

Abstract
Ribosome assembly factor URB1 is essential for ribosome biogenesis. However, its latent role in cancer remains unclear. Analysis of The Cancer Genome Atlas database and clinical tissue microarray staining showed that URB1 expression was upregulated in colorectal cancer (CRC) and prominently related to clinicopathological characteristics. Silencing of URB1 hampered human CRC cell proliferation and growth in vitro and in vivo. Microarray screening, ingenuity pathway analysis, and JASPAR assessment indicated that activating transcription factor 4 (ATF4) and X‐box binding protein 1 (XBP1) are potential downstream targets of URB1 and could transcriptionally interact through direct binding. Silencing of URB1 significantly decreased ATF4 and cyclin A2 (CCNA2) expression in vivo and in vitro. Restoration of ATF4 effectively reversed the malignant proliferation phenotype of URB1‐silenced CRC cells. Dual‐luciferase reporter and ChIP assays indicated that XBP1 transcriptionally activated ATF4 by binding with its promoter region. X‐box binding protein 1 colocalized with ATF4 in the nuclei of RKO cells, and ATF4 mRNA expression was positively regulated by XBP1. This study shows that URB1 contributes to oncogenesis and CRC growth through XBP1‐mediated transcriptional activation of ATF4. Therefore, URB1 could be a potential therapeutic target for CRC.
Keywords: ATF4, colorectal cancer, cyclin A2, ribosome assembly factor, URB1, XBP1
The classic ribosome assembly factor URB1 is overexpressed and related to unfavorable clinicopathological characteristics in colorectal cancer (CRC). URB1 silencing significantly hampered human CRC cell proliferation and decreased ATF4, CCNA2, and XBP1 expression in vivo and in vitro. Briefly, our study shows that URB1 contributes to oncogenesis and growth of CRC through XBP1‐medaited transcriptional activation of ATF4 and URB1 could be a potential therapeutic target for CRC.
1. INTRODUCTION
Colorectal carcinoma (CRC) is the most common digestive tract malignancy and has the second highest mortality rates among the 36 known cancers worldwide. 1 The global burden of CRC is expected to increase by 60% to more than 2.2 million new cases and 1.1 million deaths by 2030. 2 Although there have been improvements in diagnostic and therapeutic strategies in CRC, the overall median survival is only 42 months due to late diagnosis and severe clinicopathological characteristics. 3 Therefore, investigating the genetic landscape and exploring novel functional genes in CRC oncogenesis and progression could help ameliorate the current therapeutic limitations in CRC.
The ribosome biogenesis 1 homolog (URB1), also known as Npa1 or Nop254 in Saccharomyces cerevisiae, is a ribosome assembly factor (RAF) that is highly conserved within various eukaryotes and essential for the assembly of ribosomal subunits. 4 , 5 Urb1 associates with Urb2 and the other five RAFs to form a low‐molecular‐mass complex, which is involved in the early steps of 60S ribosomal subunit assembly. 6 It is reported that URB1 frameshift mutation resulting in a complete loss‐of‐function, like the “natural knockouts” effect, causes embryonic lethality in pigs. 7 In a previous study, we reported that RAPTOR promoted CRC proliferation by inducing mTORC1 signaling activation and upregulating URB1 and cyclin A2 (CCNA2), further suggesting an oncogenic role for URB1 in CRC. 8 PNO1 is another RAF that has been shown to contribute to CRC proliferation and progression through negative regulation of the p53 signaling pathway. 9 Considering its conserved function, similar to PNO1, and its significance in our preceding study, it is not surprising that URB1 might perform a novel function beyond ribosome assembly in tumorigenesis.
Activating transcription factor 4 (ATF4) is a member of the ATF/CREB family and belongs to the basic region‐leucine zipper family of transcription factors. Activating transcription factor 4 is increased in response to a diverse array of microenvironmental stress signals, including amino acid deprivation, hypoxia, and endoplasmic reticulum stress (ERS). 10 , 11 Of note, amino acid deprivation originating from the rapid growth of tumor cells stimulates general control non‐depressible 2 expression and activates ATF4. This promotes tumor proliferation and angiogenesis by controlling the expression of adaptive genes that give cells the capability to endure periods of stress and survive. 12 , 13 Furthermore, ATF4 plays a pivotal role in promoting osteoblast cell proliferation through ERS‐induced PERK‐EIF2a‐ATF4 signaling pathway activation. Blocking ATF4 decreased the expression of metabolism‐related genes and cell proliferation markers, such as cyclin E, cyclin D, and cell division cycle gene 2. 14 It is known that ATF4 is overexpressed in various malignant tumors and responsible for catastrophic biological behaviors due to multiple cellular stresses and dysfunctions in metabolism. 15 , 16 , 17 , 18 It also induces p53‐independent apoptosis, a mechanism by which tumor cells reduce the stress resulting from rapid proliferation and nutrient limitations inside a growing tumor mass. 19 However, the role of ATF4 and its interaction with URB1 in tumorigenesis needs to be elucidated.
In this study, we showed that URB1 is involved in CRC proliferation through activation of ATF4 transcription. Overexpression of ATF4 effectively restored the malignant proliferation phenotype in URB1‐silenced CRC cells. A further study on the regulatory mechanisms of URB1 on ATF4 could lead to identification of new biomarkers and therapeutic targets for CRC.
2. MATERIALS AND METHODS
2.1. Patient specimens and tissue microarray
Forty pairs of cancerous and matched noncancerous samples were obtained from patients with CRC who had undergone radical surgical excision between April and August 2018 at the Gansu Provincial People’s Hospital (Lanzhou, China). None of these patients had undergone radiotherapy or chemotherapy prior to surgery. The specimens were used with written informed consent from the patients, and the study was approved by the Ethics Committee of the Gansu Provincial People’s Hospital. Two tissue microarray (TMA) slides containing 80 pairs and 101 pairs of CRC tissue samples were purchased from Shanghai Outdo Biotech. Samples and TMAs were processed using routine methods for quantitative PCR or immunohistochemistry (IHC) staining. Clinicopathological characteristics, including gender, age, TNM stage, lymph node metastasis, histological grade, pathological subtype, and K‐RAS mutation status, were obtained and summarized based on medical records.
2.2. Immunohistochemistry
Immunohistochemistry for anti‐URB1 (1:50; Abcam) in TMA, and anti‐ATF4 (1:50; Abcam), anti‐CCNA2 (1:50; Abcam), and anti‐Ki‐67 (1:50; Abcam) in xenograft tissues was carried out using standard methods. The intensity and percentage of positively stained cells in each sample were evaluated by two experienced pathologists blinded to this study. For URB1 protein levels, negative expression was defined as no or weak staining in cells, whereas positive expression was defined as distinct or strong staining in more than 20% of cells. 20 The high or low expression levels were assessed as previously described. 9
2.3. Cell culture and transfection
The RKO, SW480, LoVo, and HCT116 human CRC cell lines and the NCM460 colon epithelial cell line and the fibroblast CCD‐18Co were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). HCT116 cells were maintained in McCoy’s 5A medium (ScienCell), and the other cell lines were maintained in high glucose DMEM (Gibco). All of the media contained 10% FBS (Thermo Fisher Scientific), penicillin (100 U/mL), and streptomycin (100 µg/mL). Cells were cultured in a humid atmosphere with 5% CO2 at 37°C. To test the authenticity of RKO and HCT116 cells, short tandem repeat profiling was carried out.
Two shRNAs for URB1 silencing and a nonsense control shRNA were cloned into GV115 to construct the interfering lentiviral vectors (named sh1 and sh2) and the control lentiviral vector (named nc). We cloned the URB1 coding sequence (CDS) into overexpression vector (cat. CV130) and ATF4 CDS into overexpression vector (cat. GV365) to construct the corresponding overexpression lentiviral vectors. All of the lentiviral vectors were constructed and purchased from GeneChem. The XBP1 overexpression vector (CMV‐XBP1) was constructed and purchased from Hanbio Biotech (Shanghai, China). Then the corresponding vectors were added to cultured cells according to the protocols recommended by the manufacturer. Transfection efficiency was evaluated by quantitative real time PCR and western blotting.
