

Abstract
In lung cancer, CD133+ cells represent the subset of cancer stem cells (CSC) able to sustain tumor growth and metastatic dissemination. CSC function is tightly regulated by specialized niches composed of both stromal cells and extracellular matrix (ECM) proteins, mainly represented by collagen. The relevance of collagen glycosylation, a fundamental post‐translational modification controlling several biological processes, in regulating tumor cell phenotype remains, however, largely unexplored. To investigate the bioactive effects of differential ECM glycosylation on lung cancer cells, we prepared collagen films functionalized with glucose (Glc‐collagen) and galactose (Gal‐collagen) exploiting a neoglycosylation approach based on a reductive amination of maltose and lactose with the amino residues of collagen lysines. We demonstrate that culturing of tumor cells on collagen determines a glycosylation‐dependent positive selection of CSC and triggers their expansion/generation. The functional relevance of CD133+ CSC increase was validated in vivo, proving an augmented tumorigenic and metastatic potential. High expression of integrin β1 in its active form is associated with an increased proficiency of tumor cells to sense signaling from glycosylated matrices (glyco‐collagen) and to acquire stemness features. Accordingly, inhibition of integrin β1 in tumor cells prevents CSC enrichment, suggesting that binding of integrin β1 to Glc‐collagen subtends CSC expansion/generation. We provide evidence suggesting that collagen glycosylation could play an essential role in modulating the creation of a niche favorable for the generation and selection/survival of lung CSC. Interfering with this crosstalk may represent an innovative therapeutic strategy for lung cancer treatment.
Keywords: cancer stem cells, collagen, glycans, lung cancer, tumor–extracellular matrix interactions
Glycans are well known for their involvement in recognition phenomena between cells; however, the role of small glycan epitopes in cell–extracellular matrix (ECM) interactions needs to be further elucidated. Here we exploit bioactive ECM mimetics, functionalizing type I collagen films with different glycans, to investigate the relevance of specific ECM glycosignatures in tumor‐ECM interactions. We show that in vitro culturing of lung cancer cells on glycosylated collagen films results in differential modulation of cancer stem cells (CSC), associated in vivo with an enhanced tumor initiation ability and increased metastatic potential. Interactions of CSC with glycosylated collagen are regulated through the active form of integrin β1. Interfering with integrin β1 signaling results in abrogation of CSC enrichment induced by glycosylated collagen.

Abbreviations
- CAF
cancer associated fibroblast
- CSC
cancer stem cell
- ECM
extracellular matrix
- ELDA software
extreme limiting dilution analysis
- EMT
epithelial‐to‐mesenchymal transition
- Gal
galactose
- Glc
glucose
- ITGB1
integrin β1
- MIC
metastasis‐initiating cells
- NSCLC
non‐small cell lung cancer
1. INTRODUCTION
Contribution of the microenvironment in its cellular and non‐cellular components including the ECM has been described as crucially involved in tumor development. 1
In particular, the ECM has a prominent role in dictating tumor progression and cancer cell fate through multiple mechanisms involving both biomechanical and physical properties. 2 , 3 , 4 , 5 In fact, ECM‐driven signaling relies on the interactions of ECM components with cell receptors and on mechanosensing, 6 , 7 , 8 which can both control cancer cell differentiation, proliferation and migration. 9 Collagen represents the main structural protein in the ECM, and its modifications in terms of deposition and post‐translational modifications impose profound changes in ECM features. 10 , 11 Indeed, matrix stiffening induced by increased collagen deposition and cross‐linking has been shown to promote malignant transformation in breast cancer. 12
ECM is also crucial for the maintenance of stemness properties, preservation of cell polarity and regulation of asymmetric cell division. 13 Similar to normal stem cells, CSC, which are operationally defined as the cancer cell subset endowed with stem‐like properties that can drive and sustain tumor formation, 14 also rely on specific niches which control their self‐renewal and differentiation. 15
In lung cancer, CD133+ cells represent the CSC and, within this subset, MIC have been recently defined as CD133+ CXCR4+ EpCAM– cells. 16 , 17 Stimuli from the tumor microenvironment are able to modulate this specific subset; 17 , 18 however, little information is available on the contribution of the ECM proteins and their post‐translational modification, in particular glycosylation, in dictating tumor behavior. Interestingly, some emerging evidence suggests that investigation of glycosylation in the tumor microenvironment may be crucial to advance the comprehension of cancer biology, disease progression and dissemination processes. 19 , 20
Glycosylation is critical for a wide range of biological processes including maintenance of tissue homeostasis, cancer progression and ECM remodeling. 21 , 22 Glycosylation events also regulate in bidirectional ways interactions between integrins and ECM proteins. 23
The role of ECM glycosignatures in mediating tumor cells–ECM interactions therefore appears of crucial importance.
Here, we used glucose and galactose‐functionalized collagen matrices (Glc‐collagen and Gal‐collagen) to mimic different glycosylation patterns and to better understand the role of ECM glycosignatures in the regulation of carcinogenesis and metastasis by regulating CSC phenotypes in lung cancer.
2. MATERIALS AND METHODS
2.1. Collagen preparation and neoglycosylation
Type I collagen films from bovine Achilles tendon (Sigma‐Aldrich, catalog no. C9879) were produced as previously reported. 24 Neoglycosylation of collagen films was carried out as previously reported. 24 , 25 Briefly, to obtain Glc‐collagen, 342 mg maltose was dissolved in 20 mL citrate buffer pH 6.00 followed by sequential addition of NaBH3CN. The solution was added to the collagen film (80 mg, 12 × 7 cm) and reacted overnight. Collagen film was washed with 40 mL H2O mQ for 15 minutes (×3 times), and finally with 20 mL EtOH for 20 minutes. The same protocol was used for the functionalization of the collagen films with lactose to obtain Gal‐collagen (Figure S1). 24 , 25
2.2. Cell culture on collagen films
Cell lines A549 lung adenocarcinoma, H460 large cell lung carcinoma (purchased from ATCC) and LT73 primary lung adenocarcinoma (established in our lab), were all cultured in RPMI medium supplemented with 10% fetal bovine serum‐FBS (all from Lonza) at 37°C under 5% CO2.
Collagen films were cut using a scalpel to adapt them to the size of the 6‐well plates, sterilized under UV light and rehydrated with complete medium for 2 hours before cancer cell seeding at 5 × 105 cells/well. Cells seeded on pristine (non‐functionalized) collagen or on plastic 6‐well plates were used as control. Cells were then harvested by trypsinization for downstream experiments.
For siRNA transfection, 5 × 105 tumor cells were seeded in a 6‐well plate and, the day after, Opti‐MEM + Lipofectamine 2000 (ThermoFisher) containing 20 μM esiRNA1 ITGB1 (EHU065071) or esiRNA Universal Negative Control (both from Sigma) were added. Cells were then plated on collagen films 48 hours after transfection.
For integrin β1‐blocking experiments, cells were incubated for 15 minutes at 37°C with anti‐Integrin β1 antibody 50 μg/mL (clone AIIB2 MABT409; Millipore), seeded on collagen matrices and analyzed after 4 hours.
2.3. Flow cytometry analysis
List of anti‐human antibodies: PE‐CD133/1 (Clone AC133, Miltenyi Biotec), APC‐CXCR4 (Clone 12G5), FITC‐CD44 (Clone L178), PE‐CD166 (Clone 3A6) (all from BD Bioscience) and CD29‐APC (clone TS2/16, BioLegend) were incubated with cells for 30 minutes at 4°C.
Integrin β1 antibodies: anti‐mouse CD29 (Clone 9EG7‐550531 recognizing the active form of integrin β1), anti‐human CD49b (Clone 12F1‐555668), anti‐human CD49a (clone SR84 559 594) (all from BD Bioscience) were incubated with cells for 1 hour at room temperature (RT), then washed and incubated with secondary antibody (anti‐rat IgG2 Alexa Fluor 488 and/or anti‐mouse IgG1, Alexa Fluor 647, Thermo Fisher), for 30 minutes at RT.
