

Abstract
The Hippo‐YAP pathway regulates organ size, tissue homeostasis, and tumorigenesis in mammals. In response to cell density, external mechanical pressure, and/or other stimuli, the Hippo core complex controls the translocation of YAP1/TAZ proteins to the nucleus and thereby regulates cell growth. Abnormal upregulation or nuclear localization of YAP1/TAZ occurs in many human malignancies and promotes their formation, progression, and metastasis. A key example is squamous cell carcinoma (SCC) genesis. Many risk factors and crucial signals associated with SCC development in various tissues accelerate YAP1/TAZ accumulation, and mice possessing constitutively activated YAP1/TAZ show immediate carcinoma in situ (CIS) formation in these tissues. Because CIS onset is so rapid in these mutants, we propose that many SCCs initiate and progress when YAP1 activity is sustained and exceeds a certain oncogenic threshold. In this review, we summarize the latest findings on the roles of YAP1/TAZ in several types of SCCs. We also discuss whether targeting aberrant YAP1/TAZ activation might be a promising strategy for SCC treatment.
Keywords: cervical cancer, esophageal cancer, head‐and‐neck cancer, hippo‐YAP pathway, lung cancer, squamous cell carcinomas
We summarize the latest findings on the roles of YAP1/TAZ in several types of SCCs, proposing that many SCCs initiate and progress when YAP1 activity is sustained and exceeds a certain oncogenic threshold.

1. INTRODUCTION
Stratified squamous epithelium (SSE) consists of layers of squamous epithelial cells arranged over top of a basement membrane. The outermost layers of the nasal‐oral cavity, esophagus, trachea, bronchi, cervix, vagina, and skin are all SSE. SSE constantly interacts with various stimuli, including harmful stresses. In so doing, SSE becomes the origin of several human malignancies, including esophageal squamous cell carcinoma (ESCC), head‐and‐neck SCC (HNSCC), cutaneous (skin) SCC (CuSCC), lung SCC (LSCC), and cervical SCC (CvSCC), which are among the most common forms of human cancers. This transformation is often associated with dysregulation of the Hippo‐YAP intracellular signaling pathway. In this review, we provide an overview of how Hippo‐YAP signaling drives SCC onset and development and discuss how this knowledge might lead to new therapeutic avenues for these insidious cancers.
2. GENETIC ALTERATIONS IN SCCS
The most common genomic alterations in human SCCs are summarized in Table 1. 1 , 2 , 3 , 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11 , 12 The frequencies of these mutations differ among different SCC types. CvSCCs have the lowest mutation rate (4.2 mutations/megabase), 5 with CuSCC having the highest (50‐60 mutations/megabase). 6 , 7
Table 1.
DNA alteration frequencies in various human SCCs
| Gene name | Alteration a frequency (%) | |||||
|---|---|---|---|---|---|---|
| HPV+ HNSCC 1 , 2 , 3 | HPV− HNSCC 1 , 2 , 3 | CvSCC 4 , 5 , 12 | CuSCC 6 , 7 , 12 | ESCC 8 , 9 , 12 | LSCC 10 , 11 , 12 | |
| Cell cycle control | ||||||
| TP53 | 3‐4 | 81‐84 | 5 | 65‐95 | 83‐93 | 73‐84 |
| RB1 | 6 | 3‐4 | 4‐6 | 18‐27 | 2‐9 | 7‐16 |
| MYC | 3 | 9‐14 | 5 | 5‐10 | 2‐9 | 10 |
| SRC | 2 | 5‐21 | 7 | |||
| CDKN2A | 0 | 21‐58 | 2 | 45‐46 | 5‐20 | 44 |
| CDKN2B | 0 | 43 | 1 | 3 | 6 | 28 |
| CCND1 | 31 | 3 | 3‐8 | 33‐46 | 14 | |
| FBXW7 | 4‐22 | 6 | 16 | 10‐23 | 4‐8 | 4 |
| E2F1 | 19 | 2 | 5 | 8‐18 | 14 | 5 |
| CDK6 | 0 | 8 | 2 | 3‐8 | 5 | |
| RTK | ||||||
| EGFR | 0‐6 | 12‐15 | 5 | 14‐31 | 1‐25 | 2‐9 |
| FGFR1 | 0‐1 | 2‐10 | 2 | 10‐31 | 1 | 7‐19 |
| FGFR2 | 4 | 0‐2 | 4 | 7‐15 | 1‐4 | 2‐4 |
| FGFR3 | 11‐12 | 1‐2 | 2 | 13‐31 | 33 | 2‐5 |
| DDR2 | 2‐6 | 3‐5 | 5 | 18‐51 | 7 | |
| ERBB2 | 3 | 3‐4 | 0‐6 | 18‐44 | 2‐6 | |
| IGF1R | 0 | 4 | 4 | 13‐33 | 4 | |
| EPHA2 | 2‐3 | 4‐5 | 5 | 15‐21 | 1 | 2 |
| MET | 0 | 2 | 8 | 21‐23 | 1 | 4 |
| MAPK‐PI3K pathway | ||||||
| HRAS | 0‐4 | 3‐5 | 1 | 20‐26 | 3 | |
| KRAS | 6‐11 | 0‐1 | 4 | 7‐10 | 4 | 2‐5 |
| NRAS | 0 | 1 | 1 | 5‐8 | 1 | |
| BRAF | 3 | 1 | 18‐26 | 1‐2 | 4‐5 | |
| MAPK1 | 8 | 3 | 4 | |||
| NF1 | 0‐6 | 3 | 8 | 15‐56 | 2‐4 | 11‐14 |
| PIK3CA | 22‐56 | 13‐34 | 14‐20 | 10‐23 | 5‐17 | 49 |
| PIK3R1 | 0‐3 | 1 | 3‐4 | 5‐15 | 1 | 4 |
| PIK3CG | 0 | 7 | 2 | 33‐44 | 3 | 11 |
| PIK3CB | 13 | 13 | 10 | 13‐31 | 17 | |
| PTEN | 2‐6 | 4‐12 | 6 | 7‐10 | 2‐5 | 20 |
| AKT1 | 5 | 7 | 3 | 8‐10 | 1‐16 | 4 |
| AKT2 | 5 | 3 | 4‐8 | |||
| AKT3 | 3 | 8 | 1 | 3‐16 | ||
| RICTOR | 11 | 0 | 7 | 10‐18 | 1 | 16 |
| Cell death | ||||||