The sequences used were as follows: sh1, 5′–CCGGATGAACAGATTCACCGTAAATCTCGAGATTTACGGTGAATCTGTTCATTTTTT‐3′; sh2, 5′–CCGGACACAGCCTTATCCCTTATTTCTCGAGAAATAAGGGATAAGGCTGTGTTTTTT‐3′; nc, 5′‐CCGGTTCTCCGAACGTGTCACGTTTCAAGAGAACGTGACACGTTCGGAGAATTTTT‐3′; URB1‐forward (F), GATCCAGTTTGGTTAATTAATAGTTATTAATAGTAATCAATTACGGGGTC; URB1‐reverse (R), CACACATTCCACAGGAATTCTCATTTGTCGTCATCATCCTTATAGTCC; ATF4‐F, GAGGATCCCCGGGTACCGGTCGCCACCATGACCGAAATGAGCTTCCTG; ATF4‐R, TCCTTGTAGTCCATACCGGGGACCCTTTTCTTCCCCCTTG; XBP1‐F, ATGGTGGTGGTGGCGCCCCC; and XBP1‐R, TTAGTTCATTAATGGCTTCCAGCT.
2.4. Quantitative real‐time PCR
Total RNA was extracted using TRIzol reagent (Sigma). First‐strand cDNA was synthesized using the Promega M‐MLV reagent kit (Promega; cat. M1705) according to the manufacturer’s instructions. The mRNA was quantified by real‐time PCR using SYBR Master Mixture (Takara Bio). GAPDH mRNA expression levels served as the internal control for normalization. The comparative Ct method was used to evaluate the relative mRNA expression levels of genes among different groups. The primer sequences were as follows: URB1‐F, GCGGAAACCTGACACTCTTG; URB1‐R, GACTTCGCTCGTGGGATAAA; ATF4‐F, CAGCAGCACCAGGCTCT; ATF4‐R, TCGAAGGTGTCTTTGTCGGT; CCNA2‐F, CCCAGAAGTAGCAGAGTTTGTG; CCNA2‐R, TTGTCCCGTGACTGTGTAGAG; XBP1‐F, CCGCAGCAGGTGCAGG; and XBP1‐R, GAGTCAATACCGCCAGAATCCA.
2.5. Western blot analysis
Total protein was extracted from RKO and HCT116 cells and separated by SDS‐PAGE. The extracts were transferred to nitrocellulose membranes, blocked, and incubated with a primary Ab overnight at 4°C. The immune complexes were incubated with secondary Abs and then visualized using a chemiluminescence detection system (Thermo Fisher Scientific). The band intensity was semiquantified using ImageJ software. Protein expression was normalized to the level of GAPDH. The primary Abs, including anti‐URB1 rabbit polyclonal Ab, anti‐ATF4 rabbit mAb, anti‐XBP1 rabbit mAb, and anti‐CCNA2 mouse mAb were purchased from Abcam.
2.6. Cell proliferation assay
Cells in logarithmic growth were seeded into 96‐well plates at a density of 2000 cells/well in an atmosphere of 5% CO2 at 37°C. The Celigo S Imaging Cell Cytometer (Nexcelom Bioscience) was utilized to count and record the growth of cells every day for 5 days. For MTT assays, the cells were incubated with MTT (20 μL, 5 μg/mL) every day until 4 hours prior to termination of cell culture. After aspiration of the medium, DMSO (100 μL) was used to lyse the formazan. Absorbance at 490 nm was recorded using a microplate reader.
2.7. Colony formation assay
Cells were seeded into 6‐well culture plates at a density of 500 cells/well or 1000 cells/well. The cells were cultured in a humidified incubator at 37°C with 5% CO2 for 14 days to allow for colony formation. Medium was replaced and cell status was observed every 2‐3 days. After washing with PBS and fixation with paraformaldehyde, cells were stained with crystal violet (0.1%) for 10‐20 minutes. The plates were scanned and the number of colonies was counted.
2.8. Flow cytometry assays
Cells growing in 6‐well plates were harvested and centrifuged (150 g) for 5 minutes and then washed with precooled D‐Hanks solution (pH 7.2‐7.4, 4°C) followed by binding buffer. Propidium iodide and annexin V‐APC (KeyGEN) was used to stain the cells for apoptosis analysis. Propidium iodide staining was used for cell cycle analysis. The percentage of cells was analyzed by flow cytometry (FCM) (FACSCalibur).
2.9. Microarray assay and bioinformatics analysis
Microarray assay was carried out to identify DEGs between RKO cells transfected with sh1 (sh‐URB1#1, n = 3) and nc (sh‐NC, n = 3) lentiviral vectors. This experiment was implemented by Shanghai GeneChem. In brief, total RNA was extracted from URB1 shRNA‐ and control shRNA‐transfected RKO cells. Complementary DNA was synthesized, labeled, and hybridized to the human GeneChip primeview array (Affymetrix). GeneChip Scanner 3000 and GeneChip GCOS 1.4 software were used to scan and analyze the data. The Ingenuity Pathway Analysis (IPA) system (Ingenuity Systems) was used to identify the DEGs and predict the underlying regulatory mechanisms. The JASPAR database (http://jaspar.genereg.net/) was consulted to predict the XBP1 binding sites within the promoter region of ATF4.
2.9.1. Immunofluorescence
Cells were seeded into 24‐well culture plates and treated with URB1 silencing (sh‐URB1#1) or negative control (sh‐NC) lentiviral vectors, followed by 4% paraformaldehyde fixation for 15 minutes and permeabilization with 0.3% Triton X‐100 at 37°C for 30 minutes. After blocking with 10% goat serum for 30 minutes at room temperature, the cells were incubated with fluorescent Abs against ATF4 (1:1000 dilution in PBS) and XBP1 (1:500 dilution in PBS) at 4°C overnight. Subsequently, cells were further incubated with a FITC‐conjugated secondary Ab and 5% BSA in PBS for 1 hour at room temperature in the dark. Cells were then stained with DAPI for 5 minutes. Images were acquired on a confocal microscope (Leica Microsystems).
2.9.2. Dual‐luciferase reporter assay
The Cell Genome Extraction Kit was used to extract total genomic DNA. The ATF4 promoter sequence was amplified using the primers (F, 5′‐ACGCGT CACTGGGAGCCTTGTGACTT‐3′; R, 5′‐CTCGAGAAACTCTGGAAAGGAGCCGG‐3′; product length 597 bp). The target fragment was recovered by agarose gel electrophoresis and ligated between the MluI and Xhol restriction sites of the pGLC‐basic vector to obtain the ATF4 promoter luciferase reporter vector (WT‐pGL3‐ATF4). A mutant vector of the ATF4 promoter (Mut‐pGL3‐ATF4) was constructed according to the site‐specific mutation kit instructions, and the mutation primers were as follows (F, 5′‐CGAACTACCCCAGGGAGCAAAGCGCGAAGAAAATCT‐3′; R, 5′‐AGATTTTCTTCGCGCTTTGCTCCCTGGGGTAGTTCG‐3′). The XBP1 overexpression vector (CMV‐XBP1) and the negative control vector (CMV‐Empty) were constructed by Hanbio Biotech. Lipofectamine 2000 was used for transfection and 48 hours later, firefly luciferase and Renilla luciferase activity was measured using a Dual‐Luciferase Reporter Assay System (Hanbio). Relative luciferase activity was corrected using the Renilla luciferase activity of the CMV‐Empty and normalized to the activity of the control.
2.9.3. Chromatin immunoprecipitation assay
RKO cells in logarithmic growth period were cross‐linked in 4% paraformaldehyde and the reaction was quenched with glycine. Pelleted cells were resuspended in the SDS lysis buffer and incubated on ice for 10 minutes. Chromatin DNA was sonicated and centrifuged for 10 minutes at 13 000 g at 4°C. XBP1 was immunoprecipitated from the cleared lysates by incubation overnight with a rabbit monoclonal anti‐XBP1 Ab (ab220783) at 4°C. Agarose protein A/G was added and incubated overnight with shaking at 4°C. After washing and elution, the protein‐DNA complex was reversed by heating at 65°C for 4 hours. Eluate was adjusted to 40 mmol/L Tris pH 6.8, 10 mmol/L EDTA, then incubated with RNase A, and followed by proteinase K. DNA was recovered by DNA recovery kit. Polymerase chain reaction was carried out with MasterMix from 2 Prime with the following primer sets: ATF4 (primer) promoter F, 5′‐GGCCTCGGTTTACCATTGGA‐3′; and R, 5′‐AGTCTGCATGGCTCCTCCTA‐3′.
2.10. In vivo experiment
Animal studies were undertaken in strict accordance with the Guide for the Care and Use of Laboratory Animals and approved by the Ethics Committee of the Gansu Provincial Hospital (Gansu, China). Twenty (n = 10 per group) BALB/C nude mice (4 weeks old, female) were purchased from Shanghai SLCA Laboratory Animal Co. RKO cells (1 × 106 cells in 200 μL PBS) transfected with sh‐URB1#1 or sh‐NC lentiviral vectors were injected s.c. into the right axilla of the nude mice. The weight of each mouse and xenograft length and width were measured every 2 days starting on day 10 after injection. These data were used to calculate the weight and volume of xenografts. Intraperitoneal injection of D‐Luciferin (15 mg/mL), carried out prior to anesthesia and analysis using the Fluorescence Imaging System (Perkin Elmer), was used to monitor the xenograft growth status in vivo. Xenograft tissues were evaluated macroscopically and microscopically.
