

Abstract
Pancreatic ductal adenocarcinoma(PDAC) is resistant to the PD-1/PD-L1 blockade therapy. Previously, the combination of PD-1 blockade and vaccine therapy was shown to have a modest antitumor activity in murine models of PDAC. We used a murine syngeneic model of metastatic PDAC to identify, among multiple T cell modulators tested, which therapeutic agents in combination with the GVAX cancer vaccine and an anti-PD-1 antagonist antibody(αPD-1) are able to improve the survival. We found that an anti-CD137 agonist antibody(αCD137) most significantly improved survival in the mouse PDAC model. Moreover, αPD-1 and αCD137 together in combination with vaccine therapy more significantly increased the expression of costimulatory molecules CD137 and OX40 on CD4+PD-1+ and CD8+PD-1+ T cells comparing to αPD-1 or αCD137, respectively, suggesting that T cell activation within PDACs were enhanced by a synergy of αCD137 and αPD-1. On another hand, αCD137 treatment led to an increase in effector memory T cells independent of αPD-1. Although αCD137 does not increase the cytotoxic effector T cell function, the addition of αCD137 to GVAX+αPD-1 increased expression of IFNγ in EOMES+ exhausted tumor-infiltrating T cells. Taken together, this preclinical study established the mechanism of targeting CD137 to enhance effector memory and activated T cells in PDAC. Immunohistochemistry analysis of resected human PDACs following the neo-adjuvant GVAX treatment showed increased levels of CD8+ T cells in those with high levels of CD137 expression, supporting an ongoing clinical trial of testing CD137 as a potential target in treating PDACs that are inflamed with T cells by vaccine therapy.
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) carries a very poor prognosis, with 5-year survival rates of 9% for all stages [1]. Surgical resection is currently the best curative option, but recurrence is high despite use of neoadjuvant and/or adjuvant chemotherapy and radiotherapy regimens [2, 3]. In addition, most patients have metastatic disease at diagnosis [2]. A desmoplastic stroma composed of various cell types and extracellular matrix proteins creates a drug-resistant, immunosuppressive and overall protumorigenic environment [4–7]. Due to this fibrotic stroma and a lack of anti-tumor effector T cells in the PDAC tumor microenvironment (TME), pancreatic cancer is an immunogenically “cold” tumor that does not respond to immune checkpoint inhibitors as single agents, and requires multiple agents to elicit an anti-tumor immune response [8–13].
Cancer vaccine therapy can potentially prime the immune system with an increase CD8+ T cell infiltration into the TME of PDAC and an expanded repertoire of anti-tumor CD8+ T cells. Through a neoadjuvant clinical trial, GVAX, a whole-cell granulocyte-macrophage colony-stimulating factor (GM-CSF)-secreting allogenic PDAC vaccine, was previously shown to induce formation of novel tertiary lymphoid structures in the PDAC tumors of patients and covert PDAC from an immunogenically “cold” tumor to an immunogenically “hot” one [14, 15]. While GVAX does increase T cell infiltration into the tumor, it is also associated with an increase in immunosuppressive signals [15]. GVAX treatment results in an upregulation of the programmed death-1 (PD-1) receptor and its ligand (PD-L1) in the TME[15]. Combining anti-PD-1 antagonist antibody with GVAX improved anti-tumor efficacy compared to GVAX alone in a murine model of metastatic PDAC [16]; however, the functionality of vaccine-induced effector T cells remain to be low[15]. Clinical trials are currently underway to investigate the efficacy of the combination of vaccine therapies including GVAX and listeria-based vaccine and immune checkpoint inhibitors (ICIs) including anti-PD-1 antibody in patients with both localized PDACs (NCT02451982) and metastatic PDACs. We, and others, have observed that small subgroups of patients respond to ICI-based combinations[17]; however, the response rates are low and most are transient[18]. Thus, the current challenge is to identify the additional immune inhibitory or stimulatory signals within the complex PDAC TME that require further modification to effectively enhance effector T cell function.
Beside PD-L1/PD-1, a few pathways that inhibit T cell function are particularly of interest as immunotherapeutic targets. Cytotoxic T-lymphocyte activation antigen-4 (CTLA4) expression increases on T cells after their activation, and blocks CD28 receptor signaling between T cells and antigen-presenting cells by binding to CD80 and CD86 [19]. Lymphocyte activation gene 3 (LAG3) suppresses the effector T cell function and facilitates a state of exhaustion along with PD-1 [20–22]. T cell immunoglobulin and mucin domain-containing 3 (TIM3) is similarly expressed on exhausted T cells, and is associated with CD8+ T cell populations within tumors that are not able to proliferate [21, 23]. Targeting these immunosuppressive signals on T cells in combination with anti-PD-1 antagonist therapy is actively being tested in many clinical trials including those specifically designed for PDCAs [24].
On another hand, co-stimulatory molecules such as CD137 have not been studied as targets for overcoming the resistance of PDACs to immune checkpoint inhibitors and for enhancing the anti-tumor efficacy of anti-PD-1/PD-L1 antibodies. CD137 belongs to the family of tumor necrosis factor receptors (TNFR), and is involved in regulation of immune responses [25]. Costimulatory signals from the CD137 receptor promote T cell survival and proliferation, and can lead to effective expansion of CD8+ T cell subsets [25]. A recent study showed that human melanoma-infiltrating lymphocytes, which were found to naturally harbor a subset of CD137+ T cells, responded to the in vitro treatment with a CD137 agonist that led to the proliferation of T cells with an sustained anti-tumor function [26]. Consistently, it was suggested that a CD137 agonist is a potential treatment strategy for expanding antigen-specific cytotoxic T cells in immunogenically “hot” tumors such as melanoma [27]. On another hand, a published study showed that a small repertoire of CD8+ T cells also existing in the immunogenically “cold” tumors such as PDAC are capable of responding to the CD137 agonist treatment [28]. We thus hypothesized that the CD137 agonist can be used as a treatment strategy for PDAC to activate and expand the antitumor CD8+ T cells in patients who also receive vaccine and anti-PD-1 antibody therapies that induce the intratumoral infiltration of T cells.
Therefore, we have examined a panel of T cell and myeloid cell targeting agents, whose clinical counterparts are currently in the clinical development, for their potentials in enhancing antitumor efficacy of the combination of GVAX and anti-PD-1 antibody in the preclinical model of PDAC. Previously, we explored the myeloid cell-targeting agent, more specifically, an anti-CSF-1 receptor antagonist antibody in combination with GVAX and a PD-1 antagonist antibody. In this study, we explored T cell modulators targeting LAG3, TIM3, CTLA4 and CD137 in combination with GVAX and a PD-1 antagonist antibody in a murine model of metastatic PDAC. We demonstrated that the addition of a CD137 agonist antibody to GVAX and PD-1 blockade was effective in prolonging survival in the PDAC-bearing mice and enhancing effector T cell infiltration and functionalities within the TME. These results support the use of combination immunotherapy including a CD137 agonist to treat patients with PDAC. The findings in this preclinical study has led to the addition of a new arm of patients to receive the neoadjuvant/adjuvant combination immunotherapy with GVAX, Nivolumab, and Urelumab for resectable PDACs (NCT02451982).
2. Materials and Methods
2.1. Cell Culture
The KPC cell line is an established PDAC tumor cell line originally derived from transgenic KPC mice harboring tissue-specific mutations in Kras and p53 through expression of cre recombinase under control of pancreatic-specific pdx-1 promoter. The KPC mice were previously backcrossed to the C57Bl/6 mouse background for nine generations as described[29]. Cells were grown in KPC media containing RPMI 1640 medium (Life Technologies) supplemented with 10% heat inactivated FBS (Benchmark), 2mM L-glutamine (Life Technologies), 1% MEM-NEAA (Life Technologies), 1 mM sodium pyruvate (Sigma Aldrich), and 100 units/mL penicillin and 100 μg/mL streptomycin (Life Technologies). KPC cells were maintained in culture at 37°C in 5% CO2. B78H1 cells are a murine fibroblast cell line engineered to secrete GM-CSF. B78H1 cells were grown in B78H1 media containing RPMI 1640 medium supplemented with 10% heat inactivated FBS, 1mM L-glutamine, and 100 units/mL penicillin and 100 μg/mL streptomycin. B78H1 cells were maintained in culture at 37°C in 5% CO2.
Harvested tumor-infiltrating immune cells were processed in CTL media consisting of RPMI 1640 medium, 10% heat inactivated FBS, 1% MEM-NEAA, 1mM L-glutamine, 1% HEPES (Life Technologies), 100 units/mL penicillin and 100 μg/mL streptomycin, and 50 μM 2-mercaptoethanol (Sigma).
2.2. Mouse Model
All animal experiments conformed to the Animal Care and Use Committee (IACUC) guidelines at Johns Hopkins University. Mice were monitored twice a day to ensure adherence to IACUC protocol. Mice with signs of distress such as hunched posture, lethargy, ascites and dehydration were promptly euthanized and considered to have reached the survival endpoint. Female C57Bl/6 mice, aged 7–8 weeks old, were purchased from The Jackson Laboratory for these experiments. The mice developed high burden liver metastases following the inoculation of the KPC tumor cells via hemispleen surgery and hemisplenic injection as previously described [30]. In short, mice were anesthetized, the spleen was eviscerated, clipped and divided in half. Half of the spleen was injected with 2×105 KPC tumor cells and subsequently removed (to avoid development of tumor outside the liver from residual tumor cells), while the other half was left in the animal.
