

Abstract
The deregulation of epigenetic pathways has been implicated as a critical step in tumorigenesis including in childhood brain tumor medulloblastoma. The H3K27me3 demethylase UTX/KDM6A plays important roles in development and is frequently mutated in various types of cancer. However, how UTX regulates tumor development remains largely unclear. Here, we report the generation of a UTX-deleted mouse model of SHH medulloblastoma that demonstrates the tumor suppressor functions of UTX, which could be antagonized by the deletion of another H3K27me3 demethylase JMJD3/KDM6B. Intriguingly, UTX deletion in cancerous cerebellar granule neuron precursors resulted in the impaired recruitment of host CD8+ T cells to the tumor microenvironment through a non-cell autonomous mechanism. In both mouse medulloblastoma models and in human medulloblastoma cells, we showed that UTX activates Th1-type chemokines, which are responsible for T cell migration. Surprisingly, our results showed that the depletion of cytotoxic CD8+ T cells did not affect mouse medulloblastoma growth. Nevertheless, the UTX/chemokine/T cell recruitment pathway we identified may be applied to many other cancers and may be important for improving cancer immunotherapy. In addition, UTX is required for the expression of NeuroD2 in precancerous progenitors, which encodes a potent proneural transcription factor. Overexpression of NEUROD2 in CGNPs decreased cell proliferation and increased neuron differentiation. We showed that UTX deletion led to impaired neural differentiation, which could coordinate with active SHH signaling to accelerate medulloblastoma development. Thus, UTX regulates both cell-intrinsic oncogenic processes and the tumor microenvironment in medulloblastoma. Our study provides insights into both medulloblastoma development and context dependent functions of UTX in tumorigenesis.
Keywords: SHH medulloblastoma, UTX/KDM6A, Th1-type chemokines, T cell migration, neuronal differentiation
1. Introduction
Cancer development is regulated by both intrinsic drivers in cancerous cells and their microenvironments. Mutations in precancerous cells lead to uncontrolled proliferation, reduced differentiation, decreased cell death, and increased malignant transformation. The tumor microenvironment (TME) consists of various non-cancerous cells interacting with cancer cells and plays critical roles in regulating tumor growth (1-4). The understanding of TME-tumor interactions enables novel cancer treatment strategies. Host immune cells are important components of the TME, and immunotherapy is the most exciting development in cancer treatment in recent years. One key step in T cell mediated tumor killing is cytotoxic CD8+ T cell recruitment to tumor sites. The presence of high numbers of CD8+ T cells in the tumors of cancer patients correlate with enhanced survival (5-7). Tumor-produced chemokines were shown to play critical roles in attracting immune cells to tumor sites including in medulloblastoma (5, 8, 9). It was shown recently that epigenetic mechanisms regulate tumor chemokine gene expression. In ovarian and colon cancers, methylations of histone and DNA regulate the expression of T helper 1 (Th1) type chemokine CXCL9/CXCL10 genes, which are key factors for CD8+ T cell recruitment and cancer killing (10, 11). Therefore, a therapeutic strategy that enhances specific chemokine production and CD8+ T cell recruitment may be achieved through epigenetic manipulation to improve efficacy of cancer immunotherapy.
Medulloblastomas, the most common malignant childhood brain tumor, are classified into four subgroups based on distinctive transcription profiles. These subgroups are referred to as WNT, SHH, Group 3, and Group 4 (12-16). Within these subgroups, SHH medulloblastoma composes 25% of all medulloblastoma and is characterized by abnormally elevated Sonic hedgehog (SHH) signaling in cerebellar granule neuron precursors (CGNPs). SHH medulloblastomas are highly heterogeneous in both inter- and intra-tumoral levels (17-19). Recent tumor heterogeneity studies of human and similar mouse medulloblastomas revealed SHH medulloblastoma cell contents and their potential functions in tumor development (20-22). SHH medulloblastomas contain mainly ATOH1+ CGNP-like tumor cells and NEUROD1+ differentiated granule neurons. Other minor cell populations include additional neuron types, astrocytes, fibroblasts, blood vessel cells, and various types of immune cells (23, 24). It is of interest how immune cells are recruited to medulloblastoma sites and whether the host immune response plays a role in restricting tumor growth.
Epigenetic regulation is critical in normal development and in cancer. The histone H3 lysine 27 (H3K27me3) demethylase UTX (ubiquitously transcribed tetratricopeptide repeat gene on X chromosome; also known as KDM6A: lysine-specific demethylase 6A) was identified as a potential tumor suppressor since loss-of-function mutations were repeatedly found in patients with cancers of the bladder, colon, kidney, blood, brain, and other organs (25-29). UTX activates gene expression by removing the repressive histone mark H3K27me3 and by interacting with other epigenetic activators. In a mouse model of T-cell acute lymphoblastic leukemia, UTX regulates many genes that have tumor suppressing functions (30). In human invasive breast cancer, UTX was found to be a central factor in mediating epithelial-mesenchymal transition (EMT) (31). UTX-null bladder cancer cells as well as multiple myeloma cells were more sensitive to inhibitors targeting the H3K27 methyltransferase PRC2 (32, 33). In a lung cancer mouse model, UTX deletion cooperated with the KRAS mutation to promote tumor progression (34). However, the exact molecular functions of UTX in various cancers remain largely unclear, and few genetic mouse models of UTX-mutant solid tumors have been developed for functional study. UTX in both human and mouse is located on chromosome X, which escapes X-chromosome inactivation (35). UTY is a UTX homolog on the Y chromosome, which encodes a protein displaying minimum H3K27me3 demethylase activities (36, 37). Genetic deletion of Utx in mice causes gender-specific and non-specific developmental defects in multiple tissues (36, 38, 39), suggesting enzymatic activity-dependent and –independent functions during development. Utx deletion alone does not cause tumorigenesis. It is likely that Utx deletion cooperates with mutations of other gene to function in cancer development.
One important characteristic of medulloblastoma is its significant association with mutations and abnormal levels of epigenetic regulators (40-42). Previously, we have shown that BRG1, a chromatin remodeling factor, and JMJD3 (Jumonji domain-containing protein D3; also known as KDM6B: lysine-specific demethylase 6B), another H3K27me3 demethylase, are key epigenetic co-activators in SHH signaling and positively regulate SHH medulloblastoma (43-45). The H3K27 methyltransferase PRC2 complexes and demethylase UTX are also important for medulloblastoma growth since the genes encoding the PRC2 subunits are often amplified and UTX mutations are often observed in medulloblastoma patients (12, 16, 41). UTX mutations are enriched in Group 4 medulloblastoma and are also identified with lower frequencies in SHH and Group 3 medulloblastoma (17, 41). The function of UTX in medulloblastoma development has not yet been explored.
In this study, using a mutant SmoM2-induced SHH medulloblastoma mouse model (46), we demonstrated a tumor suppressor function of UTX in medulloblastoma tumorigenesis. Deleting UTX in precancerous CGNPs caused early tumor initiation and reduced survival, which could be reversed by further deleting JMJD3. Intriguingly, we observed significantly reduced CD8+ T cells in UTX deleted medulloblastoma, which is likely caused by the reduction of UTX-activated chemokine gene expression. Using a human medulloblastoma cell line, we demonstrated the essential function of UTX in activating Th1-type chemokines and chemokine-induced T cell migration. In addition, we identified the proneural gene NeuroD2 as a direct target of UTX in medulloblastoma and impaired neural differentiation as a key process leading to the early initiation of UTX deleted medulloblastoma. Therefore, our study reveals both cell-intrinsic and non-cell autonomous functions of UTX in regulating medulloblastoma development and the TME, providing novel insights to UTX function in cancer.