2.10.1. Statistical analysis
Data for continuous variables are presented as means ± SD. An independent sample t test was used to assess the significant differences between two groups, and differences among three or more groups were compared by one‐way ANOVA. The χ2 test was used to examine the associations between URB1 expression and different clinical parameters. P < .05 was defined as statistically significant. GraphPad Prism 7.0 software and SPSS 20.0 (IBM) were used to produce all statistics.
3. RESULTS
3.1. URB1 is highly expressed and positively associates with poor clinicopathological characteristics in human CRC
By consulting and analyzing The Cancer Genome Atlas datasets (http://ualcan.path.uab.edu), we found that URB1 expression is noticeably higher in colorectal cancer tissues compared with adjacent normal tissues (Figure 1A). We then showed that URB1 mRNA expression was significantly upregulated in cancer tissues compared with paratumor tissues based on RT‐PCR detection in 40 CRC samples (Figure 1B). Furthermore, the two sheets of CRC TMA were used to analyze the correlation between URB1 expression and clinicopathological features by IHC staining. From TMA‐HColA180Su17 IHC staining, we found that 55.45% (56/101) of the cancer specimens showed a higher URB1 expression, whereas URB1 overexpression was only detected in 37.62% (38/101) of the adjacent normal specimens (Figure 1C,D). Compared with paratumor tissues, URB1 was markedly overexpressed in cancer tissues and significantly correlated with poorer tumor differentiation and later T and TNM stage (Figure 1E,G,H), but not with the other clinicopathological characteristics (Table 1). Moreover, from TMA‐HColA160Cs01 IHC staining, we observed that the URB1 expression pattern was not related to K‐RAS mutation (Figure 1F). The results uncovered that URB1 was upregulated in CRC, and stronger URB1 expression was closely correlated with more severe clinicopathological characteristics, suggesting that URB1 might be an oncogene in CRC.
FIGURE 1.
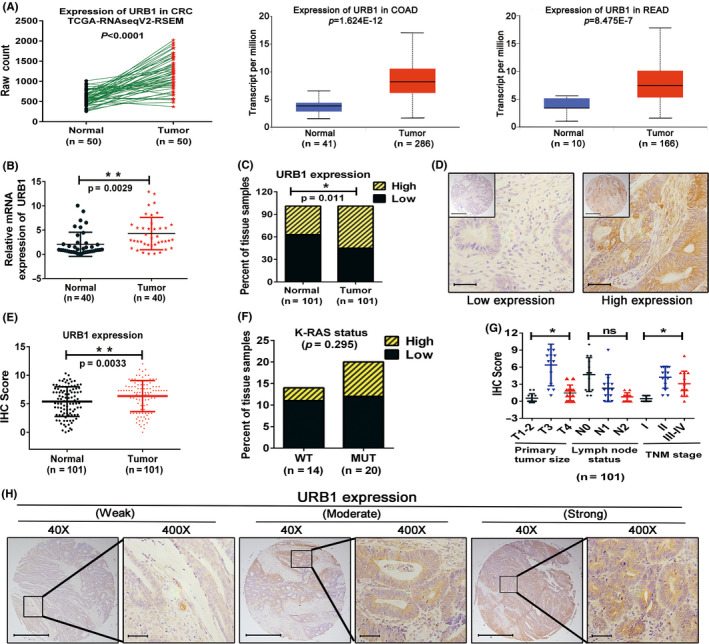
Expression and clinical correlation of URB1 in colorectal cancer (CRC). A, Comparison of URB1 expression in CRC tissues and adjacent normal tissues based on The Cancer Genome Atlas (TCGA) datasets. COAD, colon adenocarcinoma; READ, rectal adenocarcinoma. B, mRNA expression of URB1 was measured in paired clinical CRC samples using quantitative real‐time PCR (n = 40). C‐E, Immunohistochemistry (IHC) was utilized to measure the quantification of URB1 protein expression in clinical CRC tissue microarray (HcolA180Su17) (n = 101). F, Correlation between URB1 expression and K‐RAS status from CRC tissue microarray (HcolA160Cs01). G, Overexpression of URB1 was associated with greater T stage (P = .012) and TNM stage (P = .005) in CRC patients. H, Quantification and representative photomicrographs of IHC staining for URB1 protein expression in TMA of CRC. Scale bar = 100 μm (×40 magnification) and 50 μm (×400 magnification). Data are expressed as mean ± SD. *P < .05, **P < .01. ns, not significant
TABLE 1.
Correlation between URB1 expression and clinicopathologic characteristics in patients with colorectal cancer
| Characteristic | No. of cases | URB1 expression | P value a | |
|---|---|---|---|---|
| Low | High | |||
| Age (y) | ||||
| ≤60 | 22 | 9 | 13 | .697 |
| >60 | 79 | 36 | 43 | |
| Gender | ||||
| Male | 50 | 19 | 31 | .189 |
| Female | 51 | 26 | 25 | |
| Pathologic subtype | ||||
| Adenocarcinoma | 63 | 30 | 33 | .429 |
| Mucinous adenocarcinoma | 38 | 15 | 23 | |
| Pathological grading | ||||
| pG1 | 23 | 16 | 7 | .023* |
| pG2 | 62 | 23 | 39 | |
| pG3 | 16 | 6 | 10 | |
| Primary tumor | ||||
| T1, T2 | 6 | 5 | 1 | .012* |
| T3 | 75 | 36 | 39 | |
| T4 | 20 | 4 | 16 | |
| Lymph node status | ||||
| N0 | 61 | 29 | 32 | .757 |
| N1 | 30 | 12 | 18 | |
| N2 | 10 | 4 | 6 | |
| TNM stage | ||||
| I | 6 | 5 | 1 | .005** |
| II | 54 | 29 | 25 | |
| III‐IV | 41 | 11 | 30 | |
χ2 test.
P < .05.
P < .01.
3.2. URB1 silencing inhibits proliferation and induces apoptosis of CRC cells in vitro
We explored the potential effect of URB1 on the malignant biological phenotype of CRC cells. First, we utilized RT‐PCR and western blotting to measure the endogenous expression levels of URB1 in CRC cell lines (RKO, HCT116, SW480, and LOVO) and the NCM460 normal colon epithelial cell line. The results indicated that CRC cells have higher URB1 mRNA and protein expression levels than normal colon epithelial cells (Figure 2A). We designed and synthesized two shRNAs (sh1 and sh2) to knock down URB1 in RKO and HCT116 cells, and then verified that URB1 mRNA and protein expression was efficiently silenced by the two shRNAs (Figure 2B). We carried out Celigo cell counting, MTT, and colony formation assays to test the effect of URB1 on CRC cell proliferation. Results showed that URB1 silencing significantly impeded the proliferation and cloning ability of RKO and HCT116 cells (Figure 2C‐E). Furthermore, FCM cell apoptosis assay results showed that URB1 silencing significantly induced apoptosis in CRC cells (Figure 2F). Taken together, the results showed that URB1 might enhance the malignant proliferation phenotype of CRC cells, suggesting that URB1 acts as an oncogene in CRC.
FIGURE 2.