| BIRC2 | 3 | 7 | 13 | 5 | 1 | 2 |
| FADD | 6 | 32 | 4 | 3 | 15 | |
| TRAF3 | 22 | 1 | 5 | 13‐18 | 4 | |
| CASP8 | 3 | 11 | 5‐12 | 23‐36 | 4 | |
| BCL2L1 | 11 | 0 | 5 | 3‐5 | 15 | 7 |
| Cell differentiation | ||||||
| TP63 | 28 | 19 | 4‐23 | 15‐17 | 16‐41 | |
| SOX2 | 16 | 6 | 20 | 10 | 21‐48 | |
| NOTCH1 | 8‐17 | 16‐26 | 7 | 40‐64 | 9‐14 | 8 |
| NOTCH2 | 0 | 9 | 5‐20 | 50‐62 | 4‐5 | 4‐10 |
| NOTCH3 | 4‐5 | 36‐54 | 2‐6 | 6‐10 | ||
| NOTCH4 | 4‐8 | 26‐41 | 5 | |||
| MAML1 | 3 | 10‐23 | 3 | |||
| JAG1 | 11 | 3 | 8‐26 | 4 | ||
| JAG2 | 9 | 3 | 13‐33 | 5 | ||
| Transcriptional regulation | ||||||
| EZH2 | 1 | 8‐10 | 1 | 3 | ||
| EP300 | 13‐16 | 34‐38 | 8‐10 | 5 | ||
| KMT2D (MLL2) | 18 | 14 | 69‐79 | 12‐19 | 20‐24 | |
| KMT2C (MLL3) | 6 | 19 | 38‐62 | 4‐8 | 17 | |
| NSD1 | 2 | 10 | 6 | 18‐21 | 2‐4 | 8 |
| MED1 | 4‐5 | 26‐44 | 1 | 4 | ||
| DDX3X | 4‐6 | 5‐13 | 3 | |||
| Oxidative stress | ||||||
| NFE2L2 | 0 | 14 | 4‐7 | 0‐7 | 5‐10 | 15‐19 |
| KEAP1 | 0 | 5 | 3 | 8 | 3‐4 | 12‐16 |
| CUL3 | 0‐3 | 6‐10 | 3 | 3‐23 | 1‐2 | 7‐8 |
| Hippo‐YAP related pathway | ||||||
| FAT1 | 3 | 32 | 11 | 44‐62 | 11‐15 | 20 |
| FAT2 | 6 | 49‐85 | 6‐9 | 11 | ||
| FAT3 | 7 | 21‐72 | 7‐11 | 18 | ||
| FAT4 | 6 | 26‐74 | 7 | 16 | ||
| LATS1 | 4 | 13‐18 | 4 | |||
| LATS2 | 2 | 13‐26 | 2 | |||
| YAP1 | 10‐16 | 5 | 6 | 2 | ||
| WWTR1 (TAZ) | 13 | 3 | 27 | |||
| AJUBA | 0 | 7 | 2 | 18‐26 | 2‐7 | 2 |
| NF2 | 3‐5 | 10‐17 | 1 | 2 | ||
| SCRIB | 2‐6 | 23‐44 | 9 | |||
DNA alterations include somatic mutations and copy number alterations.
TP53 is the gene most frequently altered across all SCCs, except SCCs caused by human papillomavirus (HPV) infection [HPV+ SCCs] (Table 1). In general, HPV+ SCCs display fewer mutations compared with HPV− SCCs, occur in younger people, and have a better prognosis. 1 , 4 HPV− SCCs tend to occur in older individuals with a history of tobacco and/or alcohol abuse, and almost always show TP53 alterations. In addition, HPV− SCCs often bear mutations of CDKN2A and CCND1 (Table 1). HPV− CuSCCs exhibit mutations of KMT2C/D, RAS/RAF/AJUBA or NOTCH more frequently than other HPV− SCCs. All SCCs commonly show some mutation/amplification of PIK3CA/PTEN or TP63 (Table 1).
Although TP53 shows the highest alteration rate in human SCCs, mice with loss of TP53 alone never develop spontaneous SCCs. 13 Even when TP53 inactivation is coupled with K‐RAS activation in the skin 14 or with Akt activation in the oral cavity, 15 SCCs take 6‐7 mo to appear. Similarly, mice lacking TP53 or RB/p107/p130 develop carcinoma in situ (CIS) or invasive CvSCC only after estrogen exposure for 5‐6 mo. 16 , 17 Thus, other essential gene mutations and/or epigenetic alteration(s) appear to be required for SCC onset. The fact that clones with multiple mutations of the above‐mentioned genes are present not only in human tumors but also in normal SSE bolsters this hypothesis. 18 , 19
3. OVERVIEW OF HIPPO‐YAP SIGNALING
3.1. Pathway elements
Hippo‐YAP signaling (Figure 1) is triggered by changes to extracellular matrix (ECM) rigidity, external mechanical forces, high cell density, adherens junction or integrin‐SRC signaling, or G‐protein‐coupled receptor signaling induced by certain growth factors or hormones. 20 The mammalian Hippo pathway core consists of the large tumor suppressor homolog (LATS) kinases and their adaptor proteins mps 1 binder kinase activator‐1 (MOB1A/B), and the mammalian STE20‐like protein (MST) kinases with their adaptor protein salvador homolog‐1 (SAV1). 20 MOB1A/B binding to LATS kinases greatly enhances their kinase activities. The Hippo pathway terminates with the transcriptional co‐factors Yes‐associated protein‐1 (YAP1) and transcriptional co‐activator with PDZ‐binding motif (TAZ) [WW domain containing transcriptional regulator 1 (WWTR1)]. The function of YAP1 and TAZ is to bind to components of the tight and adherens junctions, including Angiomotin (AMOT), 21 α‐catenin, 22 Protein Tyrosine Phosphatase, Non‐Receptor Type 14 (PTPN14), 23 and Scribble. 24 This binding induces LATS to phosphorylate YAP1/TAZ, restricting them to the cytoplasm where E3‐ubiquitin ligase SCFβ TRCP ubiquitinates them and facilitates their proteasome‐mediated degradation. Unphosphorylated YAP1/TAZ interact principally with nuclear TEA domain transcription factors (TEADs) that activate expression of target genes controlling cell growth. 20 Therefore, YAP1/TAZ generally promote cell proliferation in a manner negatively regulated by Hippo signaling.