Monoclonal antibodies were administered intraperitoneally twice a week for four weeks after surgery in survival studies. Anti-mouse PD-1 (mIgG1-D265A, clone 4H2, Bristol-Myers-Squib) and anti-mouse CTLA4 (mIgG2a, clone 9D9, Bristol-Myers-Squib) antibodies were administered at 100 μg (5 mg/kg) per dose. This CTLA4 antibody was shown to deplete Tregs expressing CTLA4 in the TME [31]. Anti-mouse TIM3 (clone RMT3-23, BioXcell) and anti-mouse LAG3 (Clone C9B7W, BioXcell) antibodies were administered at 200 μg (10 mg/kg) per dose. Anti-mouse CD137 antibody (mIgG1, clone BMS-469452, Bristol-Myers-Squib) was administered at 20 μg (1 mg/kg) per dose. GVAX vaccine was prepared by combining equal cell number and volume of the GM-CSF-expressing by-stander B78H1 cells and the KPC PDAC tumor cells at an initial concentration of 2 × 107 cells/mL per cell line. This GVAX formula either with Panc02 or KPC tumor cells has been utilized in the past for all the preclinical GVAX studies in the murine PDAC model[16, 32–36]. The cell suspension was irradiated at 50 Gy and administered SC to three limbs (100 μL each) to deliver 3×106 cells of each cell line to the mouse. GVAX vaccine was administered subcutaneously (SC) on days 4, 7, 14 and 21 after surgery in survival studies.
2.3. Analysis of infiltrating immune cells in liver metastasis
Adhering to a shortened treatment course (GVAX on days 4 and 7; anti-PD-1 and anti-CD137 antibodies and days 4, 6, and 10), tumor-infiltrating lymphocytes (TILs) were recovered from each mouse liver on day 14 following KPC tumor cell inoculation. Each liver with diffused liver metastases throughout the liver was mechanically processed sequentially through 40-μm and 100-μm nylon filters, and the cell suspension was brought to a volume of 25 mL in CTL media. Cell suspension was centrifuged at 500 g for 5 minutes, and cell pellets were suspended in 4 mL of ACK lysis buffer (Quality Biological) for 2 minutes. Lysing was quenched by adding CTL media to a total volume of 50 mL, and cells were centrifuged again at 500 g. Cell pellets were suspended in 6 mL of 80% Percoll (GE Healthcare Life Sciences), overlaid with 6 mL of 40% Percoll, and then centrifuged for 25 minutes at 2400 g with the brake turned off. The resulting lymphocyte layer was removed from the Percoll gradient, suspended in CTL media, and subsequently washed twice with PBS (Gibco). Cells were split into two populations for staining and analysis; one to be stimulated for IFNγ production prior to flow cytometry analysis, and the other left unstimulated for characterization by flow cytometry.
2.4. TIL stimulation to induce IFNγ production
The isolated liver metastasis TILs from each mouse were plated in a 24-well plate, and incubated with CD3/CD28 stimulation beads (Dynabeads Mouse T-Activator CD3/CD28, Life Technologies) according to manufacturer instructions in CTL media for 15 hours in an incubator set to 37°C in 5% CO2. GolgiPlug (BD Biosciences) and GolgiStop (BD Biosciences) were added to the cell and bead suspension and incubated for an additional 4 hours. Following treatment with GolgiStop and GolgiPlug, the beads were removed according to manufacturer instructions, washed twice with PBS, counted and suspended at a concentration of 1×107 cells/mL.
2.5. Cell staining and flow cytometry
After isolation of the TILs from the liver, 100 μL volumes of cell suspension at a concentration of 1×107 cells/mL were plated into designated wells of a 96-well plate. Cells were first stained with Live-Dead Aqua (Invitrogen) for 30 minutes on ice, washed twice with PBS, and then blocked with rat anti-mouse Fc antibody (CD16/CD32, BD Biosciences). After blocking, cells were incubated for 1 hour with the following anti-mouse fluorophores: CD3-PerCP-Cy5.5 (Biolegend), CD4-APC-Fire (BD Biosciences), CD8a-PE/Cy7 (Biolegend), PD-1-FITC (eBioscience), OX40-APC (Biolegend), CD44-PE (Biolegend), CD62L-APC (Biolegend), and CCR7-BV421 (Biolegend). Cells were washed following staining, and suspended in Fix/Perm buffer (eBioscience) for 30 minutes. After fixation, intracellular staining with Eomes PE (Invitrogen) and IFNγ BV421 (Biolegend) was done on ice for 30 minutes. Flow cytometry was performed using the CytoFLEX flow cytometer (B eckman Coulter).
2.6. Human PDAC immunohistochemistry
According to the Johns Hopkins Medical Institution (JHMI) Institutional Review Board (IRB) approved protocol (NA_00015858), we obtained slides from 16 formalin-fixed paraffin-embedded tissues obtained from tumor samples surgically resected two weeks after a single neoadjuvant intradermal administration of GVAX in patients with resectable PDAC[15]. Antigen retrieval was done in the Citrate buffer in a Diva decloaker for 30 seconds at 125°C. The slides were pre-treated with peroxide block for 5 minutes followed by serum-free protein block for 30 min. Slides were then incubated at room temperature with mouse anti-CD137 IgG1 (clone BBK-2, Thermofisher) at the final concentration of 1μg/mL for 1 hour. Goat anti-mouse secondary antibodies (Vector Laboratories) were added for 30 minutes at room temperature. The signal was amplified and detected using the ABC Vectastain Kit (Vector Laboratories) according to the manufacturer’s instructions. The slides were developed using DAB and counterstained by hematoxylin. The density of CD137 expressing immune cells within the whole tumor area, which was previously circled on the slide by pathologists[15], was determined using HALO image analysis software (Indica Labs). To separate patients with high CD137+ cell density from low CD137+ cell density, we used the median density of CD137+ cells (82 cells/cm2) as the cutoff. The median is commonly chosen as a cutoff which would separate all the samples into two equal sized groups and which would lead to the highest power to detect the difference between two groups[37, 38]. The correlation analysis between CD137 expression and the densities of other immune cell subtypes in human PDAC tumor samples was conducted. The data on the densities of other immune cell subtypes were derived from previously published multiplex immunohistochemistry analysis[18].
2.7. Statistical analysis
All statistical analyses and graphing of results were performed using GraphPad Prism software (GraphPad Software). Kaplan-Meier curves and log-ranks tests were used to analyze survival outcomes between treatment groups. When comparing cell number or cell percentages from the FACS analysis, unpaired t-tests were used. For human PDAC samples comparing cell density and cell density ratios, the mean values were analyzed using unpaired t-test. A p-value less than 0.05 was considered statistically significant for these results.
3. Results
3.1. CD137 agonist antibody in combination with GVAX and PD-1 antagonist antibody improved survival in a murine model of metastatic PDAC
The primary goal of our study was to investigate new strategies to enhance the effects of pancreatic GVAX vaccine and anti-PD-1 antibody (αPD-1) combination therapy. We used a murine PDAC model of liver metastasis to explore the effects of four different antibodies targeting T cell signals in combination with GVAX and αPD-1. This preclinical model of liver metastatic PDAC was established in syngeneic C56Bl6 mice via the hemispleen injection of the KPC tumor cells. The KPC tumor cells derived from transgenic KPC mice that harbor KRAS and P53 knock-in mutations. The murine KPC liver metastasis model is one of few orthotopic syngeneic models of PDACs that have an immunogenically “cold” TME resembling human PDAC[16, 32, 33, 35, 39]. Mice were treated with GVAX subcutaneously on days 4, 7, 14 and 21 following implantation of KPC tumor cells (Fig. 1A). The mice were also treated twice a week for four weeks with either αPD-1, αLAG3, αTIM3, αCTLA-4, or αCD137 antibodies in combination with GVAX depending upon the treatment group. Survival was monitored in each group of mice over 90 days (Fig. 1B). On Day 90, survived mice were examined by autopsy; and all found to be free of disease. Untreated mice would all die from diffused metastases in live within a relatively narrow window of time, making the survival an objective endpoint for this tumor model. By contrast, diffused liver metastases are difficult for measurement by size or weight. Previously, we have demonstrated that the GVAX+αPD-1 double combination immunotherapy enhanced the survival of PDAC-bearing mice comparing to either GVAX or αPD-1 alone, which has a minimal antitumor activity in the same mouse model of PDAC[16]. Thus, we focused the current study on comparing between the GVAX+αPD-1 double combination immunotherapy and one of triple combination immunotherapy with the addition of αCD137 antibody, αLAG3 antibody, αTIM3 antibody or μCTLA-4 antibody are shown (Fig. 1B). The addition of αCD137 to GVAX+αPD-1 significantly prolonged survival as compared to GVAX+αPD-1. Addition of αTIM3 or αLAG3 antibodies showed no survival benefit compared to GVAX+αPD-1, suggesting further halting T cell suppressive mechanisms when PD-1 has already been blocked is not effective (Supplemental Fig. S1). The lack of efficacy is not due to absence of TIM3 and LAG3 expression on the T cells (Supplemental Fig. S1E). While αCTLA4 exhibited an improvement on survival in this mouse model, the improvement was not statistically significant in duplicated experiments. By contrast, αCD137 repeatedly showed a statistically significant improvement on survival in this model, therefore, was chosen to be a focus of this study.