Prior to FACS analysis, samples were incubated with 7‐AAD viability staining solution (10 μL/tube) (eBioscience) to exclude dead cells.
FACSCalibur and FACSCanto cell analyzers (Becton Dickinson) were used for data acquisition and FlowJo software V10 was used for data analysis.
LT73 CD133neg cell line was generated by sorting CD133 negative cells from the parental LT73 cell line using FACSAria cell sorter (BD Biosciences), as already reported. 17
Human disseminated tumor cells (DTC) were quantified in dissociated murine lung tissue, as 7AAD‐ live cells negative for anti‐mouse MHC class I (eBioscience), and their relative content of CD133+ CXCR4+ MIC was evaluated, as already detailed. 17
2.4. Cell cycle and apoptosis analysis
Single cell suspensions were incubated for 10 minutes in Flow Cytometry Fixation Buffer 1X (R&D Systems) at RT and then permeabilized with Triton solution 1X for 30 minutes. The samples were then incubated in a solution of propidium iodide (PI) 200 ng/mL and RNase 10 µg/mL (Sigma‐Aldrich) for 45 minutes and analyzed by FACS.
For analysis of apoptotic cells, cell lines cultured on collagen matrices and plastic plates for 72 hours were stained with Annexin V and PI, using the Annexin V‐FITC Kit (Miltenyi Biotec). Apoptotic cells were identified as Annexin V positive PI negative.
2.5. Immunofluorescence
Cells were plated onto an 8‐well chamber slide (Nunc Lab‐Tek), at 5 × 104 cells/well. After 24 hours, cells were fixed with Paraformaldehyde 4%, permeabilized with Triton X‐100 0.2%, blocked with BSA 2% + 3% NGS (normal goat serum) solution for 30 minutes. Cells were then incubated with rat anti‐mouse CD29 (Clone 9EG7‐BD), for 1 hour at RT, and subsequently with goat anti‐rat Alexa Fluor IgG 555 (Abcam) secondary antibody, for 45 minutes at RT. Slides were mounted with VECTASHIELD Mounting Medium, containing DAPI (Vector Laboratories) and visualized under a fluorescence microscope.
2.6. PKH‐67 labeling
A549, LT73 and H460 cells were labeled with the PKH‐67 Fluorescent Cell Linker Kit (Sigma) according to the manufacturer’s instructions. The labeling ratio was determined by flow cytometry and monitored during cell passages until small and stable fractions of PKH‐positive and PKH‐negative cells were reached, distinctive for each tested cell line.
2.7. Migration assay
Cells (50 000) previously cultured on collagen films were seeded in RPMI‐1640 1% FBS on 8 μm‐pore Transwell® cell culture inserts (BD Falcon) in 24‐well plates. The lower chamber was filled with 500 µL RPMI + 10% FBS as chemo‐attractant. After 48 hours, cells on the top of the insert were removed by cotton swab and migrated cells in the lower face of the insert were fixed with methanol and mounted on slides using the VECTASHIELD Mounting Medium containing DAPI (Vector Laboratories). For each insert, cells in four random fields were counted by fluorescence microscope visualization at 20× magnification and the values were averaged.
2.8. In vivo tumorigenic assay and limiting dilution assay
In vivo experiments were approved by the Ethics Committee for Animal Experimentation of the Fondazione IRCCS Istituto Nazionale dei Tumori, according to EU Directive 2010/63/EU.
LT73 cells were cultured for 72 hours on films of pristine collagen or Glc‐collagen, prior to sc injection in SCID mice with Matrigel (Corning; ratio 1:1) at 1 × 103, 1 × 104, 1 × 105 cells. At the end of the observation period (35 days), we evaluated tumor take as presence of growing tumor (≥100 mg). ELDA software was used to estimate the frequency of CSC in the different groups providing confidence intervals for 1/(stem cell frequency). Differences between groups in CSC frequency were estimated by chi‐squared test.
2.9. Statistical analysis
Data represent the means ± standard deviation or error (SD/SE) of replicate experiments (n = as indicated in figure legends). Statistically significant differences were determined with Student’s t tests when comparing two groups or one‐way ANOVA with Tukey post‐hoc test or two‐way ANOVA with Bonferroni post‐hoc test for multiple comparisons. Analyses were then carried out.
3. RESULTS
3.1. Cancer stem cells are selected on neoglucosylated collagen matrices
We previously reported transcriptional analysis of lung CAF showing that biological pathways linked to collagen biosynthesis and remodeling are significantly enriched in activated and pro‐tumorigenic fibroblasts. 26 These data led us to speculate on a direct involvement of collagen modifications in the regulation of tumor cell properties and of stemness phenotype.
To evaluate the effect of collagen glycosylation on the modulation of the cell subset responsible for cancer initiation (ie CSC), synthetic collagen matrices conjugated with glucose (Glc‐collagen) and galactose (Gal‐collagen) were used (Figure S1). 24 , 25 Glc‐ and Gal‐collagen matrices were obtained through a two‐step process that involves preparation of the collagen film as a first step and then functionalization with α‐glucose and β‐galactose by a reductive amination protocol with the corresponding maltose and lactose disaccharides. The collagen matrices were produced using insoluble bovine type I collagen from Achilles tendon, existing as elongated fibrils including telopeptides, used as gold standard for tissue engineering purposes and collagenase assays as a result of its ability to preserve the in vivo collagen features. Considering the insolubility of the starting powdered collagen, the films were produced by mechanical homogenization in acidic conditions and the final 2D insoluble matrices were obtained by solvent casting method with a tailored thickness of 700 nm. Conjugation of glycans in the heterogeneous phase allowed functionalization of the collagen surface, obtaining 20 nM saccharide/cm2 and maintenance of the same secondary structure of untreated collagen films, as characterized by Fourier‐transformed infrared spectroscopy (FT‐IR). Characterization of collagen matrices by atomic force microscopy (AFM) demonstrated that the conjugation step did not affect the structural organization and aggregation of collagen films. However, whereas the unmodified collagen film showed an amorphous surface with fewer organized fibrils, conjugations of glycans resulted in more organized structures depending on the conjugate glycan epitopes. Due to maintenance of the collagen secondary structure, stability of the film in aqueous condition and the ability to control the glycan exposition at the surface level only, the glycosylated matrices were proven to be affordable tools to study the effect of specific glycosignatures of collagen on different cell lines. 24 , 25
Following previous results in which differential neoglycosylation of collagen matrices has shown cell line‐specific effects, we tested different NSCLC cell lines (A549, LT73 and H460), initially culturing cancer cells on pristine/glyco‐collagen matrices for 72 hours (Figure 1A) and then analyzing the modulation of CSC subsets. Cells adherent to collagen matrices showed a more elongated fibroblast‐like phenotype compared to cells cultured on plastic plates. In particular, for each tested cell line we observed the presence of floating rounded cells unable to bind matrices, especially in glyco‐collagen, suggesting that collagen matrices may be selective for the binding of specific cancer cell subsets.
Figure 1.