Figure 1.
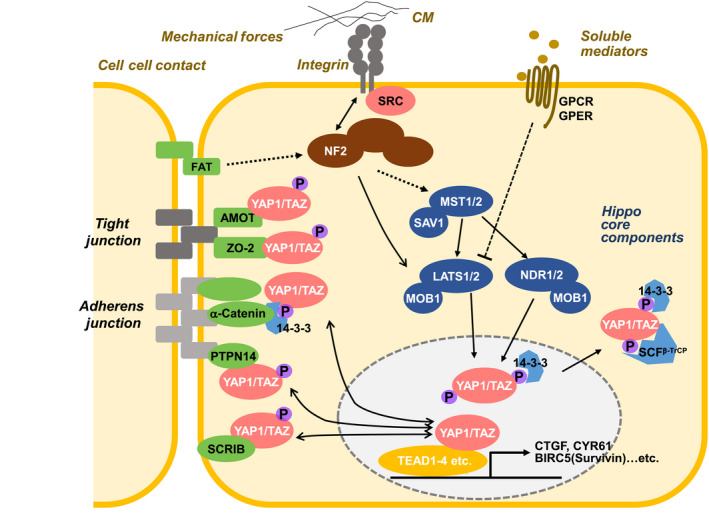
Mammalian Hippo‐YAP signaling network. Hippo‐YAP signaling is controlled by high cell density, external mechanical forces, rigidity of the ECM, integrin‐SRC signaling, and the binding of certain growth factors or hormones to G‐protein‐coupled receptors. The core components of the mammalian canonical Hippo pathway are MST kinases, LATS/NDR kinases, the SAV1 adaptor and the MOB1 adaptor. When LATS/NDR phosphorylates YAP1/TAZ, these effectors bind to 14‐3‐3 proteins and are retained in the cytoplasm. Phosphorylated YAP1/TAZ also bind to SCFβ TRCP, promoting their degradation and preventing the activation of TEAD transcription factors driving the expression of cell survival/anti‐apoptosis genes (such as CTGF, CYR61, AXL, BIRC5). Components of the tight and adherens junctions (such as AMOT, α‐Catenin, PTPN14, Scribble) also bind to phospho‐YAP1/TAZ to control their cytoplasmic localization and activity
3.2. Interaction with cancer‐related genes
Dysregulation of the Hippo‐YAP pathway has been implicated in numerous cancer types. 25 The YAP1/TAZ‐TEADs complex directly targets genes encoding growth factors and cytokines, including connective tissue growth factor (CTGF), cysteine‐rich angiogenic inducer‐61 (CYR61), and tyrosine‐protein kinase receptor gene (AXL). Other targets are involved in the epithelial‐mesenchymal transition, anti‐apoptosis (BIRC5, DDIT4, Trail, NuRD), stem cell maintenance, and cell proliferation, invasion, and/or metastasis. 26 YAP1/TAZ also bind to other transcription factors driving morphogenesis, differentiation, proliferation and apoptosis, including PAX3, SMADs, P73, T‐box transcription factor‐5 (TBX5), RUNT‐related transcription factors (RUNX1/2), erythroblastic oncogene‐B4 (ERBB4) and NK2 Homeobox‐1 (NKX2‐1). 27 Reducing YAP1/TAZ activation has thus been contemplated as a treatment for YAP1/TAZ‐driven tumors. Interestingly, however, even though the activated YAP1 protein itself frequently accumulates in tumors, actual mutation of the YAP1 and WWTR1 (TAZ) genes is uncommon in human cancers, 28 including in SCCs (Table 1).
3.3. Crosstalk with SCC‐related pathways
Hippo signaling participates in crosstalk with the WNT/β‐catenin, NOTCH, SHH, SRC, TGFβ, PI3K/Akt and other pathways crucial for SSE morphogenesis and homeostasis. 29 Several lines of evidence link this interaction with general SCC carcinogenesis:
(1) Activated nuclear YAP1 that is recruited to TCF/LEF binding sites along with β‐catenin enhances WNT signaling. Phosphorylated cytoplasmic YAP1 inhibits Disheveled (DVL), supporting β‐catenin degradation that suppresses WNT signaling. Conversely, YAP1 mRNA is upregulated by WNT signaling.
(2) SHH pathway activation upregulates YAP1 mRNA, stabilizes YAP1 protein, and promotes nuclear YAP accumulation. SHH also induces nuclear translocation of the YAP/TEAD/IRS1 complex, which regulates GLI1/2.
(3) YAP1 upregulates transcription of the NOTCH ligand Jagged‐1. Conversely, Jagged‐1 appears to activate YAP, and γ‐secretase inhibitors that abrogate NOTCH signaling also suppress YAP‐induced intestinal dysplasia in mice. 30
(4) Phosphorylated R‐SMADs bind to YAP/TAZ, linking Hippo signaling to the bone morphogenetic protein receptor (BMPR) and TGF‐β pathways. Generally speaking, nuclear YAP/TAZ enhances nuclear SMAD activity, whereas cytoplasmic YAP/TAZ limits nuclear SMAD accumulation and regulates sensitivity to TGF‐β ligands.
(5) YAP1 is directly and indirectly activated by SRC signaling. 31 , 32 Dasatinib‐mediated suppression of SRC prevents YAP1 activation. 33
(6) AKT blocks MST1/2 activity, resulting in YAP activation. 29
(7) Finally, a recent comprehensive study of genetic alterations across a range of SCCs also supports the importance of Hippo/YAP signaling in SCC carcinogenesis. 34
4. LINKS BETWEEN HIPPO‐YAP SIGNALING AND SCC ONSET
In this section, we discuss recent findings linking Hippo‐YAP signaling aberrations to specific SCC subtypes.