2. Materials and Methods
2.1. Mice
The SmoM2 mice were purchased from Jackson Laboratory (46). The Atoh1-Cre mice (47) were provided by Dr. Lin Gan (Rochester University) and Dr. Jane Johnson (University of Texas Southwestern Medical Center). The UtxF/F mice were provided by Dr. Kai Ge (NIDDK, NIH) (39). These mice contain loxP sites flanking exon 24 of the Utx gene, which encodes essential amino acids for the UTX demethylase activity. Deletion of exon 24 also caused frame shift and the loss of UTX protein. The Jmjd3F/F mice (71), which target exons 15-21 of Jmjd3, were provided by Dr. Rongfu Wang (Houston Methodist Hospital). The NOD/SCID mice were purchased from the University of Texas Southwestern Mouse Breeding Core Facility. All mice are maintained on a mixed genetic background at the University of Texas Southwestern Medical Center Animal Facility. The Utx+/+ tumor was generated by crossing SmoM2 mice with Atoh1-Cre mice, while the UtxF/F tumor was obtained by crossing UtxF/Y Atoh1-Cre males with UtxF/F SmoM2 females. All procedures were performed in accordance with the IACUC-approved protocols.
2.2. Cell cultures, lentivirus preparation and infection
The medulloblastoma cell line Daoy was purchased from ATCC and was cultured as suggested by the supplier. For packaging the lentivirus, the shUTX (CTATGAATCTCTAATCTT) (68) cloned in the lentiviral vector pLKO. 1 was mixed with psPAX2 and pMD2.G (Addgene) and was transfected into HEK 293T cells using Polyjet (Signagen). Supernatant containing the viruses was collected 48-72 hours after transfection and used for subsequent infection. Daoy cells were infected with lentiviral shUTX supernatant containing 8 ug/ml polybrene for 24 hours. In order to establish stable cell knockdowns, puromycin (1 μg/ml) was added three days after initial infection for at least 2 weeks. The knockdown efficiency was measured by Western blotting and real-time PCR.
Normal or SmoM2 CGNPs were cultured from P4-P7 cerebellum. Briefly, cerebella from normal or SmoM2 pups were dissected, and dissociated to single cells. Cells were cultured in DMEM/F12 medium containing 25 mM KCl, N2, SHH conditioned medium, and 10% FBS. pLVX-NeuroD2 or control lentivirus were prepared as described above. Supernatant containing the viruses was centrifuged at 20,000 rpm for 2 hours at 4 °C. Concentrated virus was reconstituted in PBS and used for subsequent CGNP infection during initial seeding. Cells were fixed 48 and 72 hours after seeding and used for immunostaining. For BrdU tracing experiment in cell culture, BrdU (10 μM) was added 24 hours after seeding. For BrdU proliferation assay, BrdU was added 3 hours before harvest.
2.3. Tumor transplantation
Medulloblastoma from Utx+/+ Atoh1-Cre SmoM2 and UtxF/F Atoh1-Cre SmoM2 mice were dissected and dissociated. The tumor cells (5×106) were mixed with Matrigel (BD Biosciences) and injected subcutaneously in the flank region of NOD/ SCID mice. Tumor growth was monitored daily and was harvested for analysis one month after injection.
2.4. In vivo CD8+ T cell depletion and Flow cytometry
Anti-CD8 antibodies (10 mg/kg, clone 169.4, Bio X cell) or isotype control (10 mg/kg, clone LTF-2, Bio X cell) were administered intraperitoneally every 3 days starting at postnatal day 5. Mice were closely observed for tumor formation. CD8+ T cell depletion efficiency in the blood, the spleen, and the tumor was measured by flow cytometry. The primary antibodies used were against human CD3 (FITC, Biolegend, HIT3a), human CD8 (PE, 555367, BD Pharmingen), mouse CD3 (PE, eBioscience, 145-2C11), and mouse CD8 (APC, Biolegend, 53-6.7). Propidium iodide was used to stain dead cells. Stained cells were analyzed with a BD FACSCalibur (BD Bioscience). Flowjo software was used for data analyses.
2.5. Immunoblotting
Daoy cells were lysed in RIPA buffer (50 mM Tris, pH 8, 250 mM NaCl, 0.05% SDS, 0.5% DOC, 1% NP-40). The cell lysates were separated on 12% SDS-PAGE gels. The antibodies used were against GAPDH (Sigma-Aldrich, St Louis, MO, USA) and UTX (33510, Cell Signaling Technology).
2.6. Immunostaining, image processing and BrdU tracing
Paraffin sections of brain or tumor tissues were used for hematoxylin and eosin staining as well as immunostaining. BrdU (10 mg/kg; B5002; Sigma-Aldrich) was injected subcutaneously in Utx+/+ and UtxF/F Atoh1-Cre SmoM2 mice at P13. Mice were harvested 24 hours later, and their brains were fixed in 4% PFA at 4 °C overnight. The fixed brains were subject to subsequent double immunofluorescent staining of BrdU and HuC/D. For systematic CD3 T cell quantification, every 5th section throughout the tumor tissue was used to determine the total number of T cells in each tumor sample (n=5 for Utx+/+ tumors and n=3 for UtxF/F tumors). The antibodies used were against Ki67 (eBioscience), CD3 (ab16669, Abcam), UTX (33510, Cell Signaling Technology), BrdU (G3G4, DHSB), HuC/D (ab184267, Abcam), microtubule-associated protein 2 (MAP2, M9942, Sigma), NEUROD2 (ab104430, Abcam), NeuN (ABN78, Millipore) and beta III Tubulin (TUBB3, ab78078, Abcam). Bright-field images were acquired using a Hamamatsu Nanozoomer 2.0 HT whole slide scanner at the University of Texas Southwestern Medical Center Whole Brain Microscopy Facility. Fluorescent images were acquired by a confocal system (Zeiss LSM710), and three images were taken per tumor at 200x magnification and processed using ImageJ.
2.7. Chromatin immunoprecipitation (ChIP)
The ChIP experiments were performed as described previously (43). Dounced tissue or dissociated cells were crosslinked with PFA and sonicated to fragments. The antibodies used were against H3K27me3 (07-449, Millipore). The precipitated DNA was purified and subjected to real-time PCR. The graphics shown are representative of experiments performed in triplicate. The experiments were repeated three times.
2.8. RT-PCR and qPCR
The RNA from Daoy cells or tumor tissues was extracted with TRIZOL (Invitrogen). cDNAs were synthesized by reverse transcription using Iscript (Bio-Rad), followed by PCR or quantitative PCR analysis. A Bio-Rad real-time PCR system (C1000 Thermal Cycler) was used for quantitative PCR. GAPDH was used to normalize input RNA. The standard errors were calculated according to a previously described method (43). The PCR primer sequences were listed in Table S1.
2.9. RNA-seq analyses
CD15+ cells were MACS enriched from P10 and P28 Utx+/+ or UtxF/F Atoh1-Cre SmoM2 cerebella. Dissociated cerebellar cells were incubated with anti-mouse CD15 microbeads (Miltenyi), and CD15+ cells were isolated by the AutoMACS Pro Separation System (Miltenyi Biotech) according to the manufacturer’s protocol. The bulk RNA-seq used Utx+/+ and Utx+/Y tumor mice as wildtype and UtxF/F tumor mice as mutants. Total RNAs were extracted, and RNA-seq libraries were prepared using the Illumina RNA-Seq Preparation Kit and sequenced on a HiSeq 2500 sequencer at the University of Texas Southwestern Sequencing Core Facility. The expression levels of genes were quantified by a Kallisto program (72), and the differentially expressed genes were detected by an EdgeR program (73). Genes with a count-per-Million (CPM) less than 1 in more than 2 samples were excluded. The differentially expressed genes with a fold change larger than 2 and an FDR<0.05 were selected as UTX-regulated genes. Gene ontology analysis was performed using DAVID tools (http://david.abcc.ncifcrf.gov/).
2.10. T-cell migration assay
In vitro T-cell migration assays were conducted using 24-well Costar plates with 3.0 mm polycarbonate membrane transwells (Corning Inc., Corning, NY). Cultured Daoy cells with or without UTX knockdown were treated with IFN-γ (10 ng/mL; R&D Systems) for 24 hours, and supernatants (600 μl) were removed and placed at the bottom chambers of the transwells. Human CD8+ T cells (70027, StemCell Technologies) were activated by anti-CD3/CD28-coated beads (11161D, Thermo Fisher) supplemented with 50 U/ml rhIL-2 for 5-7 days. Figure 4D shows that more than 90% of T cells used were CD3+/CD8+ T cells (human CD3: FITC, Biolegend, HIT3a and human CD8: PE, 555367, BD Pharmingen), Activated CD8+ T cells (2x105 cells) treated with human CXCR3 antibodies (MAB160, R&D Systems) or isotype control were added to the insert of the transwells. After 10 hours of incubation at 37 °C, T cells that migrated to the bottom chamber were collected and counted by flow cytometry.
