein injection once a week for four weeks. Irradiation was given 24 hours after the injections using the SAARP with delivery to the tumor bearing hemisphere. Mice were monitored daily for neurological impairment and/or a drop in weight of more than 20% of original weight.
2.3. Small molecule inhibitors
CBL0137 was obtained from Selleck Chemicals (#S8483) and was diluted in DMSO for storage of stock solutions. Working concentrations were made immediately before use and diluted in cell media or sterile saline (Pharmaceutical Grade 0.9% Sodium Chloride, Henry Schein, Pfizer Injectables) for in vitro and in vivo studies, respectively. Sterile saline served as the vehicle control for in vivo studies and DMSO at a final percentage equivalent to that of the drug suspensions served as the vehicle control for all in vitro studies.
2.4. Cell growth assays via live cell imaging
Cells were plated at 1 × 103 cells per well into Geltrex treated 96-well plates. The next day, cells were treated with CBL0137 at 150 nM or 300 nM or with vehicle control (DMSO), and left unirradiated or irradiated with 3 Gy 6 hours post-drug or vehicle treatment. Irradiation was done using a GammaCell 40 Irradiator (Best Theratronics). Sham irradiated control plates were transported to the radiation facility, but not exposed. Following irradiation, plates were immediately put into the IncuCyte ZOOM live cell system (Satroius) with phase-contrast images taken every 4 hours for a total of 72 hours for GBM NU757, GBM 3691, and GBM 08–387 or 96 hours for GBM 1016 due to its slower doubling time. Resulting images were analyzed using the IncuCyte ZOOM analysis software for label-free proliferation measurements whereby overall confluence, as a measure of cell growth, is longitudinally tracked for the duration of image acquisition.
2.5. Colony formation assays
Cells were plated at 250 cells per well into Geltrex treated 6-well plates. The next day, cells were treated with CBL0137 at 150 nM or with vehicle control (DMSO), and left unirradiated (0 Gy) or irradiated with 1, 2, 3, or 4 Gy six hours post-drug or vehicle treatment. Media was changed 24 hours later. Ten days post-treatment, cells were washed before being fixed and stained with a 0.5% crystal violet solution. Plates were imaged on the LI-COR Odyssey near infrared imaging system and analyzed via a custom ImageJ macro which counts individual colonies, allowing for unbiased quantification.
2.6. Comet assay
Single-cell gel electrophoresis under alkaline conditions was performed using manufacturer’s instructions and reagents (Trevigen). Cells were plated at 200,000 cells per well into Geltrex treated 6 cm plates. The next day, cells were given a 6 hour pretreatment of 300 nM CBL0137 or vehicle (DMSO), and then left unirradiated or irradiated with 3 Gy. One hour after irradiation, 5 × 103 cells were mixed with LMAgarose and pipetted onto CometSlides. Cells were lysed overnight at 4°C then placed in unwinding buffer for 20 minutes followed by electrophoresis for 1 hour. Slides were then washed, dried, and DNA stained with SYBR gold nucleic acid stain (Invitrogen) for 30 minutes. Images were acquired using a Leica DM5500B upright epifluorescence microscope with 20–30 images taken per slide, and with each cell line having three technical replicates performed, for a minimum number of 100 of cells to be analyzed per condition for each cell line. Quantitative comet analysis was performed using Comet Score by TriTek (version 1.6.1.22) which unbiasedly quantifies tail length as a measure of pixel distance from the center of the comet head.
2.7. Limiting dilution assay
At a maximum of two hours following subcutaneous tumor dissociation, cells were resuspended at 1 × 107 cells/mL in Neurobasal media. Viable cells, based on 4’,6-Diamidino-2-phenylindole dihydrochloride (DAPI) exclusion, were sorted using a FACS Aria II Cell Sorter (BD Biosciences) into 96 well plates at a final cell number per well of 1 (one full 96 wells/plate), 5 (36 wells/plate), 10 (36 wells/plate), or 20 (24 wells/plate). Tumorsphere formation was evaluated 10 days after sorting, and wells were scored positive or negative for the presence of at least one tumorsphere. At least three subcutaneous tumors were evaluated per treatment group. The estimated stem cell frequency was calculated using a publicly available webtool to calculate an estimated stem cell frequency and associated statistical significance (20).
2.8. Immunofluorescence
Cells were plated at 2 × 104 cells per well onto Geltrex treated coverslips which were placed into the wells of 24 well plates. The next day, cells were treated with a 6 hour pretreatment of vehicle (DMSO) or CBL0137 at 150 nM, and then left unirradiated or irradiated with 3Gy of irradiation. For siRNA studies, cells were plated at 500,000 cells per 10 cm Geltrex treated plate that contained coverslips. The next day, cells were treated with siControl, siSSRP1 MISSION esiRNA pool targeting human SSRP1 (Sigma-Aldrich, EHU015991–50UG), or siSPT16 MISSION esiRNA pool targeting human SUPTH16H (Signma-Aldrich, EHU039881–50UG) using lipofectamine RNAimax (Thermo Fisher, 13-778-030). All siRNAs were used at 2.5 nM. siSSRP1 treated cells were either left unirradiated or irradiated 24 hours post treatment. siSPT16 treated cells were either left unirradiated or irradiated 72 hours post treatment. Irradiation time points were chosen based on the time point of greatest protein depletion as evaluated by immunoblotting for SSRP1 (1:500; Biolegend, 50-170-613) and SPT16 (1:500; Biolegend, 50-170-598). Coverslips were harvested and cells fixed at 1, 6, and 24 hours post-irradiation. Cells were then immunostained for γH2AX (1:500; Millipore, 05–636 JBW301) or 53BP1 (1:100; Cell Signaling, 4937S). Secondary detection was accomplished using Alexa Fluor 647 or Alexa Fluor 488 goat anti-mouse IgG (1:500; Invitrogen, A28181). Nuclei were counterstained with Hoechst. Images were acquired using a Leica DM5500B upright epifluorescence microscope with 20–30 images taken per slide to allow for a total number of 100–150 of cells to be analyzed per condition. Cells were then scored as having more or less than 10 γH2AX or 53BP1 foci.
2.9. Western blotting
Cells were plated at 2 × 106 cells per plate onto Geltrex treated 15 cm plates. 24 hours later, cells were treated with vehicle (DMSO) or CBL0137 at 150nM, and then 6 hours later left unirradiated or irradiated with 3 Gy of irradiation. Cell pellets were collected and frozen on dry ice 1, 6, 12, and 24 hours post irradiation. Whole cell extracts were made using a 50 mM Tris pH 8.0, 120 nM NaCl, 0.5% NP-40 lysis solution supplemented with PhosSTOP Phosphatase Inhibitor Tablets (4906837001, Roche) and cOmplete, Mini EDTA-free Protease Inhibitor Cocktail Tablets (11836170001, Roche). Samples were run on 4–20% Mini-PROTEAN TGX precast gels (Bio-Rad Laboratories, Inc.) and transferred to PVDF membranes (Millipore Corp.). The membranes were blocked with 5% (wt/vol) bovine serum albumin or 5% dry milk (as per manufacture’s recommendation) in TBS-Tween-20 (TBST; 0.1–0.2% vol/vol) and probed with primary antibodies overnight at 4°C. Secondary antibodies (LI-COR Biosciences) were incubated in TBST plus 0.01–0.02% SDS and visualized with the LI-COR Odyssey near infrared imaging system. Quantification of band intensity was achieved using the associated Image Studio Ver 5.2 software. 80 μg of protein lysate was used for Olig2 (1:2500; Millipore, AB9610). 60 μg of protein lysate was used for Chk1 (2G1D5) (1:1000; Cell Signaling, 2360S), phospho-Chk1 (Ser345) (133D3) (1:1000; Cell Signaling, 2348S), Rad17 (1:1000, Bethyl Laboratory, A305–788A-M), phospho-Rad17 (Ser645) (D5H5) (1:1000, Cell Signalling, 6981S), GFAP (1:2500; Invitrogen, REP130300), Sox2 (1:500; MAB2018, R&D), and MAP2 (1:1000; Biolegend, 828101). 20 μg of protein lysate was used for p53 (1:1000; Cell Signaling, 9282S), phospho-p53 (Ser392) (1:1000; Cell Signaling, 9281S), Chk2 (1:1000; Cell Signaling, 2662S), phospho-Chk2 (Thr68) (C131C1) (1:1000; Cell Signaling, 2197S), and Nestin (1:500; Novus, NB200–265). Secondary detection was achieved via IRDye 800CW goat anti-rabbit IgG (1:20,000; Li-Cor, 926–32211), IRDye 800CW Goat anti-mouse IgG (1:20,000; 926–32210), or IRDye 800CW Goat anti-rat IgG (1:20,000; 926–32219).
2.10. Statistical analysis
Statistical analyses were conducted using Graphpad Prism 5, unless otherwise stated. The statistical test used for each experiment is listed within the corresponding Figure legend. 3 biological repeats were performed for each specimen and each biological replicate included 3–5 technical replicates. Dose enhancement factors were calculated based off protocols published by Franken et al. (21). Stem cell frequencies and associated p values were calculated using the Extreme Limiting Dilution Analysis (ELDA) online tool (20).