4.1. Head‐and‐neck SCCs
The mammalian head‐and‐neck region comprises the oral cavity, pharynx, and larynx, all of which are covered with SSE. HNSCC is the 7th most common human cancer worldwide, with tongue cancer being the most frequent subtype. About 15% of HNSCC patients are infected with HPV, making this virus a key cause of HNSCC (although the underlying molecular mechanism is not clear). 35 HPV that integrates into the host cell genome generates the viral E6 and E7 proteins known to target p53 and RB, respectively, compromising tumor suppression. 36
HPV+ HNSCC is found principally in the oropharynx. 35 These patients tend to have better prognoses and some are even cured. 36 The remaining 85% of HNSCC patients that are HPV− have poor prognoses because they do not benefit from either intensified chemo/radio therapy or current molecular targeting drugs. 36 As noted above, TP53 is mutated in 84% of HPV− HNSCC cases, with mutations of EGFR, CDKN2A/B, FADD, FAT Atypical Cadherin1 (FAT1), NFE2L2, and CCND1 also frequently observed. These mutations, including that of TP53 (3%), are rare in HPV+ HNSCC. In contrast, mutations of NOTCH, TP63, and PI3K/PTEN are common in both HPV− and HPV+ HNSCC. Because the E6 oncoprotein of HPV efficiently inactivates TP53, TP53 inhibition is likely to be a critical tumorigenic event in HNSCC genesis. However, as noted above, mice lacking TP53 alone never develop spontaneous HNSCC. 13 , 37
Several lines of evidence tie YAP1 activation to HNSCC genesis:
(1) We generated mice with tongue‐specific loss of Mob1a/b that led to constitutive activation of endogenous YAP1 in this tissue. 33 These mutants developed tongue CIS with surprising rapidity and exhibited invasive HNSCC within 4 wk of Mob1a/b deletion.
(2) YAP1 activation can be detected in human precancerous dysplasia in the tongue and is linked to decreased patient survival. 33
(3) Lastly, 8.6% of human HNSCC show amplification of the 11q22 chromosomal region containing the YAP1 locus. 38 Excess YAP1 activity is a feature of such malignant SCCs and leads to a poor prognosis. 38
4.2. Cervical SCCs
Cervical SCCs (CvSCCs) are the 4th most frequent cancer in women worldwide. CvSCC most often arises due to infection with high‐risk HPV, but the complete molecular mechanism has yet to be elucidated.
Numerous studies implicate YAP1 in human CvSCC genesis:
(1) YAP1 activation or LATS1 inactivation is linked to heightened proliferation and invasiveness of CvSCC cell lines, 39 as well as with unfavorable histologic grading, early recurrence, and lymph node metastasis in patients. 40
(2) Nuclear YAP1 protein is upregulated both in invasive CvSCCs and in precancerous high‐grade squamous intraepithelial lesions (HSIL). 41
(3) LATS1 protein is downregulated in 45% of CvSCC cases. 39
(4) Human chromosomal region 11q22 is amplified in 16% of CvSCCs. 4
In mice, uterus‐specific loss of MOB1A/B leads to cervical CIS within 1 wk in a manner dependent on YAP1. 42 Additional p53 deficiency in these mutants produces invasive tumors within 3 wk. In contrast, transgenic (Tg) mice showing modest activation of YAP (S127A), a constitutively active mutant form of YAP1, exhibit invasive CvSCC only after 6‐8 mo. 41 Tg mice that both lack YAP1 and express HPV16 under the control of the K14 promoter do not show the cervical phenotype of HPV16 Tg mice alone. 42 The HPV E6 protein prevents the YAP1 protein from undergoing proteasome‐mediated degradation. 41 HPV E7 protein binds to and targets PTPN14 for proteasome‐mediated degradation, which likely relieves the cytoplasmic retention of YAP by PTPN14. 43 , 44 This combination of HPV E6/E7‐induced accumulation of YAP protein plus its release to the nucleus leads to vigorous YAP activation that may promote human cervical epithelial cell proliferation and eventually CIS onset. 23 , 42 HPV E6 also inhibits p53, spurring the progression of CIS to invasive CvSCC.
4.3. Cutaneous SCCs
About 20%‐50% of human skin cancers are cutaneous squamous cell carcinomas (CuSCCs) arising from malignant transformation of epidermal keratinocytes in the cutaneous SSE. CuSCCs exhibit the highest mutation rate among SCCs, 6 , 7 with frequent alterations to TP53, NOTCH1, NOTCH2, KMT2D, FAT1‐4 and HRAS/BRAF/AJUBA (Table 1). Mutations of CDKN2A (INK4A/ARF) and PI3KCA/PTEN are also common, as is amplification of tyrosine kinase receptor genes (Table 1).
Normal epidermal cell development and skin homeostasis are thought to be maintained by tight and adherens junctions and catenin‐based cell adhesion structures that sense skin status via Hippo‐YAP signaling. 22 YAP1 and TAZ act redundantly to regulate the proliferation of basal layer stem/progenitor cells and balance epidermal growth. 22 , 45 When the skin suffers a wound, keratinocytes boost both their proliferation and YAP protein. 45
Little information is known about Hippo/YAP signaling in CuSCCs, but accumulating evidence suggests that YAP1 activation is involved in CuSCC genesis:
(1) LATS1 and LATS2 are mutated in 18% and 26% of CuSCCs, respectively12.
(2) YAP protein levels are elevated in human CuSCCs and correlate with disease progression. 46
(3) YAP promotes human CuSCC cell proliferation in vitro. 47
(4) Tg mice expressing constitutively active YAP have thicker epithelium compared with controls and their immature keratinocytes display increased mitogenic activity. 22
CIS forms in the skin of these mutants starting on day 9 after YAP activation and invasive CuSCC appears in 2‐4 wk. Also in these Tg mice, epithelial scratch wounding leads to synergism between the wound healing response and YAP1 activation that promotes spindle cell SCC development. 48
(5) YAP‐deficient mice show delayed wound healing and epidermal shrinkage. 45 YAP deficiency also prevents the formation of AP‐1‐dependent CuSCCs in mice. 49
4.4. Esophageal SCCs
Esophageal cancer is the 6th leading cause of cancer‐related deaths worldwide. Most patients are diagnosed only after the malignancy has become advanced because it is difficult to detect early and grows aggressively. Although esophageal adenocarcinoma (EADC) predominates in Western countries, esophageal SCC (ESCC) is the most common form in East Asia. Alcohol and smoking are key risk factors for ESCC, as are genetic polymorphisms that inactivate alcohol‐metabolizing enzymes. 50 Like other HPV− SCCs, ESCCs usually exhibit mutations of genes involved in the cell cycle, apoptosis regulation, and histone modification, including TP53 (93%), FGFR3, CCND1, NOTCH1‐3, CDKN2A, and KMT2D (Table 1).
