

Abstract
E‐cadherin, an epithelial cell–specific cell adhesion molecule, has both promoting and suppressing effects on tumor invasion and metastasis. It is often downregulated during cancer progression through gene deletion/mutation, transcriptional repression, or epigenetic silencing. We describe a novel regulatory switch to induce stimulus‐dependent downregulation of mRNA encoding E‐cadherin (CDH1 mRNA) in KRAS‐mutated cancer cells. The regulatory switch consists of ZEB1 and oncogenic K‐Ras, does not target the promoter region of CDH1, and requires an external cue to temporally downregulate E‐cadherin expression. Its repressive effect is maintained as long as the external stimulus continues and is attenuated with cessation of the stimulus. Contextual external cues that turn this regulatory switch on include activation of protein kinase C or fibroblast growth factor signaling. The mode of action is distinct from that of EPCAM repression by ZEB1, which does not require an external cue. Thus, KRAS‐mutated cancer cells acquire a novel mode of regulating E‐cadherin expression depending on ZEB1, which could contribute to phenotypic plasticity of cancer cells during malignant progression.
Keywords: E‐cadherin, fibroblast growth factor, K‐Ras, protein kinase C, ZEB1
We describe a novel regulatory switch to regulate E‐cadherin expression in KRAS‐mutated cancer cells. The regulatory switch consists of ZEB1 and oncogenic K‐Ras, and requires an external cue to temporally downregulate E‐cadherin expression. This regulatory switch could contribute to the phenotypic plasticity of cancer cells during malignant progression.

Abbreviations
- EGFP
enhanced green fluorescent protein
- EMT
epithelial‐mesenchymal transition
- FGF
fibroblast growth factor
- HDAC
histone deacetylase
- TF
transcription factor
- TGF‐β
transforming growth factor‐β
- ZEB1
zinc finger E‐box–binding homeo box 1
1. INTRODUCTION
E‐cadherin, encoded by the CDH1 gene, is a calcium‐dependent cell adhesion molecule localized in adherens junctions of epithelial cells. 1 The downregulation of E‐cadherin expression is considered one of the hallmarks of cells undergoing epithelial‐mesenchymal transition (EMT) and is closely associated with cell invasion. It has been reported that interference of E‐cadherin function by neutralizing antibodies results in the acquirement of invasive properties by nontransformed epithelial cells. 2 Moreover, an inverse correlation between invasiveness and E‐cadherin expression level was observed in various human carcinoma cell lines, and the invasiveness of E‐cadherin–negative cancer cells was prevented by its exogenous expression. 3
E‐cadherin is also a putative tumor suppressor as it suppresses the progression from adenoma to carcinoma 4 and is involved in contact inhibition, thereby suppressing excessive cell growth. 5 Furthermore, its downregulation facilitates immune escape 6 and leads to drug resistance in epidermal growth factor receptor‐mutated cancer cells. 7 Consistently, loss of E‐cadherin expression is often detected in invasive cancer cells and correlates with a poor prognosis in some types of cancers. 8 , 9 , 10 However, there is also a recent emergence of reports of contrasting effects on cancer progression. E‐cadherin expression is required for cancer‐associated fibroblast‐led collective invasion of squamous cell carcinoma cells, where the E‐cadherin–N‐cadherin heterologous cell‐cell junction is formed. 11 Formation of such a heterologous junction facilitates metastatic colonization. 12 Moreover, E‐cadherin expression is indispensable for the metastasis of breast cancers, although its downregulation enhances cancer cell invasion. 13 E‐cadherin expression affects organotropism in the metastasis of pancreatic cancer cells; cells with E‐cadherin expression metastasize to the liver, whereas those lacking E‐cadherin metastasize to the lung. 14 Therefore, E‐cadherin expression has both positive and negative effects on invasion and metastasis, 15 and temporal and reversible regulation of E‐cadherin expression appears to be crucial during cancer progression. Supporting this notion, cells with reversible EMT phenotypes have more advantages than those with stable EMT phenotypes in the formation of metastatic colonization. 16
E‐cadherin expression/function is regulated at several different levels. The function of E‐cadherin is abrogated by deletion, mutation, or epigenetic silencing due to promoter hypermethylation of CDH1. 17 , 18 , 19 , 20 , 21 Transcriptional repressors that induce EMT, collectively termed as EMT‐TFs, such as zinc finger E‐box–binding homeobox 1 (ZEB1), ZEB2, Snail, Slug, and Twist, are also involved in the downregulation of E‐cadherin expression. 22 Alternatively, miRNAs, among which miR‐9 is the best studied, posttranscriptionally repress the expression of E‐cadherin by directly targeting CDH1 3′UTR 23 . 24 , 25 E‐cadherin function is also rapidly downregulated at the protein level. It is downregulated by the ubiquitin‐proteasome system through the action of the E3 ubiquitin ligase termed Hakai. 26 Internalization or relocalization of surface E‐cadherin is involved in the regulation of cancer cell invasion and metastasis. 27 , 28 Moreover, tyrosine phosphorylation of β‐catenin, one of the E‐cadherin–associated molecules, disables E‐cadherin–mediated cell adhesion. 29 , 30
Cancer cells in the tumor microenvironment receive signals from various extracellular signaling molecules. E‐cadherin is repressed in cancer cells at the edge of tumor nests and at the invasive front, whereas its expression is retained in those at the center of tumor nests of solid tumors, 31 suggesting a possible involvement of environmental factor(s) in its downregulation. The external cues that trigger the downregulation of E‐cadherin expression include transforming growth factor‐β (TGF‐β) and Wnt ligands, 32 both of which induce or stabilize the expression of EMT‐TFs. In this study, we detected a novel regulatory switch for the downregulation of E‐cadherin expression in KRAS‐mutated cancer cells. Constitutively active mutations in KRAS are well‐known driver mutations that are frequently observed in various types of cancers. 33 The switch machinery requires persistent K‐Ras and ZEB1 function and external cues but does not require the upregulation of ZEB1 for its function. Previous studies have shown that ZEB1 was associated with E‐box sequences located in the promoter region of CDH1 gene to repress the expression of E‐cadherin 34 , 35 in a manner dependent on histone deacetylase (HDAC) activity. 36 However, this novel regulatory switch does not target the CDH1 promoter. The external cues that turn this switch on include fibroblast growth factor (FGF)‐2 that is abundantly present in the tumor microenvironment. 37 , 38 Therefore, this regulatory switch could contribute to the phenotypic plasticity of cancer cells during malignant progression.