2.11. Cell viability assay
Normal or SmoM2 CGNPs infected with NEUROD2 over-expressing lentivirus (48 or 72 hours after infection) were used to measure cell proliferation. Relative cell titer was determined using CellTiter-Glo® Luminescent Cell Viability Assay (Promega, G7572) according to the manufacture’s instruction.
2.12. Statistical analysis
The data are expressed as means ± s.d. Statistical analysis was performed by either an ANOVA post hoc t-test for multiple comparisons or a two-tailed unpaired Student’s t-test. A p-value < 0.05 was considered significant.
3. Results
3.1. UTX functions as a tumor suppressor in SHH medulloblastoma
SHH medulloblastoma is caused by abnormally active SHH signaling in CGNP cells. In order to determine the function of UTX in SHH medulloblastoma, we first deleted Utx in developing cerebella using a CGNP-specific Atoh1-Cre (47). Both male and female Utx-deleted mice were grossly normal and fertile. Gross anatomy of Utx-deleted cerebellum at postnatal day 28 (P28) did not show any observable defects as evidenced by the H&E staining (Figure 1A). Deletion of UTX did not affect the global H3K27me3 level (Figure 1A), likely due to the presence of another H3K27me3 demethylase JMJD3. Thus, UTX is not required for normal CGNP and cerebellum development. However, deleting Utx using Atoh1-Cre in a well-characterized SHH medulloblastoma mouse model, which is created through a Cre-induced constitutively active mutant SmoM2 (46), significantly reduced the survival of tumor-bearing mice from a median of 78 days to a median of 57 days for males and 46 days for females (Figure 1B, 1C). This result indicates that UTX acts as a tumor suppressor in SHH medulloblastoma if deleted in tumor progenitors. The differences between male and female mutant mice suggest that the enzymatically inactive UTY protein in male mice partially compensates for the loss of the Utx gene. Thus, in addition to the H3K27me3 demethylase activity of UTX, its enzymatic activity-independent function may also contribute to its tumor suppressor function. In the subsequent experiment, we only used female mice to perform the medulloblastoma studies to exclude the possible interference from UTY.
Figure 1. UTX plays a tumor suppressor role in SHH medulloblastoma.

A. H&E, UTX and H3K27me3 staining of sagittal sections of cerebella from P28 Atoh1-Cre Utx+/+ and Atoh1-Cre UtxF/F mice. UTX was deleted in the differentiated granule neurons.
B. H&E and UTX staining of sagittal sections of cerebella from P28 Utx+/+ and UtxF/F Atoh1-Cre SmoM2 mice. UTX was deleted in the tumor cells.
C. Survival curves of mice harboring SmoM2 medulloblastoma with indicated Utx genotypes.
D. UTX deletion increased tumor cell proliferation as indicated by increased BrdU staining at both early tumor development (P28) and later tumor stage. Quantification is shown on the right (n=5).
E. Survival curves of mice harboring SmoM2 medulloblastoma with indicated Utx and Jmjd3 genotypes.
Student’s t-test, **, p<0.01.
UTX deletion resulted in an earlier formation of medulloblastoma and faster proliferation in both early and late stage tumors compared with those observed in controls. In Atoh1-Cre SmoM2 mice, the increased SHH activity in early postnatal cerebella causes over-proliferation of precancerous CGNPs, most of which differentiate into granule neurons (48). At P28, some CGNPs transform to cancer cells. These mice showed limited proliferative foci mostly in the posterior cerebellum. On the contrary, the cerebellum of UtxF/F Atoh1-Cre SmoM2 mice showed extensive proliferation as shown by the BrdU staining (Figure 1D). The quantification of BrdU+ tumor cells in both P28 and later stage SmoM2 cerebella showed that UTX deletion significantly increased tumor cell proliferation (Figure 1D). These data indicate that UTX suppresses SHH medulloblastoma initiation and proliferation.
Previously, we have shown that another H3K27me3 demethylase JMJD3 plays a central role in coordinating an epigenetic co-activator complex to activate SHH target gene expression. JMJD3 knockdown in medulloblastoma cells inhibited tumor cell growth (44). Consistently, deleting JMJD3 in SmoM2 medulloblastoma model significantly delayed tumor growth and extended survival (Figure 1E). Interestingly, deleting UTX and JMJD3 simultaneously led to an unchanged survival curve compared to wildtype SmoM2 mice (Figure 1E). This result showed that JMJD3 and UTX regulate SHH medulloblastoma in opposite directions, likely through controlling different pathways.
3.2. UTX deletion in medulloblastoma led to decreased T cell numbers in tumor microenvironment
To understand the function of UTX in medulloblastoma, we performed RNA-seq comparing the gene expression profiles in the control and in the Utx-deleted SmoM2 medulloblastoma tumors. Our RNA-seq data showed that the majority of differentially expressed genes were downregulated in Utx-deleted tumors (Figure 2A), which confirms the function of UTX in activating gene expression. Surprisingly, UTX did not regulate the gene sets that specifically control SHH medulloblastoma growth such as the SHH pathway genes and the tumor identity genes (Figure 2B, 2C). Strikingly, the gene ontology analyses indicated a major defect in the tumor-induced host immune response (Figure 2B, Table S2). The most significantly reduced genes in Utx-deleted tumors were those involved in T-cell development and function (Figure 2C). These included most CD8+ T cell markers such as Cd3, Cd8, Cd28, and many other T cell genes, but not Cd4 (Figure 2C). Markers for other types of immune cells such as NK cells (Ncr1) macrophages (Mac-1), and dendritic cells (Cd11c, Cd123) were not significantly different. We confirmed these changes by RT-qPCR (Figure 2D). Since UTX was only deleted in tumor cells but not in immune cells, the decrease of genes related to T cells suggests a change of T cell numbers in Utx-deleted tumors. Differences in T cell numbers were quantified by the systemic immunohistochemistry quantification of CD3+ cells in P28 and late stage SmoM2 tumors (Figure 2E). We also performed flow cytometry analyses of SmoM2 tumors for CD8+ T cells (Figure 2F). Our results indicate that UTX deletion in the medulloblastoma cells significantly reduced CD8+ T cell numbers in the tumor microenvironment, possibly by affecting T cell migration and recruitment.
Figure 2. UTX deletion in medulloblastoma led to decreased T cell numbers in the tumor environment.

A. Heatmap of RNA-seq results shows that the majority of differentially expressed genes were downregulated in UTX-deleted SmoM2 tumors compared to wildtype tumors (n=2 per group).
B. Gene ontology analysis shows genes involved in immune response was overrepresented in UTX regulated genes.
C. Heatmap of examples of SHH medulloblastoma specific genes, immune genes, chemokine, and chemokine receptor genes in the RNA-seq tumor samples.
D. RT-qPCR confirmation of T cell markers in RNA-seq data using tumor samples (n=18 for Utx+/+ tumors and n=14 for UtxF/F tumors).
E. (Left) Representative IHC images of CD3 staining of medulloblastoma sections at P28. (Right) Quantification of CD3 cells by IHC staining (P28, n=5 for Utx+/+ tumor and n=3 for UtxF/F tumor) and later tumor stage (n=4 for Utx+/+ tumor and n=3 for UtxF/F tumor).
F. (Left) Representative flow cytometry images of CD3 and CD8 staining at late tumor stage. (Right) Flow cytometric quantification of CD8+ T cells (n=5).
Student’s t-test, *, p<0.05; **, p<0.01.