The risk of ESCC in Eastern China has been associated with certain polymorphisms of the FAT4 gene, which acts upstream of the Hippo core. YAP1 protein is often overexpressed in ESCC 51 and correlates positively with ESCC histological grade, stage, and diameter, as well as with overall and disease‐free patient survival. 51 YAP1 knockdown promotes the apoptosis of ESCC cells or suppresses their proliferation and invasion. 52 Fewer ESCC cells show properties of cancer stem cells in the absence of YAP1 activation. 53 Among human ESCCs, 65% show LATS downregulation and 62% display TAZ upregulation. 54 This profile correlates positively with histological grade, clinical stage, and lymph node metastasis of ESCCs as well as with decreased overall and progression‐free patient survival. High TAZ expression is also associated with low LATS2 in human ESCCs. To date, whether YAP and TAZ differ in their roles in ESCC onset and development is unknown. In mice, loss of YAP1 specifically in the esophagus decreases esophageal progenitor cell proliferation and stratification. Conversely, Tg mice with constitutive Yap1 expression show enhanced cell proliferation that induces abnormal stratification of esophageal layers at E18.5. 55 It is unclear if they would go on to develop esophageal CIS or invasive ESCC, but it is quite likely that these animals would die during the perinatal period before any malignancy could progress.
4.5. Lung SCCs
Lung cancer is the 2nd most common cancer in humans and the most lethal. A lung cancer is classified as either a small‐cell lung cancer (SCLC) or a non–small‐cell lung cancer (NSCLC). NSCLCs occur more frequently compared with SCLCs and have a worse prognosis. About 30% of NSCLCs are lung SCCs (LSCCs). LSCC is essentially incurable due to a current lack of effective treatments. Although lung adenocarcinomas (LADCs) often exhibit EGFR mutations or ALK fusions, LSCCs are not driven by these oncogenic changes and so rarely respond to agents targeting them. 56 Almost all LSCCs feature TP53 alterations as well as mutations of oxidative stress response genes (KEAP1, NFE2L2), PI3K/PTEN pathway genes, squamous differentiation genes (TP63, SOX2), and cell cycle‐related genes (CDKN2A/B) (Table 1).
In a mouse model, the trans‐differentiation of LADCs into LSCCs was associated with LKB1 inactivation and led to malignant progression as well as drug resistance. 57 YAP activation levels differ between mouse LADCs and LSCCs, with the former demonstrating YAP1 hyperactivation. Indeed, forced YAP activation largely abolishes the LADC‐to‐LSCC transition, 58 suggesting that YAP functions may differ between these NSCLC subtypes. Work in mouse models and human LADC cell lines has confirmed that YAP hyperactivation promotes malignant progression of LADC. 59 In striking contrast, YAP activation in LSCCs downregulates GPX2 in a ΔNp63‐dependent manner. 60 As a result, excessive intracellular reactive oxygen species (ROS) accumulate and kill the LSCC cells. Analyses of human cancer patient‐derived xenograft (PDX) models have shown that higher YAP activation correlates positively with suppression of LSCC growth and negatively with lymph node metastasis. 61 These observations were unexpected, and why YAP activation would promote LADC growth but suppress LSCCs is still unknown. Importantly, these findings indicate that caution should be exercised in administering YAP inhibitors to LSCC patients.
5. PUTATIVE MECHANISM BY WHICH HIPPO/YAP SIGNALING DRIVES SCC ONSET AND DEVELOPMENT
The above body of work establishes that MOB1 deletion or YAP1 activation induces CIS formation within 5‐9 d in the context of HNSCCs, CvSCCs, and CuSCCs. This timeline is unusually rapid, given that most cancers initiate over a long period of multi‐step carcinogenesis in which several mutated genes act additively or synergistically. However, it should be noted that: (a) a CIS diagnosis in these SCCs is obtained only after the dysplastic cells in the basal layer rise to the top layer of the epidermis; and (b) it takes about 7 d for MOB1 protein to be completely lost in conditional mutants. 33 Thus, dysplastic SCC cells may be created immediately after YAP1 activation. These factors have led us to propose that CIS in almost any SSE tissue is initiated when YAP1 activity is sustained and exceeds a certain oncogenic threshold (Figure 2). The resulting dysplastic cells are able to expand uncontrollably without any other genetic alterations, setting the stage for eventual SCC progression.
Figure 2.

Proposed mechanism for SCC genesis using HNSCC as an example. Both HPV+ HNSCCs and HPV− HNSCCs may be caused by an accumulation of YAP1 activity that is driven by either mutation of the indicated genes, HPV infection, or risk factors such as smoking. When the oncogenic threshold of YAP1 activity is surpassed, transformation ensues. Each rounded rectangle represents a factor promoting HNSCC genesis. The height of each rectangle represents relative YAP1 activation intensity, 33 and the width represents the frequency at which this gene or factor contributes to HNSCC malignancies
Studies of SCCs in the aforementioned tissues have pinpointed ongoing exposure to carcinogens such as smoking, HPV infection, and excessive epidermal growth factor (EGF) or estrogen, as well as abrasion of the mucosa due to mechanical irritation, as major causes of SCCs. 35 Each of these factors has been shown to activate YAP1 directly in 1 or more SSE cell types. 62 , 63 , 64 In addition, as detailed above, loss‐of‐function or gain‐of‐function mutations of genes that result in YAP activation are linked to SCCs. 62 , 65 , 66 , 67 When these gene mutations combine with carcinogens, YAP1 activity rises even higher. 33 , 42 In short, there is substantial evidence supporting our hypothesis that, for many SCCs, the original CIS is caused by a simple sustaining of YAP1 activity over a particular threshold. When additional genetic and epigenetic alterations occur, the already elevated level of YAP activation accelerates SCC development. Depending on the degree of YAP1 activation induced by 1 alteration, the oncogenic threshold may be overcome with relatively few additional mutations. This scenario may explain why patients with HPV+ HNSCC exhibit favorable therapeutic responses and have better prognoses. In the case of HNSCC, the HPV E6/E7 proteins are strong activators of YAP1, meaning that the threshold is exceeded by few other deleterious mutations to trigger the transition to SCC. 35 Considering that, in mice, YAP1 inhibition not only prevents SCC onset but also delays its progression, YAP1 may be an appealing target for molecular therapy of these devastating malignancies.