2. MATERIALS AND METHODS
2.1. Reagents and antibodies
The reagents used in this study were as follows: recombinant human TGF‐β1 (from R&D Systems or Pepro Tech); recombinant human FGF basic‐TS (thermostable; Proteintech); phorbol 12‐myristate 13‐acetate (PMA; Adipogen Life Sciences); cholera toxin (CTx) solution, actinomycin D (ActD), and U0126 (FUJIFILM Wako Pure Chemical Cooperation); bisindolylmaleimide I (BIM I) and Gö 6983 (Cayman Chemical); cycloheximide (CHX; Nacalai Tesque); LY294002 (Sigma‐Aldrich); and SB431542 (Calbiochem).
The antibodies used in this study are listed in Table S1.
2.2. Cell culture
Human cancer cells (PANC‐1, A549, SUIT‐2, HepG2/C3A, and SAS), HaCaT human keratinocyte cells, and MDCK‐I canine kidney epithelial cells were obtained and cultured in DMEM (Nacalai Tesque) supplemented with 4.5 g/L glucose, 10% FBS, 50 U/mL penicillin, and 50 μg/mL streptomycin as previously described. 39 , 40 , 41 , 42 NMuMG normal murine mammary gland epithelial cells were cultured in the same medium additionally supplemented with 10 μg/mL insulin. 40 All cells were cultured at 37°C under a 5% CO2 atmosphere. The concentrations of reagents added to the cell culture were as follows: TGF‐β1, 1 ng/mL; FGF basic‐TS, 10 ng/mL; PMA, 100 nmol/L, unless otherwise noted; U0126, 10 µmol/L; and BIM I, 1 µmol/L.
2.3. Plasmid construction and lentivirus production
Human K‐RasG12D and H‐RasG12V were subcloned into pEGFP‐C1 vector (Clontech) to obtain expression plasmids coding for the enhanced green fluorescent protein (EGFP) fusion proteins for stably expressing cell lines. EGFP‐K‐Ras wt expression plasmid was generated by point mutagenesis. Lentiviral vector encoding full‐length mouse ZEB1 was generated by Gateway technology (Invitrogen). Lentivirus production was performed as previously described. 39 Cells transfected with plasmid vectors or infected with lentiviral vectors were cloned.
2.4. Immunoblotting and immunofluorescence
Immunoblotting was performed as previously described, 41 except that cells were lysed in RIPA buffer consisting of 20 mmol/L Tris‐HCl (pH 7.4), 150 mmol/L NaCl, 1% Nonidet P‐40, 5 mmol/L EDTA, 0.5% sodium deoxycholate, 0.1% SDS, and a protease inhibitor cocktail (Nacalai Tesque). As a loading control, α‐tubulin was used.
The procedures for immunofluorescence labeling were as follows: Cells were seeded in 8‐well culture slides, fixed in 1:1 acetone‐methanol solution, permeabilized with 0.2% Triton X‐100 in PBS, blocked with Blocking One solution (Nacalai Tesque), and incubated with primary antibodies (Table S1) for 1 hour at room temperature. The cells were washed and then incubated with secondary antibodies for 1 hour at room temperature. The secondary antibodies and the dilution rates are shown in Table S1. For nuclear staining, the cells were incubated with TO‐PRO‐3 (Invitrogen Molecular Probes) for 30 minutes. Fluorescence was examined using an Olympus FV1000 confocal microscope.
2.5. Quantitative real‐time PCR
Quantitative real‐time PCR was performed as previously described, 43 with the exception that cDNA was synthesized using oligo (dT) primer. The relative expression level of each mRNA was normalized against the expression level of GAPDH mRNA. The primer sequences are shown in Table S2.
2.6. RNA interference
Transfection of siRNAs was performed using Lipofectamine RNAiMAX transfection reagent (Invitrogen). The final concentration of siRNA was 5 nmol/L. The stealth RNAi siRNA against human ZEB1 (ID: #1 HSS110548, #2 HSS110549), siRNA Negative Control Low GC Duplex #2 (12935110), siRNA Negative Control Med GC Duplex #2 (12935112), silencer select siRNAs against human K‐Ras (ID: #1 s7939, #2 s7940) and human Snail (ID: s13187), and negative control No.1 siRNA (4390843) were obtained from Invitrogen.
2.7. Luciferase assay
Luciferase assay was performed as previously described. 43 The promoter region of CDH1 (−991/+57) was inserted into pGL4‐MLP and used. Data are presented as mean ± SD of triplicate determination.
2.8. Statistical analysis
Student's t test was used for the results of luciferase assay, and one‐way ANOVA with Tukey's test was used for those of quantitative real‐time PCR to determine the significance of differences among experimental groups.