3. Results
3.1. CBL0137 in combination with irradiation led to decreased GBM cell growth and clonogenic survival
To determine if CBL0137 enhances the cellular sensitivity to irradiation, we first performed longitudinal cell growth studies. We had previously determined the EC50 for CBL0137 to be approximately 150 nM using a panel of GBM CSCs (8). Therefore, 150 nM and/or 300 nM were the concentrations used for all of our studies. We used both primary patient specimens GBM NU757 and GBM 1016 grown in stem promoting media conditions as well as CSCs that were first prospectively sorted based on the CD133 epitope from GBM 3691 and GBM 08–387 and then grown in stem promoting conditions as a means to include both of the current methodologies used for propagating and studying CSCs (5, 22–24). The stem state for the CSCs from GBM 3691 and GBM 08–387 were validated by the positive protein expression of the stem cell markers Sox2, Olig2, and Nestin and low/no signal for the more differentiated markers GFAP and MAP2 (Supplementary Fig. S1). Cells were exposed to CBL0137 or vehicle for 6 hours prior to receiving 3 Gy or mock irradiation. Cell confluence, as a metric of cell growth, was then monitored via live-cell imaging for 72–96 hours. For all four specimens, the greatest decrease in cell growth was seen for the combination treatment of 150 nM CBL0137 and irradiation. Combination treatment for GBM NU757 and GBM 08–387 reached statistical significance as compared to either individual treatment, 3691 reached statistical significance for the combination treatment as compared to CBL0137 alone but not irradiation, and GBM 1016 did not reach statistical significance (Fig. 1a–d). At 300 nM, both the combination treatment and CBL0137 as a monotherapy caused a decrease in proliferation (Supplementary Fig. S2). All four GBM specimens reached statistical significance for the combination as compared to irradiation with GBM 3691 and GBM 08–387 also reaching statistical significance for combination treatment as compared to CBL0137 alone (Supplementary Fig. S2). Next, we performed colony formation assays using GBM NU757 and GBM 3691 using a 6 hour pre-treatment of 150 nM CBL0137 and 0–4 Gy of irradiation with a media change 24 hours post-irradiation. Clonogenic survival was reduced for both specimens with a resulting dose enhancement factor (DEF at surviving fraction 0.5 with a DEF greater than 1 indicating a synergistic effect) of 1.72 for GBM NU757 and 1.22 for GBM 3691 (Fig. 2a–b). Together, these data indicate that CBL0137 treatment prior to irradiation radiosensitized GBM CSCs.
Figure 1. Sustained exposure to CBL0137 in combination with irradiation decreased GBM cell growth.
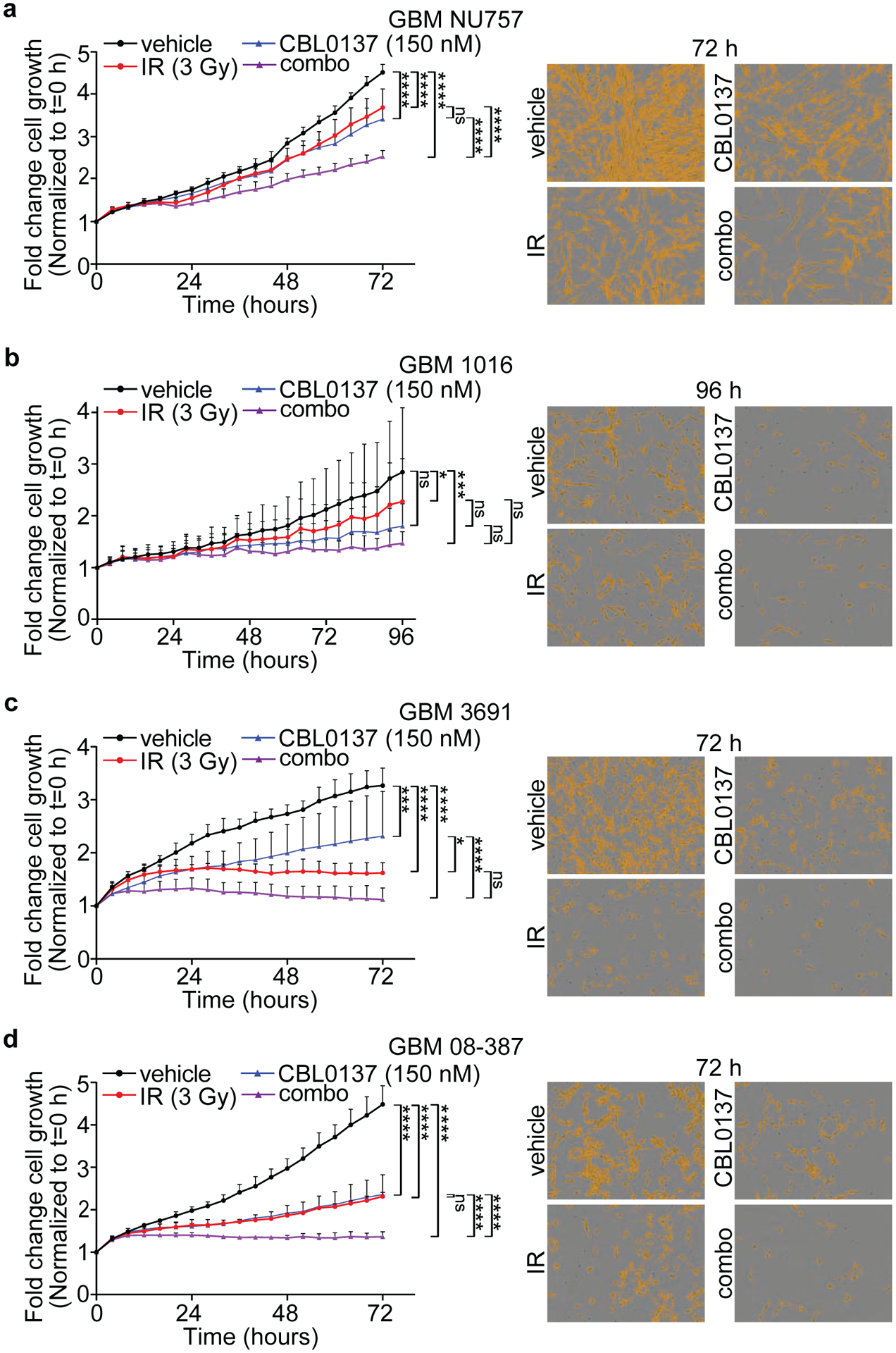
GBM NU757 (a), GBM 1016 (b), GBM 3691 (c), and GBM 08–387 (d) were treated with vehicle (DMSO), CBL0137 (150 nM), irradiation (IR, 3 Gy) or CBL0137 and IR (combo; 6 hour pre-treatment with CBL0137). Cell growth was monitored on the IncuCyte ZOOM live-cell imaging system for 72 hours (GBM NU757, GBM 3691, and GBM 08–387) or 96 hours (1016) and fold change in cell growth via confluence for vehicle (black line), CBL0137 (blue line), IR (red line), and combo (purple line) was normalized to t = 0 hours (h) and graphed. Representative images are shown to the right with the mask overlay (yellow) that was used to quantify the percent confluence. Each experiment was repeated 3 times per GBM specimen with 5 technical replicates per repeat. Data were analyzed via a 2-way ANOVA with a Tukey’s post-hoc multiple comparison test and significance at endpoint is shown. Error bars represent standard deviation; ns, no significance; *, p < 0.05; ***, p < 0.001; ****, p < 0.0001.
Figure 2. Combination treatment compromised clonogenic survival.