3.3. UTX activates chemokine gene expression through H3K27me3 demethylation
From the RNA-seq data, we observed that the expression of various chemokine genes was significantly downregulated in Utx-deleted tumors (Figure 2C). Using RT-qPCR, we confirmed the decreased levels of the T helper 1 (Th1) type chemokine gene Cxcl9 and other chemokine genes such as Cxcl13 and Ccl21a (Figure 3A). However, another Th1-type chemokine gene Cxcl10 as well as the SHH target gene Gli1 were not significantly changed when UTX was deleted in SmoM2 tumors (Figure 3A). Thus, UTX activates the expression of a selective set of chemokine genes in SmoM2 medulloblastoma. UTX could activate genes by removing repressive H3K27me3 marks locally. We performed H3K27me3 ChIP-qPCR at the promoter regions of specific chemokine genes and observed increased levels of H3K27me3 marks at the promoters of Cxcl9, Cxcl13, and Ccl21a in Utx-deleted tumors (Figure 3B). GAPDH and UTX target gene p16INK4a (49, 50) were used as negative and positive controls, respectively (Figure 3B). These results indicate that UTX directly activates chemokine gene expression by removing H3K27me3 marks from the regulatory regions.
Figure 3. UTX activates chemokine gene expression through H3K27me3 demethylation.
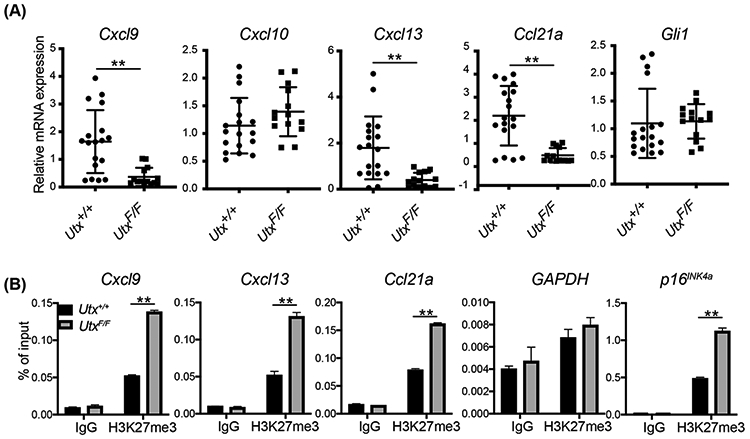
A. RT-qPCR analyses of chemokine gene expression in Utx+/+ and UtxF/F Atoh1-Cre SmoM2 medulloblastoma tumors (n=18 for Utx+/+ tumors and n=14 for UtxF/F tumors).
B. UTX deletion in SmoM2 tumors led to increased local H3K27me3 at the promoter regions of the chemokine genes as analyzed by ChIP-qPCR. GAPDH and p16INK4A were used as negative and positive control, respectively (n=3).
Student’s t-test, **, p<0.01.
3.4. UTX activates Th1-type chemokine gene expression in human medulloblastoma cell lines and enhances T cell migration
It has been shown that the human PRC2 complex inhibits CD8+ cytotoxic T cell recruitment to colon and ovarian cancers by repressing the expression of Th1-type chemokine genes CXCL9/CXCL10 in cancer cells (10, 11). We then determined whether UTX activates Th1-type chemokine genes in human medulloblastoma cells and facilitates T cell migration. In the human SHH medulloblastoma cell line Daoy (51), the shRNA-mediated UTX knockdown significantly reduced the expression of both the CXCL9 and CXCL10 genes as measured by RT-qPCR (Figure 4A). Furthermore, H3K27me3 ChIP-qPCR experiments showed that H3K27me3 was present at the promoter regions of the CXCL9 and CXCL10 genes and was increased upon shRNA-mediated UTX knockdown (Figure 4B). Thus, UTX is required for the basal expression levels of the CXCL9 and CXCL10 genes in Daoy cells, likely by antagonizing PRC2 activities and removing H3K27me3. In cancer cells, Th1-type chemokine genes are stimulated by IFN-γ (11). In the presence of IFN-γ, both CXCL9 and CXCL10 were highly induced in Daoy cells after 24 hours of treatment, whereas the induction levels were significantly reduced when UTX levels were reduced by shRNA mediated knockdown (Figure 4C). Thus, UTX is also required for IFN-γ induced CXCL9/CXCL10 activation.
Figure 4. UTX in human medulloblastoma cells enhance T cell migration through Th1-type chemokine activation.

A. UTX knockdown by RNAi inhibition decreased the expression of CXCL9 and CXCL10 in Daoy cells as shown by RT-qPCR (n=4). Western blots (right panel) show UTX protein levels.
B. UTX knockdown increased local H3K27me3 levels at the promoter regions of CXCL9 and CXCL10 as shown by ChIP-qPCR (n=3).
C. UTX knockdown reduced the IFN-γ-induced expression of CXCL9 and CXCL10 in Daoy cells as shown by RT-qPCR (n=3).
D. Human CD8+ T cell migration assay. (Left top) schematic drawing of the migration assay. Activated human CD8+ T cells were placed in the insert in the transwell and the T cells migrated to the conditioned media were counted. (Left bottom) The majority of the activated T cells were human CD8 T cells. (Right) Number of CD8+ T cells treated with or without anti-CXCR3 antibodies migrated to the conditioned media from UTX knockdown or control Daoy cells (n=4). IFN-γ (10 ng/mL) was present in all cultures. Unused culture media were used as negative controls.
E. Average expression levels of CD28 genes in human SHH medulloblastoma RNA microarray samples separated by different levels of UTX and EZH2 (n=76). UHEH, UTXhigh EZH2high, UHEL, UTXhigh EZH2low, ULEH, UTXlow EZH2high ULEL UTXlow EZH2low.
Student’s t-test, *, p<0.05; **, p<0.01.
In order to further investigate the function of UTX in cancer cell induced T cell recruitment, we performed an in vitro T cell migration assay using a transwell system. Human CD8+ T cells activated by anti-CD3/CD28 antibodies were plated in the insert, which was placed in conditioned media collected from Daoy cells transfected with UTX shRNA or control lentiviral vectors and cultured in the presence of IFN-γ (10 ng/ml). The T cell count in the conditioned media from different sources showed that control Daoy cells could secrete factors to attract T cell migration, whereas UTX knockdown in Daoy cells led to significantly reduced T cell migration from inserts to conditioned media (Figure 4D). To determine whether the UTX activated CXCL9/CXCL10 Th1-type chemokines in Daoy cells are responsible for attracting T cell migration, we added antibodies blocking CXCR3, the specific CXCL9/10 receptor expressed in T cell. The CXCR3 blockade exerted a similar reduction of T cell migration as UTX knockdown did. Importantly, treating T cells with anti-CXCR3 antibodies did not further decrease T cell migration to conditioned media from Daoy cells with UTX knockdown (Figure 4D). Thus, UTX in medulloblastoma cells activates the expression of Th1-type chemokines to recruit CD8+ T cells. Consistently, an examination of the active T cell marker CD28 in 76 human SHH medulloblastoma RNA microarray samples (52) showed that tumor groups containing high levels of UTX and low levels of H3K27me3 methyltransferase EZH2 have a significantly higher expression level of CD28 (Figure 4E). The results suggest that high UTX/low EZH2 activities in SHH medulloblastoma may facilitate the recruitment of CD28+ T cell to the tumor environment.
3.5. Depletion of CD8+ T cells did not accelerate tumor development in mouse SHH medulloblastoma
We demonstrated a non-cell autonomous function of UTX in medulloblastoma in recruiting cytotoxic T cells to the tumor environment. However, it is not clear whether these CD8+ T cells play any roles in restricting medulloblastoma development. We therefore performed a CD8+ T cell depletion experiment. In Atho1-Cre SmoM2 mice, we injected anti-CD8 antibodies every three days starting from postnatal day 5 until tumor formation, which was indicated by the presence of hydrocephalus (Figure 5A). It has been reported that this approach could effectively deplete CD8+ T cells from various tissues of pups including their brain (53). Indeed, CD8+ T cells were largely depleted from the spleen, peripheral blood, and the tumor site when examined at the end of the depletion experiment as shown by FACS analyses (Figure 5B). To our surprise, depletion of CD8+ T cells did not accelerate tumor development, and no significant effect was observed on the survival of tumor harboring mice (Figure 5C). Therefore, although SHH medulloblastoma in mice recruits cytotoxic T cells to the tumor environment, they have little effects on tumor development. To further determine whether a UTX-promoted host immune response plays inhibiting roles in medulloblastoma tumorigenesis, we transplanted same amounts of Utx+/+ or UtxF/F SmoM2 tumor cells into immunodeficient NOD/SCID mice and still observed significantly faster growth of the UtxF/F tumor allografts (Figure 5D). These results suggest that UTX could suppress tumor growth independent of the host immune system, likely through cell-autonomous mechanisms.