Additional details of materials and methods are described in Document S1.
3. RESULTS
3.1. Protein kinase C signaling downregulates E‐cadherin expression in PANC‐1 cells through a process distinct from EMT
Cancer cells receive various extracellular signals from the tumor microenvironment and change their characteristics. As it is difficult to identify the specific signals that affect tumor malignancy in vivo, we attempted to identify them through in vitro studies. We first focused on protein kinases A and C, because they are activated by diverse extracellular signals and involved in various cellular functions.
E‐cadherin plays a vital role in epithelial cell adhesion. Its downregulation promotes cell growth, invasiveness, and drug resistance and is also associated with poor prognosis of several cancers. 15 Therefore, in this study, we investigated the effects of CTx and PMA, activators of protein kinases A and C, respectively, on E‐cadherin expression in PANC‐1 pancreatic ductal adenocarcinoma cells (Figure 1A). We observed that PMA treatment induced the downregulation of E‐cadherin expression, whereas CTx did not. Unlike TGF‐β, a well‐known inducer of EMT, PMA failed to upregulate N‐cadherin expression. Therefore, this process appears to be distinct from a typical EMT, which accompanies “cadherin switching” (downregulation of E‐cadherin and upregulation of N‐cadherin). Next, we examined the time course of PMA‐induced downregulation of E‐cadherin mRNA (CDH1) and protein expression (Figure 1B,C). The downregulation of CDH1 mRNA expression was initiated from 6 hours and reached the minimum at 24 hours after stimulation, which preceded the downregulation of E‐cadherin protein. These findings suggested that the downregulation of E‐cadherin protein is due to that of CDH1 mRNA.
FIGURE 1.
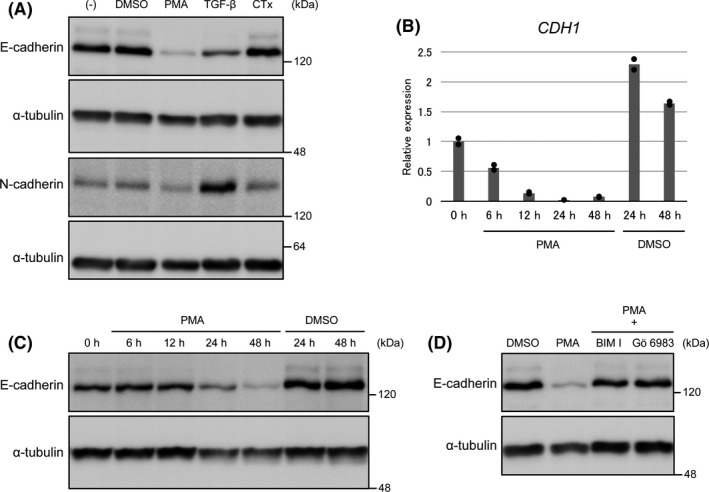
Protein kinase C (PKC) signaling downregulates E‐cadherin expression in PANC‐1 cells. A, PANC‐1 cells were treated with phorbol 12‐myristate 13‐acetate (PMA), transforming growth factor‐β (TGF‐β), and cholera toxin (CTx) (100 ng/mL) for 48 h. Expressions of E‐cadherin and N‐cadherin were analyzed by immunoblotting. B and C, Time course of PMA‐induced E‐cadherin downregulation. PANC‐1 cells were treated with PMA for 6‐48 h. B, Expression of E‐cadherin mRNA (CDH1 mRNA) was analyzed by quantitative RT‐PCR (duplicate determination). The value at 0 h is shown as “1.” CDH1 expression was upregulated during culture of PANC‐1 cells (24 and 48 h), irrespective of the presence of DMSO. C, Expression of E‐cadherin protein was analyzed by immunoblotting. D, PANC‐1 cells were treated with PMA with or without the PKC inhibitors BIM I and Gö6983 (1 µmol/L) for 48 h. Expression of E‐cadherin was analyzed by immunoblotting
PMA exerts its effect by mimicking a lipid second messenger, diacylglycerol, which activates not only protein kinase C (PKC) but also other signaling molecules. 44 E‐cadherin downregulation induced by PMA was attenuated in the presence of PKC inhibitors, BIM I and Gö6983 that target conventional PKCs (α, β, and γ), indicating that PKC signaling actually downregulates E‐cadherin expression in PANC‐1 cells (Figure 1D). Similar results were obtained in A549 lung adenocarcinoma cells (Figure S1). Although PMA is known to induce TGF‐β expression, 45 the PMA‐induced downregulation of E‐cadherin expression was not inhibited by SB431542, an inhibitor of TGF‐β type I receptors, thus excluding the possibility that it is mediated by TGF‐β signaling (Figure S2).
3.2. E‐cadherin downregulation by PKC signaling is dependent on oncogenic K‐Ras
We then analyzed the PMA‐induced downregulation of E‐cadherin expression in cell lines other than PANC‐1 and A549 and observed that SUIT‐2 pancreatic ductal adenocarcinoma cells exhibited this downregulation, whereas other cancer (HepG2/C3A hepatoblastoma cells and SAS oral squamous cell carcinoma cells) and normal cell lines (NMuMG normal murine mammary gland epithelial cells, HaCaT human keratinocyte cells, and MDCK‐I canine kidney epithelial cells) exhibited only marginal downregulation (Figure 2A).
FIGURE 2.