Figure 1. Targeting two checkpoint pathways in combination with GVAX can improve survival in a murine liver-metastatic pancreatic cancer model.
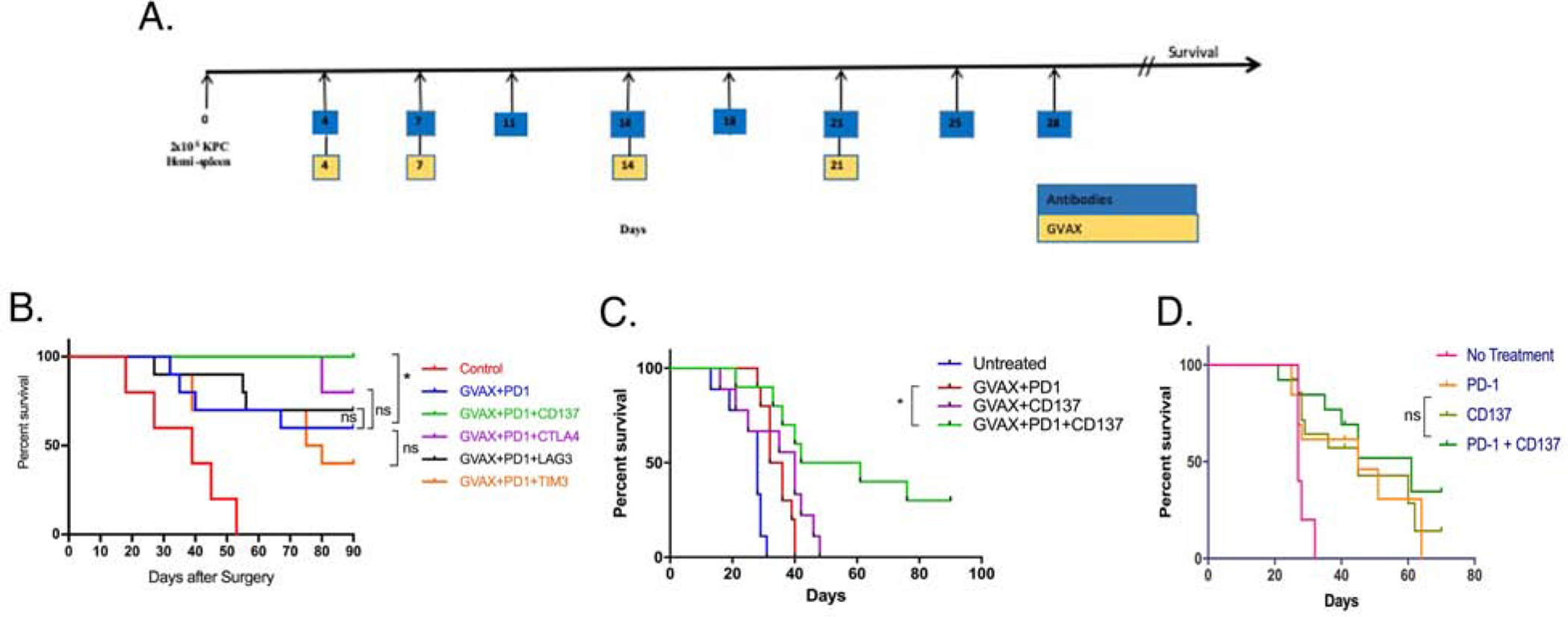
(A.) Schematic representation of treatment following inoculation of 2 × 105 KPC tumor cells by the hemispleen surgery. (B.) Kaplan-Meier curves for treatments following dosing regimen in Figure 1A. Mice were treated with GVAX and αPD-1 antibodies as indicated, in addition to αCD137, αLAG3, αTIM3 and αCTLA4 in the appropriate groups. Control groups received corresponding isotype IgG control antibody. Treated mice were monitored for survival till Day 90 following the tumor inoculation in all six treatment groups to investigate the benefits of αCD137, αLAG3, αTIM3 and αCTLA4 in addition to GVAX and αPD-1. *p<0.05. ns, not significant. (C.) A separate experiment following the schema in A. was performed. “Untreated” group received isotype control IgGs. Survival statistical analysis was done by using log-rank test. *p<0.05. ns, not significant. GVAX+αPD-1+αCD137 vs. GVAX+αCD137, p=0.0291. GVAX+αPD-1+αCD137 vs. GVAX+αCD137, p=0.0371; GVAX+αPD-1+αCD137 vs. GVAX+αPD-1, p=0.0064. (D.) Kaplan-Meier curve for treatments following dosing regimen in Figure 1A, but without GVAX. Survival statistical analysis was done by using log-rank test. αPD-1+αCD137 vs. αCD137 or αPD-1+αCD137 vs. αPD-1, p>0.05. n=10 per treatment groups in all the experiments. Data represents one representative experiment that was repeated twice.
A subsequent survival experiment was performed to further validate the effects of combining αCD137 agonist antibody with GVAX and αPD-1 blocking antibody. We increased the disease burden of the liver metastasis murine model by using a subclone of KPC tumor cells that were growing more rapidly than the cells used in the previously described survival study. This experiment which served as a validation with a second KPC tumor cell line (designated aggressive variant) showed that GVAX+αPD-1+αCD137 increased survival significantly compared to both GVAX+αPD-1 and GVAX+αCD137 treatment groups, respectively (Fig. 1C). The mice in the GVAX+αPD-1+αCD137 group that remained alive at the experiment endpoint (Day 90) were free of liver metastases. By using the same aggressive variant KPC tumor cell line, in the absence of GVAX, αPD-1 antagonist, αCD137 agonist or both did not result in any significant improvement in survival, suggesting that a vaccine-primed TME was pre-requisite for these immunotherapeutic antibodies to be effective in PDAC (Fig. 1D). In this second model, consistent with the results of clinical experience[40] of immune checkpoint inhibitors as single agents, none of the mice treated with αPD-1 alone or GVAX+αPD-1 were disease free at the experiment endpoint. We also tested different dosing schedules of the αCD137 antibody (Supplemental Fig. S2). Mice treated with GVAX+αPD-1 and 20 μg of αCD137 twice a week for four weeks had significantly better survival than those treated with one single dose of 100 μg of αCD137 given three days post tumor implantation. These results suggested that the survival benefit is particularly associated with adding αCD137 to the GVAX+αPD-1 treatment in repeated cycles. In all these experiments, we did not obverse any obvious sign of toxicity such as weight loss in mice treated with αCD137 or its based combinations, suggesting αCD137 is safe in treating PDAC-bearing mice.
3.2. High CD137 expression in human PDAC treated with vaccine therapy was associated with increased CD8+ T cell density
Above experiments supported an antitumor activity of αCD137 in the murine PDAC tumor model that resembles Kras-mutated human PDAC. We then sought to explore the role of CD137 in human PDACs. To this end, we investigated the effects of high versus low density of CD137+ cells in human PDAC tumors resected after a single neoadjuvant treatment of GVAX in the above described clinical trial by using multiplex immunohistochemistry [14, 15]. We obtained slides from 16 formalin-fixed paraffin-embedded PDAC tissues obtained from this clinical trial[15] and stained with anti-CD137 antibody. PDACs with high CD137+ cell density were separated from those with low CD137+ cell density by using the median density of CD137+ cells (82 cells/cm2) as the cutoff (Fig. 2A). The data on the densities of other immune cell subtypes were derived from previously published multiplex immunohistochemistry analysis[18]. When compared to tumors with low CD137+ cell density, those with high CD137+ cell density were associated with a significant increase in the density of CD8+ T cells as defined by multiplex immunohistochemistry with CD45, CD3, CD8 markers in the tumor area (Fig. 2B). No significant differences were observed in the density of T-helper cells, regulatory T cells, myeloid cells and mature and immature dendritic cells between tumors with high and low CD137+ cell density(Supplemental Fig. S3). There was a significant increase in the density of CD3+CD8+T cells that express the eomesodermin (Eomes) T cell exhaustion marker in tumors with a high density of CD137+ cells compared to those with low density of CD137+ cells (Fig. 2C). There was a trend towards an increased density of both Eomes+ and Eomes− CD3+CD8+PD-1+ T cells in tumors with a high density of CD137+ cells compared to those with a low density of CD137+ cells (Fig. 2C&D). Thus, the association of the high CD137+ cell density with an increased CD8+ T cell infiltration in vaccine-primed PDAC supported CD137 as a target for immunotherapy to enhance the CD8+ T cell function in vaccine-primed PDACs. However, the association of the high CD137+ cell density with a higher level of exhausted CD8+ T cells in vaccine-primed PDACs suggested that the immunotherapy targeting CD137+ cells in PDACs should mechanistically aim at re-invigorating T cells from an exhausted status to an activated, effector status. It is possible that increased densities of CD8+ T cells and CD137+ T cells are a result of response to the vaccine therapy, which warrants further investigation in the human clinical trials. Here, we examined below whether αCD137 in combination with GVAX and αPD-1 can enhance the activation status of effector T cells in the murine model of PDAC.
Figure 2. High CD137 expression in human PDAC treated with vaccine therapy was associated with increased CD8+ T cell density.