One possible reason for the frequent and early onset of SCCs in mice bearing mutated elements of the Hippo pathway is the activation of ΔNp63, the master regulator of proliferation, differentiation, and adhesion of SSE cells. By binding directly to ΔNp63 protein, activated YAP1 stabilizes this regulator. 68 Thus, YAP1 hyperactivation leading to abundant stabilized ΔNp63 may drive SSE cell differentiation and proliferation, possibly promoting 1st CIS and then SCC development. A 2nd reason for early SCC onset in Hippo‐mutant mice may be elevated production of BMP4, a soluble growth factor that also regulates ΔNp63. 69 Indeed, we detected increased BMP4 in Mob1‐deficient tongue and cervical epithelial cells compared to controls. 33 , 42 A 3rd reason for our findings may be potential positive feedback between YAP1 and EGF signaling. YAP1 promotes the transcription of EGF receptors such as EGFR and ERBB3 as well as that of various EGF‐like ligands (NRG1, NRG2, HBEGF, AREG, TGFα). 62 Conversely, all EGF‐like ligands can apparently activate YAP1, 62 so that an autocrine loop related to YAP1‐EGF/EGFR signaling may stimulate SCC genesis and progression. A 4th contributor to our results may be an interaction between YAP1 and cell adhesion genes. Excessive YAP activation reduces the expression of integrin receptor genes and epithelial cell adhesion genes such as CDH1, removing critical negative regulators of SCC growth. 70 , 71 The proliferation of SSE cells, which are tightly connected to one another, is greatly influenced by cell adhesion, ΔNp63 signaling, and EGF‐like ligands. Thus, all 4 of the above mechanisms may in fact contribute to the genesis of most SCCs (except LSCC), perhaps explaining why the phenotype is so strong in SSE cells showing YAP1/TAZ activation.
6. CONCLUSION
An oncogenic driver gene for CIS formation in SCCs has been difficult to identify. We propose that, because YAP hyperactivation induces immediate CIS onset in mouse models, YAP must be the key molecule driving CIS with no need for any other gene alteration. We therefore advance a new concept positing that human CIS can simply initiate when sustained YAP1 activity exceeds an oncogenic threshold. SCCs are generally refractory to conventional treatments, and so remain largely incurable. Thus, targeting the Hippo‐YAP pathway may be a promising strategy for suppressing the growth of most SCCs (except LSCCs, for the reasons elaborated above). In addition, our mouse models featuring mutations in the Hippo‐YAP pathway currently constitute the world's fastest spontaneous cancer onset models. Cancer progression is synchronized in these mutant animals, and the tumors are easily visualized on the mouse exterior (especially in the HNSCC and CuSCC models). These characteristics are ideal for cancer research and the development of novel anti‐cancer drugs, and we humbly take pride in sharing these new tools with the international scientific community to devise fresh approaches to treating human SCCs.
DISCLOSURE
The authors have no conflicts of interest.
ACKNOWLEDGMENTS
We are grateful for funding provided by the Japan Society for the Promotion of Science (JSPS) grants 17H01400, 26114005, 26640081, 18K06132; and the Japan Agency for Medical Research and Development (AMED) P‐CREATE grant JP19cm0106114.
Maehama T, Nishio M, Otani J, Mak TW, Suzuki A. The role of Hippo‐YAP signaling in squamous cell carcinomas. Cancer Sci. 2021;112:51–60. 10.1111/cas.14725
Contributor Information
Tomohiko Maehama, Email: tmaehama@med.kobe-u.ac.jp.
Akira Suzuki, Email: suzuki@med.kobe-u.ac.jp.
REFERENCES
- 1. Cancer Genome Atlas N . Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015;517:576‐582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lechner M, Frampton GM, Fenton T, et al. Targeted next‐generation sequencing of head and neck squamous cell carcinoma identifies novel genetic alterations in HPV+ and HPV‐ tumors. Genome Med. 2013;5:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Seiwert TY, Zuo Z, Keck MK, et al. Integrative and comparative genomic analysis of HPV‐positive and HPV‐negative head and neck squamous cell carcinomas. Clin Cancer Res. 2015;21:632‐641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cancer Genome Atlas Research N, Albert Einstein College of M, Analytical Biological S , et al. Integrated genomic and molecular characterization of cervical cancer. Nature. 2017;543:378‐384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ojesina AI, Lichtenstein L, Freeman SS, et al. Landscape of genomic alterations in cervical carcinomas. Nature. 2014;506:371‐375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pickering CR, Zhou JH, Lee JJ, et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin Cancer Res. 2014;20:6582‐6592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. South AP, Purdie KJ, Watt SA, et al. NOTCH1 mutations occur early during cutaneous squamous cell carcinogenesis. J Invest Dermatol. 2014;134:2630‐2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gao Y‐B, Chen Z‐L, Li J‐G, et al. Genetic landscape of esophageal squamous cell carcinoma. Nat Genet. 2014;46:1097‐1102. [DOI] [PubMed] [Google Scholar]
- 9. Song Y, Li L, Ou Y, et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature. 2014;509:91‐95. [DOI] [PubMed] [Google Scholar]
- 10. Cancer Genome Atlas Research N . Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012;489:519‐525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim Y, Hammerman PS, Kim J, et al. Integrative and comparative genomic analysis of lung squamous cell carcinomas in East Asian patients. J Clin Oncol. 2014;32:121‐128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. http://www.cbioportal.org/
- 13. Supsavhad W, Dirksen WP, Martin CK, Rosol TJ. Animal models of head and neck squamous cell carcinoma. Vet J. 2016;210:7‐16. [DOI] [PubMed] [Google Scholar]