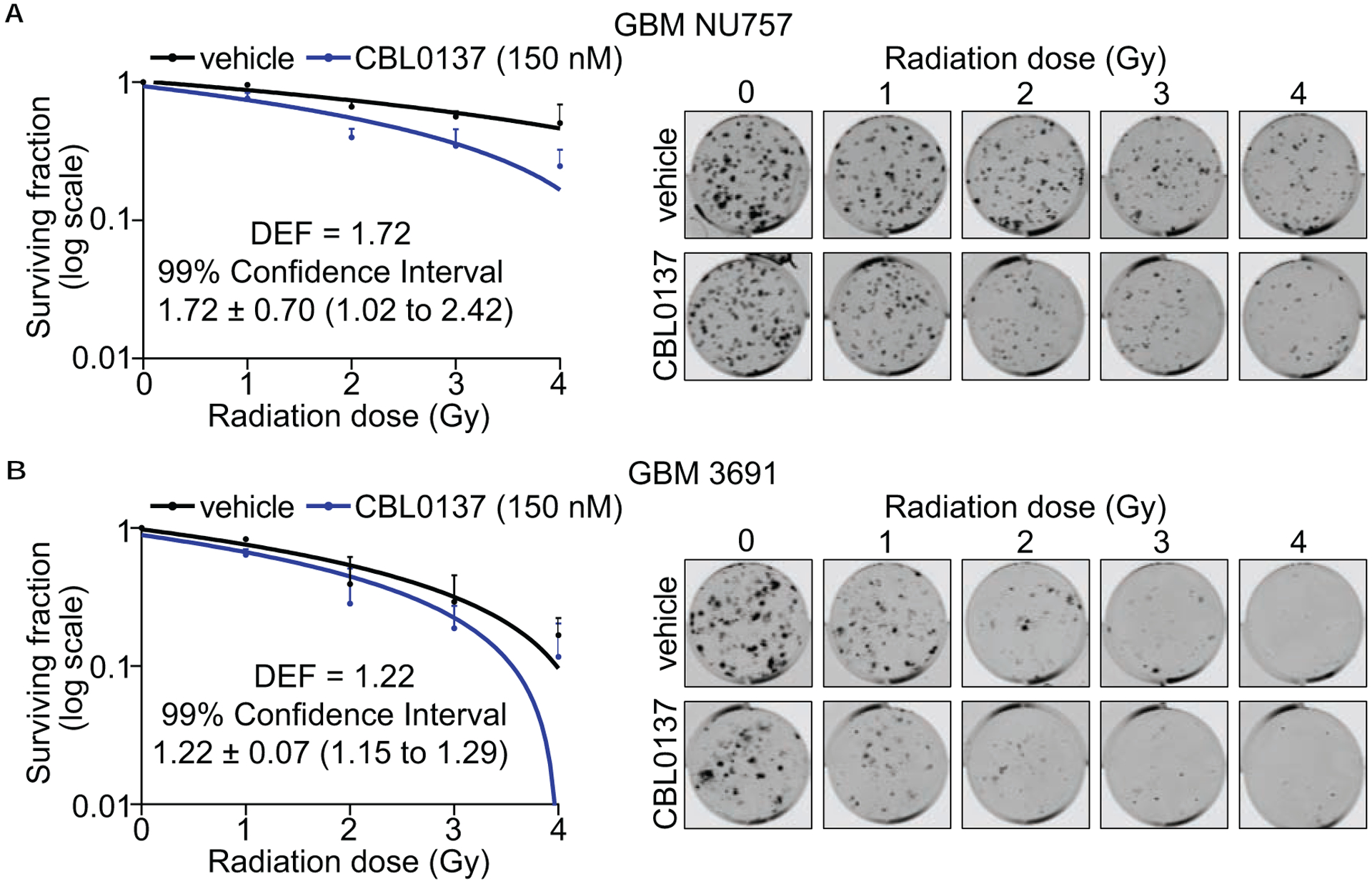
GBM NU757 (a) and GBM 3691 (b) were treated with vehicle (DMSO), CBL0137 (150 nM), and then irradiated (0–4 Gy, 6 hour pre-treatment with CBL0137). Colonies were quantified 10 days after treatment. Colonies per well were normalized to 0 Gy and linear regression was used to model the effect of radiation on survival. Data for vehicle (black line) and CBL0137 (blue line) were graphed on log10 scale. Representative images are shown to the right. Each experiment was repeated 3 times per GBM specimen with 3 technical replicates per repeat. Error bars represent standard deviation. Dose enhancement factors (DEFs) were calculated by comparing doses at which the surviving fraction was 0.5 and 99% confidence interval showed a DEF of above 1.
3.2. Combination treatment led to enhanced and prolonged DNA damage in GBM cells
To determine if CBL0137 alters the DNA damage response (DDR) after irradiation, we performed immunolabeling with γH2AX or 53BP1 in order to mark and track the resolution of foci that are mainly indicative of DNA double strand breaks. GBM NU757 and GBM 3691 were exposed to vehicle, 150 nM CBL0137, 3 Gy of irradiation, or the combination of CBL0137 and irradiation. Cells were collected 1 ,6 ,or 24 hours post irradiation, immunolabled for γH2AX or 53BP1, then scored for greater or less than 10 foci per cell. For both specimens, CBL0137, alone and in combination with irradiation, led to significantly more DNA damage compared to the vehicle treatment (Fig. 3; Fig. 4; Supplementary Fig. S3). Although CBL0137 alone has not previously been demonstrated to induce DNA damage, it has recently been reported that depletion of the FACT component SSRP1 via siRNA directly led to DNA damage (25). To evaluate if this occurs in our system, and to highlight that the damage we see with CBL0137 treatment may in part be due to disruption of FACT function, we quantified 53BP1 foci following treatment of our cells with siRNA pools to SSRP1 or SPT16 with or without irradiation. As demonstrated in previous studies, siRNA to one subunit also led to decreased expression of the other subunit (Supplementary Fig. S4a) (26). Results indicated that siRNA, alone and in combination with irradiation, led to significantly more DNA damage compared to the vehicle treatment, akin to CBL0137 (Supplementary Fig. S4b). These results indicate that GBM CSCs either retain or continue to accumulate more DNA damage following combination treatment as well as highlight the ability of CBL0137 as a monotherapy to induce DNA damage in GBM CSCs.
Figure 3. Combination treatment reduced γH2AX foci resolution.
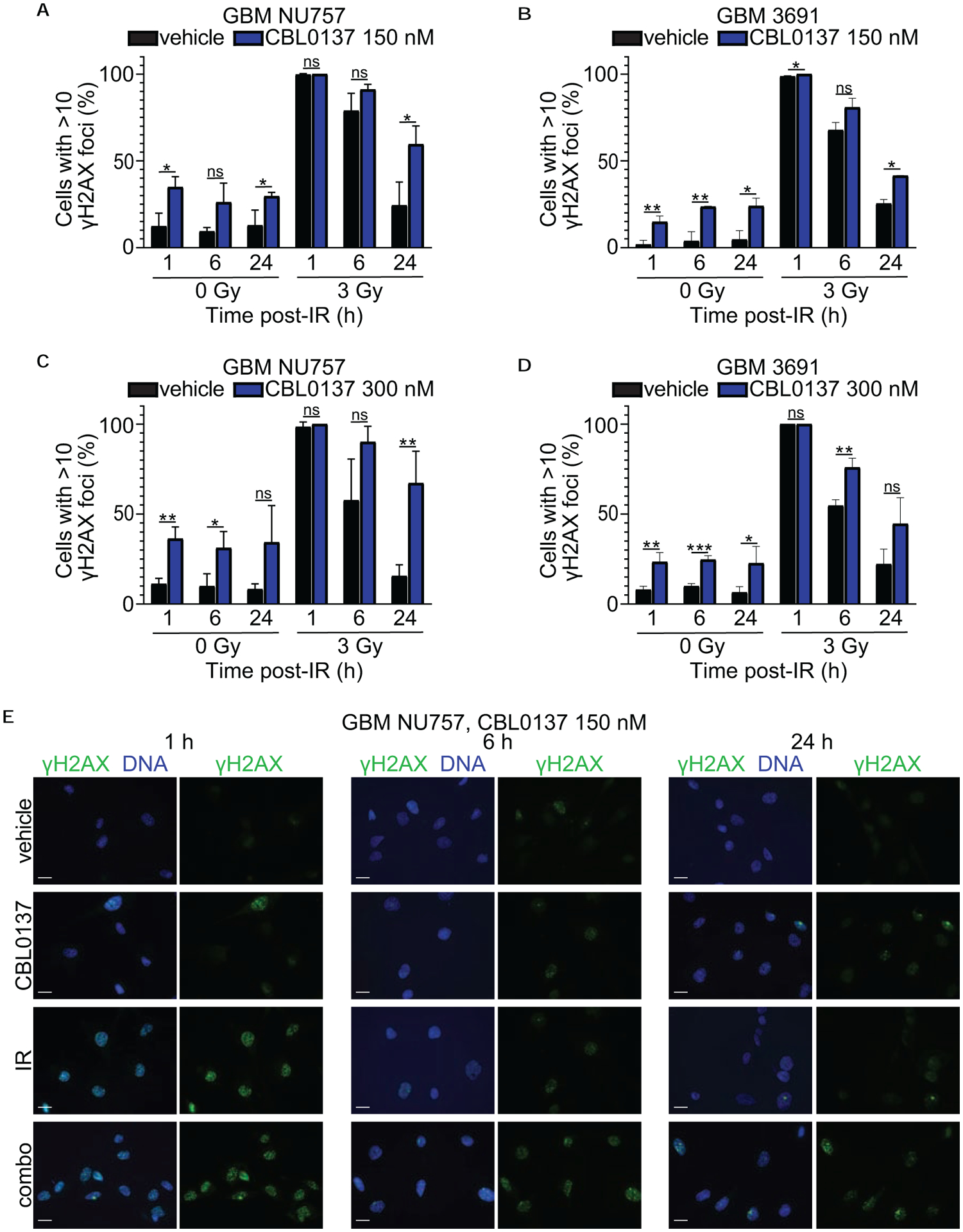
GBM NU757 (a, c) and GBM 3691 (b, d) were treated with vehicle (DMSO) and CBL0137 (150 nM or 300 nM). After a 6 hour pretreatment, cells were either left unirradiated (0 Gy) or irradiated (IR, 3 Gy). Cells were fixed at 1 hour (h), 6 h, and 24 h post irradiation and immunolabeled for γH2AX (green) with the DNA counter-stained with Hoechst (blue). The percentage of cells with greater than 10 foci was quantified and graphed for vehicle (black bar) and CBL0137 (blue bar) +/− IR. Each experiment was repeated 3 times per GBM specimen, with each experiment having over 100 cells per condition and time point analyzed. Error bars represent standard deviation. Data were analyzed via an unpaired Student t test and resulting significance is shown. Error bars represent standard deviation; ns, no significance; *, p < 0.05; **, p < 0.01; ***, p < 0.001. (e) Representative images for NU757 (150 nM CBL0137) at 63× magnification. Scale bars represent 20 μm.