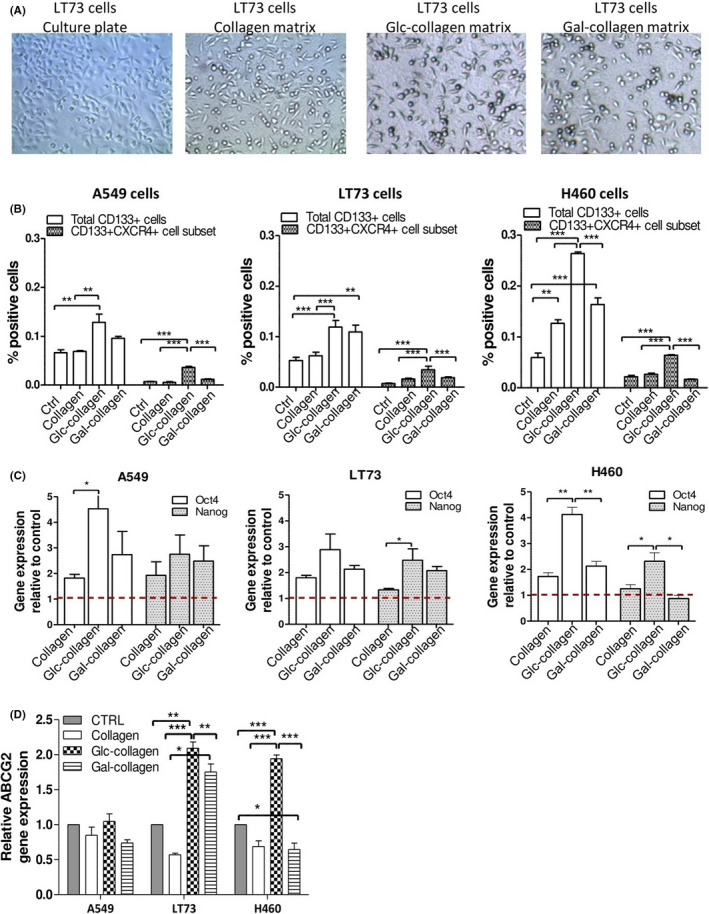
Collagen films induce cancer stem cell (CSC) enrichment. A, Morphology of LT73 cancer cells grown on culture plates, pristine collagen matrices and Glc‐collagen and Gal‐collagen matrices respectively (72 h). B, FACS analysis for the expression of CD133 and CXCR4 in A549, LT73 and H460 cell lines cultured on plastic culture plates (Control), pristine and Glyco‐collagens for 72 h. Mean ± SEM of n = 4 experiments are shown. One‐way ANOVA was used for statistical analyses; ***P < 0.001, **P < 0.01. C, Relative expression of Oct4/Nanog stemness genes and (D) ABCG2 drug transporter in A549, LT73 and H460 cell lines cultured on different collagen films for 72 h. Cells cultured on plastic culture plates were used as calibrator. Bars represent the mean fold‐change of n = 3 independent experiments. ANOVA was used for statistical analyses; *P < 0.05; **<0.01
In all tested cell lines, we observed that cancer cells grown on collagen films were generally enriched for the fraction of CD133+ cells compared to cells cultured on plastic culture plates. Notably, this increase was markedly evident when cells were cultured on Gal‐collagen (1.9 fold‐change; P = 0.0029) and especially on Glc‐collagen (2,7 fold‐change compared to control; P < 0.0001) (Figure 1B). In particular, Glc‐collagen was the most proficient at increasing the subset of CD133+ CXCR4+ MIC (2,8 fold‐change compared to control; P < 0.0001; Figure 1B and Figure S2). We also assessed the modulation of other stemness markers known to identify subsets of CSC in NSCLC, such as CD144 27 , 28 and CD166. 29 Notably, we confirmed that collagen film was able to select and enrich for the fraction of CD44+ cells (2,1 fold‐change; P = 0.04) and CD166+ cells (1,5 fold‐change; P = 0.03) compared to control. Glc‐collagen showed an even higher ability to enrich for CD44 and CD166 positive cells (respectively 2,5 and 1,5 fold‐change; P = 0.001 and P = 0.02), even if no statistical difference was observed among collagen and glyco‐collagen, suggesting that enrichment for these cell subsets may be mainly driven by collagen binding rather than related to matrix glycosylation (Figure S3).
The effect on the CSC population induced by collagen films was also confirmed by upregulation of two stemness‐related genes (Oct4/Nanog), coherently with the differential modulation of CD133+ CSC induced by glyco‐collagen (Figure 1C). Interestingly, differential modulation of MIC caused by collagen matrices was also confirmed by a similar trend in gene expression modulation of ABCG2 (Figure 1D), a drug transporter that we previously reported to be particularly expressed by chemoresistant MIC. 16 Altogether, these data indicate that glyco‐collagen may play an important role in the modulation of stemness phenotype and that specific glycans can differentially trigger such an effect.
3.2. Neoglucosylated collagen specifically selects for slow‐dividing cells enriched in CSC
We next investigated the impact of glyco‐collagen matrices on tumor cell proliferation and viability. After 72 hours of culture on collagen films, we observed a 2,4‐fold reduction of number of viable tumor cells grown on pristine collagen compared to culture plates (Figure 2A); interestingly, tumor cells cultured on glyco‐collagen matrices showed a greater cell number decrease (3,4 fold‐change; P = 0.02).
Figure 2.
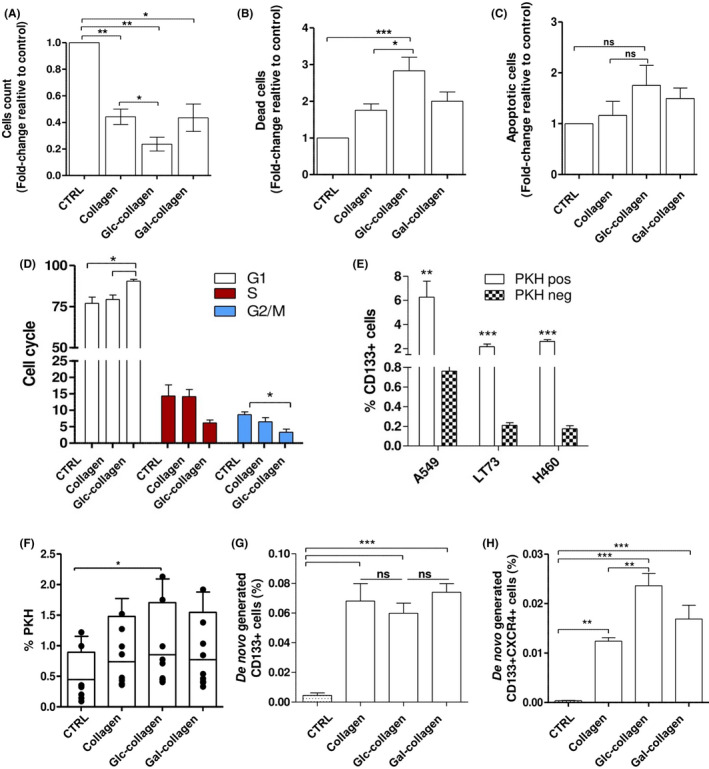
Collagen films select slow‐dividing cancer stem cells (CSC) and promote their de novo generation. A, Count of vital cells (Trypan blue exclusion method) in A549, LT73 and H460 cells grown for 72 h on collagen films. Bars represent the median fold‐change of percentage of dead cells relative to culture plate condition ± SEM. n = 3 replicates for each cell line. One‐way ANOVA was used for statistical analyses. *P < 0.05; **P < 0.01. B, Quantification of dead cells (7‐AAD vital staining) and (C) apoptotic cells (Annexin V+, propidium iodide [PI]–) in A549, LT73 and H460 cells grown for 72 h on collagen films. Bars represent the fold‐change of percentage of dead cells relative to culture plate condition ± SEM. n = 3 replicates for each cell line. One‐way ANOVA was used for statistical analyses *P < 0.05; ***P < 0.001. D, Cell cycle analysis (PI staining) of A549, LT73 and H460 cells grown for 72 h on collagen films or in culture plates. Bars are the median value of n = 2 replicates for each cell line. One‐way ANOVA was used for statistical analyses. *P < 0.05. E, CD133 content within PKHpos and PKHneg cells. Student’s t test was used for statistical analyses; **P < 0.01; ***P < 0.001. F, FACS analysis for PKH+ cells inA549, LT73 and H460 3 weeks post PKH staining. Cells were cultured on collagen films or on plastic culture plates for 72 h prior to analysis. Mean ± SEM of n = 2 replicates for each cell line is shown. One‐way ANOVA was used for statistical analyses. *P < 0.05. G, FACS analysis for the expression CD133 in LT73 CD133neg cell line cultured for 72 h on collagen films. Bars are the % of CD133+ cells. n = 4 experiments. H, FACS analysis for the expression CD133+ and CXCR4+ in LT73 CD133neg cell line as in (G)
Quantification of dead cells, according to 7‐AAD viability staining, showed that the highest mortality rate among all tested cell lines was observed on Glc‐collagen (2,5 increase compared to control; P = 0.03) whereas pristine collagen or Gal‐collagen showed a similar increase in cell death compared to cells grown on culture plates (respectively 1,6 and 1,8 fold‐change, Figure 2B). Mean values of dead cells were 10% in control conditions and 22% on collagen films. FACS analysis revealed a slight increase in the percentage of apoptotic cells in cell lines cultured on matrices compared to control (mean value: 2,7% in control and 3,8% in collagen films, P > 0.05) (Figure 2C).