Figure 5. Depletion of CD8+ T cells did not accelerate medulloblastoma growth in mice.

A. CD8+ T cell depletion paradigm by injecting anti-mouse CD8 antibodies or control IgG (10 mg/kg). Antibodies were injected every three days starting from postnatal day 5 until the mice showed symptoms of hydrocephalus.
B. Flow cytometric analyses of the efficiency of CD8+ T cell depletion in the spleen, peripheral blood, and the tumor after antibody injection.
C. Survival curves of Atoh1-Cre SmoM2 mice after injection of anti-CD8 antibodies or control IgG.
D. Comparison of Utx+/+ and UtxF/F Atoh1-Cre SmoM2 tumors transplanted subcutaneously to immunodeficient NOD/SCID mice (n=7). Representative images of transplanted tumors are shown on the top.
Student’s t-test, **, p<0.01.
3.6. The proneural gene NeuroD2 is a direct target of UTX in medulloblastoma
To understand the cell intrinsic tumor suppressor function of UTX, we turned to early medulloblastoma development stages. In Atoh1-Cre SmoM2 cerebella, at early postnatal stage (P0-P14), SmoM2 CGNPs had over-proliferated but were precancerous. Most had differentiated into granule neurons by P28, and some became cancer cells (48, 54). The frequency of cancer cells was significantly higher in the Utx mutant than in wildtype Atoh1-Cre SmoM2 cerebella at P28 (Fig. 1D). To identify target genes of UTX that mediate its function in suppressing cancer initiation, we performed another RNA-seq to compare transcriptomes of early tumors from Utx+/+ and UtxF/F SmoM2 mice (n=2 per group at P10; n=3 per group at P28). To ensure sample comparability and to exclude differentiated cells, we sorted tumor-propagating cells with the progenitor cell-surface marker CD15 using magnetic-activated cell sorting (MACS) (55, 56). We compared transcriptomes of CD15+ SmoM2 cells at P28 and at P10; the differentially expressed genes (DEGs) regulated by age are potentially involved in the transition from precancerous CGNPs to cancer cells (Figure 6A, green and blue bars, 761 down, 223 up, in P28, FDR<0.05, fold change >2). The DEGs between Utx+/+ and UtxF/F SmoM2 samples at P28 revealed UTX-regulated genes that potentially encode factors that mediate UTX tumor-suppressing activities (Figure 6A, red and blue bars, 634 down, 125 up, in mutant). An intersection of these two sets of genes revealed 176 genes that were regulated by both cancer developmental stages and UTX levels in cancer propagating cells (Figure 6A, blue bar). Since UTX is a transcription activator, we further analyzed these common DEGs and focused on the genes that were decreased from P10 to P28 in Utx+/+ CD15+ cells, whose expression was further reduced in P28 Utx mutants (Figure 6B, Quadrant III, 78 genes). These genes may include key UTX regulated genes that are also important in the transformation from pre-cancerous CGNPs to medulloblastoma cancer cells. Interestingly, we found that neural differentiation genes are enriched, including NeuroD2, which was among the most differentially expressed genes (Figure 6B, C). NeuroD2 encodes a key proneural transcription factor (57) that functions to induce cell cycle exit and differentiation in neural progenitors and to promote the expression of genes important for neuronal maturation (58, 59). We missed NeuroD2 from our previous bulk RNA-seq (Figure 1A) likely due to the late tumor stages used and its high expression level in neurons, which masked the differences in progenitors. RT-qPCR confirmed that NeuroD2 RNA was reduced during the transition from precancerous CGNPs in P10 mice to early cancer cells in P28 mice. UTX deletion led to the reduction of NeuroD2 at both stages (Figure 6D). Immunohistochemistry staining demonstrated the decrease of NEUROD2 protein levels from P14 CGNPs to P28 cancer cells, which were further decreased in Utx-deleted cancer cells (regions shown contain mostly proliferative precancerous CGNPs and cancer cells as indicated by Ki67 staining, Figure 6E). H3K27me3 ChIP-qPCR analyses using CD15+ cells from P28 Utx+/+ and UtxF/F SmoM2 tumors showed the presence of H3K27me3 in the NeuroD2 promoter region that was significantly increased in Utx-deleted cancer cells (Figure 6F). Therefore, NeuroD2 and the downstream neural differentiation pathway are likely directly regulated by UTX during medulloblastoma development.
Figure 6. Proneural gene NeuroD2 is a direct target of UTX in medulloblastoma.

A. Heatmap of all DEGs identified from RNA-seq using CD15+ cells sorted from Utx+/+ and UtxF/F Atoh1-Cre SmoM2 cerebella at P10 (n=2) and P28 (n=3). The colored bars on the left represent DEG groups categorized by comparisons between P10 and P28 Utx+/+ cells and between P28 Utx+/+ and UtxF/F samples.
B. Dot plot of differentially expressed genes comparing P10 and P28 Utx+/+ samples (x axis); and comparing Utx+/+ and UtxF/F samples at P28 (y axis). Colored dots are common DEGs in both comparisons.
C. Gene ontology analysis of enriched pathways in UTX activated genes at P28.
D. RT-qPCR of NeuroD2 expression in Utx+/+ and UtxF/F Atoh1-Cre SmoM2 CD15+ cells at P10 and P28 (n=6).
E. Immunostaining of NEUROD2, Ki67, and UTX on the consecutive sections of Utx+/+ and UtxF/F Atoh1-Cre SmoM2 tumors at P14 and P28.
F. ChIP-qPCR analysis revealed Ut deletion in SmoM2 tumors led to increased local H3K27me3 at the promoter region of NeuroD2 gene (n=3).
Student’s t-test, **, p<0.01.
3.7. NEUROD2 over-expression decreased cell proliferation and increased neuron differentiation in CGNPs
In order to examine the potential tumor suppressor function of NEUROD2, we over-expressed NEUROD2 in mouse CGNPs to investigate its role in cell proliferation and neuron differentiation. Lentivirus mediated expression of NEUROD2 in cultured CGNPs can be observed 48 hours after viral infection (Figure 7A). NEUROD2 over-expression led to decreased CGNP proliferation as indicated by decreased BrdU incorporation (Figure 7B). We quantified total cell growth in culture using Cell Viability Assay, which showed decreased cell numbers at both 48 and 72 hours after NEUROD2 over-expression (Figure 7C). Using a similar system, we confirmed NEUROD2 proliferation inhibitory effects in SmoM2 CGNP cultures (Figure 7C). In BrdU tracing experiments where BrdU was added to the cultures 48 hours before analyses, a double staining of the neuronal markers NeuN and BrdU indicates newly differentiated neurons. We observed that more BrdU-incorporated cells were NeuN positive in NEUROD2 over-expressed CGNPs compared with control CGNPs (92% versus 76%) (Figure 7D). Thus, NEUROD2 inhibits cell proliferation and increase neuronal differentiation in CGNPs and may mediate the tumor suppressor function of UTX in medulloblastoma.
Figure 7. NEUROD2 over-expression decreased cell proliferation and increased neuron differentiation in CGNPs.

A. NEUROD2 staining showed that NEUROD2 was over-expressed in CGNPs 48 hours after lentiviral infection.
B. NEUROD2 over-expression decreased cell proliferation as indicated by decreased BrdU staining in CGNPs. BrdU quantification is shown on the right (n=4).
C. Cell viability assay showed that NEUROD2 over-expression decreased cell proliferation in both normal and SmoM2 CGNPs 48 and 72 hours after virus infection (n=3 per group). Representative bright-field images on the right were taken 72 hours after virus infection.
D. Co-immunostaining of BrdU and NeuN. BrdU tracing (48 hours) experiment revealed that NEUROD2 over-expression increased neuron differentiation in CGNPs. The neuron differentiation rate was quantified as the percentage of NeuN+ BrdU+ cells in total BrdU+ cells.
Student’s t-test, **, p<0.01.