Figure 4. Combination treatment reduced 53BP1 foci resolution.
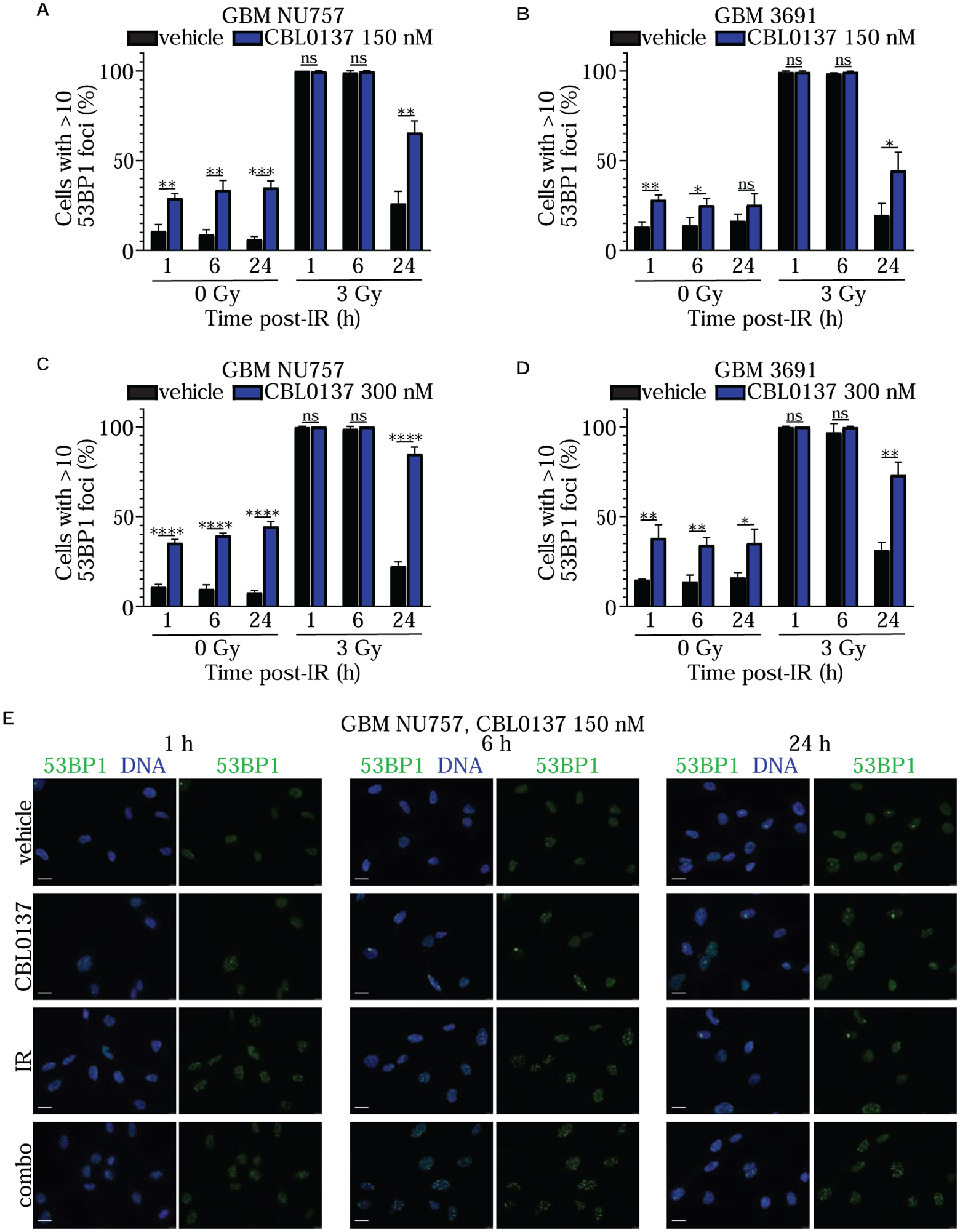
GBM NU757 (a, c) and GBM 3691 (b, d) were treated with vehicle (DMSO) and CBL0137 (150 nM or 300 nM). After a 6 hour pretreatment, cells were either left unirradiated (0 Gy) or irradiated (IR, 3 Gy). Cells were fixed at 1 hour (h), 6 h, and 24 h post irradiation and immunolabeled for 53BP1 (green) with the DNA counter-stained with Hoechst (blue). The percentage of cells with greater than 10 foci was quantified and graphed for vehicle (black bar) and CBL0137 (blue bar) +/− IR. Each experiment was repeated 3 times per GBM specimen, with each experiment having over 100 cells per condition and time point analyzed. Error bars represent standard deviation. Data were analyzed via an unpaired Student t test and resulting significance is shown. Error bars represent standard deviation; ns, no significance; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. (e) Representative images for NU757 (150 nM CBL0137) at 63× magnification. Scale bars represent 20 μm.
Next, in order to further investigate how CBL0137 impacted the DDR alone and in combination with irradiation, we performed alkaline comet assays using GBM NU757, GBM 1016, GBM 3691, and GBM 08–387. The alkaline comet assay allows for comet tails to include DNA fragments resulting mainly from single strand breaks but also double strand breaks to some extent (27–30). Cells were exposed to a 6 hour pre-treatment of vehicle or 300 nM CBL0137 then left unirradiated or exposed to 3 Gy of irradiation and collected 1 hour post irradiation to measure comet tail length. For all four specimens, tail lengths were significantly longer, and hence more DNA damage was present, in the tumor cells treated with the combination (Fig. 5). In support of the DNA damage foci data, CBL0137 alone also led to an increase in comet tail length for all four specimens. Together these data indicate that CBL0137 treatment prior to irradiation significantly increased the level of DNA damage immediately post irradiation, as well as led to sustained damage over time.
Figure 5. Combination treatment increased DNA damage.
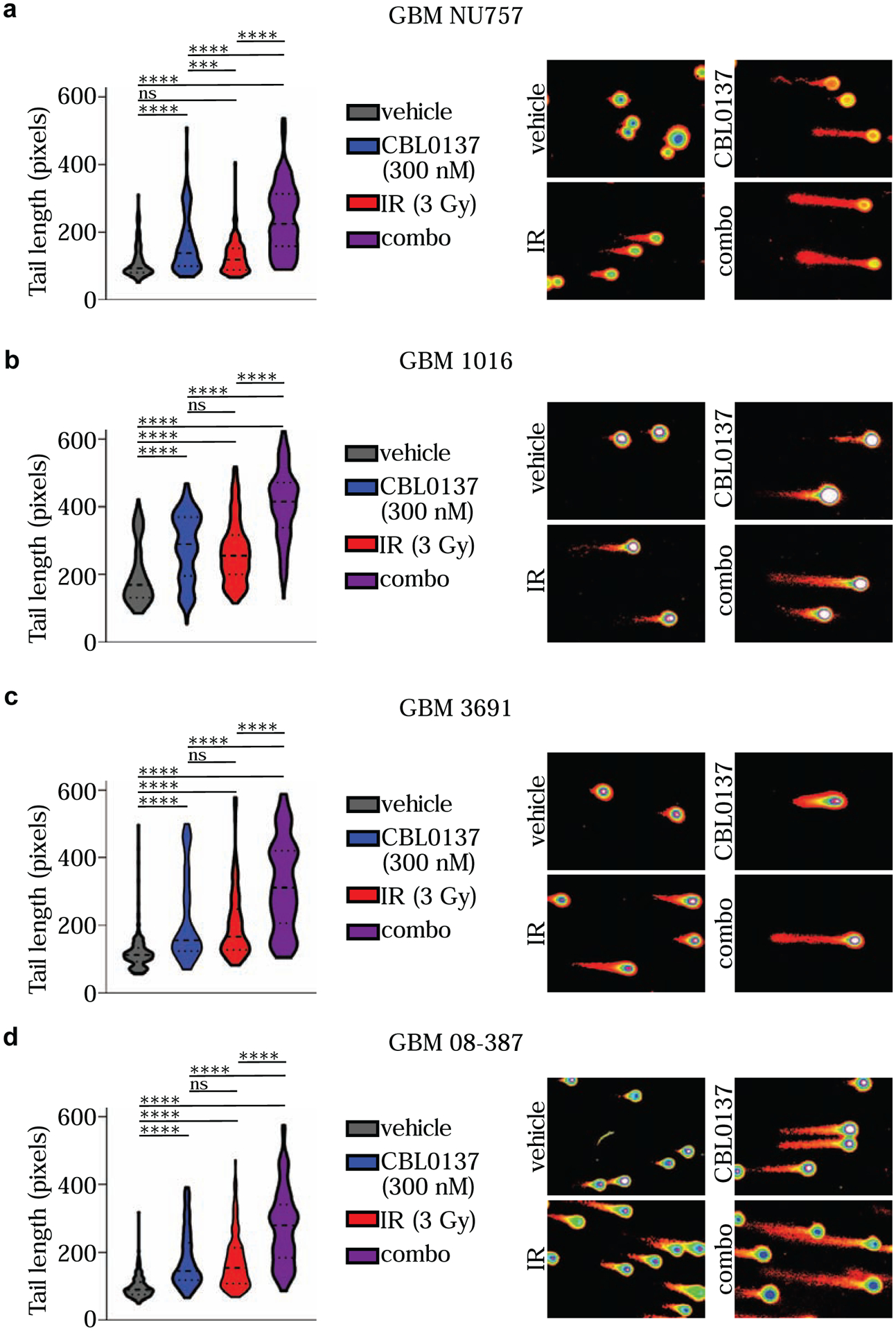
GBM NU757 (a), GBM 1016 (b), GBM 3691 (c), and GBM 08–387 (d) were treated with vehicle (DMSO), CBL0137 (300 nM), irradiation (IR, 3 Gy) or CBL0137 and IR (combo; 6 hour pre-treatment with CBL0137). Comet assays were performed 1 hour post-IR and tail length (measured in pixels) was measured by CometScore and graphed for vehicle (grey), CBL0137 (blue), IR (red), and combo (purple). Representative images for each condition as processed in CometScore are shown to the right. Each experiment was repeated 3 times per GBM specimen. Data were analyzed via a mixed-error component model with a Tukey’s post-hoc multiple comparison test. Median and quartiles are indicated by the dashed lines. ns, not significant; ***, p < 0.001; ****, p < 0.0001.