- 14. Caulin C, Nguyen T, Lang GA, et al. An inducible mouse model for skin cancer reveals distinct roles for gain‐ and loss‐of‐function p53 mutations. J Clin Invest. 2007;117:1893‐1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moral M, Segrelles C, Lara MF, et al. Akt activation synergizes with Trp53 loss in oral epithelium to produce a novel mouse model for head and neck squamous cell carcinoma. Cancer Res. 2009;69:1099‐1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shin MK, Sage J, Lambert PF. Inactivating all three rb family pocket proteins is insufficient to initiate cervical cancer. Cancer Res. 2012;72:5418‐5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shai A, Pitot HC, Lambert PF. p53 Loss synergizes with estrogen and papillomaviral oncogenes to induce cervical and breast cancers. Cancer Res. 2008;68:2622‐2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Martincorena I, Roshan A, Gerstung M, et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348:880‐886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tomasetti C, Vogelstein B, Parmigiani G. Half or more of the somatic mutations in cancers of self‐renewing tissues originate prior to tumor initiation. Proc Natl Acad Sci USA. 2013;110:1999‐2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nakatani K, Maehama T, Nishio M, et al. Targeting the Hippo signalling pathway for cancer treatment. J Biochem. 2017;161:237‐244. [DOI] [PubMed] [Google Scholar]
- 21. Chan SW, Lim CJ, Chong YF, Pobbati AV, Huang C, Hong W. Hippo pathway‐independent restriction of TAZ and YAP by angiomotin. J Biol Chem. 2011;286:7018‐7026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schlegelmilch K, Mohseni M, Kirak O, et al. Yap1 acts downstream of alpha‐catenin to control epidermal proliferation. Cell. 2011;144:782‐795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang W, Huang J, Wang X, et al. PTPN14 is required for the density‐dependent control of YAP1. Genes Dev. 2012;26:1959‐1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Skouloudaki K, Puetz M, Simons M, et al. Scribble participates in Hippo signaling and is required for normal zebrafish pronephros development. Proc Natl Acad Sci USA. 2009;106:8579‐8584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nishio M, Maehama T, Goto H, et al. Hippo vs. Crab: tissue‐specific functions of the mammalian Hippo pathway. Genes Cells. 2017;22:6‐31. [DOI] [PubMed] [Google Scholar]
- 26. Fu V, Plouffe SW, Guan KL. The Hippo pathway in organ development, homeostasis, and regeneration. Curr Opin Cell Biol. 2017;49:99‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nishio M, Goto H, Suzuki M, Fujimoto A, Mimori K, Suzuki A. The hippo signaling pathway: a candidate new drug target for malignant tumors In: Nakao K, Minato N, Uemoto S, eds. Innovative Medicine: Basic Research and Development. Tokyo, 2015; 79‐94. [PubMed] [Google Scholar]
- 28. Yu FX, Zhao B, Guan KL. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell. 2015;163:811‐828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nishio M, Otsubo K, Maehama T, Mimori K, Suzuki A. Capturing the mammalian Hippo: elucidating its role in cancer. Cancer Sci. 2013;104:1271‐1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Camargo FD, Gokhale S, Johnnidis JB, et al. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol. 2007;17:2054‐2060. [DOI] [PubMed] [Google Scholar]
- 31. Li P, Silvis MR, Honaker Y, Lien WH, Arron ST, Vasioukhin V. alphaE‐catenin inhibits a Src‐YAP1 oncogenic module that couples tyrosine kinases and the effector of Hippo signaling pathway. Genes Dev. 2016;30:798‐811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Si Y, Ji X, Cao X, et al. Src Inhibits the Hippo Tumor Suppressor Pathway through Tyrosine Phosphorylation of Lats1. Cancer Res. 2017;77:4868‐4880. [DOI] [PubMed] [Google Scholar]
- 33. Omori H, Nishio M, Masuda M, et al. YAP1 is a potent driver of the onset and progression of oral squamous cell carcinoma. Sci Adv. 2020;6:eaay3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang Y, Xu X, Maglic D, et al. Comprehensive Molecular Characterization of the Hippo Signaling Pathway in Cancer. Cell Rep. 2018;25:1304‐1317.e1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Leemans CR, Snijders PJF, Brakenhoff RH. The molecular landscape of head and neck cancer. Nat Rev Cancer. 2018. [DOI] [PubMed] [Google Scholar]
- 36. Kobayashi K, Hisamatsu K, Suzui N, Hara A, Tomita H, Miyazaki T. A review of HPV‐related head and neck cancer. J Clin Med. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Opitz OG, Harada H, Suliman Y, et al. A mouse model of human oral‐esophageal cancer. J Clin Invest. 2002;110:761‐769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Eun YG, Lee D, Lee YC, et al. Clinical significance of YAP1 activation in head and neck squamous cell carcinoma. Oncotarget. 2017;8:111130‐111143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Deng J, Zhang W, Liu S, An H, Tan L, Ma L. LATS1 suppresses proliferation and invasion of cervical cancer. Mol Med Rep. 2017;15:1654‐1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu T, Liu Y, Gao H, Meng F, Yang S, Lou G. Clinical significance of yes‐associated protein overexpression in cervical carcinoma: the differential effects based on histotypes. Int J Gynecol Cancer. 2013;23:735‐742. [DOI] [PubMed] [Google Scholar]
- 41. He C, Lv X, Huang C, et al. A Human Papillomavirus‐Independent Cervical Cancer Animal Model Reveals Unconventional Mechanisms of Cervical Carcinogenesis. Cell Rep. 2019;26:2636 ‐ 2650.e2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nishio M, To Y, Maehama T, et al. Endogenous YAP1 Activation Drives Immediate Onset of Cervical Carcinoma In Situ in Mice. Cancer Sci. 2020;111:3576‐3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. White EA, Munger K, Howley PM. High‐risk human papillomavirus E7 proteins target PTPN14 for degradation. MBio. 2016;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hatterschide J, Bohidar AE, Grace M, et al. PTPN14 degradation by high‐risk human papillomavirus E7 limits keratinocyte differentiation and contributes to HPV‐mediated oncogenesis. Proc Natl Acad Sci USA. 2019;116:7033‐7042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Elbediwy A, Vincent‐Mistiaen ZI, Spencer‐Dene B, et al. Integrin signalling regulates YAP and TAZ to control skin homeostasis. Development. 2016;143:1674‐1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jia J, Li C, Luo S, et al. Yes‐associated protein contributes to the development of human cutaneous squamous cell carcinoma via activation of RAS. J Invest Dermatol. 2016;136:1267‐1277. [DOI] [PubMed] [Google Scholar]