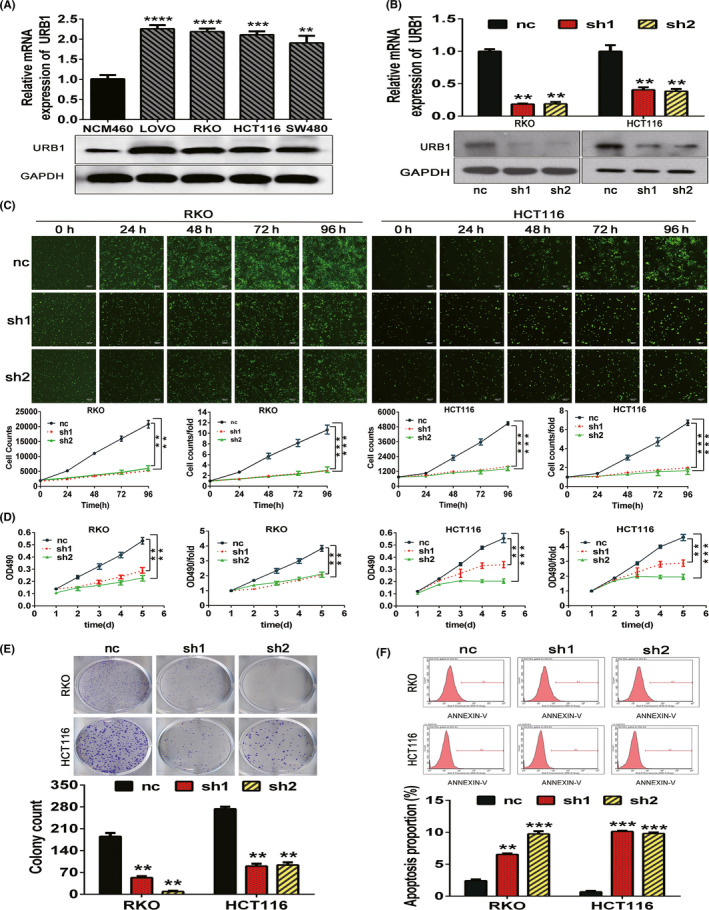
Silencing of URB1 inhibits proliferation and induces apoptosis in colorectal cancer (CRC) cells. A, URB1 mRNA and protein expression in a normal human colon epithelial cell line (NCM460) and the human CRC cell lines (LoVo, RKO, HCT116, and SW480) were assessed by quantitative real‐time PCR (qRT‐PCR) and western blotting. B, Interference efficiency of two specific shRNAs (sh1 and sh2) to URB1 in CRC cells was measured by qRT‐PCR and western blotting. C, D, Celigo cell counting assays and MTT assays indicated that URB1 silencing inhibited proliferation in CRC cells. E, Colony formation in CRC cells was repressed by URB1 silencing. F, Apoptosis in CRC cells was increased by URB1 silencing. Scale bar = 50 μm (×100 magnification). Data are expressed as mean ± SD (n = 3). **P < .01, ***P < .001, ****P < .0001. nc, control lentiviral vector; ns, not significant
3.3. Identification of ATF4 as a downstream target of URB1 in CRC
Microarray screening experiments based on three pairs of URB1 silenced (sh‐URB1#1) and negative control (sh‐NC) RKO cells and IPA were used to investigate DEGs and their related regulatory mechanisms. Pearson’s correlation analysis and principal component analysis collectively implied that the extracted samples fully met the quality inspection standards (Figure 3A,B). The volcano plot analysis identified that, compared with the sh‐NC group, there were 376 upregulated and 441 downregulated DEGs in the sh‐URB1#1 group (Figure 3C). Hierarchical clustering analysis was used to compare the expression patterns and the functional clustering of DEGs (Figure 3D). The IPA indicated that the DEGs were enriched in biological functions including cell cycle regulation and cancer, and in the canonical pathways including cell cycle control, tRNA charging, and p53 signaling (Figure 3E,F). The heatmap of DEGs and IPA upstream regulator analysis showed that ATF4 was a downstream target of URB1 and located upstream of other DEGs (Figure 3G,H). Finally, we verified that URB1 positively regulated ATF4 and CCNA2 expression in vitro (Figure 3I), which in turn supported the microarray and IPA results.
FIGURE 3.
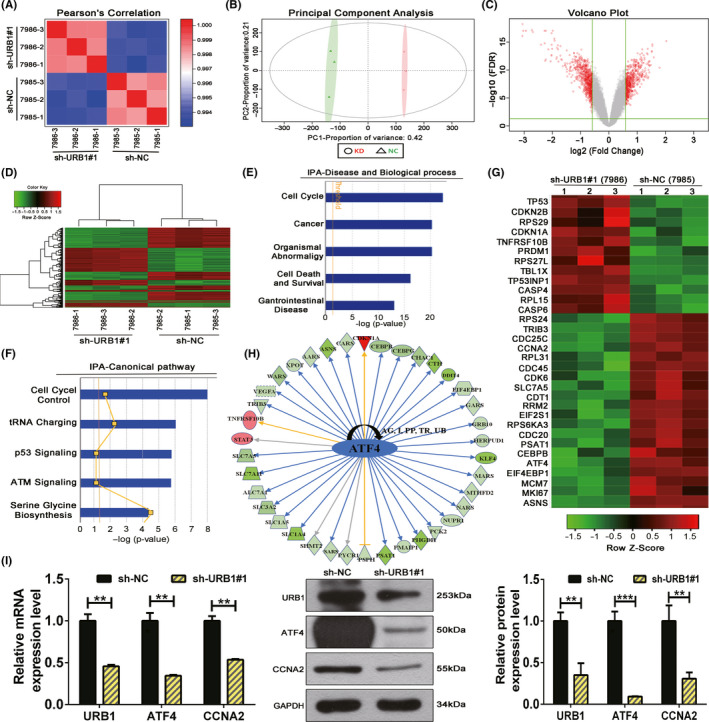
Microarray screening and Ingenuity Pathway Analysis (IPA) suggest an oncogenic role for URB1 and predicted the differentially expressed genes (DEGs) involved in URB1‐related biological processes. A, B, Pearson’s correlation (A) and principal component analysis (B) were used to affirm the correlation and variation of experimental samples. C, Volcano plot was used to compare DEG expression profiles. D, Hierarchical clustering heatmap showing the mRNA expression profiles in the negative control (sh‐NC) and silencing (sh‐URB1#1) groups. E, F, Enrichment of the top five cell biological processes and canonical signaling pathways based on IPA analysis suggested the biological functions of URB1. G, Heatmap of 32 selected DEGs highly related to the biological processes and canonical pathways analyzed in E and F. H, Upstream regulator analysis of IPA revealed that the displayed DEGs can be demonstrably regulated by URB1 through a downstream target, activating transcription factor 4 (ATF4). I, Quantitative RT‐PCR and western blotting verified the positive regulation of ATF4 by URB1. Data are expressed as mean ± SD (n = 3). **P < .01, ***P < .001. CCNA2, cyclin A2
3.4. Overexpression of ATF4 effectively restores the malignant proliferation phenotype of URB1‐silenced CRC cells
We transfected corresponding lentiviral vectors into RKO cells and utilized RT‐PCR and western blot assays to detect the recovery effect of the ATF4 overexpression vector (OE‐ATF4) on ATF4 expression in URB1‐silenced (KD‐URB1) RKO cells. The results indicated that, compared with the KD (URB1) + NC (ATF4) group, ATF4 expression was significantly higher in the KD (URB1) + OE (ATF4) group, indicating that ATF4 acts downstream of URB1 regulation (Figure 4A). Furthermore, MTT and colony formation data revealed that, compared with the NC (URB1) + NC (ATF4) group, proliferation and cloning ability was inhibited visibly in the KD (URB1) + NC (ATF4) group, but was restored effectively in the KD (URB1) + OE (ATF4) group (Figure 4B,C). Additionally, the FCM assay suggested that overexpression of ATF4 markedly reversed the increased apoptotic capacity of URB1‐silenced RKO cells (Figure 4D). These results clarify that ATF4 is a downstream target of URB1 and is involved in the oncogenic role of URB1 in CRC.
FIGURE 4.