Finally, we wanted to evaluate pathway activation for the DDR in cell lysates that matched the time course for immunofluorescence and also included an additional, intermediate time point of 12 hours post-irradiation (Fig. 6). We started by looking at activation of the DDR effector kinases, Chk1 and Chk2. Specifically, we looked at phosphorylation of Chk2 at Threonine68 (pChk2 T68) and phosphorylation of Chk1 at Serine345 (pChk1 S345). We did not see activation of Chk2 for CBL0137 alone but did see similar activation for irradiation only and combination treatment across the time course. For Chk1, irradiation only and combination treatment again showed similar activation across the time course. However, we also saw a slight increase in pChk1S345 at 6 and 12 hours post-irradiation for the CBL0137 only treatment group. Given that Chk1 has a prominent role for the DDR within S phase, and more specifically to replication stress, we evaluated phosphorylation of a key protein involved in the response to such DNA damage, Rad17. Specifically, we evaluated phosphorylation of Rad17 at Serine645 (pRad17 S645). Again we saw a similar increase in phosphorylation for the irradiation and combination treatment groups across the time course with a slight increase for CBL0137 alone at 12 hours post-irradiation. We also evaluated phosphorylation of p53 at the Serine392 (p-p53 S392), which was previously reported to be increased following treatment with CBL0137 (14). There was variable increase in phosphorylation for the earlier time points, but all three treatment groups had an increase in p-p53 S392 at 24 hours post-irradiation. Together, these data indicate a potential increase in the DNA damage response within S phase for CBL0137 treatment, although combination treatment did not lead to an increase in any of the phosphorylation events evaluated over this time course.
Figure 6. Combination treatment activates certain DNA damage response proteins.
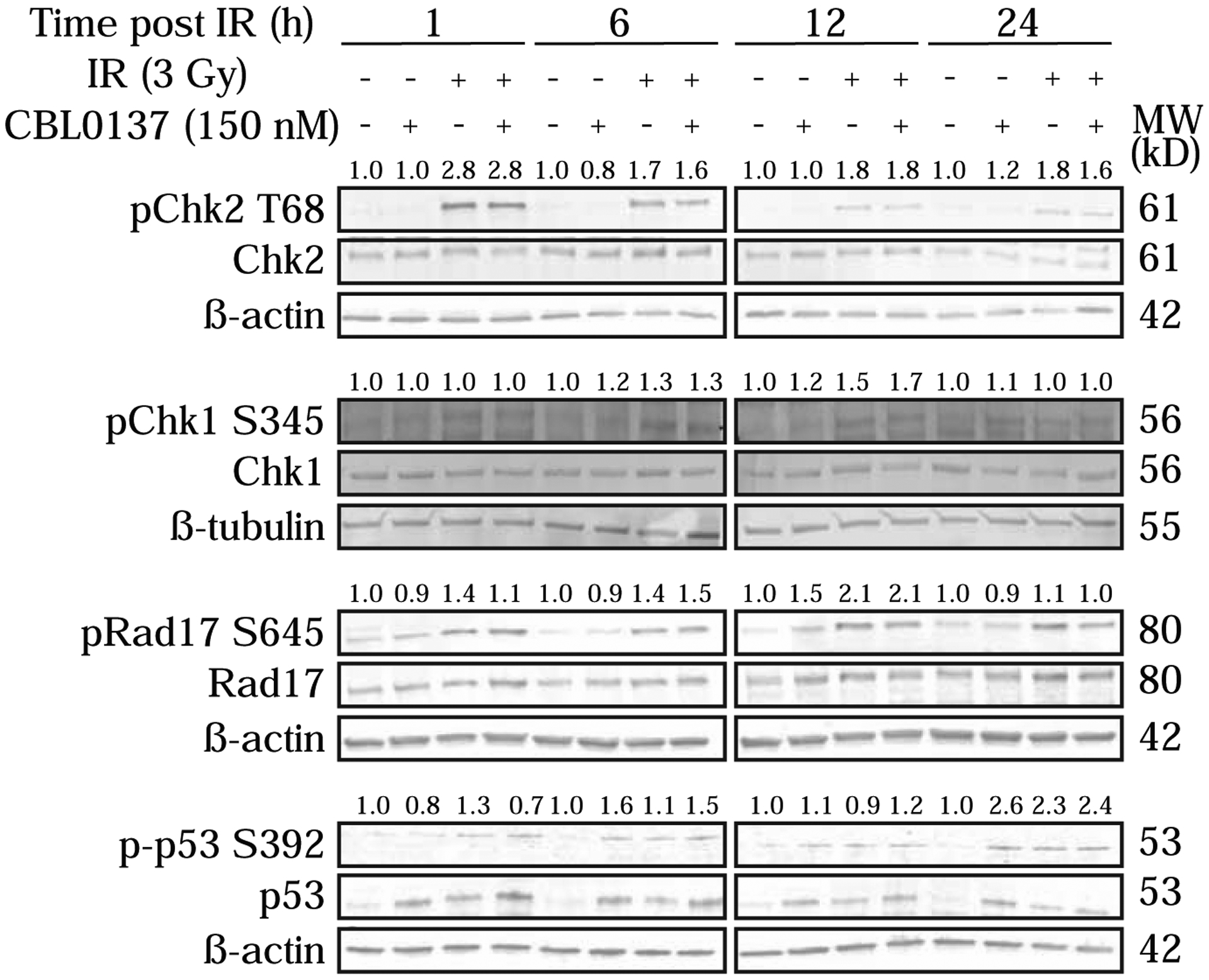
GBM 3691 was treated with vehicle (DMSO), CBL0137 (150nM), irradiation (IR, 3Gy), or CBL0137 and IR (6 hour pre-treatment with CBL0137). Cells were collected 1 hour (h), 6 h, 12 h, and 24 h post-irradiation. Immunoblots were probed for pChk2(T68)/Chk2, pChk1(S345)/Chk1, pRad17(S645)/Rad17, and p-p53(S392)/p53. Band intensities were quantified and phospho-proteins were normalized to their respective total protein, and relative ratios were calculated by normalizing conditions to vehicle within each time point. Relative ratios are shown above phospho-protein bands. β-tubulin or β-actin served as loading controls.
3.3. Combination treatment increased in vivo survival in orthotopic murine GBM models
To evaluate the in vivo efficacy of CBL0137 treatment in combination with irradiation, mice with intracranially implanted GBM 1016 or GBM 3691 cells were given DMSO (vehicle), CBL0137 (70 mg/kg, q7 days × 4), irradiation (2.5 Gy, q7 days × 4), or combo (70 mg/kg, q7 days × 4; 2.5 Gy, q7 days × 4) and monitored for overall survival (Fig. 7a–b). The treatment design was based on the published optimal treatment schedule for CBL0137 with radiotherapy given only within an acute window following drug treatment to best evaluate any potential impact of combination therapy on overall survival (9). For GBM 1016, mice in the vehicle group survived a median of 35.5 days and mice in the CBL0137 or irradiation only groups had a modest increase in survival with a median of 37 or 48 days, respectively. The combination treatment increased the median survival to 60 days. For GBM 3691, the mice in the vehicle group survived a median of 34.5 days, treatment with CBL0137 alone minimally increased the median survival to 35 days, and irradiation only led to a median survival of 44.5 days. The combination treatment led to a median survival of 48.5 days. For both cell lines, combination treatment showed a significant increase in survival over all individual treatments. These data indicate that the new therapeutic paradigm of treating GBM with CBL0137 and irradiation together led to significantly increased survival in vivo.
Figure 7. Combination treatment increased in vivo survival and reduced stem cell frequency.