In all tested cell lines, cells cultured on pristine collagen did not show any significant perturbation in cell cycle phases, whereas after exposure to Glc‐collagen matrices we observed a 1,2‐fold higher percentage of cells in the G1 phase together with a concomitant decrease of cells in S/G2 compared to the control culture plate, suggesting cell proliferation arrest (Figure 2D).
These results show that Glc‐collagen causes increased tumor cell growth arrest and cell death, overall determining the observed decrease in total cell number.
We next investigated whether the growth arrest and the increase of CD133+ CSC subsets induced by glyco‐collagen matrices could be linked to a specific selection for quiescent tumor stem cells.
Exploiting a label‐retaining assay based on PKH fluorescent membrane vital dye, we were able to identify after labeling and prolonged culturing: (i) slow‐proliferating/quiescent tumor cells retaining the dye (PKHpos); and (ii) active proliferating cells that progressively diluted the fluorescent dye during cell division (PKHneg). 16 , 30 In all tested cell lines, we verified that PKHpos slow‐dividing cells are 10‐fold enriched in CD133+ CSC compared to PKHneg proliferating cells (P < 0.0001) (Figure 2E and Figure S4A), as we previously demonstrated in the LT73 cell line. 30
A general increase for the fraction of PKHpos cells cultured on films was observed (1,7 fold‐change), which was statistically significant after exposure to Glc‐collagen (two‐fold increase) (Figure 2F).
Preferential retention of PKHpos slow‐cycling cells by collagen films is in line with the observed general increase for the CD133+ CSC subset and enrichment in G1‐arrested cells, in particular in Glc‐collagen.
3.3. Glyco‐collagen matrices trigger de novo generation of CSC
We next investigated another mechanism possibly contributing to CSC increase triggered by collagen films that is the de novo generation of stem‐like tumor cells through the conversion of non‐CSC cells into CD133+ CSC. To explore this hypothesis we took advantage of the LT73 CD133neg cell line, generated by depletion of CD133+ cells from LT73 parental cell line through FACS sorting. 17 , 18
As we previously demonstrated, 17 we confirmed that LT73 CD133neg cells cultured on a plastic plate remained negative for the expression of CD133 marker during in vitro culturing. Interestingly, culturing LT73 CD133neg cell for 72 hours on collagen matrices resulted in de novo generation of the CD133+ subset (Figure 2G and Supplementary Figure S4B). The effect of glyco‐collagen did not differ from pristine collagen in generating CD133+ CSC (frequency of de novo generated CD133+ CSC: 0.065% on pristine collagen; 0.06% on Glc‐collagen; 0.074% on Gal‐collagen, P > 0.05), suggesting that collagen binding can be the driver of de novo generation of CSC, whereas glycans minimally impact on the conversion of non‐CSC into CD133+ CSC. Interestingly, immunophenotypic analysis of de novo‐generated CD133+ cells demonstrated that Glc‐collagen was significantly more prone than pristine collagen to trigger the conversion of non‐CSC into the subset of metastatic CD133+ CXCR4+ cells (1.9 fold‐increase compared to pristine collagen, Figure 2H).
Our results suggest relevance of collagen interactions in modulating CSC phenotypes and, in particular, we prove that exposed glucose residues can trigger the expansion/de novo generation of metastasis‐initiating cells.
3.4. Glc‐collagen fosters in vivo tumorigenic potential
Having assessed by FACS analysis that glyco‐collagen and, in particular, Glc‐collagen proficiently expanded the fraction of CSC, we tested the functional relevance of such modulation.
In vitro migration assays on different collagen films showed an effective increase in migration potential of cells cultured on Glc‐collagen compared to both pristine collagen and plastic plates (respectively 1,8 and 1,6 fold‐increase, P < 0.001), data consistent with the higher enrichment in CD133+ CXCR4+ metastatic CSC subset (Figure 3A).
Figure 3.
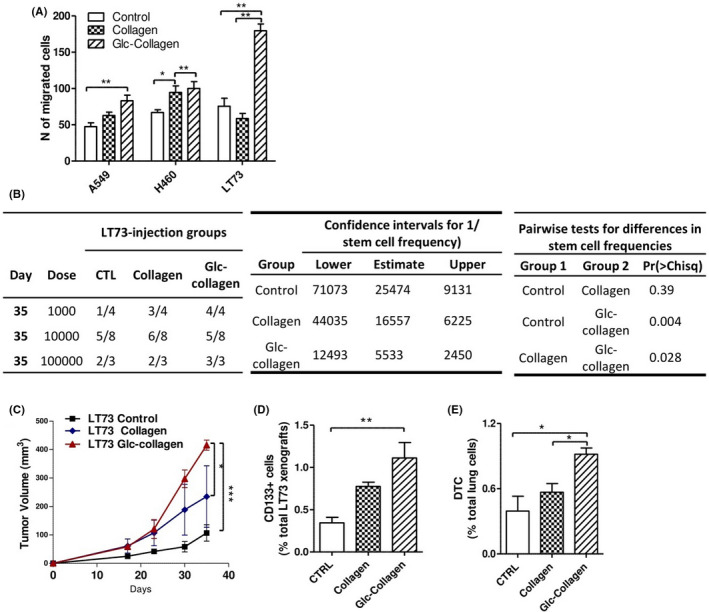
Cancer cells grown on Glc‐collagen matrix are more tumorigenic and disseminating. A, Migration assay carried out on A549, H460 and LT73 cell lines after cell culturing on collagen films or on plastic culture plates (control) for 72 h. Cells were chemoattracted with 10% FBS. Bars are the mean ± SD of the number of migrated cells counted in 4 random fields of the transwell inserts. One‐way ANOVA was used for statistical analyses *P < 0.05; **P < 0.001. B, LT73 cells grown for 72 h on plate (CTRL) collagen and Glc‐collagen were injected at serial dilutions (1 × 103, 1 × 104, 1 × 105) in the flanks of SCID mice. Tumor take rate, indicated in the table on the left, was evaluated as presence of growing tumor ≥100 mg, 35 days from s.c. injection. ELDA (extreme limiting dilution analysis) software was used to estimate the frequency of cancer stem cells (CSC) after treatments providing confidence intervals for 1/(stem cell frequency) (central table). Pairwise chi‐squared test was carried out between groups to evaluate difference in stem cell frequencies. C, Tumor growth curves of LT73 xenograft generated from s.c. injection of 1 × 105 cells after culturing on collagen films. n = 4 mice/group ***P < 0.0001 calculated by two‐way ANOVA. D, Relative content of CD133+ CSC evaluated by FACS within dissociated LT73 xenografts. Bars are the mean ± SD, n = 4 mice/group, each in duplicate technical analysis. One‐way ANOVA was used for statistical analyses. **P < 0.001. E, Number of disseminated tumor cells (DTC) in lungs of mice bearing LT73 xenografts analyzed in (C). One‐way ANOVA was used for statistical analyses *P < 0.05. DTC were identified as mouse vital, mouse H2Kneg cells by FACS on dissociated lung tissue, n = 4 mice/group
Next, we carried out an in vivo serial dilution assay, the gold standard to estimate the effective number of CSC, by injecting in SCID mice LT73 cells cultured on the different collagen matrices. Tumor take rate, evaluated as presence of growing tumors (TW ≥100 mg) 1 month post injection, demonstrated that LT73 cells grown on Glc‐collagen had a higher frequency of CSC able to initiate tumors compared to cells cultured on both plastic plates and pristine collagen (respectively 4,6 and 3 fold‐change; P = 0.004 and P = 0.028) (Figure 3B). In vivo evidence functionally correlated with the increase of CD133+ CSC evaluated by FACS in cells exposed to Glc‐collagen before injection. Among engrafted xenografts, those from the Glc‐collagen group grew significantly faster and bigger than controls (P = 0.0002) and pristine collagen (P = 0.05), whereas tumors from pristine collagen demonstrated a slightly increased tumorigenic potential compared to controls (Figure 3C).