3.8. UTX deletion led to decreased neuron differentiation during medulloblastoma development
We identified NeuroD2 as a UTX target gene in medulloblastoma. To determine whether decreased neural differentiation process contributes to UTX tumor suppressor function, we examined precancerous stages of SmoM2 cerebella at P7 and P14, when the neural differentiation process is most active. Indeed, we observed significantly reduced staining of neuronal markers NeuN, HuC/D, MAP2, and TUBB3 in UtxF/F cerebella than Utx+/+ ones (Figure 8A). To examine neuron differentiation directly, we performed BrdU tracing experiment. At P13, Utx+/+ and UtxF/F Atoh1-Cre SmoM2 pups were injected with BrdU. Cerebellum samples were collected 24 hours after BrdU injection. A double staining of the neuronal markers HuC/D and BrdU indicates newly differentiated neurons. The results showed significantly more neuron differentiation in Utx+/+ Atoh1-Cre SmoM2 cerebella than UtxF/F ones as shown by the ratio of HuC/D/BrdU double positive cells to all BrdU+ cells (Figure 8B). Therefore, reduced NEUROD2 and weakened differentiation caused by UTX deletion may coordinate with active SHH signaling to accelerate medulloblastoma formation from precancerous CGNPs.
Figure 8. UTX deletion in medulloblastoma led to impaired neural differentiation.

A. NeuN, HuC/D, MAP2, and TUBB3 staining of P7 and P14 Utx+/+ and UtxF/F Atoh1-Cre SmoM2 cerebella showed significantly reduced neuron numbers in UTX deleted samples.
B. BrdU tracing (24 hours) experiments revealed decreased differentiation in UtxF/F Atoh1-Cre SmoM2 precancerous CGNPs at P14 than the wildtype CGNPs. The neuron differentiation rate was quantified as the percentage of HuCD+ BrdU+ cells in total BrdU+ cells.
Student’s t-test, **, p<0.01.
4. Discussion
In this study, we have revealed a novel tumor suppressor function of UTX in SHH medulloblastoma and confirmed an oncogenic function of the homologous JMJD3. UTX regulates both intrinsic tumor development and the tumor microenvironment. We showed that UTX is required for the tumor to produce chemokines that potentiate T cell recruitment to the tumor microenvironment. Although the host immune response may not play a major role in this SHH medulloblastoma mouse model, we have discovered an epigenetic pathway in tumors that induces T cell recruitment. In addition, we identified NeuroD2 and its mediated neuronal differentiation as a direct UTX target that possibly mediates the tumor suppressor function of UTX. This study will provide molecular insights into the complex roles of UTX in cancer development.
For effective treatment with immunotherapy, a sufficient number of T cells present in tumor microenvironment is a prerequisite. Thus, our model provides a novel mechanism of how epigenetic regulators in tumors play a role in inducing T cell migration. It has been reported that the H3K27me3 methyltransferase PRC2 complex suppresses the expression of the Th1-type chemokines CXCL9 and CXCL10 and subsequently regulates T cell migration in human ovarian (10) and colon cancer (11) models. We showed that UTX activates the expression of Th1-type chemokines in both mouse and human medulloblastoma cells. Our RNA-seq showed that this effect is specific to CD8+ T cells since markers for other immune cells such as CD4+ T cells, NK cells, macrophages, or dendritic cells were not significantly changed. The CXCR3 antibody blocking experiments (Figure 4D) showed that the CXCL9/CXCL10 chemokines specifically mediate the UTX function in tumor cells to recruit CD8+ T cells. PRC2 and UTX are a candidate oncogene and tumor suppressor, respectively, in many cancers. It is possible that this chemokine-induced T cell recruitment pathway could be regulated bidirectionally by H3K27me3 methyltransferase and demethylase epigenetically in these cancers. Cancer immunotherapy requires sufficient T cell infiltration as a starting material (60). Our data and others have shown that epigenetic regulation of chemokine expression can promote T cell trafficking, which potentially can be used as a treatment target to improve the efficacy of cancer immunotherapy.
Besides CD8+ T cell recruitment, an important question is whether the infiltrated T cells in tumor microenvironments restrict tumor growth. Cytotoxic CD8+ T cells are one of the major anti-tumor immune cells. Surprisingly, CD8+ T cell depletion in our tumor mouse model did not lead to changes in the overall survival of the tumor harboring mice. Several factors may have contributed to the lack of effect of T cell depletion on tumor growth in the mice. First, the medulloblastoma model is known to be an aggressive tumor, which happens in 100% of mice and causes lethality within 3 months. The tumor growth may overwhelm the immune response. Second, the brain is protected by blood brain barrier, which prevents the massive infiltration of immune cells. Even though we observed T cells in the tumor microenvironment, the number of infiltrated CD8+ T cells (less than 0.03%) may be far from enough to inhibit tumor growth given the massive number of tumor cells. In medulloblastoma, it was recently reported that additional immune cells such as bone marrow derived macrophages and microglia could be recruited to medulloblastoma through the CCL2/CCR2 chemokine pathway and exhibit anti-tumor properties (61). Third, these infiltrated T cells may be inactive due to the lack of neo-antigen presentation and hence cannot recognize tumor cells, or their functions may be suppressed (62). It has been reported that medulloblastoma-produced TGF-β could reduce T cell expansion and activation, limiting T cell anti-tumor cytotoxic activities (63). Nevertheless, we could not exclude the possibility of cytotoxic CD8+ T cells having a tumor killing function in human medulloblastoma, as the level of CD28 was higher in UTXhighEZH2low SHH medulloblastoma samples than in all other variants (Fig. 4E). An early study also showed that IFN-γ levels produced in T cells correlate well with medulloblastoma patient survival (64). Future studies are needed to uncover the interactions between medulloblastoma and infiltrated immune cells.
UTX plays a tumor suppressor role in many cancers with multiple mechanisms, including activating different tumor suppressor genes (30, 65, 66). We showed that UTX regulates neural differentiation through the activation of the proneural gene NeuroD2, which could mediate the tumor suppressor function of UTX in medulloblastoma. During SHH medulloblastoma development, the expression of SmoM2 activates the SHH signaling pathway and causes initial over-proliferation of CGNPs. Precancerous CGNPs differentiate, while a small percentage of cells transform to cancer cells (48). However, the key process that coordinates with the SHH pathway to enable the transformation is not clear. Our approach, which focuses on the common pathways that are changed during transformation and are activated by UTX, identified NEUROD2-mediated neural differentiation as a key pathway that normally inhibits SHH medulloblastoma and is activated by UTX. In addition, our analyses using the cancer propagating CD15+ cells for RNA-seq analyses removed the noise brought in by neuronal cells, which made it possible to uncover neural progenitor-expressed NeuroD2 as a UTX target gene despite its high level of expression in neurons. Mutations of NeuroD2 have been identified in glioma (COSMIC), and it has been proposed to have a tumor-suppressor function (67). We further demonstrated the function of NEUROD2 in inhibiting cell proliferation and promoting neuron differentiation in CGNPs (Figure 7), which supports the hypothesis that NEUROD2 functions as a tumor suppressor in brain tumors. Although the defect in the UTX/NEUROD2/neural differentiation pathway is not sufficient to cause abnormal development and cancer on its own, it cooperates with activated SHH signaling to accelerate medulloblastoma development. Loss-of-function mutations in UTX may shift the balance between undifferentiated stem cell-like precursor cells and differentiated neurons leaning towards the former population, which is likely to increase the transformation incidence to promote cancer development.
Our study demonstrated a tumor suppressor function of UTX in SHH medulloblastoma when UTX is deleted in cancer progenitors. In human SHH medulloblastoma patients, UTX mutations were identified, albeit with low frequencies (52). The lack of frequent mutation suggests that UTX, like many other epigenetic regulators, may play additional oncogenic or tumor maintenance functions during different tumor development stages. Indeed, using a previously developed genetic system (45), we deleted UTX after tumor formation, but observed reduced tumor growth instead (Yi et al., unpublished data). Therefore, UTX may have stage-specific functions in SHH medulloblastoma development, likely explaining the low UTX mutation rate in SHH medulloblastoma.