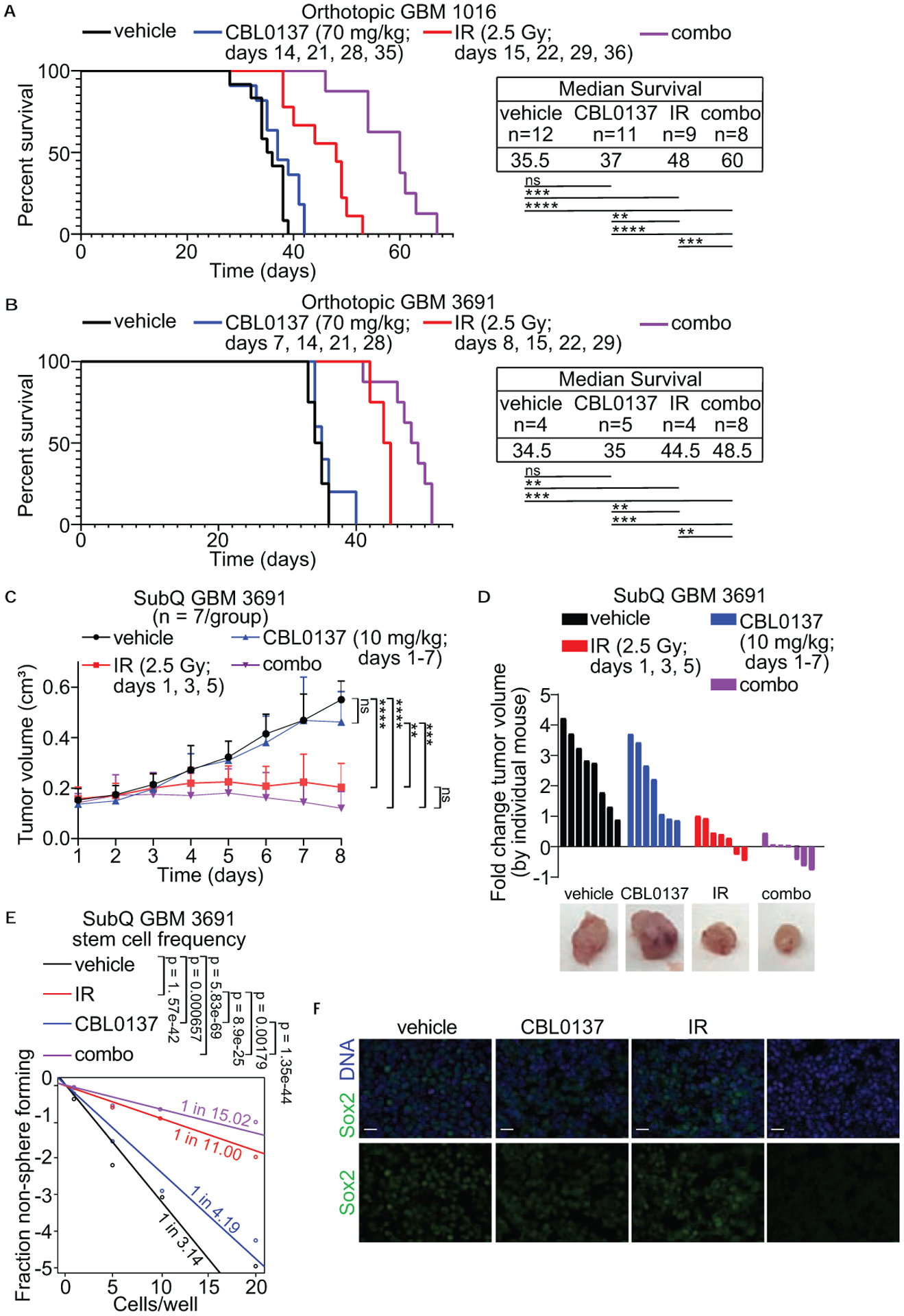
GBM 1016 (a) and GBM 3691 (b) orthotopic tumor bearing mice were treated with vehicle (saline), CBL0137 (70 mg/kg), irradiation (IR, 2.5 Gy) or CBL0137 and IR (combo) on the indicated days. Kaplan-Meier survival curves were generated for vehicle (black line), CBL0137 (blue line), IR (red line), and combo (purple line). The median survival and number of mice per group for each condition is indicated. Data were analyzed via independent log-rank (Mantel-Cox) tests between groups with a Bonferroni’s post-hoc multiple comparison test. ns, not significant; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. (c) Stem cell frequency following drug treatment was evaluated using a GBM 3691 subcutaneous (subQ), flank model. Once tumors reached approximately 0.12 cm3 mice were treated with vehicle (saline), CBL0137 (10mg/kg), IR (2.5 Gy), or CBL0137 + IR (combo) on the indicated days over a 7 day treatment course. Each group included 7 mice. Tumors were measured daily and graphed for vehicle (black line), CBL0137 (blue line), IR (red line), and combo (purple line). Data were analyzed via a 2-way ANOVA with a Tukey’s post-hoc multiple comparison test and significance at endpoint is shown. Error bars represent standard deviation; ns, not significant; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001. (d) Fold change in tumor growth for each individual mouse was calculated and graphed for vehicle (black bars), CBL0137 (blue bars), IR (red bars), and combo (purple bars). Representative images for each condition are shown to the right. (e) Tumors from three separate subQ tumors from each treatment group were dissociated and sorted for an in vitro limiting dilution assay. Cells (1, 5, 10 or 20) were plated in each well of a 96-well plate. Ten days later, each well was checked for the presence of a tumorsphere. Stem cell frequency was calculated and is indicated for vehicle (black line), CBL0137 (blue line), IR (red line), and combo (purple line). (f) Representative images for each treatment group immunolabeled for the cancer stem cell marker Sox2 (green) with the DNA counter-stained with Hoechst (blue) at 63× magnification. Scale bars represent 20 μm.
3.4. Acute exposure to CBL0137 and irradiation reduced cancer stem cell frequency in vivo
CSCs have self-renewal properties and one measure for this self-renewal is the ability for a single CSC to form a tumorsphere in in vitro assays. Here we assessed the ability of combination treatment to shift the frequency of CSCs in flank xenograft tumors after in vivo exposure to treatment in a limiting dilution assay. Limiting dilution assays are a surrogate for self-renewal and allow for the quantification of CSC frequency within a given sample. We injected GBM 3691 subcutaneously in the flanks of mice and once tumors reached approximately 0.12 cm3, mice were randomized into a 7 day, acute treatment course as we have previously done (31). Mice were treated with DMSO (vehicle), CBL0137 (IP, 10 mg/kg, for all 7 days), irradiation (2.5Gy, every other day for a total of 3 fractions), and combo (10 mg/kg, for all 7 days; 2.5Gy, every other day for a total of 3 fractions). To track any changes in tumor size during the acute study, tumors were measured daily to calculate volume. Tumor volume decreased in the irradiation only group with an even greater decrease in the combination treatment group (Fig. 7c). In order to further investigate the differences between irradiation only and combination treatment, we graphed the fold change in volume by individual mouse which highlighted the greatest number of mice with reduced or no change in tumor volume within the combination group (Fig. 7d). At the end of the treatment course, tumors from each condition were dissociated and sorted, based on viability, for a limiting dilution assay to quantify any changes in the estimated stem cell frequency among the treatment groups. Combination treatment led to the greatest shift in cancer stem cell frequency with a 1 in 15.02 chance of a CSC being present (Fig. 7e). We also saw a decrease in Sox2 positive cells within the flank xenograft tumors in the combination treatment group (Fig. 7f). Here we show that combining irradiation with CBL0137 led to not only significantly increased survival, but also decreased the radioresistant CSC subpopulation.
4. Discussion
GBM is one of the most radioresistant tumors, yet radiotherapy is still a central part of standard of care (1,3,32,33). Patient tumors will respond initially but local recurrence is inevitable (3,34). This is in part due to CSCs, the subpopulation of cancer cells most resistant to radiotherapy and most chemotherapies. CSCs are capable of self-renewal and can drive tumor regrowth, even after irradiation (5). Hence, new therapeutic strategies must inclusively target the CSC population in order to reduce recurrence, and efforts to identify new modalities to radiosensitize CSCs are ongoing and critical. The data presented here confirm that pretreatment with CBL0137 followed by irradiation led to significantly decreased cell viability for both GBM patient samples grown in stem promoting conditions and GBM patient samples sorted for CSCs based on the CD133 surface epitope. The combination of CBL0137 and irradiation led to increased levels of initial DNA damage as well as reduced damage resolution over time. In vivo, CBL0137 combined with irradiation led to significantly increased survival as well as a decrease in the frequency of CSCs within tumors.
The main target of CBL0137 is reported to be FACT, a histone chaperone that plays a role in tumor signaling pathways and the stem phenotype of CSCs. Our previous work demonstrated that acute disruption of FACT function led to a decrease in CSC associated gene expression and the stem-cell phenotype whereas prolonged exposure led to a decrease in viability (8). Here we validate that adding irradiation led to an even further reduction in cell growth as well as clonogenic cell survival. This could be in part due to the ability of CBL0137 to shift the stem phenotype of CSCs to a state where they are more sensitive to irradiation. Together, these data indicate that CBL0137 sensitizes CSCs to irradiation, at least in part, by forcing the stem cells to a more radio-sensitive state through the inhibition of CSCs-associated gene expression.