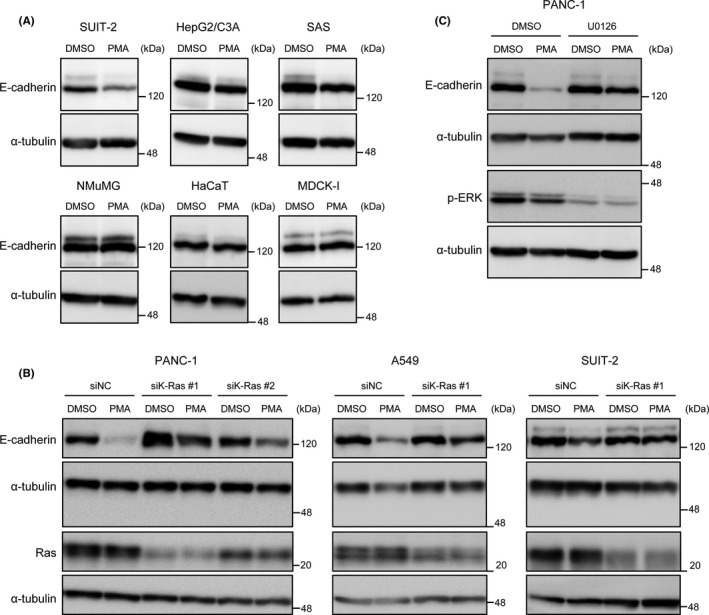
Protein kinase C (PKC)‐induced E‐cadherin downregulation is dependent on oncogenic K‐Ras. A, Cancer (SUIT‐2, HepG2/C3A, and SAS) and normal (NMuMG, HaCaT, and MDCK‐I) cell lines were treated with phorbol 12‐myristate 13‐acetate (PMA) for 48 h. Expression of E‐cadherin was analyzed by immunoblotting. B, PANC‐1, A549, and SUIT‐2 cells were transfected with siRNA against K‐Ras or nonspecific control siRNA (NC). After 72 h of transfection, cells were treated with PMA for 48 h. Expressions of E‐cadherin and Ras were analyzed by immunoblotting. C, PANC‐1 cells were treated with PMA with or without U0126 for 48 h. Expressions of E‐cadherin and p‐ERK were analyzed by immunoblotting
Intriguingly, all the three cell lines in which PMA efficiently downregulated E‐cadherin expression were KRAS‐mutated (G12D in PANC‐1 and SUIT‐2, while G12S in A549 cells). This finding suggested that oncogenic Ras signaling is required for the PMA‐induced downregulation of E‐cadherin expression. Supporting this notion, K‐Ras knockdown was found to attenuate the downregulation of E‐cadherin expression in PANC‐1, A549, and SUIT‐2 cells (Figure 2B).
We further investigated the effects of inhibitors of MEK (U0126) and phosphoinositide 3‐kinase (LY294002), which suppress two principal downstream pathways of Ras, on the downregulation of E‐cadherin expression. We observed that U0126 treatment inhibited the PMA‐induced downregulation of E‐cadherin expression in PANC‐1 cells, whereas LY294002 did not (Figure 2C and data not shown). These findings indicate that the downstream signaling pathway of K‐Ras that cooperates with PKC in the downregulation of E‐cadherin expression could be the MEK‐ERK pathway.
3.3. ZEB1 mediates E‐cadherin downregulation induced by PKC and K‐Ras
E‐cadherin expression is known to be often downregulated by EMT‐TFs. 22 We referred to the RNA‐seq data (GSM2884282) to identify the EMT‐TFs expressed in PANC‐1 cells. 46 Among the “core EMT‐TFs,” the expression of ZEB1 and Snail (encoded by SNAI1) was detected at FPKM (fragments per kilobase of exon per million mapped sequence reads) higher than 1 (Table S3). Therefore, we investigated the mRNA expression of ZEB1 and Snail after PMA stimulation in PANC‐1 cells (Figure 3A). Our results showed that ZEB1 expression was transiently upregulated from 6 to 24 hours after stimulation. The expression of Snail was also upregulated but only transiently at 6 hours after stimulation. The upregulation of ZEB1 and Snail was also detected at protein levels (Figure 3B). However, knockdown of ZEB1 inhibited the PMA‐induced E‐cadherin downregulation in PANC‐1, A549, and SUIT‐2 cells (Figure 3C), whereas Snail knockdown did not affect the downregulation (Figure S3). Therefore, we focused on the role of ZEB1.
FIGURE 3.