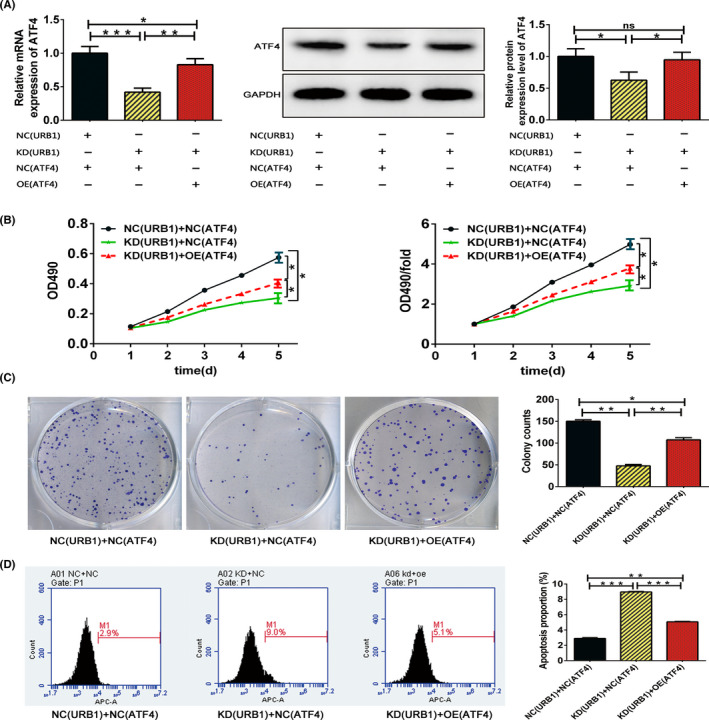
Activating transcription factor 4 (ATF4) overexpression effectively rescues the malignant phenotype of URB1‐silenced RKO cells. A, Confirmed by quantitative RT‐PCR and western blotting, the ATF4 overexpression lentiviral vector (OE ATF4) restored ATF4 expression in URB1‐silenced RKO cells. B, C, MTT and colony formation assays were used to compare the proliferation and cloning ability of RKO cells between the different transfection groups. D, Flow cytometry indicated that ATF4 overexpression reduced apoptosis in URB1‐silenced RKO cells. Data are expressed as mean ± SD (n = 3). *P < .05, **P < .01, ***P < .001. KD, knock down; NC, negative control; ns, not significant
3.5. Transcriptional regulation of ATF4 by URB1 is mediated by XBP1
Through microarray screening and in vitro experiments, we verified that XBP1, a classical transcription factor, can be positively regulated by URB1 (Figure 5A,B). By consulting the JASPAR database (http://jaspar.genereg.net/), we forecasted that XBP1 has a binding site within the 2000 bp promoter region of ATF4 (Figure 5C). Furthermore, the WT and mutant sequences of the ATF4 promoter region containing the XBP1 binding site were constructed and cloned into a luciferase reporter vector (Figure 5D). We then carried out dual‐luciferase reporter experiments and observed that the XBP1 overexpression vector prominently upregulated the luciferase activity of the WT promoter sequence but not the mutant promoter sequence of ATF4. Furthermore, CHiP assay indicated that the ATF4 promoter region can be bound physically by XBP1 protein. This indicated that XBP1 might activate ATF4 transcription by directly binding to its promoter sequence (Figure 5E,H). Moreover, we confirmed that ATF4 mRNA expression was dramatically increased by the XBP1 overexpression plasmid (Figure 5F). In addition, immunofluorescence staining was used to measure the direct interaction of XBP1 and ATF4. The data indicated that XBP1 and ATF4 colocalized in the nucleus in RKO cells and that URB1 silencing synchronously decreased the fluorescent expression of the XBP1 and ATF4 proteins (Figure 5G). In brief, the results revealed the feasibility of a URB1‐XBP1‐ATF4 regulatory mechanism in CRC cells.
FIGURE 5.
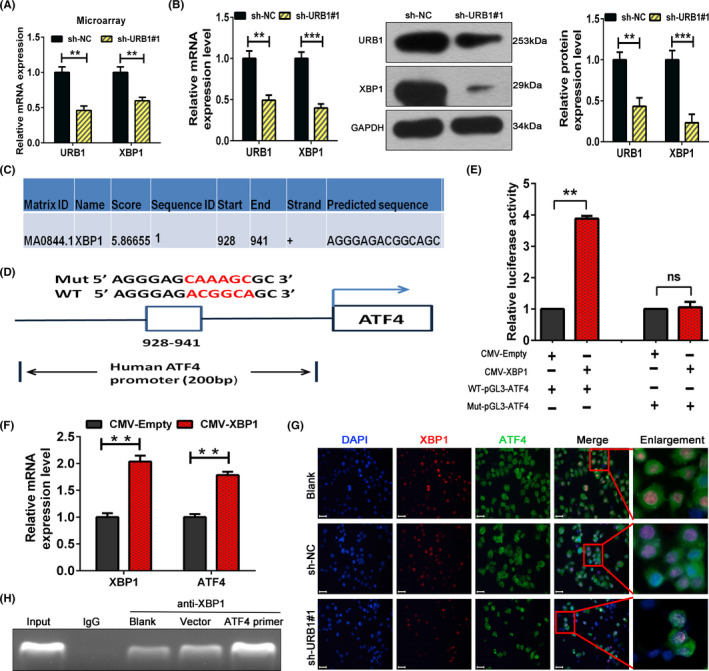
URB1 regulates activating transcription factor 4 (ATF4) through the transcriptional control of X‐box binding protein 1 (XBP1). A, B, Microarray screening, RT‐PCR, and western blotting were used to investigate the potential regulation of XBP1 by URB1. C, XBP1 binding site on the ATF4 promoter region was predicted by consulting the JASPAR database. D, Schematic illustration of the XBP1 binding site on WT and mutant (Mut) ATF4 promoter sequences. E, F, Dual luciferase reporter assays and RT‐PCR confirmed the specific binding of XBP1 with the ATF4 promoter and the activation of ATF4 mRNA transcription. G, Subcellular colocalization of XBP1 and ATF4 in RKO cells was evaluated by immunofluorescence staining. H, Interaction of XBP1 with ATF4 promoter region was validated by CHiP assay. Scale bar = 50 μm (×100 magnification). Data are expressed as mean ± SD (n = 3). **P < .01, ***P < .001. ns, not significant; sh‐NC, negative control
3.6. URB1 overexpression promotes proliferation of CRC cells and fibroblast cells in vitro
We successfully overexpressed URB1 in three CRC cells and one fibroblast to verify the oncogenic role of URB1 (Figure 6A,E). The MTT, colony formation, and FCM assays indicated that URB1 overexpression markedly promoted the proliferation of SW480 cells and inhibited the apoptosis of HCT116 cells, but with little effect to the others (Figure 6B‐D). Moreover, we found that URB1 overexpression significantly accelerated cell cycle progression and promoted proliferation in fibroblast cells (Figure 6F,G). In addition, western blotting data showed that URB1 overexpression prominently upregulated XBP1 and ATF4 protein expression levels in RKO cells (Figure 6H), implying the role of the URB1‐XBP1‐ATF4 axis in tumorigenesis. The results indicated that URB1 might be an oncogene in CRC.
FIGURE 6.
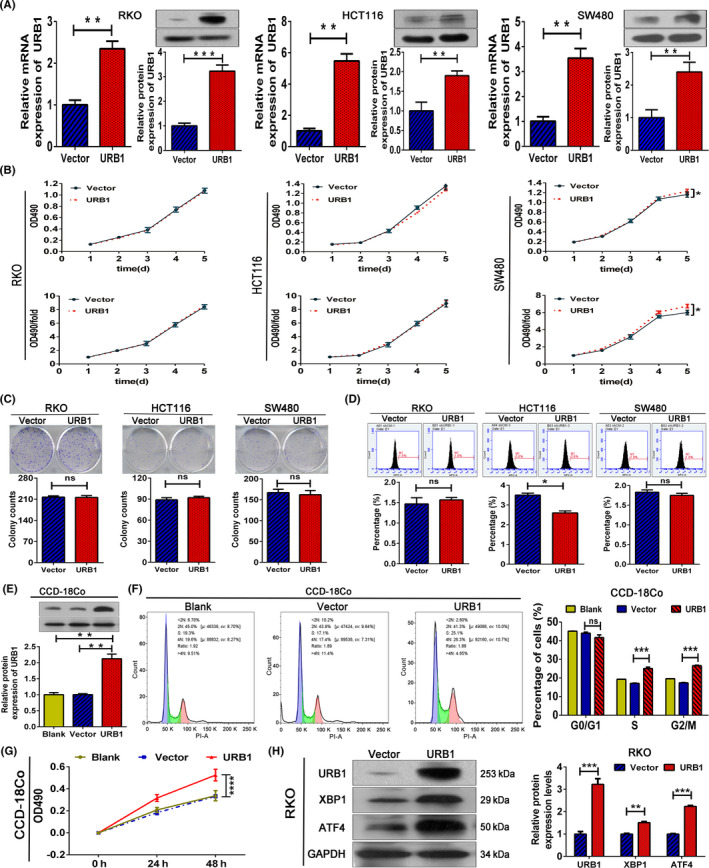
URB1 overexpression promotes proliferation in colorectal cancer (CRC) cells and fibroblasts. A, URB1 overexpression efficiency in CRC cells was measured by quantitative RT‐PCR and western blotting. B‐D, MTT, colony formation, and flow cytometry (FCM) assays were used to evaluate the effect of URB1 overexpression on CRC cells. E, URB1 overexpression efficiency in fibroblasts assessed by western blotting. F, G, FCM and MTT assays indicated that URB1 overexpression promotes cell cycle progression and proliferation in fibroblasts. Blank, blank control group; URB1, URB1 overexpression group; Vector, negative control group. H, Regulation of URB1 on X‐box binding protein 1 (XBP1) and activating transcription factor 4 (ATF4) in CRC cells was measured by western blotting. Data are expressed as mean ± SD (n = 3). *P < .05, **P < .01, ***P < .001, ****P < .0001. ns, not significant
3.7. URB1 silencing suppresses the growth of human CRC xenografts and inhibits ATF4 and CCNA2 expression in vivo
We then explored whether URB1 contributes to CRC carcinogenesis and growth in vivo. The s.c. human CRC xenograft model was established in BALB/c nude mice using URB1‐silenced (sh‐URB1#1) RKO cells and the corresponding negative control (sh‐NC) cells. The results revealed that the sh‐URB1#1 group showed a lower tumor weight, smaller tumor size, and a slower increase in tumor volume compared with the sh‐NC group (Figure 7A‐E). Fluorescence imaging in vivo was executed to observe the growth status of CRC xenografts in tumor‐bearing nude mice. Fluorescein intensity detection showed a conspicuous disparity in xenograft cell proliferation between the sh‐URB1#1 and sh‐NC groups (Figure 7F). Immunohistochemistry staining images showed that URB1, ATF4, CCNA2, and Ki‐67 protein expression levels were observably lower in the URB1‐silenced xenografts than in controls (Figure 7G), which was consistent with the conclusions in vitro. In brief, these in vivo results provide evidence that URB1 substantially contributes to tumorigenesis and growth in CRC by activating ATF4.