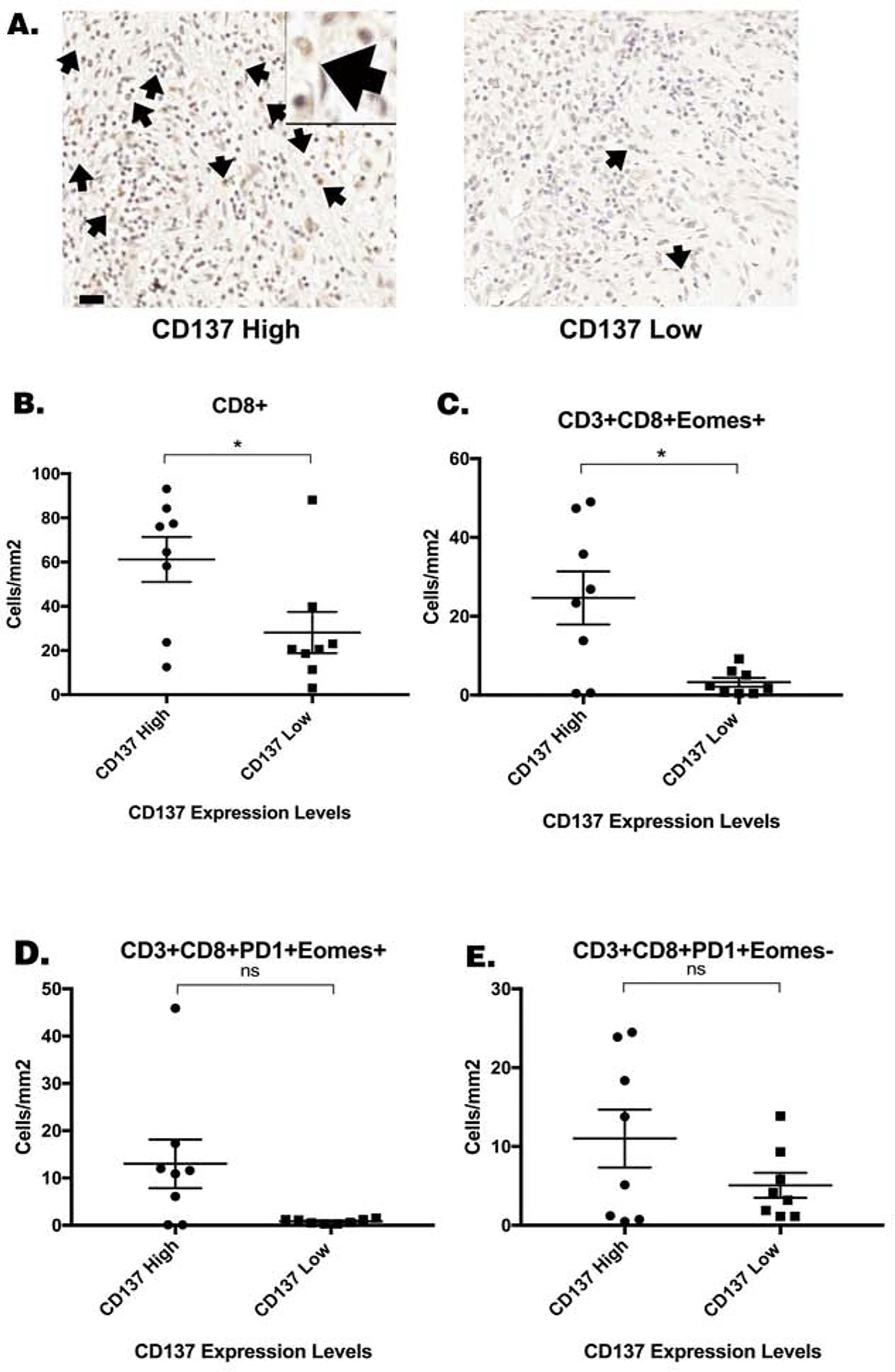
(A.) Sixteen formalin-fixed paraffin-embedded human PDAC specimens were obtained following surgical resection two weeks after one neoadjuvant dose of GVAX were analyzed by multiplex immunohistochemistry as previously described [18]. Immunohistochemistry staining of CD137 was performed on slides from each of these 16 FFPE blocks. CD137+ cells was quantified and the median density of CD137+ cells (81.6 cells/cm2) was used as the cutoff to group high CD137+ cell density (greater than 81.6 cells/cm2) and low CD137+ cell density (less than 81.6 cells/cm2) patients. Representative cases with high CD137+ cell density and low CD137+ cell density, respectively, were shown. Representative CD137+ cells were indicated by arrows. One representative CD137+ cell were shown in a higher power field. Scale bar, 20 μm. (B.) The correlation between the density of CD8+ T cells as previously determined[18] by multiplex immunohistochemistry and the density of CD137+ cells in the PDAC tumors was analyzed in this study. The densities of (C.) CD3+CD8+Eomes+ cells (D.) CD3+CD8+PD-1+Eomes+ cells, and (E.) CD3+CD8+PD-1+Eomes− T cells in the tumor area of PDAC patients as previously determined[18] by multiplex immunohistochemistry, respectively, were correlated with the density of CD137+ cells. Unpaired t tests were done. * p<0.05. ns, not significant.
3.3. Mice treated with GVAX, PD-1 antagonist and CD137 agonist exhibit increased infiltration of CD4+ and CD8+ cells into the TME
We first examined whether αCD137 can quantitatively influences the T cells infiltrating the PDAC tumors. Mice were inoculated with the KPC tumor cells on day 0. Tumor-bearing mice were treated with GVAX, αPD-1 and αCD137 on days 4 and 7, followed by another dose of antibodies on day 11. On day 14 when the antitumor effect of the treatments was not obvious, livers with metastases, which were developed at similar extents in all mice among different treatment groups, were harvested and processed to obtain TILs. Tumors were disseminated diffusely throughout the liver and not separable; and thus, the whole liver was processed for flow cytometry analysis and representative flow cytometry images were shown in Supplemental Fig. S4. The numbers of T cells and their subtypes in the TME were normalized by the total cell numbers in the liver in the experiments here and below. The results demonstrated increases in the numbers of both CD4+ and CD8+ T cells within the TME of mice treated with GVAX+αPD-1+αCD137 comparing to other treatment groups and the increase being particularly significant for CD4+ T cells comparing to the GVAX+αPD-1 GVAX+αCD137 groups (Fig. 3A&D).
Figure 3. Treatment with GVAX, αPD-1 and αCD137 increased the number of CD4+ and CD8+ cells within the tumor, along with decreased PD-1 expression on CD8+ T cells.
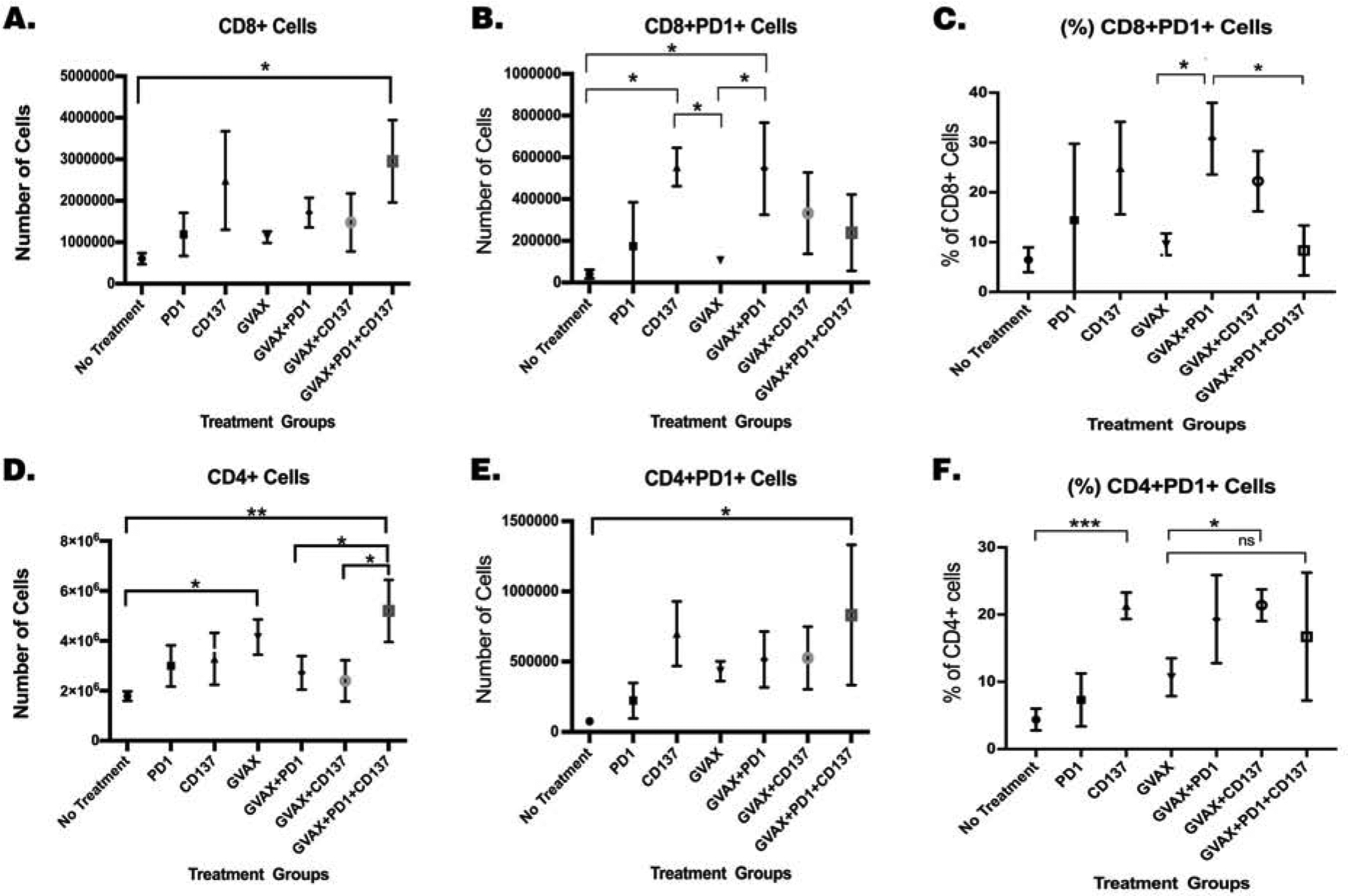
Mice were inoculated with 2×105 KPC cells via the hemispleen surgery, treated and then sacrificed 14 days after KPC cell inoculation for isolation of tumor-infiltrating immune cells for flow cytometry analysis. Normalized numbers of (A.) CD8+ and (D.) CD4+ T cells. Normalized numbers of (B.) CD8+PD-1+ and (E.) CD4+PD-1+ T cells. Percentages of (C.) CD8+PD-1+ and (F.) CD4+PD-1+ T cells among CD8 cells and CD4 cells, respectively. Unpaired t tests were done. * p<0.05; ** p<0.01; *** p<0.001; ns, not significant. Numbers of T cells were normalized by the total cell counts of the single cell suspension of the livers. Data represents mean ±SEM in one representative experiment (n=3 per treatment group per experiment, repeated twice).