Interestingly, FACS analysis revealed that xenografts generated by injection of cells exposed to collagen matrices maintained an increased content of CD133+ CSC cells compared to control (Glc‐collagen 3‐fold change, P = 0.01; Collagen 2,3 fold‐change, P > 0.05) (Figure 3D). In particular, xenografts generated from cells cultured on Glc‐collagen, enriched in MICs subsets, also showed the highest ability to disseminate to murine lungs compared to xenografts from culture plates or pristine collagen‐cultured cells (respectively 2,3 and 1,6 fold‐change P = 0.01) (Figure 3D).
Taken together, these data functionally suggest a trend for Glc‐collagen in increasing tumor‐initiating ability to an even higher extent compared to collagen alone.
3.5. Activated form of integrin β1 dictates tumor cell binding to Glc‐collagen matrices
We then investigated the mechanism underlying CSC modulation induced by binding of cancer cells to collagen matrices.
Collagen binding is mediated by cell membrane expression of integrin β1 in heterodimeric complexes with α1 or α2 subunits. 31 , 32 FACS analysis of total protein integrin β1 (CD29, clone TS2/16) showed that all tested cell lines express very high cell membrane level of integrin β1 (mean: 99% of total cells). However, as integrin β1 can exist in different affinity states for collagen ligand, we focused our analyses on the high‐affinity activated conformation state of integrin β1, identified by the antibody clone 9EG7. In this conformational state, integrin β1 can bind collagen by forming heterodimers with integrin α1 or α2 subunits. 31 , 33
LT73 cell line showed the highest membrane expression of the activated form of integrin β1 (Figure 4A), both alone and in co‐expression with integrin α1/2. Notably, FACS analysis of all cell lines revealed that CD133+ CSC express a higher level of activated integrin β1, both as total and in its heterodimeric form with integrin α1, compared to the bulk population, subtending that CSC may have a higher affinity to bind collagen (Figure 4A).
Figure 4.

Expression of activated integrin β1 correlates with tumor cell binding to Glc‐collagen matrices. A, FACS analysis of the membrane expression of the activated form of integrin β1 (clone 9EG7) alone (left panel) and in co‐expression with integrin alpha1 (central panel) or alpha2 subunits (right panel) in A549, LT73 and H460 cell lines, both in the bulk population and within the gated CD133+ subset. Bars are the mean ± SEM of n = 4 replicates for each cell line. ANOVA was used for statistical analysis. *P < 0.05; **P < 0.001. B, Time‐course analysis of A549, LT73 and H460 cell line adhesion to culture plates, collagen and Glc‐collagen matrices. At each indicated time point, adherent cells were detached and counted by the Trypan blue exclusion method. Two‐way ANOVA was used for statistical analyses. *P < 0.05; *** P < 0.0001
We also assessed in NSCLC cell lines the expression of α3/α6 integrin subunits that are known to form heterodimers with integrin β1 to bind laminin, another abundant protein of the ECM. Notably, CD133+ CSC show a similar or even lower (in LT73 cell line) expression of integrin α3/β1and α6/β1 heterodimers compared to the bulk population (Figure S5A). This differential pattern of integrin expression might suggest a possible preferential CSC affinity for collagen.
Time‐course analysis to assess the kinetics of cell binding to culture plates and collagen films showed that different cell lines adhere to culture plates with a distinctive kinetic, which is independent of the expression of integrin β1 (as expected) and significantly more efficient than adhesion to collagen films, for A549 and H460 cells. Conversely, the LT73 cell line that expresses the highest level of activated form of integrin β1 among tested cell lines was able to efficiently and rapidly bind collagen, especially Glc‐collagen matrix, with a faster kinetic than for the culture plates adhesion. This observation may suggest that the glucose exposed on Glc‐collagen could be directly involved in modulating the interaction between collagen and the activated form of integrin β1. In all tested cell lines, after 4 hours post‐seeding, almost all cells competent to bind collagen adhered to the matrices and after 24 hours the cell number remained similar, as collagen matrices selected for slow‐proliferating cells as previously shown (Figure 4B).
3.6. Collagen‐induced CSC increase is due to specific selection of CD133+ cells and subsequent CSC de novo generation
We next monitored the modulation of CSC induced by collagen in relation to the kinetics of tumor cells binding to collagen.
Modulation of CD133 expression revealed that soon after the initial phase of collagen binding, an increase for CD133+ cells can be observed and it is significantly higher in cells cultured on glyco‐collagen (Figure 5A). Notably, in LT73, showing the highest expression of integrin β1 and consequent faster binding to Glc‐collagen, a significant increase of CD133+ CSC compared to the culture plate was already evident soon after cell seeding (30 min), suggesting a link between the propensity of tumor cells to adhere to glyco‐collagen and CD133+ cell selection (Figure 5A). Conversely, in H460 and A549 cells cultured on Glc‐collagen, there was a general gradual increase in CD133+ cells compared with initial phases of cell adhesion, with a significant increase of CSC 24 hours post‐seeding.
Figure 5.
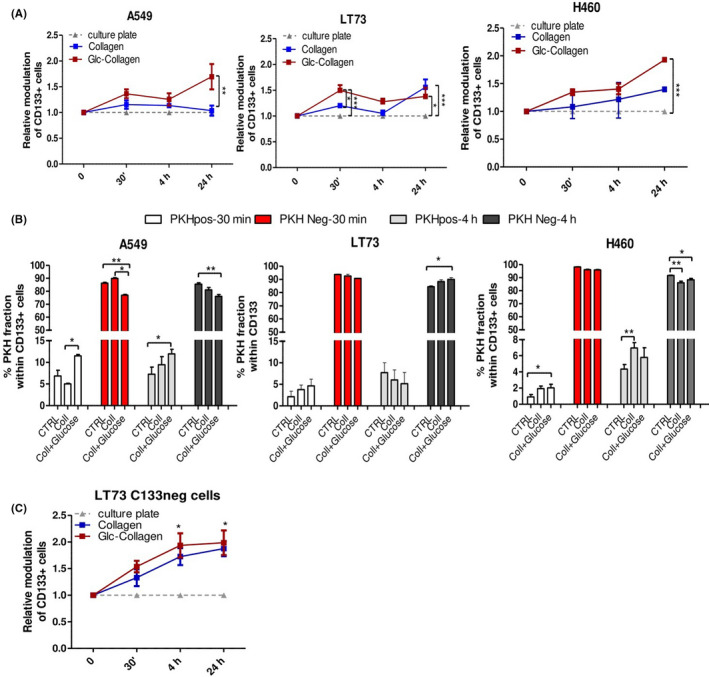
Time‐course analysis of CD133+ cancer stem cell (CSC) modulation by collagen films. A, Time‐course analysis of modulation of CD133+ cells, evaluated by FACS, during A549, LT73 and H460 cell adhesion to collagen films and neoglucosylated collagen films. The fold‐change relative to culture plates in CD133+ CSC content are reported after 30 min, 4 h and 24 h post cell seeding on films. n = 3 replicates/time point for each cell line. Two‐way ANOVA was used for statistical analyses. *P < 0.05; **P < 0.01; ***P < 0.001. B, Evaluation of the content of PKH positive and PKH negative cells within gated CD133+ CSC subset in A549, LT73 and H460 cell lines after 30 min and 4 h post cell seeding on collagen films and culture plates (CTRL). Mean ± SEM. ANOVA was used for statistical analyses. *P < 0.05; **P < 0.01. C, Generation of CD133+ CSC in LT73 CD133neg cell line, plated on collagen films compared to culture plates. n = 4 independent experiments were carried out for each time point. Two‐way ANOVA was used for statistical analyses. *P < .05 refers to difference between Glc‐collagen and collagens vs control plate
Based on these data, we investigated whether the increase of CD133+ cells can be preferentially driven by the selection/expansion of pre‐existing CSC subsets or their de novo generation in relation to specific kinetics of tumor cell lines binding to the different matrices.