FIGURE 7.
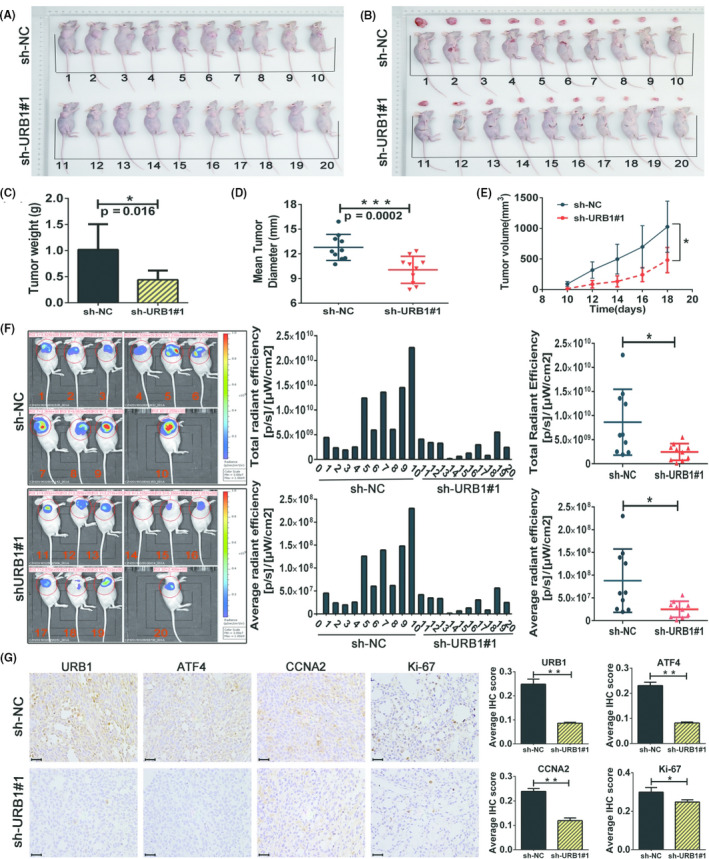
Silencing of URB1 inhibits colorectal cancer cell proliferation in vivo. A, B, Images of s.c. xenografts from URB1 silenced (sh‐URB1#1) and negative control (sh‐NC) groups (n = 10). C‐E, Comparison of tumor weight (C), mean tumor diameter (D), and tumor volume growth curves (E) for s.c. xenografts of the indicated groups. F, In vivo fluorescence imaging (left panel) and GFP intensity analysis (middle and right panels) show the different growth status of xenografts between the indicated groups (n = 10). G, Immunohistochemistry staining to detect the regulatory effect of URB1 on activating transcription factor 4 (ATF4), cyclin A2 (CCNA2), and Ki‐67 expression in vivo. Scale bar = 50 μm (×400 magnification). Data are expressed as mean ± SD (n = 5). *P < .05, **P < .01, ***P < .001
4. DISCUSSION
The most important finding of this study was that URB1, one of the RAFs, plays a pivotal role in tumorigenesis. Overexpression of URB1 in CRC was highly related to more severe clinicopathological characteristics. Silencing of URB1 curtailed carcinogenesis and CRC growth in vitro and in vivo through inhibition of proliferation and induction of apoptosis through the transcriptional regulation of ATF4. In addition, overexpression of ATF4 restored the malignant phenotype of CRC. Furthermore, we showed that the transcriptional regulation of ATF4 by URB1 was mediated by the direct binding of the transcription factor XBP1 to the ATF4 promoter. Additionally, we found that the expression patterns of CCNA2 and ATF4 were decreased following URB1 depletion, suggesting that CCNA2 might be involved in the URB1‐XBP1‐ATF4 axis in the development and progression of CRC (Figure 8).
FIGURE 8.
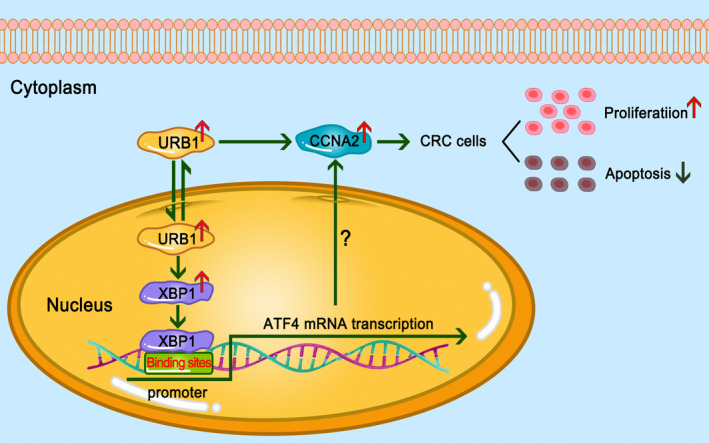
Schematic representation of the main processes of URB1 targeting the X‐box binding protein 1 (XBP1)‐activating transcription factor 4 (ATF4) regulatory axis involved in colorectal cancer (CRC) proliferation. Upregulation of URB1 activates the transcription factor XBP1, which further stimulates ATF4 mRNA transcription and ultimately promotes CRC cell proliferation. Cyclin A2 (CCNA2) could be a potential executor of the URB1‐XBP1‐ATF4 axis in CRC
Ribosome biogenesis is a classical hallmark of cell growth and proliferation and is intimately associated with tumorigenesis and progression. 21 , 22 , 23 , 24 High‐efficiency ribosome biogenesis also fuels tumor epithelial‐mesenchymal transition to program cellular plasticity, dedifferentiation, and cancer progression. 25 It was found that RAFs, including RIO1 and NOB1, are extensively upregulated in various malignant tumors. 26 , 27 One type of RAF, PNO1, is critical for ribosome biogenesis and contributes to CRC cell proliferation by negatively regulating the p53/p21 pathway. 9 These findings imply that RAF could act independently of the ribosome assembly and could be a highly conserved driver of tumor initiation and progression.
It is well known that URB1 is essential for ribosome assembly. Previous studies have revealed that Npa1 acts as the backbone of the RAFs complex to actuate the assembly of the 60S ribosome subunit. 4 , 6 , 28 , 29 Npa1, which interacts with Rrp5 and would be expected to be present in 90S particles, is also required for 40S subunit synthesis. 30 , 31 The uncontrolled growth and rapid proliferation of cancer cells require accelerated protein synthesis to fuel cell division and metabolism. Initially, we suspected that URB1 might be activated by cancer cells to increase ribosome biogenesis to adapt to alterations in the cellular environment. Interestingly, through our investigation, a novel role for URB1 in CRC oncogenesis has gradually emerged.
The prominent oncogenic function of URB1 in CRC encouraged us to explore its downstream molecular mechanism. Through a combination of microarray screening, IPA bioinformatics methods, and lentiviral‐mediated gene loss of function, we showed that ATF4 was significantly regulated by URB1 and located upstream of other DEGs such as CEBPB, CDKN1A, VEGFA, and TRIB3 (Figure 3H). In addition, restoration of ATF4 significantly reversed the malignant proliferation of URB1‐silenced CRC cells, which indicates a potential driving force for the URB1‐ATF4 regulatory mechanism in CRC initiation and progression. Activating transcription factor 4 is a stress‐induced transcription factor that is frequently activated in multiple malignant tumors and regulates the cellular stress response to benefit tumor cell adaptation to the limited availability of nutrients resulting from rapid cell proliferation. 32 , 33 , 34 Uncontrolled cancer cell growth and proliferation requires extensive protein synthesis, which can result in two states. First, a large amount of amino acids are consumed to supply protein synthesis, which leads to amino acid deprivation and simultaneous acceleration of ribosome biogenesis. Second, immoderate protein synthesis and misfolded proteins stimulate the unfolded protein response (UPR). Activating transcription factor 4 is the common downstream effector of these two responses. 35 In the present study, URB1 silencing impeded proliferation, induced apoptosis, and downregulated ATF4 expression in CRC cells. We speculated that step‐down ribosome biogenesis is likely to ameliorate immoderate protein synthesis and decrease ATF4 due to the mitigation of UPR. Therefore, we sought to identify the underlying mechanism behind URB1 regulation of ATF4.