There was a significant increase in the number of intratumoral CD8+PD-1+ T cells and the percentage of CD8+PD-1+ T cells among CD8+ T cells in the TME when mice were treated with GVAX+αPD-1 compared to no treatment and GVAX alone (Fig. 3D&C). However, there was a significant decrease in the percentage of CD8+ T cells that express PD-1 as well as a trend of decrease in the absolute number of CD8+ T cells that express PD-1 in the GVAX+αPD-1+αCD137 treatment group compared to the GVAX+αPD-1 group(Fig. 3B&C). It should be noted that the total number of CD8+ T cells was increased in the tumors treated with GVAX+αPD-1+αCD137 (Fig. 3A), suggesting that the decrease in the PD-1+ subtype of CD8+ T cells would not affect the overall number of effector T cells in these tumors as well as the number of PD-1- subtype of CD8+ T cells. By contrast, there was no substantial increase in the total number of PD-1+CD4+ T cells, nor the percent of CD4+ T cells that express PD-1, within the TME when mice were treated with the GVAX+αPD-1+αCD137 triple combination compared to the GVAX+αPD-1 double combination (Fig. 3E,F). These results suggest the survival benefit gained by adding αCD137 to the GVAX+αPD-1 combination treatment may be due to an increase in CD8+ T cell tumor infiltration, as well as a decrease in the percentage of CD8+ T cells that express PD-1.
3.4. Increased activated T cells and effector memory T cells were observed within the TME following treatment with GVAX, PD-1 antagonist and CD137 agonist
We further investigated the mechanistic effects of αCD137 on T cells within the TME by examining the expression of markers involved in T cell activation in the same liver metastasis murine KPC model. As shown in Fig. 3, the number and/or the percentage of CD8+ and CD4+ T cells expressing PD-1 increases following the GVAX+αPD-1 treatment compared to the GVAX alone or αPD-1 alone, suggesting that T cells induced by the GVAX+αPD-1 treatment may still exhibit a suppressed status. However, following the addition of αCD137, the normalized number of intratumoral CD4+PD-1+ T cells expressing costimulatory activation molecules, OX40 or CD137, increased in the GVAX+αPD-1+αCD137 treatment group compared to the GVAX+αPD-1 group or the GVAX+αCD137 group(Fig. 4A&B). Similarly, the number of CD8+PD-1+ T cells expressing OX40 or CD137 increased modestly when αPD-1 or αCD137 was added to GVAX, and increased significantly with the addition of both αCD137 and αPD-1 to GVAX (Fig. 4C&D). Although OX40 or CD137 was only expressed in a small percentage of CD4 or CD8 cells likely due to their transient expression nature, essentially all the OX40+ or CD137+ T cells were those PD-1+ T cells. Therefore, these results suggested that the combination of GVAX, αPD-1, and αCD137 enhances the activation of the PD-1+ T cells infiltrating the TME. Nevertheless, following treatment with GVAX, αPD-1, αCD137 or any of their combinations, we did not observe a statistically significant difference in the percentage of intratumoral CD8+PD-1+ or CD4+PD-1+ T cells expressing OX40 or CD137 among CD8+ or CD4+ T cells, respectively (Supplemental Fig. S5). This is likely due to the decrease of intratumoral CD8+PD-1+ or CD4+PD-1+ T cells in the αCD137-associated treatment groups.
Figure 4. The activation and memory status of T cells was enhanced following treatment with αCD137 agonist, GVAX and αPD-1.
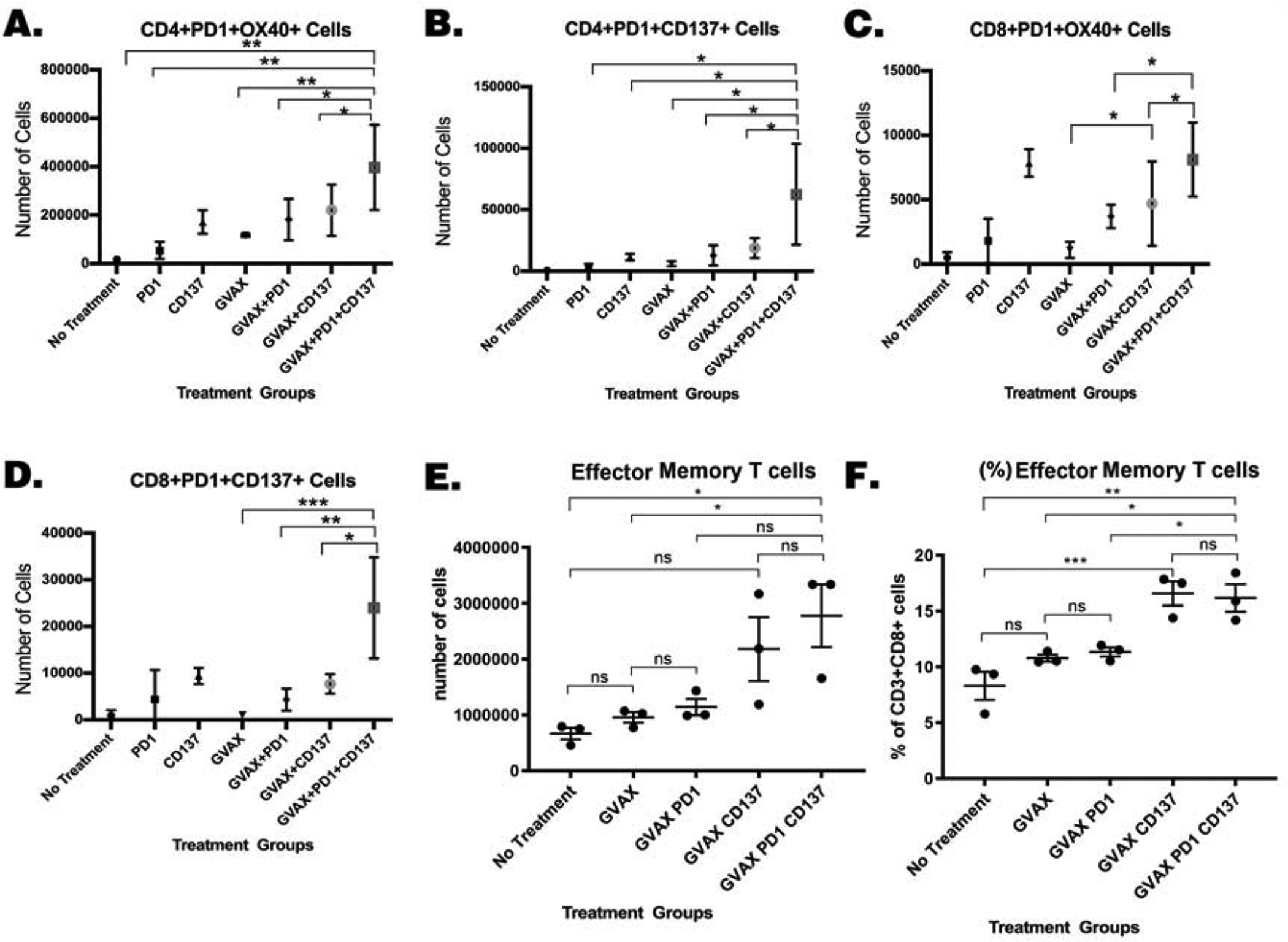
Mice were inoculated with 2×105 KPC cells via the hemispleen surgery, treated and sacrificed 14 days after KPC cell inoculation for isolation of tumor-infiltrating immune cells for flow cytometry analysis. Number of (A.) CD4+PD-1+OX40+, (B.) CD4+PD-1+CD137+, (C.) CD8+PD-1+OX40+ and (D.) CD8+PD-1+CD137+ T cells. Numbers of (E.) effector memory T-cells: CD8+CD44+ CD62L−CCR7− and percentages of (F.). effector memory T-cells among CD8+ T cells. Unpaired t tests were done. * p<0.05; ** p<0.01; *** p<0.001. Numbers of T cells were normalized by the total cell counts of the single cell suspension of the livers. Data represents mean ±SEM in one representative experiment (n=3 per treatment group per experiment, repeated twice).