UTX and JMJD3 are the only two known H3K27me3 demethylases. It has been shown that they play overlapping but specific roles during development and in cancer (68-70). In this study, we showed that UTX and JMJD3 function in opposite directions in medulloblastoma development. Previously, we have shown that JMJD3 is required for SHH induced gene activation by facilitating an epigenetic switch at target gene promoters (44). Here, we showed that JMJD3 deletion led to delayed tumor growth and extended survival (Figure 1E), which is consistent with its role in SHH pathway activation. On the contrary, UTX is not involved in SHH signaling, but regulates the neural differentiation pathway and the tumor microenvironment, which suppress SHH medulloblastoma. Therefore, the apparent opposing functions of UTX and JMJD3 in medulloblastoma development are likey through regulating different signaling pathways. Deleting both enzymes in medulloblastoma phenotypically reversed the effects of an individual deletion and resulted a similar survival curve as the mice harboring wildtype tumors (Figure 1E). Thus, these H3K27me3 demethylases regulate specific target gene sets and have distinct functions in medulloblastoma.
In summary, we used a SHH medulloblastoma model to study the tumor suppressor function of the H3K27me3 demethylase UTX. We elucidated the function of UTX in activating chemokine expression and T cell recruitment in mouse and human medulloblastoma cells. We further identified the neuronal differentiation factor NEUROD2 as a novel target of UTX that could mediate UTX’s tumor suppressor function. Our study demonstrates context-dependent functions of a histone demethylase in tumor development, which will provide insights into medulloblastoma development and shed light on UTX functions in many other cancers.
Supplementary Material
UTX functions as a tumor suppressor in SHH-subgroup medulloblastoma
UTX and JMJD3 play opposite roles in medulloblastoma tumorigenesis
UTX activates Th-1 type chemokine expression, and enhances T cell recruitment
UTX activates NeuroD2 expression and neuronal differentiation
5. Acknowledgements
We thank Drs. Gan Lin and Jane Johnson for providing the Atoh1-Cre mice, Dr. Kai Ge for providing the Utx conditional knockout mice and Dr. Rongfu Wang for providing the Jmjd3 conditional knockout mice. We thank Drs. Heyu Chen and Xiaoye Liu for their help in FACS analyses. We thank the UTSW Sequencing Facility for performing the next-generation sequencing. We thank Mr. Huaxia Dong for technical help and mouse breeding. This work was supported by grants from the Welch Foundation (I-1940-20170325 to J.W.) and NIH (R01NS09606 and R21NS104596 to J.W.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare that there is no conflict of interest.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
7. References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 2.Maman S, Witz IP. A history of exploring cancer in context. Nat Rev Cancer. 2018;18:359–76. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–22. [DOI] [PubMed] [Google Scholar]
- 4.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oelkrug C, Ramage JM. Enhancement of T cell recruitment and infiltration into tumours. Clin Exp Immunol. 2014;178:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Franciszkiewicz K, Boissonnas A, Boutet M, Combadiere C, Mami-Chouaib F. Role of chemokines and chemokine receptors in shaping the effector phase of the antitumor immune response. Cancer Res. 2012;72:6325–6332. [DOI] [PubMed] [Google Scholar]
- 9.Salsman VS, Chow KK, Shaffer DR, Kadikoy H, Li X-N, Gerken C, et al. Crosstalk between medulloblastoma cells and endothelium triggers a strong chemotactic signal recruiting T lymphocytes to the tumor microenvironment. PloS one. 2011. ;6:e20267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. 2015;527:249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nagarsheth N, Peng D, Kryczek I, Wu K, Li W, Zhao E, et al. PRC2 epigenetically silences Th1-type chemokines to suppress effector T-cell trafficking in colon cancer. Cancer research. 2016;76:275–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones DT, Jager N, Kool M, Zichner T, Hutter B, Sultan M, et al. Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012;488:100–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Northcott PA, Shih DJ, Peacock J, Garzia L, Morrissy AS, Zichner T, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. 2012;488:49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC, et al. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011;331:435–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J, et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2012;488:106–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, Ding L, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012;488:43–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kool M, Jones DT, Jager N, Northcott PA, Pugh TJ, Hovestadt V, et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell. 2014;25:393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Northcott PA, Hielscher T, Dubuc A, Mack S, Shih D, Remke M, et al. Pediatric and adult sonic hedgehog medulloblastomas are clinically and molecularly distinct. Acta neuropathologica. 2011;122:231–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morrissy AS, Cavalli FM, Remke M, Ramaswamy V, Shih DJ, Holgado BL, et al. Spatial heterogeneity in medulloblastoma. Nature genetics. 2017;49:780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang L, He X, Liu X, Zhang F, Huang LF, Potter AS, et al. Single-Cell Transcriptomics in Medulloblastoma Reveals Tumor-Initiating Progenitors and Oncogenic Cascades during Tumorigenesis and Relapse. Cancer Cell. 2019;36:302–18 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hovestadt V, Smith KS, Bihannic L, Filbin MG, Shaw ML, Baumgartner A, et al. Resolving medulloblastoma cellular architecture by single-cell genomics. Nature. 2019;572:74–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vladoiu MC, El-Hamamy I, Donovan LK, Farooq H, Holgado BL, Sundaravadanam Y, et al. Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature. 2019;572:67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yao M, Ventura PB, Jiang Y, Rodriguez FJ, Wang L, Perry JS, et al. Astrocytic trans-Differentiation Completes a Multicellular Paracrine Feedback Loop Required for Medulloblastoma Tumor Growth. Cell. 2020;180:502–20. e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bockmayr M, Mohme M, Klauschen F, Winkler B, Budczies J, Rutkowski S, et al. Subgroup-specific immune and stromal microenvironment in medulloblastoma. Oncoimmunology. 2018;7:e1462430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van der Meulen J, Speleman F, Van Vlierberghe P. The H3K27me3 demethylase UTX in normal development and disease. Epigenetics. 2014;9:658–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang L, Shilatifard A. UTX Mutations in Human Cancer. Cancer Cell. 2019;35:168–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463:360–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nickerson ML, Dancik GM, Im KM, Edwards MG, Turan S, Brown J, et al. Concurrent alterations in TERT, KDM6A, and the BRCA pathway in bladder cancer. Clinical Cancer Research. 2014;20:4935–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, et al. Comprehensive characterization of cancer driver genes and mutations. Cell. 2018;173:371–85. e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ntziachristos P, Tsirigos A, Welstead GG, Trimarchi T, Bakogianni S, Xu L, et al. Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia. Nature. 2014;514:513–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi HJ, Park JH, Park M, Won HY, Joo Hs, Lee CH, et al. UTX inhibits EMT-induced breast CSC properties by epigenetic repression of EMT genes in cooperation with LSD1 and HDAC1. EMBO reports. 2015;16:1288–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ler LD, Ghosh S, Chai X, Thike AA, Heng HL, Siew EY, et al. Loss of tumor suppressor KDM6A amplifies PRC2-regulated transcriptional repression in bladder cancer and can be targeted through inhibition of EZH2. Science translational medicine. 2017;9:eaai8312. [DOI] [PubMed] [Google Scholar]
- 33.Ezponda T, Dupéré-Richer D, Will CM, Small EC, Varghese N, Patel T, et al. UTX/KDM6A loss enhances the malignant phenotype of multiple myeloma and sensitizes cells to EZH2 inhibition. Cell reports. 2017;21:628–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu Q, Tian Y, Zhang J, Tong X, Huang H, Li S, et al. In vivo CRISPR screening unveils histone demethylase UTX as an important epigenetic regulator in lung tumorigenesis. Proceedings of the National Academy of Sciences. 2018;115:E3978–E86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Greenfield A, Carrel L, Pennisi D, Philippe C, Quaderi N, Siggers P, et al. The UTX gene escapes X inactivation in mice and humans. Human molecular genetics. 1998;7:737–42. [DOI] [PubMed] [Google Scholar]