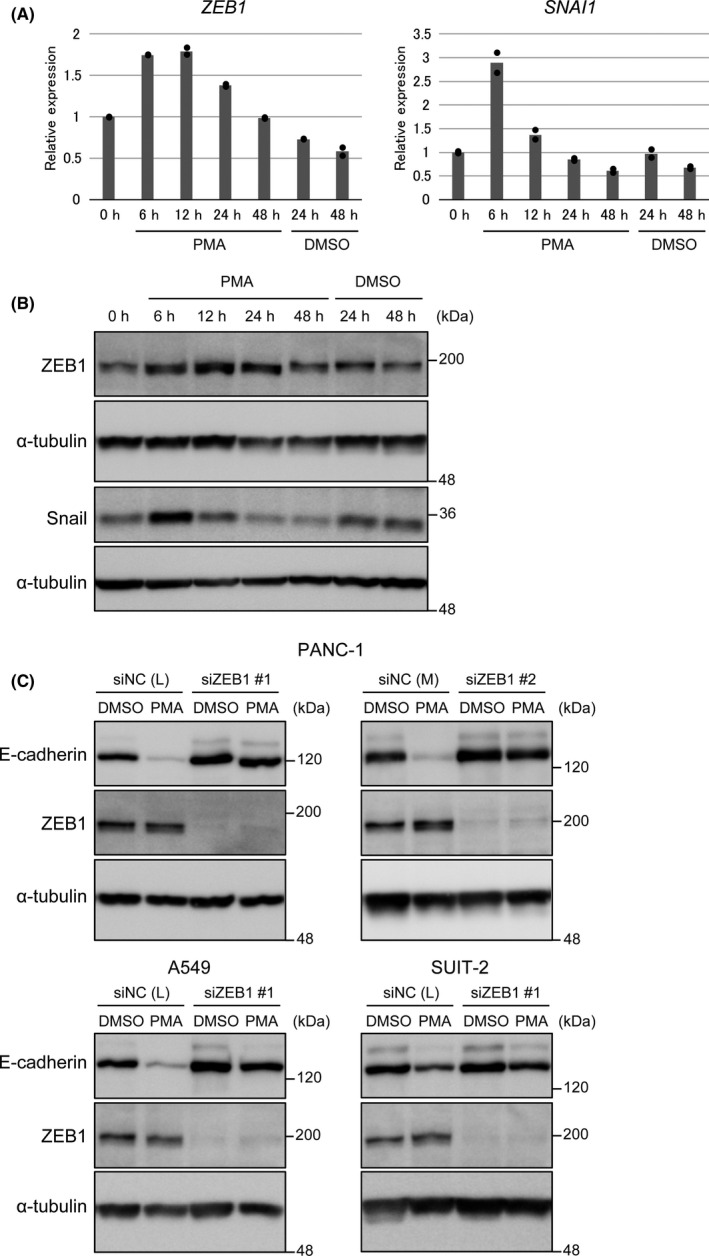
Zinc finger E‐box–binding homeobox 1 (ZEB1) is required for E‐cadherin downregulation by protein kinase C (PKC) and K‐Ras. A and B, Expressions of ZEB1 and Snail after phorbol 12‐myristate 13‐acetate (PMA) stimulation. PANC‐1 cells were treated with PMA for 6‐48 h. A, Expressions of ZEB1 and SNAI1 mRNA were analyzed by quantitative RT‐PCR (duplicate determination). The value at 0 h is shown as “1.” B, Expressions of ZEB1 and Snail protein were analyzed by immunoblotting. C, PANC‐1, A549, and SUIT‐2 cells were transfected with siRNA against ZEB1 or nonspecific control siRNA (NC). After 24 h of transfection, cells were treated with PMA for 48 h. Expressions of E‐cadherin and ZEB1 were analyzed by immunoblotting
3.4. Ectopic expression of ZEB1 and oncogenic K‐Ras permits E‐cadherin downregulation by PKC in nontransformed epithelial cells
To further examine the requirement of ZEB1 and K‐Ras for PKC‐induced E‐cadherin downregulation, we used MDCK‐I cells that lack endogenous ZEB1 expression to establish cells ectopically expressing ZEB1 at the level equivalent to that in PANC‐1 cells (MDCK‐ZEB1). These cells retained E‐cadherin expression, which was not downregulated by PMA treatment (Figure 4A,B; Figure S4A). In this experimental setting, PMA upregulated ectopic ZEB1 expression probably due to the presence of the PMA‐responsive activator protein‐1 motif in the promoter region of the lentiviral vector CSII‐EF‐RfA.
FIGURE 4.
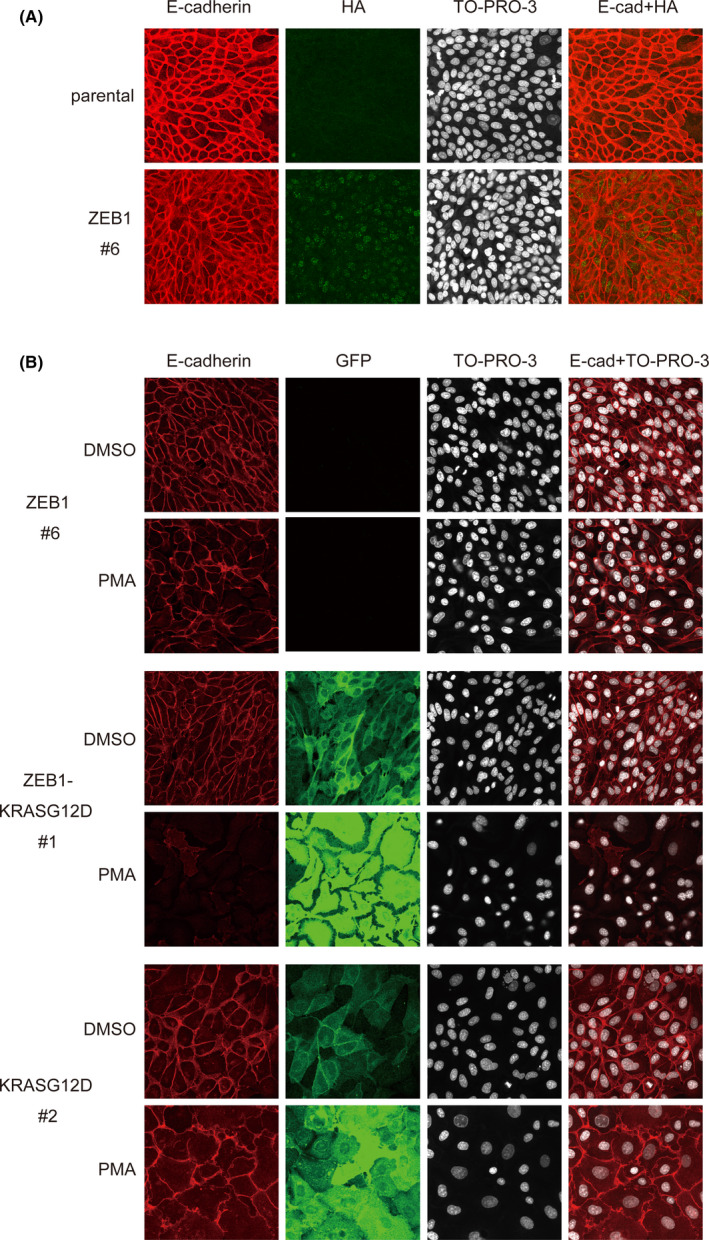
Ectopic expression of zinc finger E‐box–binding homeobox 1 (ZEB1) and oncogenic K‐Ras permits E‐cadherin downregulation by protein kinase C (PKC) in MDCK‐I cells. A, MDCK‐I cells and those stably expressing HA‐tagged ZEB1 (MDCK‐ZEB1) were immunostained with anti‐E‐cadherin and anti‐HA antibodies and stained with TO‐PRO‐3 to detect nuclei. B, MDCK‐ZEB1 and MDCK‐I cells stably expressing EGFP‐KRASG12D (MDCK‐KRASG12D) or ZEB1 and EGFP‐KRASG12D (MDCK‐ZEB1‐KRASG12D) were treated with phorbol 12‐myristate 13‐acetate (PMA) (10 nmol/L) for 48 h, immunostained with anti‐E‐cadherin and anti‐GFP antibodies, and stained with TO‐PRO‐3 to detect nuclei