To further investigate the mechanism of αCD137 in enhancing intratumoral T cell function, we examined the profile of naïve T cells by FACS surface markers (CD8+CD44−CD62L+CCR7+), central memory T cells by FACS surface markers (CD8+CD44+CD62L+CCR7+), and effector memory T cells by FACS surface marker (CD8+CD44+CD62L−CCR7−) as previously described [41]. Shown in Fig. 4E&F, αCD137-associated combination treatment groups, including the GVAX+αCD137 group and the GVAX+αPD-1+αCD137 group, had significantly higher percentages of intratumoral effector memory T cells among CD8+ T cells and had a trend toward higher normalized numbers of effector memory T cells infiltrating the TME comparing to the GVAX alone or GVAX+αPD-1 treatment groups. By contrast, the GVAX+αCD137 group and the GVAX+αPD-1+αCD137 group do not differ significantly (Fig. 4E&F); and αCD137 does not appear to influence the numbers and percentages of naïve T cells and central memory T cells among CD8+ T cells infiltrating the TME (Supplemental Fig. S5). These results suggest that CD137 agonist, but not PD-1 blockade, can enhance the effector memory T cell status in PDAC of this murine liver metastasis model.
3.5. The addition of αCD137 agonist increased expression of IFNγ in exhausted tumor-infiltrating T cells
To further investigate how the treatments with GVAX, αPD-1, and/or αCD137 play a role in the T cell exhaustion, we assessed the expression of T cell exhaustion marker [42], Eomes, on CD4+ and CD8+ T cells in the same liver metastasis murine KPC model. TILs isolated from liver metastases were stained for various surface and intracellular cell markers and analyzed by flow cytometry. The expression of Eomes on CD4+ T cells was in less than 2% of intratumoral CD4+ T cells at baseline (Supplemental Fig. S6E) and remained in low percentages in all treatment groups. The Eomes expression on CD8+ T cells was in approximately 10% of intratumoral CD8+ T cells at baseline and increased to approximately 23% in the GVAX+αCD137 treatment group (Supplemental Fig. S6F). Nevertheless, no statistically significant changes in Eomes expression were observed in CD4+ or CD8+ T cells among all treatment groups (Supplemental Fig. S6). Both the percentage of PD-1+Eomes+ CD4+ T cells among intratumoral CD4+ T cells and that of PD-1+Eomes+ CD8+ T cells among intratumoral CD8+ T cells were low among all treatment groups (Fig. 5A and Supplemental Fig. S6). Taken together, these results suggested that T cell exhaustion may be primarily mediated by non-Eomes mechanisms in the intratumoral CD4+PD-1+ or CD8+PD-1+ T cells that were induced by the GVAX or GVAX+αPD-1 treatments.
Figure 5. The addition of αCD137 agonist increased expression of IFNγ in exhausted tumor-infiltrating T cells.
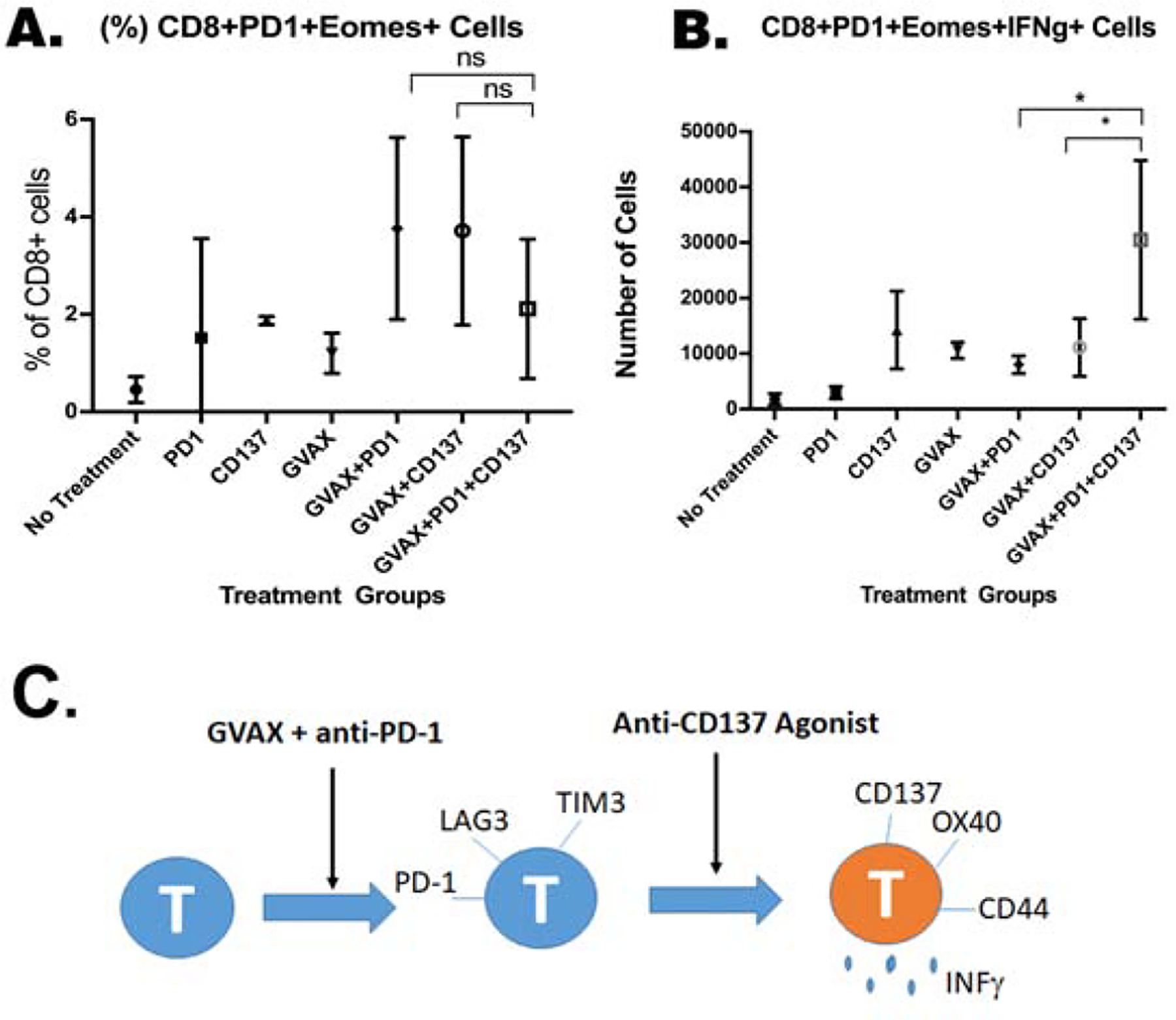
Mice were inoculated with 2×105 KPC cells via the hemispleen surgery, treated and sacrificed 14 days after KPC cell inoculation for isolation of tumor-infiltrating immune cells for flow cytometry analysis. (A.) Percentage of CD8+PD-1+Eomes+ T cells among CD8+ T cells. In a separate experiment, TILs were stimulated with mouse T-activator CD3/CD28 beads following isolation and then further characterized by flow cytometry. (B.) Number of IFNγ-expressing CD8+PD-1+Eomes+ T cells. Unpaired t tests were done. * p<0.05; ** p<0.01; ns, not significant. Numbers of T cells were normalized by the total cell counts of the single cell suspension of the livers. Data represents mean ±SEM in one representative experiment (n=3 per treatment group per experiment, repeated twice). (C,) A schematic model shows the role of anti-CD137 agonist antibody in regulating T cell function in pancreatic cancer.
To investigate how the treatments with GVAX, αPD-1, and/or αCD137 play a role in the effector T cell function, we assessed the ability of TILs from different treatment groups to produce IFNγ. TILs were stimulated with the CD3/CD28 T cell activation beads. Following activation, we determined the expression of IFNγ in T cells by flow cytometry. Consistent with our prior publication, the two GVAX+αPD-1 treatment groups including both the GVAX+αPD-1 and GVAX+αPD-1+αCD137 groups exhibited the highest numbers of intratumoral CD4+, CD4+PD-1+, CD4+PD-1+OX40+, CD8+, CD8+PD-1+, or CD8+PD-1+OX40+ T cells that express IFNγ among all treatment groups (Supplemental Fig. S7), suggesting that αCD137 does not play a major role in regulating the cytotoxic T cell function. Interestingly, even though the subgroup of exhausted CD8+ T cells, characterized by the expression of PD-1 and Eomes, is a minor subgroup as above suggested, the GVAX+αPD-1+αCD137 treatment significantly increased the IFNγ expression in this subgroup of exhausted CD8+ T cells comparing to any other treatment including GVAX+αPD-1 (Fig. 5B). Consistently, we found CD4+ T cells that express other T cell exhaustion markers LAG3 and TIM3 significantly decreased in PDAC treated by the GVAX+αPD-1+αCD137 treatment compared to the GVAX+αPD-1 treatment or the GVAX+αCD137 treatment (Supplemental Fig. S8). This result further suggested that combining αCD137 with the GVAX+αPD-1 treatment may play a role in re-invigorating the exhausted T cells (Fig. 5C). This mechanism may also account for the increased survival of PDAC-bearing mice following the GVAX+αPD-1+αCD137 treatment.