- 47. Walko G, Woodhouse S, Pisco AO, et al. A genome‐wide screen identifies YAP/WBP2 interplay conferring growth advantage on human epidermal stem cells. Nat Commun. 2017;8:14744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vincent‐Mistiaen Z, Elbediwy A, Vanyai H, et al. YAP drives cutaneous squamous cell carcinoma formation and progression. Elife. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zanconato F, Forcato M, Battilana G, et al. Genome‐wide association between YAP/TAZ/TEAD and AP‐1 at enhancers drives oncogenic growth. Nat Cell Biol. 2015;17:1218‐1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tanaka F, Yamamoto K, Suzuki S, et al. Strong interaction between the effects of alcohol consumption and smoking on oesophageal squamous cell carcinoma among individuals with ADH1B and/or ALDH2 risk alleles. Gut. 2010;59:1457‐1464. [DOI] [PubMed] [Google Scholar]
- 51. Muramatsu T, Imoto I, Matsui T, et al. YAP is a candidate oncogene for esophageal squamous cell carcinoma. Carcinogenesis. 2011;32:389‐398. [DOI] [PubMed] [Google Scholar]
- 52. Zhao S, Zhao J, Li X, et al. Effect of YAP1 silencing on esophageal cancer. Onco Targets Ther. 2016;9:3137‐3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang L, Zhang Z, Yu X, et al. Unbalanced YAP‐SOX9 circuit drives stemness and malignant progression in esophageal squamous cell carcinoma. Oncogene. 2019;38:2042‐2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gao Y, Yi J, Zhang K, et al. Downregulation of MiR‐31 stimulates expression of LATS2 via the hippo pathway and promotes epithelial‐mesenchymal transition in esophageal squamous cell carcinoma. J Exp Clin Cancer Res. 2017;36:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bailey DD, Zhang Y, van Soldt BJ, et al. Use of hPSC‐derived 3D organoids and mouse genetics to define the roles of YAP in the development of the esophagus. Development. 2019;146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lindeman NI, Cagle PT, Beasley MB, et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J Thorac Oncol. 2013;8:823‐859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Han X, Li F, Fang Z, et al. Transdifferentiation of lung adenocarcinoma in mice with Lkb1 deficiency to squamous cell carcinoma. Nat Commun. 2014;5:3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gao Y, Zhang W, Han X, et al. YAP inhibits squamous transdifferentiation of Lkb1‐deficient lung adenocarcinoma through ZEB2‐dependent DNp63 repression. Nat Commun. 2014;5:4629. [DOI] [PubMed] [Google Scholar]
- 59. Lau AN, Curtis SJ, Fillmore CM, et al. Tumor‐propagating cells and Yap/Taz activity contribute to lung tumor progression and metastasis. EMBO J. 2014;33:468‐481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Huang H, Zhang W, Pan Y, et al. YAP suppresses lung squamous cell carcinoma progression via deregulation of the DNp63‐GPX2 axis and ROS accumulation. Cancer Res. 2017;77:5769‐5781. [DOI] [PubMed] [Google Scholar]
- 61. Won KY, Kim HK, Kim GY, Song MJ, Lim SJ. Hippo pathway and tumoral FOXP3 expression correlate with tumor growth in squamous cell carcinoma of the lung. Pathol Res Pract. 2020;216:153003. [DOI] [PubMed] [Google Scholar]
- 62. He C, Mao D, Hua G, et al. The Hippo/YAP pathway interacts with EGFR signaling and HPV oncoproteins to regulate cervical cancer progression. EMBO Mol Med. 2015;7:1426‐1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhao Y, Zhou W, Xue L, Zhang W, Zhan Q. Nicotine activates YAP1 through nAChRs mediated signaling in esophageal squamous cell cancer (ESCC). PLoS One. 2014;9:e90836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhou X, Wang S, Wang Z, et al. Estrogen regulates Hippo signaling via GPER in breast cancer. J Clin Invest. 2015;125:2123‐2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Martin D, Degese MS, Vitale‐Cross L, et al. Assembly and activation of the Hippo signalome by FAT1 tumor suppressor. Nat Commun. 2018;9:2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. García‐Escudero R, Segrelles C, Dueñas M, et al. Overexpression of PIK3CA in head and neck squamous cell carcinoma is associated with poor outcome and activation of the YAP pathway. Oral Oncol. 2018;79:55‐63. [DOI] [PubMed] [Google Scholar]
- 67. Chai Y, Li Q, Zhao H, et al. SOX2 antagonizes WWC1 to drive YAP1 activation in esophageal squamous cell carcinoma. Cancer Med. 2019;8:7055‐7064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yuan M, Luong P, Hudson C, Gudmundsdottir K, Basu S. c‐Abl phosphorylation of DeltaNp63alpha is critical for cell viability. Cell Death Dis. 2010;1:e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tribulo C, Guadalupe Barrionuevo M, Aguero TH, Sanchez SS, Calcaterra NB, Aybar MJ. DeltaNp63 is regulated by BMP4 signaling and is required for early epidermal development in Xenopus. Dev Dyn. 2012;241:257‐269. [DOI] [PubMed] [Google Scholar]
- 70. Ishihara E, Nagaoka Y, Okuno T, et al. Prostaglandin E2 and its receptor EP2 trigger signaling that contributes to YAP‐mediated cell competition. Genes Cells. 2020;25:197‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Nardone G, Oliver‐De La Cruz J, Vrbsky J, et al. YAP regulates cell mechanics by controlling focal adhesion assembly. Nat Commun. 2017;8:15321. [DOI] [PMC free article] [PubMed] [Google Scholar]