- 36.Shpargel KB, Sengoku T, Yokoyama S, Magnuson T. UTX and UTY demonstrate histone demethylase-independent function in mouse embryonic development. PLoS genetics. 2012;8:e1002964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walport LJ, Hopkinson RJ, Vollmar M, Madden SK, Gileadi C, Oppermann U, et al. Human UTY (KDM6C) is a male-specific Nϵ-methyl lysyl demethylase. Journal of Biological Chemistry. 2014;289:18302–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Welstead GG, Creyghton MP, Bilodeau S, Cheng AW, Markoulaki S, Young RA, et al. X-linked H3K27me3 demethylase Utx is required for embryonic development in a sex-specific manner. Proc Natl Acad Sci U S A. 2012;109:13004–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang C, Lee J-E, Cho Y-W, Xiao Y, Jin Q, Liu C, et al. UTX regulates mesoderm differentiation of embryonic stem cells independent of H3K27 demethylase activity. Proceedings of the National Academy of Sciences. 2012;109:15324–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Batora NV, Sturm D, Jones DT, Kool M, Pfister SM, Northcott PA. Transitioning from genotypes to epigenotypes: why the time has come for medulloblastoma epigenomics. Neuroscience. 2014;264:171–85. [DOI] [PubMed] [Google Scholar]
- 41.Dubuc AM, Remke M, Korshunov A, Northcott PA, Zhan SH, Mendez-Lago M, et al. Aberrant patterns of H3K4 and H3K27 histone lysine methylation occur across subgroups in medulloblastoma. Acta neuropathologica. 2013;125:373–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jones DT, Northcott PA, Kool M, Pfister SM. The role of chromatin remodeling in medulloblastoma. Brain Pathol. 2013;23:193–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhan X, Shi X, Zhang Z, Chen Y, Wu JI. Dual role of Brg chromatin remodeling factor in Sonic hedgehog signaling during neural development. Proceedings of the National Academy of Sciences. 2011;108:12758–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shi X, Zhang Z, Zhan X, Cao M, Satoh T, Akira S, et al. An epigenetic switch induced by Shh signalling regulates gene activation during development and medulloblastoma growth. Nature communications. 2014;5:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi X, Wang Q, Gu J, Xuan Z, Wu J. SMARCA4/Brg1 coordinates genetic and epigenetic networks underlying Shh-type medulloblastoma development. Oncogene. 2016;35:5746–5758. [DOI] [PubMed] [Google Scholar]
- 46.Mao J, Ligon KL, Rakhlin EY, Thayer SP, Bronson RT, Rowitch D, et al. A novel somatic mouse model to survey tumorigenic potential applied to the Hedgehog pathway. Cancer research. 2006;66:10171–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang H, Xie X, Deng M, Chen X, Gan L. Generation and characterization of Atoh1-Cre knock-in mouse line. genesis. 2010;48:407–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dey J, Ditzler S, Knoblaugh SE, Hatton BA, Schelter JM, Cleary MA, et al. A distinct Smoothened mutation causes severe cerebellar developmental defects and medulloblastoma in a novel transgenic mouse model. Molecular and cellular biology. 2012;32:4104–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bracken AP, Kleine-Kohlbrecher D, Dietrich N, Pasini D, Gargiulo G, Beekman C, et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes & development. 2007;21:525–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Agger K, Cloos PA, Rudkjær L, Williams K, Andersen G, Christensen J, et al. The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A–ARF locus in response to oncogene-and stress-induced senescence. Genes & development. 2009;23:1171–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ivanov DP, Coyle B, Walker DA, Grabowska AM. In vitro models of medulloblastoma: choosing the right tool for the job. Journal of biotechnology. 2016;236:10–25. [DOI] [PubMed] [Google Scholar]
- 52.Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T, et al. The whole-genome landscape of medulloblastoma subtypes. Nature. 2017;547:311–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bantug GR, Cekinovic D, Bradford R, Koontz T, Jonjic S, Britt WJ. CD8+ T lymphocytes control murine cytomegalovirus replication in the central nervous system of newborn animals. The Journal of Immunology. 2008;181:2111–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schuller U, Heine VM, Mao J, Kho AT, Dillon AK, Han YG, et al. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell. 2008;14:123–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Read TA, Fogarty MP, Markant SL, McLendon RE, Wei Z, Ellison DW, et al. Identification of CD15 as a marker for tumor-propagating cells in a mouse model of medulloblastoma. Cancer Cell. 2009;15:135–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ward RJ, Lee L, Graham K, Satkunendran T, Yoshikawa K, Ling E, et al. Multipotent CD15+ cancer stem cells in patched-1-deficient mouse medulloblastoma. Cancer Res. 2009;69:4682–90. [DOI] [PubMed] [Google Scholar]
- 57.Olson JM, Asakura A, Snider L, Hawkes R, Strand A, Stoeck J, et al. NeuroD2 is necessary for development and survival of central nervous system neurons. Dev Biol. 2001;234:174–87. [DOI] [PubMed] [Google Scholar]
- 58.Lee J, Hollenberg S, Snider L, Turner D, Lipnick N, Weintraub H. NeuroD, a new basic helix-loop-helix protein, can convert Xenopus ectoderm into neurons. Science. 1995;268:836–44. [DOI] [PubMed] [Google Scholar]
- 59.Farah MH, Olson JM, Sucic HB, Hume RI, Tapscott SJ, Turner DL. Generation of neurons by transient expression of neural bHLH proteins in mammalian cells. Development. 2000;127:693–702. [DOI] [PubMed] [Google Scholar]
- 60.Tang H, Wang Y, Chlewicki LK, Zhang Y, Guo J, Liang W, et al. Facilitating T cell infiltration in tumor microenvironment overcomes resistance to PD-L1 blockade. Cancer cell. 2016;29:285–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maximov V, Chen Z, Wei Y, Robinson MH, Herting CJ, Shanmugam NS, et al. Tumour-associated macrophages exhibit anti-tumoural properties in Sonic Hedgehog medulloblastoma. Nature communications. 2019;10:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gate D, Danielpour M, Rodriguez J, Kim G-B, Levy R, Bannykh S, et al. T-cell TGF-β signaling abrogation restricts medulloblastoma progression. Proceedings of the National Academy of Sciences. 2014;111:E3458–E66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wiegering V, Eyrich M, Rutkowski S, Wölfl M, Schlegel PG, Winkler B. TH1 predominance is associated with improved survival in pediatric medulloblastoma patients. Cancer Immunology, Immunotherapy. 2011;60:693–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zheng L, Xu L, Xu Q, Yu L, Zhao D, Chen P, et al. Utx loss causes myeloid transformation. Leukemia. 2018;32:1458–65. [DOI] [PubMed] [Google Scholar]
- 66.Wang JK, Tsai M-C, Poulin G, Adler AS, Chen S, Liu H, et al. The histone demethylase UTX enables RB-dependent cell fate control. Genes & development. 2010;24:327–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Agrawal R, Garg A, Benny Malgulwar P, Sharma V, Sarkar C, Kulshreshtha R. p53 and miR-210 regulated NeuroD2, a neuronal basic helix-loop-helix transcription factor, is downregulated in glioblastoma patients and functions as a tumor suppressor under hypoxic microenvironment. Int J Cancer. 2018;142:1817–28. [DOI] [PubMed] [Google Scholar]
- 68.Agger K, Cloos PA, Christensen J, Pasini D, Rose S, Rappsilber J, et al. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature. 2007;449:731–4. [DOI] [PubMed] [Google Scholar]
- 69.Arcipowski KM, Martinez CA, Ntziachristos P. Histone demethylases in physiology and cancer: a tale of two enzymes, JMJD3 and UTX. Current opinion in genetics & development. 2016;36:59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xiao Z, Shen J, Zhang L, Li L, Li M, Hu W, et al. The Roles of Histone Demethylase UTX and JMJD3 (KDM6B) In Cancers: Current Progress and Future Perspectives. Current medicinal chemistry. 2016;23:3687–3696. [DOI] [PubMed] [Google Scholar]
- 71.Zhao W, Li Q, Ayers S, Gu Y, Shi Z, Chen Y, et al. Jmjd3 negatively regulates reprogramming through histone demethylase Activity-Dependent and-Independent pathways. Cell. 2013;152:1037–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nature biotechnology. 2016;34:525–7. [DOI] [PubMed] [Google Scholar]
- 73.McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic acids research. 2012;40:4288–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.