4. Discussion
This study demonstrated that through the rational combination of the αCD137 agonist antibody with GVAX and αPD-1 blocking antibody, survival was prolonged significantly in a murine model of metastatic PDAC. Among T cell modulating agents tested in this study, the αCD137 appears to be the only immune-oncology agent that adds significant benefit on top of the combination of GVAX+αPD-1 in our preclinical studies of PDAC. This study further established the mechanism of targeting CD137 to enhance the effector memory and activation status of T cells in PDAC in a vaccine therapy-dependent manner. Through human PDAC specimen analysis, this study demonstrated that PDACs with higher densities of CD8+ T cells including exhausted T cells following the GVAX treatment is associated with higher density of CD137+ cells, making CD137 a potential target to enhance the efficacy of GVAX.
Thus, a new arm of testing the combination of GVAX, Nivolumab and Urelumab (human αCD137 agonist antibody) has been added to the ongoing clinical trial of neoadjuvant immunotherapy for resectable PDACs (NCT02451982). Till today, nine patients were enrolled to the new cohort and treated with the combination of GVAX, Nivolumab and Urelumab. All nine evaluable patients, after receiving one combination treatment of GVAX, Nivolumab and Urelumab, underwent surgical resection of their PDACs successfully. Although toxicity, particularly the liver toxicity, is a concern for the clinical development of CD137 agonist[43], the liver toxicity has not been observed in this cohort of resectable PDAC patients (Zheng et al. Late Breaking Abstract #812, the 35th Annual Meeting of the Society of ImmunoTherapy for Cancer in 2020). It is possible that resectable PDAC, which is not associated with liver metastasis, would less likely develop liver toxicity with the CD137 agonist treatment. Therefore, localized PDAC may be more suitable for the αCD137-based combination therapies. Nevertheless, the liver metastases-bearing mice in this study also did not demonstrate any sign of toxicity with αCD137-based treatments. Therefore, further clinical testing of αCD137-based treatments in different stages of PDAC is warranted.
In this preclinical study, further investigation into the status of effector T cells in the TME revealed increased expression of CD137 and OX40 co-stimulatory molecules on PD-1+CD4+ and PD-1+CD8+ T cells when αCD137 was added. Belonging to the TNFR superfamily, CD137 and OX40 are able to stimulate and generate survival signals in effector T cells [23, 44, 45]. The increase in CD137 and OX40 within the tumor of mice that received GVAX+αCD137+αPD-1 therapy suggested that their survival benefit was due to an enhanced T cell activation in addition to the quantitative increase of effector T cells. Consistently, αCD137 appears to be responsible for the increase of effector memory T cells in the PDAC tumors. Interestingly, the percentage of Eomes+ cells among PD-1+CD8+ T cells was low and not statistically different between the various treatment groups, suggesting that Eomes is not a major T cell exhaustion pathway in this model. Nevertheless, the percentage of the Eomes+PD-1+CD8+ T cells in the TME that express IFNγ was significantly increased by the treatment with the GVAX+αPD-1+αCD137 triple combination than any other combination treatment. This result suggested that even the T cells with an exhaustion phenotype were receiving activation signals from αCD137 and other downstream factors elicited by the triple combination therapy to sustain an activated status and maintain a high cytotoxic T cell function.
Taken together, this study suggests that the addition of αCD137 to GVAX +αPD-1 results in a more sustained activation status and memory status in effector T cells. It is possible that αCD137 enhances the trafficking of activated, effector memory T cells into the TME. More likely, the combination of αCD137 and αPD-1 not only re-invigorates the intratumoral effector T cells, which were induced by GVAX and would also be in an exhausted status due to the induction of PD-L1[15], but also converts the status of these effector T cells into a sustained activation and memory status. This study has several limitations. First, in the mouse model used in this study, PDAC tumors were implanted although the resulted liver metastases were spontaneously developed. Second, metastases formed diffusely in the liver and thus were not separable for isolation. We cannot exclude that a small portion of immune cells in the single cell suspension of the liver were resident normal immune cells. Third, the sample size in each treatment group within one individual experiment was kept small due to the large time consumption of processing tumor infiltrating immune cells. However, each experiment was repeated at least twice and was found to be reproducible. Nevertheless, the aforementioned ongoing clinical trial (NCT02451982), which is testing the combination of Nivolumab, αCD137 antibody Urelumab, and GVAX in surgically resectable PDACs, will provide an opportunity of validating the mechanistic role of CD137 agonist as a part of this combination therapy to enhance the effector memory and activation status of T cells in human PDACs.
Supplementary Material
Highlights.
CD137 agonist, PD-1 antagonist, and vaccine form a rational combination for pancreatic cancer
Targeting CD137 increases activated, effector memory T cells in pancreatic cancer
Targeting CD137 converts T cells from an exhausted status to an activated status
The combination of CD137 agonist and PD-1 blockade re-invigorates effector T cells
Acknowledgement
This study is supported by a BMS II-ON grant (LZ), NIH grant R01 CA169702 (LZ), NIH grant R01 CA197296 (LZ), The Viragh Foundation and the Skip Viragh Pancreatic Cancer Center at Johns Hopkins (LZ), National Cancer Institute Specialized Programs of Research Excellence in Gastrointestinal Cancers grant P50 CA062924 (LZ), Sidney Kimmel Comprehensive Cancer Center grant P30 CA006973 (LZ), NIH T32 CA126607 (ABB, LZ), ASCO Young Investigator Award (MTS) and NIH T32 CA 9071-36 (MTS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
LZ receives grant support from Bristol-Meyer Squibb, Merck, Astrazeneca, iTeos, Amgen, NovaRock, Inxmed, and Halozyme. LZ is a paid consultant/Advisory Board Member at Biosion, Alphamab, NovaRock, Akrevia/ Xilio, Datarevive, QED, Natera, and Mingruizhiyao. LZ holds shares at Alphamab and Mingruizhiyao. No potential conflicts of interest were disclosed by the other authors.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References:
- [1].Siegel RL, Miller KD, Jemal A, Cancer statistics, 2019, CA Cancer J Clin, 69 (2019) 7–34. [DOI] [PubMed] [Google Scholar]
- [2].Wolfgang CL, Herman JM, Laheru DA, Klein AP, Erdek MA, Fishman EK, Hruban RH, Recent progress in pancreatic cancer, CA Cancer J Clin, 63 (2013) 318–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kang MJ, Jang JY, Kim SW, Surgical resection of pancreatic head cancer: What is the optimal extent of surgery?, Cancer Lett, 382 (2016) 259–265. [DOI] [PubMed] [Google Scholar]
- [4].Rucki AA, Zheng L, Pancreatic cancer stroma: understanding biology leads to new therapeutic strategies, World J Gastroenterol, 20 (2014) 2237–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Takahashi K, Ehata S, Koinuma D, Morishita Y, Soda M, Mano H, Miyazono K, Pancreatic tumor microenvironment confers highly malignant properties on pancreatic cancer cells, Oncogene, 37 (2018) 2757–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lunardi S, Muschel RJ, Brunner TB, The stromal compartments in pancreatic cancer: are there any therapeutic targets?, Cancer Lett, 343 (2014) 147–155. [DOI] [PubMed] [Google Scholar]
- [7].Kota J, Hancock J, Kwon J, Korc M, Pancreatic cancer: Stroma and its current and emerging targeted therapies, Cancer Lett, 391 (2017) 38–49. [DOI] [PubMed] [Google Scholar]
- [8].Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, Wigginton JM, Safety and activity of anti-PD-L1 antibody in patients with advanced cancer, N Engl J Med, 366 (2012) 2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Feng M, Xiong G, Cao Z, Yang G, Zheng S, Song X, You L, Zheng L, Zhang T, Zhao Y, PD-1/PD-L1 and immunotherapy for pancreatic cancer, Cancer Lett, 407 (2017) 57–65. [DOI] [PubMed] [Google Scholar]
- [10].Kleponis J, Skelton R, Zheng L, Fueling the engine and releasing the break: combinational therapy of cancer vaccines and immune checkpoint inhibitors, Cancer Biol Med, 12 (2015) 201–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, Sherry RM, Topalian SL, Yang JC, Lowy I, Rosenberg SA, Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma, J Immunother, 33 (2010) 828–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Banerjee K, Kumar S, Ross KA, Gautam S, Poelaert B, Nasser MW, Aithal A, Bhatia R, Wannemuehler MJ, Narasimhan B, Solheim JC, Batra SK, Jain M, Emerging trends in the immunotherapy of pancreatic cancer, Cancer Lett, 417 (2018) 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Foley K, Kim V, Jaffee E, Zheng L, Current progress in immunotherapy for pancreatic cancer, Cancer Lett, 381 (2016) 244–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jaffee EM, Hruban RH, Biedrzycki B, Laheru D, Schepers K, Sauter PR, Goemann M, Coleman J, Grochow L, Donehower RC, Lillemoe KD, O’Reilly S, Abrams RA, Pardoll DM, Cameron JL, Yeo CJ, Novel allogeneic granulocyte-macrophage colony-stimulating factor-secreting tumor vaccine for pancreatic cancer: a phase I trial of safety and immune activation, J Clin Oncol, 19 (2001) 145–156. [DOI] [PubMed] [Google Scholar]