Then, we additionally introduced oncogenic K‐Ras (G12D) fused to EGFP into MDCK‐ZEB1 cells (MDCK‐ZEB1‐KRASG12D). Here, the EGFP fusion protein was used for easy detection of cells expressing the transgene. We observed that MDCK‐ZEB1‐KRASG12D cells still retained E‐cadherin expression, but PMA treatment induced its downregulation (Figure 4B; Figure S4B). In contrast, PMA failed to downregulate E‐cadherin expression in MDCK‐ZEB1 cells expressing wt K‐Ras fused to EGFP (MDCK‐ZEB1‐KRASwt; Figure S4B), indicating that constitutively active Ras signaling is indispensable. Exogenous K‐Ras (G12D) alone was not sufficient for the PKC‐mediated E‐cadherin downregulation (Figure 4B; Figure S4B), although it induced spreading morphology of MDCK‐I cells, which was further enhanced by PMA stimulation, as previously reported. 47 , 48 In these experiments, it was observed that PMA upregulated ectopic Ras expression probably due to the presence of the activator protein‐1 motif in the promoter region of the expression vector pEGFP‐C1. Of note, PMA‐induced downregulation of E‐cadherin expression was observed when oncogenic H‐Ras (G12V) fused to EGFP was introduced into MDCK‐ZEB1 cells (Figure S5), indicating that oncogenic Ras signaling required in the regulation is not limited to K‐Ras. We concluded that ZEB1 and oncogenic Ras constitute a regulatory switch for E‐cadherin downregulation that is triggered by PKC signaling.
3.5. ZEB1 function is temporarily modulated by Ras and PKC signaling
We next elucidated the mechanism by which ZEB1, oncogenic Ras, and PKC signaling integrate to downregulate the expression of E‐cadherin. Intriguingly, the increased expression of ZEB1 after PMA stimulation returned to basal level after 48 hours and thereafter, whereas the expression of E‐cadherin remained repressed (Figure 5A). In addition, PMA stimulation did not upregulate ZEB1 expression in A549 cells (Figure S6). These findings suggested that the activation of ZEB1 function rather than the increased expression level by PKC signaling is important for the downregulation of E‐cadherin expression. To explore this possibility, we established ZEB1‐overexpressing PANC‐1 cell clones. The expression levels of E‐cadherin and exogenous ZEB1 were not inversely correlated in PANC‐1 cells; we isolated several clones that retained E‐cadherin expression despite high ZEB1 levels (Figure S7). We also observed that PMA treatment again induced E‐cadherin downregulation in these cell clones, indicating that a regulatory switch for E‐cadherin downregulation is maintained to be functional.
FIGURE 5.