By exploiting the PKH label‐retaining assay, we observed at early time points in all cell lines a significant enrichment for the fraction of CD133+ PKHpos driven by collagen binding and, in particular, by Glc‐collagen matrices (Figure 5B), consistent with a specific selection of the pre‐existing fraction of slow‐dividing CSC. Conversely, the fraction of PKHneg cells was counter selected by collagen binding, indicating that the initial increase of CD133+ cells was mainly due to the specific selection of already existing CSC expressing high levels of integrin β1 (Figure 5B).
Analysis of CD133+ cells 4 hours post tumor cell seeding showed in the LT73 cell line a significant increase of CD133+ PKHneg cells: as slowly proliferative CD133+ PKHpos cells are not expected to divide and consequently dilute PKH dye within 4 hours post cell seeding, the observed increase of the CD133+ PHKneg cell subset prompted us to speculate a de novo generation of CSC subset (CD133+) within the compartment of non‐CSC PKHneg cells. Conversely, in H460 and A549 cell lines, a similar modulation of PKHpos and neg compartments within CD133+ cells was maintained compared to early time points, consistent with the selection of pre‐existing CSC (Figure 5B).
These different modulations of CD133+ cells in response to collagen binding in LT73 cells can be explained by the high propensity of cells to rapidly bind collagen and also by the ability of this cell line among others to efficiently modulate and de novo generate CSC in response to microenvironmental stimuli, as we previously reported. 18 To confirm the triggering of de novo generation of CSC induced by collagen, we monitored the appearance of CD133+ cells induced in LT73 CD133neg cells after binding to collagen. We found that de novo generation of CD133+ cells was significantly evident 4 hours post‐seeding and similarly maintained after 24 hours (Figure 5C). De novo generation of CSC at 4 hours was coherent with the enrichment of the CD133+ PKHneg fraction observed in the LT73 parental cell line (Figure 5B). No significant difference in the ability to generate CSC was detected between pristine collagen and glyco‐collagens, confirming end‐point data at 72 hours (Figure 2F).
3.7. Collagen‐induced increase of CD133+ CSC is mediated by integrin β1
In time‐course analyses, we observed that the collagen‐induced increase of CD133+ cells at different time points was constantly associated with a reduced membrane expression of activated integrin β1, likely due to integrin internalization and activation of downstream transduction pathways (Figure 6A). Interestingly, we observed that 24 post‐seeding, cells unable to bind collagen (shown in Figure 1A) demonstrated depletion in the CSC subset and a significantly lower expression of the activated form of integrin β1 compared to cells competent to adhere to the collagen matrix. These data suggest that cancer cell collagen binding and collagen selection of CD133+ CSC can be mediated by activated integrin β1 (Figure S6A).
Figure 6.
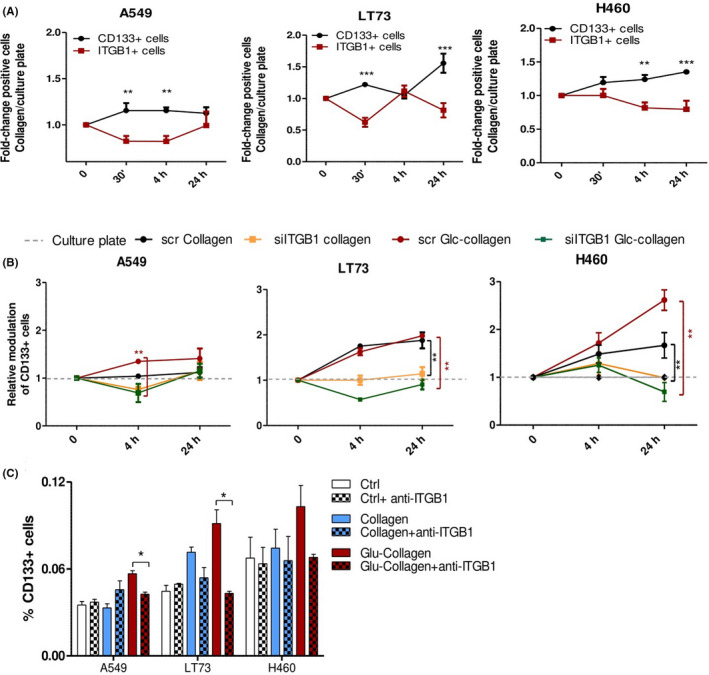
Integrin β1 mediated collagen‐induced cancer stem cell (CSC) enrichment. A, Time‐course analysis for modulation of CD133+ cells and integrin β1 active form during A549, LT73 and H460 cell line adhesion to pristine and neoglucosylated collagen films. Fold‐change of positive cells relative to culture plates are reported after 30 min, 4 h and 24 h post cell seeding on films. n = 3 replicates/time point for each cell line. Two‐way ANOVA was used for statistical analyses, **P < 0.01; ***P < 0.001. B, Time‐course analysis of modulation of CD133+ cells during adhesion to collagen films of A549, LT73 and H460 cell lines, transfected with scrambled siRNA or siITGB1.Fold‐change in the percentage of CD133+ positive cells relative to culture plates are reported after 4 h and 24 h post cell seeding on films. n = 2 experiments for each cell line. Two‐way ANOVA was used for statistical analyses. **P < 0.01. C, FACS analysis for CD133 expression in cancer cell lines cultured for 4 h on collagen matrices or culture plate in presence of integrin β1 blocking antibody (50 μg/mL). Bars are the mean value ± SD of n = 2 independent experiments performed for each cell line, in technical duplicate. ANOVA was used for statistical analyses. *P < 0.05
Next, to establish the role of integrin β1 in mediating collagen‐induced modulation of CD133+ subsets, we transiently knocked down ITGB1 gene expression in different cell lines trough siRNA‐mediated silencing and we repeated time‐course analyses of collagen binding. Compared to scrambled siRNA control, siITGB1 cells showed a 7,7‐fold (± 5 SD) reduction of gene expression and a 50% reduction in protein levels of active integrin β1 (Figure S6B‐D).
ITGB1 gene silencing did not affect CSC subpopulations (data not shown) indicating that in standard 2D culture conditions, integrin β1 is not essential for CD133+ CSC maintenance. However, ITGB1 knock‐down was able to prevent the increase of CD133+ cells on pristine and Glc‐collagen matrices, both in the initial phase of binding and 24 hours post cell seeding (Figure 6B). The effect of integrin β1 knock‐down was even more effective in counteracting Glc‐collagen matrix effects, stressing the essential role of integrin β1 binding and downstream pathway activation in mediating signaling specifically triggered by Glc‐collagen.
We validated these results by incubating cancer cell lines with an integrin β1 blocking antibody (AIIB2) before culturing on collagen matrices (Figure 6C). Four hours post cell seeding, we confirmed that blocking of integrin β1 did not directly affect the CD133+ CSC compartment but significantly prevented the selection/generation of this subpopulation, in particular that induced by Glc‐collagen.
Overall, our data suggest that integrin β1 is a mediator of the modulation of the CSC subset triggered by collagen and its targeting may prevent the enrichment in CSC caused by specific ECM glycosylation patterns.