- [15].Lutz ER, Wu AA, Bigelow E, Sharma R, Mo G, Soares K, Solt S, Dorman A, Wamwea A, Yager A, Laheru D, Wolfgang CL, Wang J, Hruban RH, Anders RA, Jaffee EM, Zheng L, Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation, Cancer Immunol Res, 2 (2014) 616–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Soares KC, Rucki AA, Wu AA, Olino K, Xiao Q, Chai Y, Wamwea A, Bigelow E, Lutz E, Liu L, Yao S, Anders RA, Laheru D, Wolfgang CL, Edil BH, Schulick RD, Jaffee EM, Zheng L, PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors, J Immunother, 38 (2015) 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Popovic A, Jaffee EM, Zaidi N, Emerging strategies for combination checkpoint modulators in cancer immunotherapy, J Clin Invest, 128 (2018) 3209–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tsujikawa T, Kumar S, Borkar RN, Azimi V, Thibault G, Chang YH, Balter A, Kawashima R, Choe G, Sauer D, El Rassi E, Clayburgh DR, Kulesz-Martin MF, Lutz ER, Zheng L, Jaffee EM, Leyshock P, Margolin AA, Mori M, Gray JW, Flint PW, Coussens LM, Quantitative Multiplex Immunohistochemistry Reveals Myeloid-Inflamed Tumor-Immune Complexity Associated with Poor Prognosis, Cell Rep, 19 (2017) 203–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chen L, Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity, Nat Rev Immunol, 4 (2004) 336–347. [DOI] [PubMed] [Google Scholar]
- [20].Andrews LP, Marciscano AE, Drake CG, Vignali DA, LAG3 (CD223) as a cancer immunotherapy target, Immunol Rev, 276 (2017) 80–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Turnis ME, Andrews LP, Vignali DA, Inhibitory receptors as targets for cancer immunotherapy, Eur J Immunol, 45 (2015) 1892–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bauer C, Kuhnemuth B, Duewell P, Ormanns S, Gress T, Schnurr M, Prevailing over T cell exhaustion: New developments in the immunotherapy of pancreatic cancer, Cancer Lett, 381 (2016) 259–268. [DOI] [PubMed] [Google Scholar]
- [23].Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC, Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity, J Exp Med, 207 (2010) 2187–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Osipov A, Murphy A, Zheng L, From immune checkpoints to vaccines: The past, present and future of cancer immunotherapy, Adv Cancer Res, 143 (2019) 63–144. [DOI] [PubMed] [Google Scholar]
- [25].Shuford WW, Klussman K, Tritchler DD, Loo DT, Chalupny J, Siadak AW, Brown TJ, Emswiler J, Raecho H, Larsen CP, Pearson TC, Ledbetter JA, Aruffo A, Mittler RS, 4-1BB costimulatory signals preferentially induce CD8+ T cell proliferation and lead to the amplification in vivo of cytotoxic T cell responses, J Exp Med, 186 (1997) 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ye Q, Song DG, Poussin M, Yamamoto T, Best A, Li C, Coukos G, Powell DJ Jr., CD137 accurately identifies and enriches for naturally occurring tumor-reactive T cells in tumor, Clin Cancer Res, 20 (2014) 44–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wolfl M, Kuball J, Ho WY, Nguyen H, Manley TJ, Bleakley M, Greenberg PD, Activation-induced expression of CD137 permits detection, isolation, and expansion of the full repertoire of CD8+ T cells responding to antigen without requiring knowledge of epitope specificities, Blood, 110 (2007) 201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sakellariou-Thompson D, Forget MA, Creasy C, Bernard V, Zhao L, Kim YU, Hurd MW, Uraoka N, Parra ER, Kang Y, Bristow CA, Rodriguez-Canales J, Fleming JB, Varadhachary G, Javle M, Overman MJ, Alvarez HA, Heffernan TP, Zhang J, Hwu P, Maitra A, Haymaker C, Bernatchez C, 4-1BB Agonist Focuses CD8(+) Tumor-Infiltrating T-Cell Growth into a Distinct Repertoire Capable of Tumor Recognition in Pancreatic Cancer, Clin Cancer Res, 23 (2017) 7263–7275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Foley K, Rucki AA, Xiao Q, Zhou D, Leubner A, Mo G, Kleponis J, Wu AA, Sharma R, Jiang Q, Anders RA, Iacobuzio-Donahue CA, Hajjar KA, Maitra A, Jaffee EM, Zheng L, Semaphorin 3D autocrine signaling mediates the metastatic role of annexin A2 in pancreatic cancer, Sci Signal, 8 (2015) ra77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Soares KC, Foley K, Olino K, Leubner A, Mayo SC, Jain A, Jaffee E, Schulick RD, Yoshimura K, Edil B, Zheng L, A preclinical murine model of hepatic metastases, J Vis Exp, (2014) 51677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD, Peggs KS, Ravetch JV, Allison JP, Quezada SA, Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma, J Exp Med, 210 (2013) 1695–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Blair AB, Kleponis J, Thomas DL 2nd, Muth ST, Murphy AG, Kim V, Zheng L, IDO1 inhibition potentiates vaccine-induced immunity against pancreatic adenocarcinoma, J Clin Invest, 129 (2019) 1742–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Soares KC, Rucki AA, Kim V, Foley K, Solt S, Wolfgang CL, Jaffee EM, Zheng L, TGF-beta blockade depletes T regulatory cells from metastatic pancreatic tumors in a vaccine dependent manner, Oncotarget, 6 (2015) 43005–43015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Blair AB, Kim VM, Muth ST, Saung MT, Lokker N, Blouw B, Armstrong TD, Jaffee EM, Tsujikawa T, Coussens LM, He J, Burkhart RA, Wolfgang CL, Zheng L, Dissecting the Stromal Signaling and Regulation of Myeloid Cells and Memory Effector T Cells in Pancreatic Cancer, Clin Cancer Res, 25 (2019) 5351–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Saung MT, Muth S, Ding D, Thomas DL 2nd, Blair AB, Tsujikawa T, Coussens L, Jaffee EM, Zheng L, Targeting myeloid-inflamed tumor with anti-CSF-1R antibody expands CD137+ effector T-cells in the murine model of pancreatic cancer, J Immunother Cancer, 6 (2018) 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Leao IC, Ganesan P, Armstrong TD, Jaffee EM, Effective Depletion of Regulatory T Cells Allows the Recruitment of Mesothelin-Specific CD8 T Cells to the Antitumor Immune Response Against a Mesothelin-Expressing Mouse Pancreatic Adenocarcinoma, Clin Transl Sci, 1 (2008) 228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Adachi M, Taki T, Huang C, Higashiyama M, Doi O, Tsuji T, Miyake M, Reduced integrin alpha3 expression as a factor of poor prognosis of patients with adenocarcinoma of the lung, J Clin Oncol, 16 (1998) 1060–1067. [DOI] [PubMed] [Google Scholar]
- [38].Muller CB, de Barros RL, Castro MA, Lopes FM, Meurer RT, Roehe A, Mazzini G, Ulbrich-Kulczynski JM, Dal-Pizzol F, Fernandes MC, Moreira JC, Xavier LL, Klamt F, Validation of cofilin-1 as a biomarker in non-small cell lung cancer: application of quantitative method in a retrospective cohort, J Cancer Res Clin Oncol, 137 (2011) 1309–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Blair AB, Kim VM, Muth ST, Saung MT, Lokker N, Blouw B, Armstrong TD, Jaffee EM, Tsujikawa T, Coussens LM, He J, Burkhart RA, Wolfgang CL, Zheng L, Dissecting the Stromal Signaling and Regulation of Myeloid Cells and Memory Effector T Cells in Pancreatic Cancer, Clin Cancer Res, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, Waterhouse D, Ready N, Gainor J, Aren Frontera O, Havel L, Steins M, Garassino MC, Aerts JG, Domine M, Paz-Ares L, Reck M, Baudelet C, Harbison CT, Lestini B, Spigel DR, Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer, N Engl J Med, 373 (2015) 123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Sallusto F, Geginat J, Lanzavecchia A, Central memory and effector memory T cell subsets: function, generation, and maintenance, Annu Rev Immunol, 22 (2004) 745–763. [DOI] [PubMed] [Google Scholar]
- [42].Probst S, Arnold SJ, Eomesodermin-At Dawn of Cell Fate Decisions During Early Embryogenesis, Curr Top Dev Biol, 122 (2017) 93–115. [DOI] [PubMed] [Google Scholar]
- [43].Qi X, Li F, Wu Y, Cheng C, Han P, Wang J, Yang X, Optimization of 4-1BB antibody for cancer immunotherapy by balancing agonistic strength with FcgammaR affinity, Nat Commun, 10 (2019) 2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lee SJ, Rossi RJ, Lee SK, Croft M, Kwon BS, Mittler RS, Vella AT, CD134 Costimulation Couples the CD137 Pathway to Induce Production of Supereffector CD8 T Cells That Become IL-7 Dependent, J Immunol, 179 (2007) 2203–2214. [DOI] [PubMed] [Google Scholar]
- [45].Virani NA, Thavathiru E, McKernan P, Moore K, Benbrook DM, Harrison RG, Anti-CD73 and anti-OX40 immunotherapy coupled with a novel biocompatible enzyme prodrug system for the treatment of recurrent, metastatic ovarian cancer, Cancer Lett, 425 (2018) 174–182. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.