Given that ATF4 was verified as a downstream target of URB1 in CRC, we searched for its upstream transcription factor by consulting the JASPAR database. Results showed that XBP1 had a high score for binding with the ATF4 promoter region (Figure 5C). Interestingly, microarray results had shown that XBP1 was one of the DEGs influenced by URB1, and through the JASPAR database, we verified that XBP1 was positively regulated by URB1. Through a series of experiments, we confirmed that XBP1 directly binds with the ATF4 promoter sequence and activates ATF4 mRNA transcription in vitro, suggesting that XBP1 mediates the transcriptional regulation of ATF4 by URB1. It is widely known that XBP1 is similar to ATF4, also belongs to the bZIP protein family, is a downstream target of the IRE/XBP1 signaling pathway, and is the most conserved response effector of ERS. 36 X‐box binding protein 1 mediates the proper folding and degradation of misfolded proteins by transcriptionally regulating multiple downstream target genes of UPR, thus balancing ERS to protect cells from death. 37 , 38 At present, XBP1 has been found to be significantly overexpressed in a variety of malignancies and to increase cell survival, proliferation, invasion, and metastasis in the harsh tumor microenvironment. 39 , 40 , 41 , 42 As a highly conserved intracellular zinc finger structural transcription factor and executor of UPR, XBP1 regulates UPR‐related processes by directly binding to the promoter of its downstream target gene. Given that ATF4 is a noted target gene of UPR, it is likely that XBP1 and ATF4 jointly participate in cell stress regulation. As URB1 is crucial to protein synthesis, it contributes to tumorigenesis through its extraribosomal function. We speculated that URB1 overexpression in tumor cells might overspeed protein synthesis, arouse UPR, and further activate XBP1 expression, to balance the stress environment and promote cell survival and proliferation.
More interestingly, URB1 silencing notably downregulated ATF4 and the cell cycle promoting factor CCNA2. Located downstream of the p53/p21 pathway, CCNA2 most likely controls cell cycle progression, cellular senescence, and determines cell fate. 43 , 44 , 45 , 46 An increasing number of studies suggest that CCNA2 is aberrantly expressed in various tumors and participates in multiple steps of tumor progression. 44 , 47 , 48 , 49 While it responds to diverse therapies, CCNA2 is also important to different tumor states, including proliferation and susceptibility to apoptosis. 50 , 51 , 52 In accordance with expectations, the current study showed that CCNA2 plays a key role in CRC. We showed that URB1 silencing significantly inhibited proliferation and induced apoptosis in CRC cells by downregulating ATF4 and CCNA2. This further suggested that CCNA2 might be an executor of the URB1‐XBP1‐ATF4 regulatory axis in CRC.
In conclusion, this is the first exploration of the ribosome assembly factor URB1 in tumorigenesis. We showed that URB1 acts as an oncogenic driver in CRC through transcriptional activation of ATF4. Moreover, the transcriptional regulation of ATF4 by URB1 was mediated by the transcription factor XBP1. We have also shown that CCNA2, a downstream target of the p53/p21 pathway, was positively regulated by the URB1‐XBP1‐ATF4 axis and determined the fate of CRC cells. Therefore, our findings suggest that identification of inhibitors of URB1 could be a novel therapeutic strategy in CRC.
ETHICAL CONSIDERATION STATEMENTS
The study was approved by the Ethics Committee of the Gansu Provincial People’s Hospital.
CONFLICT OF INTEREST
There are no conflicts of interest to declare.
ACKNOWLEDGMENTS
This study was supported by grants from the National Natural Science Foundation of China (81660157), the Gansu Provincial Health Committee (GSWSKY‐2019‐69), the Gansu Provincial Nature Science Foundation for Young Scientists and Technology Planning Project (18JR3RA058), and the Gansu Provincial People’s Hospital (19SYPYB‐6, 19SYPYB‐7, 18GSSY4‐2, and 18GSSY3‐1). We thank Hongling Li, Xiaoling Gao, Min Zhang, Xinhai Pei, and Guoqing Ji for helpful advice and discussions. We thank LetPub for its linguistic assistance during the preparation of this manuscript.
Wang T, Li L‐Y, Chen Y‐F, et al. Ribosome assembly factor URB1 contributes to colorectal cancer proliferation through transcriptional activation of ATF4. Cancer Sci. 2021;112:101–116. 10.1111/cas.14643
Funding information National Natural Science Foundation of China, Grant/Award Number: 81660157; Gansu Provincial Health Committee, Grant/Award Number: GSWSKY‐2019‐69; Gansu Provincial Nature Science Foundation for Young Scientists & Technology Planning Project, Grant/Award Number: 18JR3RA058; Gansu Provincial People’s Hospital, Grant/Award Numbers: 19SYPYB‐6, 19SYPYB‐7, 18GSSY4‐2, 18GSSY3‐1.
Contributor Information
Xiang‐Yong Hao, Email: haoxy-2008@163.com.
Tian‐Kang Guo, Email: tiankangguo59@hotmail.com.
REFERENCES
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018; 68(6): 394‐424. [DOI] [PubMed] [Google Scholar]
- 2. Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global patterns and trends in colorectal cancer incidence and mortality. Gut. 2017;66(4):683‐691. [DOI] [PubMed] [Google Scholar]
- 3. Magaji BA, Moy FM. Survival rates and predictors of survival among colorectal cancer patients in a Malaysian tertiary hospital. BMC Cancer. 2017;17(1):339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rosado IV, de la Cruz J. Npa1p is an essential trans‐acting factor required for an early step in the assembly of 60S ribosomal subunits in Saccharomyces cerevisiae . RNA. 2004;10(7):1073‐1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sekiguchi T, Hayano T, Yanagida M, Takahashi N, Nishimoto T. NOP132 is required for proper nucleolus localization of DEAD‐box RNA helicase DDX47. Nucleic Acids Res. 2006;34(16):4593‐4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rosado IV, Dez C, Lebaron S, Caizergues‐Ferrer M, Henry Y, de la Cruz J. Characterization of Saccharomyces cerevisiae Npa2p (Urb2p) reveals a low‐molecular‐mass complex containing Dbp6p, Npa1p (Urb1p), Nop8p, and Rsa3p involved in early steps of 60S ribosomal subunit biogenesis. Mol Cell Biol. 2007;27(4):1207‐1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Derks MFL, Gjuvsland AB, Bosse M, et al. Loss of function mutations in essential genes cause embryonic lethality in pigs. PLoS Genet. 2019;15(3):e1008055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang T, Zhang WS, Wang ZX, et al. RAPTOR promotes colorectal cancer proliferation by inducing mTORC1 and upregulating ribosome assembly factor URB1. Cancer Med. 2020; 9(4): 1529‐1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shen A, Chen Y, Liu L, et al. EBF1‐mediated upregulation of ribosome assembly factor PNO1 contributes to cancer progression by negatively regulating the p53 signaling pathway. Can Res. 2019;79(9):2257‐2270. [DOI] [PubMed] [Google Scholar]
- 10. Ameri K, Harris AL. Activating transcription factor 4. Int J Biochemi Cell Biol. 2008;40(1):14‐21. [DOI] [PubMed] [Google Scholar]
- 11. Singleton DC, Harris AL. Targeting the ATF4 pathway in cancer therapy. Expert Opin Ther Targets. 2012;16(12):1189‐1202. [DOI] [PubMed] [Google Scholar]
- 12. Wortel IMN, van der Meer LT, Kilberg MS, van Leeuwen FN. Surviving stress: modulation of ATF4‐mediated stress responses in normal and malignant cells. Trends Endocrinol Metab. 2017;28(11):794‐806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang Y, Ning YU, Alam GN, et al. Amino acid deprivation promotes tumor angiogenesis through the GCN2/ATF4 pathway. Neoplasia. 2013;15(8):989‐997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang K, Wang M, Li Y, et al. The PERK‐EIF2alpha‐ATF4 signaling branch regulates osteoblast differentiation and proliferation by PTH. Am J Physiol Endocrinol Metab. 2019;316(4):E590‐E604. [DOI] [PubMed] [Google Scholar]