4. DISCUSSION
The ECM is in direct contact with tumor cells providing them with soluble factors and mechanical cues able to regulate tumor development and progression. 34 Contribution of ECM glycosylation to the modulation of cancer cell niches is still poorly investigated as a result of the lack of adequate systems able to mimic ECM glycosignatures. Our aim was to provide proof of concept that glycosylation of collagen could play a crucial biological role in the generation of specific stem cell niches in lung cancer.
Glycans control tumor cell adhesion, motility, and invasiveness through the modulation of receptor/ligand interactions between cells and ECM‐binding proteins. 22 , 35 Aberrant cell‐surface and basal lamina glycosylation affects tumor cell–ECM adhesion triggering downstream signaling pathways used also for the survival and outgrowth of disseminated tumor cells during the metastatic cascade. 36 , 37
Cancer stem cells, functionally defined as tumor‐initiating/propagating cells, 14 reside in specialized niches composed by stromal cells and ECM, essential for maintenance of stemness properties and regulation of asymmetric cell division/proliferation. 13 In lung cancer, CD133+ cells represent CSC and are responsible for tumor initiation, chemoresistance and distant metastasis. 38 In particular, the subset of CD133+ cells co‐expressing the chemokine receptor CXCR4 is endowed with the highest metastasis initiation potential (MIC). 16 , 17
Despite some previous evidence describing the ability of stromal soluble factors to modulate lung CSC phenotype, 17 , 18 , 38 , 39 this work provides the first demonstration of the ability of ECM modifications to dictate stemness phenotype in lung cancer and highlights the essential role of collagen glycosylation in controlling the interaction of lung CSC with the ECM.
For our study we used glyco‐collagen matrices 25 that have been shown capable of triggering biological responses able to dictate cell fate in different cellular systems. 24 , 40
The significance of collagen glycosylation by α‐d‐Glc‐(1‐2)‐β‐d‐Gal(1‐O) disaccharide has already been highlighted and correlated with embryonic lethality in past studies. 41 However, the contribution of glycosylation degree and the epitope exposed is different depending on cell type. 42 We show here that culture of lung cancer cells on pristine collagen films compared to culture plates was able to induce an increase of CD133+ CSC with a concomitant upregulation of expression of stemness‐related genes. Similar modulation of stemness phenotype was also observed in glioblastoma and colon cancer cells, cultured on collagen‐coated plates. 43 , 44
Analysis of specific effects induced by different glyco‐collagen films proved that glucose residues had the greatest ability to increase the CD133+ fraction, especially the metastatic subset of CD133+ CXCR4+ cells. In Glc‐collagen matrices, the expansion of CSC subsets was correlated with an overall enhanced cell mortality and cell growth arrest, suggesting that stem‐like cells may have a preferential binding affinity or survival advantage on this substrate compared to proliferating and differentiated tumor cells.
Interestingly, results obtained from label‐retaining assays corroborated this hypothesis demonstrating that culture of PKH‐labeled cells on Glc‐collagen resulted in enrichment for the slow‐proliferating PKHpos cells which contain CSC subsets. 30
As the effects on CSC modulation triggered by Gal‐collagen were more similar to pristine collagen than to Glc‐collagen, we suggest a specific role of glucose in regulation of CSC and MIC rather than a generic response to collagen glycosylation.
Cancer‐associated fibroblasts are able to induce de novo generation of CSC 17 and here we show that ECM components can also exert a similar effect in modulating CD133+ subsets. Culture of LT73 CD133neg cells on collagen determined the appearance of CD133+ CSC and this effect was elicited by collagen exposure, irrespective of glycosylation.
Finally, in vivo limiting dilution assays demonstrated that modulation of lung CSC subsets induced by Glc‐collagen is functionally correlated with an increased tumorigenic potential and enhanced dissemination ability compared to cells cultured on pristine collagen or control cells.
Altogether, these findings support the role of collagen as a fundamental component of the lung CSC‐niche. Notably, aberrant glycosylation of both cancer cells and basal membrane are associated with enhanced invasion and metastasis formation in breast cancer. 37 , 45 It may be possible that in lung cancer a fibrotic environment caused by deposition of aberrant glycosylated collagen could represent the most favorable soil for MIC selection/survival, representing a novel promising target to impair metastasis formation.
Integrin receptors respond to particular biochemical and physical characteristics of ECM by initiating a cascade of events that affect cell proliferation, differentiation and survival. 31 , 32 , 33 , 46 Here we also report that expression of the activated form of integrin β1 is significantly overexpressed in the CSC subset, alone or in the heterodimeric form with integrin α1, which is coherent with a higher proficiency of CD133+ cells to adhere to collagen, in particular to Glc‐collagen, than bulk cells. We also analyzed the expression of other integrin subunits, α3 and α6, that form heterodimer complexes with β1 integrin to bind laminin, another ECM protein whose function is strictly regulated by glycosylation. 47 Notably, CSC show a similar or even lower expression of α3/β1and α6/β1 integrin heterodimers compared to the bulk tumor population indicating a potential preferential affinity of CSC to bind collagen rather than other structural components of the ECM. Mechanistically, we prove that integrin β1 is one of the mediators of CSC modulation induced by collagen, as knock‐down of ITGB1 gene expression and the use of an integrin β1 blocking antibody interrupted CSC interaction with ECM and prevented both the initial selection of pre‐existing CD133+ CSC, preferentially dictated by Glc‐collagen, and their subsequent expansion/de novo generation.
In conclusion, the present study although preliminary and based on synthetic glycomimetics has brought new insights into the unexplored role of ECM glycosylation in controlling the behavior of lung CSC and MIC, suggesting a possible new mechanism by which CSC adhesion, survival and growth could be regulated by glycosylated collagen.
Our approach could provide innovative tools to study the mechanism underlying disseminated tumor cells quiescence and reactivation, a process so far poorly understood. The ability to understand the functions of ECM glycans in tumor progression is going to represent a potential breakthrough for elucidation of the complexity of microenvironment regulation of carcinogenesis and metastasis.
DISCLOSURE
The authors have no conflicts of interest to declare.
Supporting information
Fig S1‐S6
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Italian Ministry of Health [grant number RF‐2016‐02362946 to L.R]; Associazione Italiana per la Ricerca sul Cancro [grant number IG21431 to L.R].
Gardelli C, Russo L, Cipolla L, et al. Differential glycosylation of collagen modulates lung cancer stem cell subsets through β1 integrin‐mediated interactions. Cancer Sci. 2021;112:217–230. 10.1111/cas.14700
Giulia Bertolini and Luca Roz contributed equally to this work.
Contributor Information
Laura Russo, Email: laura.russo@unimib.it.
Luca Roz, Email: luca.roz@istitutotumori.mi.it.