Interestingly, we observed that CBL0137 alone led to an increase in DNA damage in GBM CSCs which had not been observed in other cancer cell types (14,16). In both the foci studies and comet assay there was increased DNA damage with CBL0137 as a monotherapy. It is possible that GBM cells are particularly sensitive to the intercalation of CBL0137 into the DNA and/or inhibiting FACT might prevent an essential role for this complex in the DDR in GBM cells, as the siRNA data would indicate. FACT has been shown to have a role in the DDR so it is possible that trapping of FACT by CBL0137 in GBM cells prevents a pool of FACT from being able to respond to DNA damage (12,13,15,25). Given the inherent level of DNA damage reported for GBM CSCs, a direct impact by CBL0137 and/or a reliant role on FACT for the DDR may contribute to the increased impact on overall viability we observed (5,23,35). There is also the possibility that trapping of FACT by CBL0137 prevents its reported role in replication and hence there is an increase in DNA damage during S phase (25,36,37). This model is particularly intriguing given the recent reports highlighting replication stress and components of the replication machinery as contributory to the radioresistant phenotype of GBM CSCs (35,38,39). The precise mechanism of CBL0137 induced DNA damage in CSCs is an intriguing area for future exploration that could elucidate additional points of fragility for GBM CSCs.
Importantly, our in vivo studies demonstrated that combination treatment significantly increased survival compared to either treatment alone, and this longer survival is likely in part due to CBL0137 sensitizing the patient GBM specimens to irradiation. In particular, the acute flank tumor study validated that in vivo exposure to the drug does impact the CSC subpopulation. In our survival studies, we irradiated the tumors 24 hours after administration of CBL0137, but modified treatment paradigms may improve survival even further. Overall, our data support a new paradigm for GBM treatment whereby CBL0137 is given in combination with irradiation. GBM has an extremely high rate of recurrence due to the presence of CSCs that are resistant to current standard of care treatment. Our novel finding that CBL0137 sensitizes GBM CSCs to irradiation, leading to tumor cell death and increased survival in vivo, presents an exciting new treatment paradigm for an otherwise bleak tumor prognosis.
Supplementary Material
Supplemental Figure 1. Validation of cancer stem cell marker expression in sorted cancer stem cell specimens. GBM 3691 and GBM 08–387 were validated to have high expression of the cancer stem cell markers Nestin, Sox2, and Olig 2 versus low/no expression of the differentiation markers GFAP and MAP2. β-tubulin or β-actin served as loading controls.
Supplemental Figure 2. Sustained exposure to combo treatment decreased GBM cell growth. GBM NU757 (a), GBM 1016 (b), GBM 3691 (c), and GBM 08–387 (d) were treated with vehicle (DMSO), CBL0137 (300 nM), irradiation (IR, 3 Gy) or CBL0137 and IR (combo; 6 hour pre-treatment with CBL0137). Cell growth was monitored on the IncuCyte ZOOM live-cell imaging system and fold change in cell growth via confluence for vehicle (black line), CBL0137 (blue line), IR (red line), and combo (purple line) was normalized to t = 0 hours (h) and graphed. Each experiment was repeated 3 times per GBM specimen with 5 technical replicates per repeat. Data were analyzed via a 2-way ANOVA with a Tukey's post-hoc multiple comparison test and significance at endpoint is shown. Error bars represent standard deviation; ns, no significance; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
Supplemental Figure 3. Representative images of foci for GBM 3691. GBM 3691 were treated with vehicle (DMSO) and CBL0137 (300 nM), radiation (IR, 3 Gy), or both CBL0137 and IR (combo). Cells were fixed at 1 hour (h), 6 h, and 24 h post irradiation and immunolabeled for (a) γH2AX (green) or (b) 53BP1 (green) with the DNA counter-stained with Hoechst (blue). Representative images for 3691 at 63× magnification. Scale bars represent 20 μm.
Supplemental Figure 4. siRNA depletion of FACT subunits reduced 53BP1 foci resolution. (a) GBM 3691 was treated with siControl, siSSRP1, or siSPT16 with depletion validated by immunoblot. β-tubulin served as the loading control. (b) siRNA treated cells were either left unirradiated (0 Gy) or irradiated (IR, 3 Gy). Cells were fixed at 1 hour (h), 6 h, and 24 h post irradiation and immunolabeled for 53BP1 with the DNA counter-stained with Hoechst. The percentage of cells with greater than 10 foci were quantified and graphed for siControl (black bar), siSSRP1 (yellow bar), or siSPT16 (green bar) +/− IR. Each experiment was repeated 3 times with each experiment having over 100 cells per condition and time point analyzed. Error bars represent standard deviation. Data were analyzed via an unpaired Student t test and resulting significance is shown. Error bars represent standard deviation; ns, no significance; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
Highlights.
CBL0137 radiosensitizes glioblastoma cancer stem cells.
CBL0137 in combination with irradiation increases DNA damage in cancer stem cells.
Combination treatment increases survival in mouse models of glioblastoma.
Cancer stem cells are targeted following in vivo exposure to combination therapy.
Acknowledgements
The authors thank Dr. Jeremy Rich (UC San Diego) for the kind gift of the GBM 3691 and GBM 08-387 cells, Dr. Anita Hjelmeland (University of Alabama) for the kind gift of the GBM 1016 cells, and Dr. Craig Horbinski and the Northwestern University Nervous System Tumor Bank for the kind gift of the GBM NU757 cells. We also thank members of the Venere laboratory for insightful discussion and constructive comments on the manuscript.
Funding: This work was supported by the National Cancer Institute of the National Institutes of Health under award number R03CA227206, a Research Scholar Grant RSG-18-066-01-TBG from the American Cancer Society, and funds from The Ohio State University Comprehensive Cancer Center/Department of Radiation Oncology (MV); the National Institute of General Medical Sciences of the National Institutes of Health under award number 2T32GM068412-11A1 (MMT); an Ohio State University Graduate School Dean’s Distinguished University Fellowship (AAZ); the Pelotonia Fellowship Program (AD); an American Brain Tumor Association Basic Research Fellowship supported by an Anonymous Corporate Donor (MSS); and the National Institute of General Medical Sciences of the National Institutes of Health under award numbers R01GM112895 and R01GM108743 (MKS). Research reported in the publication was supported by The Ohio State University Comprehensive Cancer Center and the National Institutes of Health under grant number P30 CA016058. We specifically thank members of the Target Validation Shared Resource, Small Animal Imaging Core, Analytic Cytometry Shared Resource, Genomics Shared Resource, and the Biostatistics Shared Resource. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
The authors declare no potential conflicts of interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Stupp R, Hegi ME, Mason WP, Bent MJvd, Taphoorn MJB, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. The Lancet 2009;10. [DOI] [PubMed] [Google Scholar]
- 2.Ostrom QT, Gittleman H, Liao P, Rouse C, Chen Y, Dowling J, et al. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2007–2011. Neuro-Oncology 2014;16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tamura K, Aoyagi M, Wakimoto H, Ando N, Nariai T, Yamamoto M, et al. Accumulation of CD133-positive glioma cells after high-dose irradiation by Gamma Knife surgery plus external beam radiation. Journal of Neurosurgery 2010;113:310–8 [DOI] [PubMed] [Google Scholar]
- 4.Yu VY, Nguyen Dan, Pajonk Frank, Kupelian Patrick, Kaprealian Tania, Selch Michael, et al. Incorporating Cancer Stem Cells in Radiation Therapy Treatment Response Modeling and the Implication in Glioblastoma Multiforme Treatment Resistance. International Journal of Radiation Oncology*Biology*Physics 2015;91:866–75 [DOI] [PubMed] [Google Scholar]
- 5.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006;444:756–60 [DOI] [PubMed] [Google Scholar]
- 6.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a Cancer Stem Cell in Human Brain Tumors. Cancer Research 2003;63:5821–8 [PubMed] [Google Scholar]
- 7.Chen J, Li Y, Yu T-S, McKay RM, Burns DK, Kernie SG, et al. A restricted cell population propagates glioblastoma growth following chemotherapy. Nature 2012: 522–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dermawan JK, Hitomi M, Silver DJ, Wu Q, Sandlesh P, Sloan AE, et al. Pharmacological Targeting of the Histone Chaperone Complex FACT Preferentially Eliminates Glioblastoma Stem Cells and Prolongs Survival in Preclinical Models. Cancer Res 2016;76:2432–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barone TA, Burkhart CA, Safina A, Haderski G, Gurova KV, Purmal AA, et al. Anticancer drug candidate CBL0137, which inhibits histone chaperone FACT, is efficacious in preclinical orthotopic models of temozolomide-responsive and - resistant glioblastoma. Neuro Oncol 2017;19:186–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Formosa T The Role of FACT in Making and Breaking Nucleosomes. BBiochim Biophys Acta 2012:247–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orphanides G, Wu W-H, Lane WS, Hampsey M, Reinberg D. The chromatin-specic transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature 1999;400:284–8 [DOI] [PubMed] [Google Scholar]