- 15. Ojha R, Leli NM, Onorati A, et al. ER translocation of the MAPK pathway drives therapy resistance in BRAF‐mutant melanoma. Cancer Discov. 2019;9(3):396‐415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Adam I, Dewi DL, Mooiweer J, et al. Upregulation of tryptophanyl‐tRNA synthethase adapts human cancer cells to nutritional stress caused by tryptophan degradation. OncoImmunology. 2018;7(12):e1486353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gonzalez‐Gonzalez A, Munoz‐Muela E, Marchal JA. Activating transcription factor 4 modulates TGFbeta‐induced aggressiveness in triple‐negative breast cancer via SMAD2/3/4 and mTORC2 signaling. Clin Cancer Res. 2018;24(22):5697‐5709. [DOI] [PubMed] [Google Scholar]
- 18. Matassa DS, Amoroso MR, Agliarulo I, et al. Translational control in the stress adaptive response of cancer cells: a novel role for the heat shock protein TRAP1. Cell Death Dis. 2013;4:e851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ishizawa JO, Kojima K, Chachad D, et al. ATF4 induction through an atypical integrated stress response to ONC201 triggers p53‐independent apoptosis in hematological malignancies. Sci Signal. 2016;9(415):ra17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dong T, Liu Z, Zhao S, et al. The expression of CD9 and PIK3CD is associated with prognosis of follicular lymphoma. J Cancer. 2015;6(12):1222‐1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhou X, Liao WJ, Liao JM, Liao P, Lu H. Ribosomal proteins: functions beyond the ribosome. J Mol Cell Biol. 2015;7(2):92‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brighenti E, Calabrese C, Liguori G, et al. Interleukin 6 downregulates p53 expression and activity by stimulating ribosome biogenesis: a new pathway connecting inflammation to cancer. Oncogene. 2014;33(35):4396‐4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. de Las H‐R, Perucho L, Paciucci R, Vilardell J, LLeonart ME. Ribosomal proteins as novel players in tumorigenesis. Cancer Metastasis Rev. 2014;33(1):115‐141. [DOI] [PubMed] [Google Scholar]
- 24. Donati G, Montanaro L, Derenzini M. Ribosome biogenesis and control of cell proliferation: p53 is not alone. Can Res. 2012;72(7):1602‐1607. [DOI] [PubMed] [Google Scholar]
- 25. Prakash V, Carson BB, Feenstra JM, et al. Ribosome biogenesis during cell cycle arrest fuels EMT in development and disease. Nat Commun. 2019;10(1):2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Berto G, Ferreira‐Cerca S. The Rio1 protein kinases/ATPases: conserved regulators of growth, division, and genomic stability. Curr Genet. 2019;65(2):457‐466. [DOI] [PubMed] [Google Scholar]
- 27. Liu K, Chen HL, Wang S, et al. High expression of RIOK2 and NOB1 predict human non‐small cell lung cancer outcomes. Sci Rep. 2016;6:28666. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28. Joret C, Capeyrou R, Belhabich‐Baumas K, et al. The Npa1p complex chaperones the assembly of the earliest eukaryotic large ribosomal subunit precursor. PLoS Genet. 2018;14(8):e1007597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dez C, Froment C, Noaillac‐Depeyre J, Monsarrat B, Caizergues‐Ferrer M, Henry Y. Npa1p, a component of very early pre‐60S ribosomal particles, associates with a subset of small nucleolar RNPs required for peptidyl transferase center modification. Mol Cell Biol. 2004;24(14):6324‐6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schafer T, Strauss D, Petfalski E, Tollervey D, Hurt E. The path from nucleolar 90S to cytoplasmic 40S pre‐ribosomes. EMBO J. 2003;22(6):1370‐1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Grandi P, Rybin V, Bassler J, et al. 90S pre‐ribosomes include the 35S pre‐rRNA, the U3 snoRNP, and 40S subunit processing factors but predominantly lack 60S synthesis factors. Mol Cell. 2002;10(1):105‐115. [DOI] [PubMed] [Google Scholar]
- 32. Gao S, Ge A, Xu S, et al. PSAT1 is regulated by ATF4 and enhances cell proliferation via the GSK3beta/beta‐catenin/cyclin D1 signaling pathway in ER‐negative breast cancer. J Exp Clin Cancer Res. 2017;36(1):179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zeng P, Sun S, Li R, Xiao ZX, Chen H. HER2 upregulates ATF4 to promote cell migration via activation of ZEB1 and downregulation of E‐Cadherin. Int J Mol Sci. 2019;20(9):2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lim JKM, Delaidelli A, Minaker SW, et al. Cystine/glutamate antiporter xCT (SLC7A11) facilitates oncogenic RAS transformation by preserving intracellular redox balance. Proc Natl Acad Sci USA. 2019;116(19):9433‐9442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rozpedek W, Pytel D, Mucha B, Leszczynska H, Diehl JA, Majsterek I. The role of the PERK/eIF2alpha/ATF4/CHOP signaling pathway in tumor progression during endoplasmic reticulum stress. Curr Mol Med. 2016;16(6):533‐544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hassan H, Tian X, Inoue K, et al. Essential role of X‐box binding protein‐1 during endoplasmic reticulum stress in podocytes. J Am Soc Nephrol. 2016;27(4):1055‐1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brodsky JL, Merz A, Serio T. Organelle and proteome quality control mechanisms: how cells are able to keep calm and carry on. Mol Biol Cell. 2014;25(6):733‐734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP‐1. Nat Immunol. 2003;4(4):321‐329. [DOI] [PubMed] [Google Scholar]
- 39. Spaan CN, Smit WL, van Lidth de Jeude JF, et al. Expression of UPR effector proteins ATF6 and XBP1 reduce colorectal cancer cell proliferation and stemness by activating PERK signaling. Cell Death Dis. 2019;10(7):490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen X, Iliopoulos D, Zhang Q, et al. XBP1 promotes triple‐negative breast cancer by controlling the HIF1alpha pathway. Nature. 2014;508(7494):103‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sun Y, Jiang F, Pan Y, et al. XBP1 promotes tumor invasion and is associated with poor prognosis in oral squamous cell carcinoma. Oncol Rep. 2018;40(2):988‐998. [DOI] [PubMed] [Google Scholar]
- 42. Pommier A, Anaparthy N. Unresolved endoplasmic reticulum stress engenders immune‐resistant, latent pancreatic cancer metastases. Science. 2018;360(6394):eaao4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xu S, Wu W, Huang H, et al. The p53/miRNAs/Ccna2 pathway serves as a novel regulator of cellular senescence: Complement of the canonical p53/p21 pathway. Aging Cell. 2019;18(3):e12918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ma Q. MiR‐219‐5p suppresses cell proliferation and cell cycle progression in esophageal squamous cell carcinoma by targeting CCNA2. Cell Mol Biol Lett. 2019;24:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang Z, Yang B, Zhang M, et al. lncRNA epigenetic landscape analysis identifies EPIC1 as an oncogenic lncRNA that interacts with MYC and promotes cell‐cycle progression in cancer. Cancer Cell. 2018;33(4):706‐720.e709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li J, Ying Y, Xie H, et al. Dual regulatory role of CCNA2 in modulating CDK6 and MET‐mediated cell‐cycle pathway and EMT progression is blocked by miR‐381‐3p in bladder cancer. FASEB J. 2019;33(1):1374‐1388. [DOI] [PubMed] [Google Scholar]
- 47. Dong S, Huang F, Zhang H, Chen Q. Overexpression of BUB1B, CCNA2, CDC20, and CDK1 in tumor tissues predicts poor survival in pancreatic ductal adenocarcinoma. Biosci Rep. 2019;39(2):BSR20182306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gao T, Han Y, Yu L, Ao S, Li Z, Ji J. CCNA2 is a prognostic biomarker for ER+ breast cancer and tamoxifen resistance. PLoS One. 2014;9(3):e91771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wu Y, Li H, Wang H, Zhang F, Cao H, Xu S. MSK2 promotes proliferation and tumor formation in squamous cervical cancer via PAX8/RB‐E2F1/cyclin A2 axis. J Cell Biochem. 2019;120(7):11432‐11440. [DOI] [PubMed] [Google Scholar]
- 50. Gan Y, Li Y, Li T, Shu G, Yin G. CCNA2 acts as a novel biomarker in regulating the growth and apoptosis of colorectal cancer. Cancer Manag Res. 2018;10:5113‐5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Peng X, Pan K, Zhao W, et al. NPTX1 inhibits colon cancer cell proliferation through down‐regulating cyclin A2 and CDK2 expression. Cell Biol Int. 2018;42(5):589‐597. [DOI] [PubMed] [Google Scholar]
- 52. Li X, Ma XL, Tian FJ, et al. Downregulation of CCNA2 disturbs trophoblast migration, proliferation, and apoptosis during the pathogenesis of recurrent miscarriage. Am J Reprod Immunol. 2019;82(1):e13144. [DOI] [PubMed] [Google Scholar]