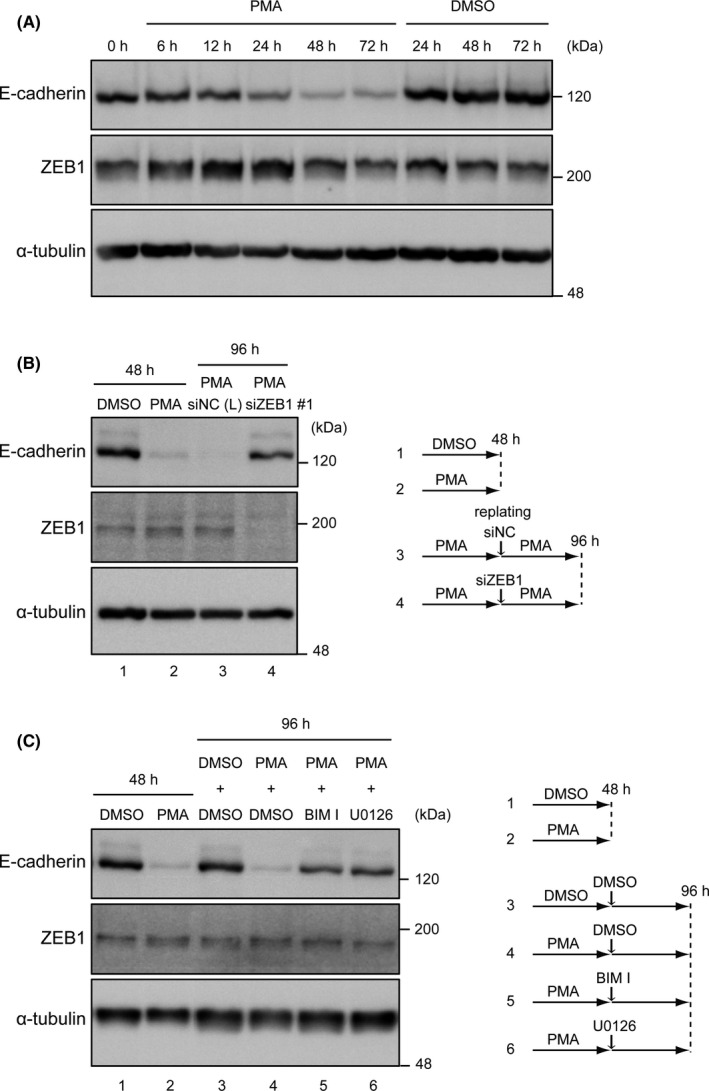
Zinc finger E‐box–binding homeobox 1 (ZEB1) function is temporally modulated by Ras and protein kinase C (PKC) signaling for the downregulation of E‐cadherin. A, Immunoblotting analysis of the expression of E‐cadherin and ZEB1 after phorbol 12‐myristate 13‐acetate (PMA) stimulation. PANC‐1 cells were treated with PMA for 6‐72 h. B, PANC‐1 cells were treated with PMA (10 nmol/L) for 48 h. Cells were then replated, transfected with siRNA against ZEB1 or nonspecific control siRNA (NC), and cultured in the presence of PMA for another 48 h. Expressions of E‐cadherin and ZEB1 were analyzed by immunoblotting. The experimental design is shown on the right. C, PANC‐1 cells were treated with PMA (10 nmol/L) for 48 h and then treated with or without BIM I or U0126 for another 48 h. Expressions of E‐cadherin and ZEB1 were analyzed by immunoblotting. The experimental design is shown on the right
We next examined whether ZEB1 expression is persistently required for the downregulation of E‐cadherin expression (Figure 5B). PMA stimulation of cells showed effective downregulation of E‐cadherin expression at 48 hours. We then transfected siZEB1 and cultured these cells for another 48 hours, which restored the E‐cadherin expression. In contrast, when cells were transfected with siNC (negative control siRNA) instead of siZEB1, E‐cadherin expression remained repressed in the presence of PMA. Furthermore, the decreased expression of E‐cadherin (at 48 hours after PMA stimulation) was reversed after treating cells with PKC and MEK inhibitors (Figure 5C). Altogether, these findings indicate that ZEB1, K‐Ras (Erk signaling), and PKC signaling are required for persistent E‐cadherin downregulation.
3.6. ZEB1‐Ras regulatory switch downregulates E‐cadherin expression through a novel mechanism
Previous studies have reported that ZEB1 represses E‐cadherin expression through its association with the CDH1 promoter region. 34 , 35 Consistently, the knockdown of ZEB1 enhanced the activity of luciferase construct containing −991/+57 of the CDH1 promoter region (Figure 6A; Figure S8A) and upregulated the mRNA expression of CDH1 around twofold (Figure 6B; Figure S8B). These results indicated that ZEB1 in unstimulated cells partially repressed the expression of E‐cadherin by targeting the promoter region. However, PMA stimulation did not suppress but enhanced the reporter activity, although it downregulated the mRNA expression (Figure 6B,C; Figure S8B,C). Under PMA stimulation, ZEB1 could effectively downregulate E‐cadherin expression through a genomic region distinct from the promoter.
FIGURE 6.
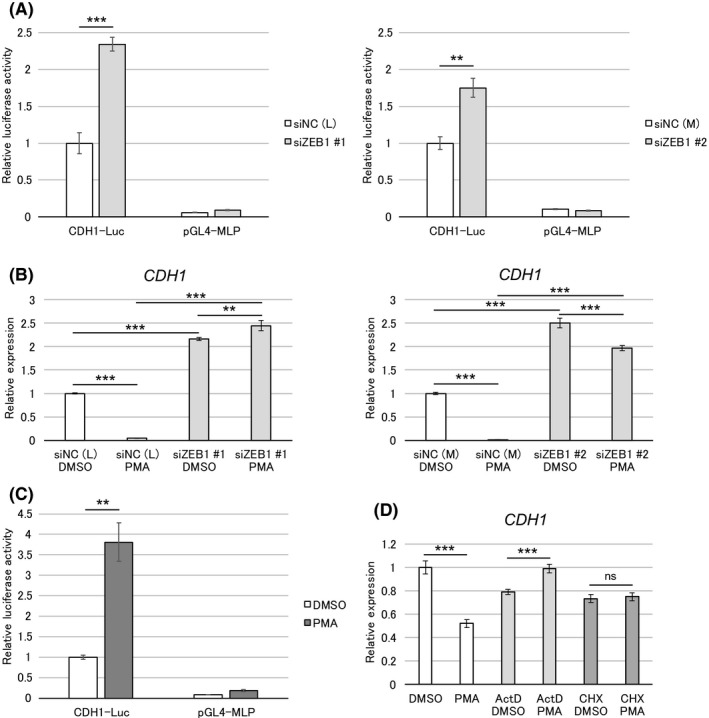
Zinc finger E‐box–binding homeobox 1 (ZEB1)‐Ras regulatory switch downregulates E‐cadherin expression through a novel mechanism. A, PANC‐1 cells were transfected with siRNA against ZEB1 or nonspecific control siRNA (NC). After 24 h of siRNA transfection, cells were transfected with a CDH1 promoter‐reporter construct (CDH1‐Luc) or control vector pGL4‐MLP. Luciferase activity was measured at 27 h after reporter transfection. B, PANC‐1 cells were transfected with siRNA against ZEB1 or nonspecific control siRNA (NC). After 24 h of transfection, cells were treated with phorbol 12‐myristate 13‐acetate (PMA) (10 nmol/L) for 48 h. C, PANC‐1 cells were transfected with CDH1‐Luc or pGL4‐MLP. After 9 h of transfection, cells were treated with PMA (10 nmol/L) for 18 h, and then luciferase activity was measured. D, PANC‐1 cells were treated with PMA (10 nmol/L) with or without actinomycin D (1 µg/mL) or cycloheximide (50 µg/mL) for 8 h. B and D, Expression of CDH1 mRNA was analyzed by quantitative RT‐PCR (triplicate determination). The value in DMSO control is shown as “1.” Data are presented as mean ± SD. **P < 0.01; ***P < 0.001; ns, not significant
Treatment with ActD and CHX repressed the PMA‐induced E‐cadherin downregulation, indicating the need for de novo mRNA and protein synthesis (Figure 6D; Figure S8D). Although the underlying mechanism remains to be elucidated in detail, we conclude that this regulatory switch downregulates E‐cadherin expression through a novel mechanism.