- 12.Piquet S, Le Parc F, Bai SK, Chevallier O, Adam S, Polo SE. The Histone Chaperone FACT Coordinates H2A.X-Dependent Signaling and Repair of DNA Damage. Mol Cell 2018;72:888–901 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gao Y, Li C, Wei L, Teng Y, Nakajima S, Chen X, et al. SSRP1 Cooperates with PARP and XRCC1 to Facilitate Single-Strand DNA Break Repair by Chromatin Priming. Cancer Research 2017;77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gasparian AV, Burkhart CA, Purmal AA, Brodsky L, Pal M, Saranadasa M, et al. Curaxins: anticancer compounds that simultaneously suppress NF-kappaB and activate p53 by targeting FACT. Sci Transl Med 2011;3:95ra74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Safina A, Cheney P, Pal M, Brodsky L, Ivanov A, Kirsanov K, et al. FACT is a sensor of DNA torsional stress in eukaryotic cells. Nucleic Acids Research 2017;45:1925–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carter DR, Murray J, Cheung BB, Gamble L, Koach J, Tsang J, et al. Therapeutic targeting of the MYC signal by inhibition of histone chaperone FACT in neuroblastoma. Sci Transl Med 2015;7:312ra176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De S, Lindner DJ, Coleman CJ, Wildey G, Dowlati A, Stark GR. The FACT Inhibitor CBL0137 Synergizes With Cisplatin in Small-Cell Lung Cancer by Increasing NOTCH1 Expression and Targeting Tumor-Initiating Cells. Cancer Research 2018:2396–406 [DOI] [PubMed] [Google Scholar]
- 18.Venere M, Horbinski C, Crish JF, Jin X, Vasanji A, Major J, et al. The mitotic kinesin KIF11 is a driver of invasion, proliferation, and self-renewal in glioblastoma. Sci Transl Med 2015;7:304ra143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boyd NH, Walker K, Fried J, Hackney JR, McDonal PC, Benavides GA, et al. Addition of carbonic anhydrase 9 inhibitor SLC-0111 to temozolomide treatment delays glioblastoma growth in vivo. JCI Insight 2017;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu Y, Smyth GK. ELDA: Extreme limiting dilution analysis for comparing depleted andenriched populations in stem cell and other assays. Journal of Immunological Methods 2009;347:70–8 [DOI] [PubMed] [Google Scholar]
- 21.Franken NAP, Rodermond HM, Stap J, Haveman J, Bree Cv. Clonogenic assay of cells in vitro. Nature Protocols 2006;1:2315–9 [DOI] [PubMed] [Google Scholar]
- 22.Li A, Walling J, Kotliarov Y, Center A, Steed ME, Ahn SJ, et al. Genomic Changes and Gene Expression Profiles Reveal That Established Glioma Cell Lines Are Poorly Representative of Primary Human Gliomas. Mol Cancer Res 2008;6:21–30 [DOI] [PubMed] [Google Scholar]
- 23.Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006;9:391–403 [DOI] [PubMed] [Google Scholar]
- 24.Pollard SM, Yoshikawa K, Clarke ID, Danovi D, Stricker S, Russell R, et al. Glioma Stem Cell Lines Expanded in Adherent Culture Have Tumor-Specific Phenotypes and Are Suitable for Chemical and Genetic Screens. Cell Stem Cell 2009;4:568–80 [DOI] [PubMed] [Google Scholar]
- 25.Prendergast L, Hong E, Safina A, Poe D, Gurova K. Hisone chaperone FACT is essential to overcome replciation stress in mamalian cells. Oncogene 2020;39:5124–5137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Safine A, Garcia H, Commane M, Guryanova O, Degan S, Kolesnikova K, et al. Complex mutual regulation of facilitates chromatin transcription (FACT) subunits on both mRNA and protein levels in human cells. Cell Cycle 2013;12:2423–2434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olive PL. DNA damage and repair in individual cells: appliactions of the comet assay in radiobiology. Int J Radiat Biol 1999;75:395–405 [DOI] [PubMed] [Google Scholar]
- 28.Cortes-Gutierrez EI, Fernandex JL, Davila-Rodriguez MI, Lopez-Fernandez C, Gosalvez J. Two-tailed Comet Assay (2T-Comet): Simultaneous Detection of DNA Single and Double Strand Breaks. Springer Protocol 2017;1560. [DOI] [PubMed] [Google Scholar]
- 29.Peycheva E, Georgieva M, Miloshev G. Comparison between alkaline and neutral vairants of yeast comet assay. Biotechnology & Biotechnological Equipment 2009;1090–1092 [Google Scholar]
- 30.Moller P The alkaline comet assay: Towards Validation in Biomonitoring of DNA damaging Exposures. Basic Clin Pharmacol Toxicol 2006;98:336–45 [DOI] [PubMed] [Google Scholar]
- 31.Venere M, Hamerlik P, Wu Q, Rasmussen RD, Song LA, Vasanji A, et al. Therapeutic targeting of constitutive PARP activation compromises stem cell phenotype and survival of glioblastoma-initiating cells. Cell Death & Differentiation 2013;21:258–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vlashi E, McBride WH, Pajonk F. Radiation Responses of Cancer Stem Cells. Journal of Cellular Biochemistry 2009;108:339–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corso CD, Bindra RS, Mehta MP. The role of radiation in treating glioblastoma: here to stay. Journal of Neurooncology 2017;134:479–85 [DOI] [PubMed] [Google Scholar]
- 34.Milano MT, Okunieff P, Donatello RS, Mohile NA, Sul J, Walter KA, et al. Patterns and Timing of Recurrence After Temozolomide-Based Chemoradiation for Glioblastoma. International Journal of Radiation Oncology*Biology*Physics 2010;78:1147–55 [DOI] [PubMed] [Google Scholar]
- 35.Carruthers R, Ahmed S, Ramachandran S, Strathdee K, Kathreena Kurian, Hedley A, et al. Replication Stress Drives Constitutive Activation of the DNA Damage Response and Radioresistance in Glioblastoma Stem-like Cells. Cancer Research 2018;17:5060–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abe T, Sugimura K, Hosono Y, Takami Y, Akita M, Yoshimura A, et al. The Histone Chaperone Facilitates Chromatin Transcription (FACT) Protein Maintains Normal Replication Fork Rates. The Journal of Biological Chemistry 2011:30504–305012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang J, Zhang X, Feng J, Leng H, Li S, Xiao J, et al. The Histone Chaperone FACT Contributes to DNA Replication-Coupled Nucleosome Assembly. Cell Reports 2016;16:3414. [DOI] [PubMed] [Google Scholar]
- 38.Dungey FA, Löser DA, Chalmers AJ. Replication-dependent radiosensitization of human glioma cells by inhibition of poly(ADP-Ribose) polymerase: mechanisms and therapeutic potential. Int J Radiat Oncol Biol Phys 2008;4:1188–97 [DOI] [PubMed] [Google Scholar]
- 39.Pedersen H, Anne Adanma Obara E, Juul Elbæk K, Vitting-Serup K, Hamerlik P. Replication Protein A (RPA) Mediates Radio-Resistance of Glioblastoma Cancer Stem-Like Cells. Int J Mol Sci 2020;5 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Validation of cancer stem cell marker expression in sorted cancer stem cell specimens. GBM 3691 and GBM 08–387 were validated to have high expression of the cancer stem cell markers Nestin, Sox2, and Olig 2 versus low/no expression of the differentiation markers GFAP and MAP2. β-tubulin or β-actin served as loading controls.
Supplemental Figure 2. Sustained exposure to combo treatment decreased GBM cell growth. GBM NU757 (a), GBM 1016 (b), GBM 3691 (c), and GBM 08–387 (d) were treated with vehicle (DMSO), CBL0137 (300 nM), irradiation (IR, 3 Gy) or CBL0137 and IR (combo; 6 hour pre-treatment with CBL0137). Cell growth was monitored on the IncuCyte ZOOM live-cell imaging system and fold change in cell growth via confluence for vehicle (black line), CBL0137 (blue line), IR (red line), and combo (purple line) was normalized to t = 0 hours (h) and graphed. Each experiment was repeated 3 times per GBM specimen with 5 technical replicates per repeat. Data were analyzed via a 2-way ANOVA with a Tukey's post-hoc multiple comparison test and significance at endpoint is shown. Error bars represent standard deviation; ns, no significance; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
Supplemental Figure 3. Representative images of foci for GBM 3691. GBM 3691 were treated with vehicle (DMSO) and CBL0137 (300 nM), radiation (IR, 3 Gy), or both CBL0137 and IR (combo). Cells were fixed at 1 hour (h), 6 h, and 24 h post irradiation and immunolabeled for (a) γH2AX (green) or (b) 53BP1 (green) with the DNA counter-stained with Hoechst (blue). Representative images for 3691 at 63× magnification. Scale bars represent 20 μm.
Supplemental Figure 4. siRNA depletion of FACT subunits reduced 53BP1 foci resolution. (a) GBM 3691 was treated with siControl, siSSRP1, or siSPT16 with depletion validated by immunoblot. β-tubulin served as the loading control. (b) siRNA treated cells were either left unirradiated (0 Gy) or irradiated (IR, 3 Gy). Cells were fixed at 1 hour (h), 6 h, and 24 h post irradiation and immunolabeled for 53BP1 with the DNA counter-stained with Hoechst. The percentage of cells with greater than 10 foci were quantified and graphed for siControl (black bar), siSSRP1 (yellow bar), or siSPT16 (green bar) +/− IR. Each experiment was repeated 3 times with each experiment having over 100 cells per condition and time point analyzed. Error bars represent standard deviation. Data were analyzed via an unpaired Student t test and resulting significance is shown. Error bars represent standard deviation; ns, no significance; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.