REFERENCES
- 1. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423‐1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol. 2012;196:395‐406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Montagner M, Dupont S. Mechanical forces as determinants of disseminated metastatic cell fate. Cells. 2020;9(1):250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Burgstaller G, Oehrle B, Gerckens M, White ES, Schiller HB, Eickelberg O. The instructive extracellular matrix of the lung: basic composition and alterations in chronic lung disease. Eur Respir J. 2017;50(1):1601805. [DOI] [PubMed] [Google Scholar]
- 5. Socovich AM, Naba A. The cancer matrisome: From comprehensive characterization to biomarker discovery. Semin Cell Dev Biol. 2019;89:157‐166. [DOI] [PubMed] [Google Scholar]
- 6. Seguin L, Desgrosellier JS, Weis SM, Cheresh DA. Integrins and cancer: regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol. 2015;25:234‐240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Broders‐Bondon F, Nguyen Ho‐Bouldoires TH, Fernandez‐Sanchez ME, Farge E. Mechanotransduction in tumor progression: The dark side of the force. J Cell Biol. 2018;217:1571‐1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Deville SS, Cordes N. The Extracellular, Cellular, and Nuclear Stiffness, a Trinity in the Cancer Resistome‐A Review. Front Oncol. 2019;9:1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pickup MW, Mouw JK, Weaver VM. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014;15:1243‐1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gelse K, Poschl E, Aigner T. Collagens–structure, function, and biosynthesis. Adv Drug Deliv Rev. 2003;55:1531‐1546. [DOI] [PubMed] [Google Scholar]
- 11. Bretaud S, Nauroy P, Malbouyres M, Ruggiero F. Fishing for collagen function: About development, regeneration and disease. Semin Cell Dev Biol. 2019;89:100‐108. [DOI] [PubMed] [Google Scholar]
- 12. Levental KR, Yu H, Kass L, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891‐906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Du J, Chen X, Liang X, et al. Integrin activation and internalization on soft ECM as a mechanism of induction of stem cell differentiation by ECM elasticity. Proc Natl Acad Sci USA. 2011;108:9466‐9471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea–a paradigm shift. Cancer Res. 2006;66:1883‐1890. [DOI] [PubMed] [Google Scholar]
- 15. Cabarcas SM, Mathews LA, Farrar WL. The cancer stem cell niche–there goes the neighborhood? Int J Cancer. 2011;129:2315‐2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bertolini G, Roz L, Perego P, et al. Highly tumorigenic lung cancer CD133+ cells display stem‐like features and are spared by cisplatin treatment. Proc Natl Acad Sci USA. 2009;106:16281‐16286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bertolini G, D’Amico L, Moro M, et al. Microenvironment‐Modulated Metastatic CD133+/CXCR4+/EpCAM‐ Lung Cancer‐Initiating Cells Sustain Tumor Dissemination and Correlate with Poor Prognosis. Cancer Res. 2015;75:3636‐3649. [DOI] [PubMed] [Google Scholar]
- 18. Andriani F, Bertolini G, Facchinetti F, et al. Conversion to stem‐cell state in response to microenvironmental cues is regulated by balance between epithelial and mesenchymal features in lung cancer cells. Mol Oncol. 2016;10(2):253‐271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Peixoto A, Relvas‐Santos M, Azevedo R, Santos LL, Ferreira JA. Protein Glycosylation and Tumor Microenvironment Alterations Driving Cancer Hallmarks. Front Oncol. 2019;9:380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat. Rev. Cancer. 2015;15:540‐555. [DOI] [PubMed] [Google Scholar]
- 21. Bertozzi CR, Kiessling LL. Chemical glycobiology. Science. 2001;291:2357‐2364. [DOI] [PubMed] [Google Scholar]
- 22. Ohtsubo K, Marth JD. Glycosylation in cellular mechanisms of health and disease. Cell. 2006;126:855‐867. [DOI] [PubMed] [Google Scholar]
- 23. Marsico G, Russo L, Quondamatteo F, Pandit A. Glycosylation and Integrin Regulation in Cancer. Trends Cancer. 2018;4:537‐552. [DOI] [PubMed] [Google Scholar]
- 24. Russo L, Sgambato A, Lecchi M, et al. Neoglucosylated collagen matrices drive neuronal cells to differentiate. ACS Chem Neurosci. 2014;5(4):261‐265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Russo L, Gautieri A, Raspanti M, et al. Carbohydrate‐functionalized collagen matrices: design and characterization of a novel neoglycosylated biomaterial. Carbohydr Res. 2014;389:12‐17. [DOI] [PubMed] [Google Scholar]
- 26. Gandellini P, Andriani F, Merlino G, D'Aiuto F, Roz L, Callari M. Complexity in the tumour microenvironment: Cancer associated fibroblast gene expression patterns identify both common and unique features of tumour‐stroma crosstalk across cancer types. Semin Cancer Biol. 2015;35:96‐106. [DOI] [PubMed] [Google Scholar]
- 27. Leung EL, Fiscus RR, Tung JW, et al. Non‐small cell lung cancer cells expressing CD44 are enriched for stem cell‐like properties. PLoS One. 2010;5:e14062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu J, Xiao Z, Wong SK, et al. Lung cancer tumorigenicity and drug resistance are maintained through ALDH(hi)CD44(hi) tumor initiating cells. Oncotarget. 2013;4:1698‐1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang WC, Shyh‐Chang N, Yang H, et al. Glycine decarboxylase activity drives non‐small cell lung cancer tumor‐initiating cells and tumorigenesis. Cell. 2012;148:259‐272. [DOI] [PubMed] [Google Scholar]
- 30. Moro M, Bertolini G, Pastorino U, Roz L, Sozzi G. Combination treatment with all‐trans retinoic acid prevents cisplatin‐induced enrichment of CD133+ tumor‐initiating cells and reveals heterogeneity of cancer stem cell compartment in lung cancer. J Thorac Oncol. 2015;10:1027‐1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Banno A, Ginsberg MH. Integrin activation. Biochem Soc Trans. 2008;36:229‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Takada Y, Ye X, Simon S. The integrins. Genome Biol. 2007;8:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285:1028‐1032. [DOI] [PubMed] [Google Scholar]
- 34. Chen YC, Hsu HS, Chen YW, et al. Oct‐4 expression maintained cancer stem‐like properties in lung cancer‐derived CD133‐positive cells. PLoS One. 2008;3:e2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Laubli H, Borsig L. Altered Cell Adhesion and Glycosylation Promote Cancer Immune Suppression and Metastasis. Front Immunol. 2019;10:2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Varki A, Kannagi R, Toole B, Stanley P. Glycosylation Changes in Cancer. 2015;597‐609. [PubMed] [Google Scholar]
- 37. Reticker‐Flynn NE, Bhatia SN. Aberrant glycosylation promotes lung cancer metastasis through adhesion to galectins in the metastatic niche. Cancer Discov. 2015;5:168‐181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Eramo A, Lotti F, Sette G, et al. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008;15:504‐514. [DOI] [PubMed] [Google Scholar]
- 39. Chen WJ, Ho CC, Chang YL, et al. Cancer‐associated fibroblasts regulate the plasticity of lung cancer stemness via paracrine signalling. Nat Commun. 2014;5:3472. [DOI] [PubMed] [Google Scholar]
- 40. Russo L, Battocchio C, Secchi V, et al. Thiol‐ene mediated neoglycosylation of collagen patches: a preliminary study. Langmuir. 2014;30:1336‐1342. [DOI] [PubMed] [Google Scholar]
- 41. Basak T, Vega‐Montoto L, Zimmerman LJ, Tabb DL, Hudson BG, Vanacore RM. Comprehensive characterization of glycosylation and hydroxylation of basement membrane collagen IV by high‐resolution mass spectrometry. J Proteome Res. 2016;15:245‐258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bauvois B, Roth S. A collagen:glucosyltransferase at the surface of malignant fibroblasts. J Cell Physiol. 1985;124:213‐218. [DOI] [PubMed] [Google Scholar]
- 43. Motegi H, Kamoshima Y, Terasaka S, Kobayashi H, Houkin K. Type 1 collagen as a potential niche component for CD133‐positive glioblastoma cells. Neuropathology. 2014;34:378‐385. [DOI] [PubMed] [Google Scholar]
- 44. Kirkland SC. Type I collagen inhibits differentiation and promotes a stem cell‐like phenotype in human colorectal carcinoma cells. Br J Cancer. 2009;101:320‐326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Barkan D, Green JE, Chambers AF. Extracellular matrix: a gatekeeper in the transition from dormancy to metastatic growth. Eur J Cancer. 2010;46:1181‐1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Roskelley CD, Bissell MJ. Dynamic reciprocity revisited: a continuous, bidirectional flow of information between cells and the extracellular matrix regulates mammary epithelial cell function. Biochem Cell Biol. 1995;73:391‐397. [DOI] [PubMed] [Google Scholar]
- 47. Kariya Y, Kato R, Itoh S, et al. N‐Glycosylation of laminin‐332 regulates its biological functions. A novel function of the bisecting GlcNAc. J Biol Chem. 2008;283:33036‐33045. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S6
Supplementary Material