3.7. FGF signaling triggers the ZEB1‐Ras regulatory switch for E‐cadherin downregulation
After observing that PMA induced E‐cadherin downregulation by activating the PKC pathway, we explored the growth factors present in the tumor microenvironment and replaced PMA in the induction of E‐cadherin downregulation. We observed that epidermal growth factor and hepatocyte growth factor only weakly downregulated the expression of E‐cadherin in PANC‐1 cells (data not shown). However, FGF‐2, which is abundantly present in tumor microenvironment including pancreatic cancer, 38 induced E‐cadherin downregulation in a ZEB1– and K‐Ras–dependent manner (Figure 7A,B). Thus, FGF‐2 signaling can also turn on the ZEB1‐Ras regulatory switch for downregulating E‐cadherin expression. Unexpectedly, the PKC inhibitor did not inhibit this downregulation, whereas the MEK inhibitor U0126 did (Figure 7C). In the case of FGF‐2 signaling, some pathway(s) other than the PKC pathway function to trigger E‐cadherin downregulation. These findings indicate that ZEB1 and oncogenic Ras represent indispensable components of the regulatory switch required for E‐cadherin downregulation, but PKC signaling can be replaced by other stimuli.
FIGURE 7.
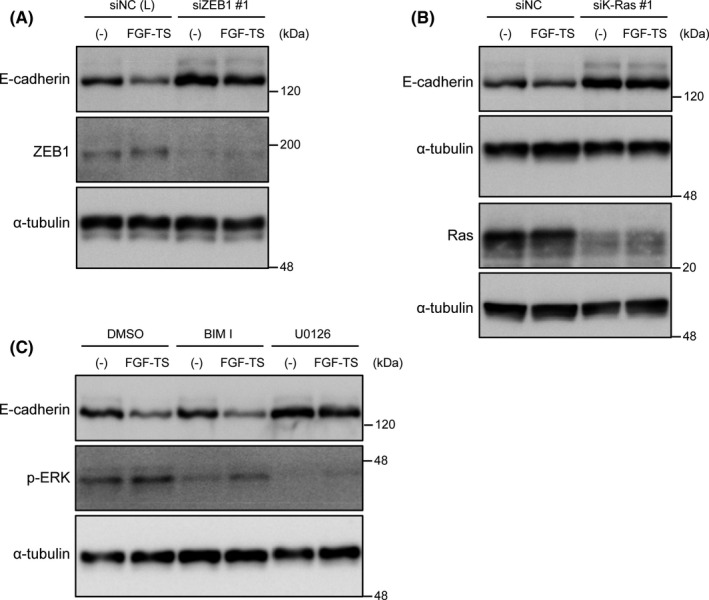
FGF signaling induces downregulation of E‐cadherin dependent on zinc finger E‐box–binding homeobox 1 (ZEB1) and K‐Ras. A, PANC‐1 cells were transfected with siRNA against ZEB1 or nonspecific control siRNA (NC). After 24 h of transfection, cells were treated with FGF basic‐TS for 48 h. Expressions of E‐cadherin and ZEB1 were analyzed by immunoblotting. B, PANC‐1 cells were transfected with siRNA against K‐Ras or nonspecific control siRNA (NC). After 72 h of transfection, cells were treated with FGF basic‐TS for 48 h. Expressions of E‐cadherin and Ras were analyzed by immunoblotting. C, PANC‐1 cells were treated with FGF basic‐TS with or without BIM I or U0126 for 48 h. Expressions of E‐cadherin and p‐ERK were analyzed by immunoblotting
3.8. Effects of the regulatory switch on other epithelial genes
We finally examined whether this regulatory switch affected the expression of other epithelial genes in PANC‐1 cells. We observed that EPCAM and ESRP2 were downregulated by PMA treatment, although not extensively, which was attenuated by the knockdown of ZEB1 (Figure 8A) or K‐Ras (Figure 8B). Of note, basal expression of EPCAM was strikingly upregulated by ZEB1 knockdown. This result indicates that ZEB1 can effectively repress the expression of EPCAM independently of PKC signaling in unstimulated cells. In contrast, ZEB1 represses ESRP2 expression only when PKC signaling is active. Therefore, this regulatory switch also affects other genes associated with epithelial cell phenotypes in PANC‐1 cells.
FIGURE 8.
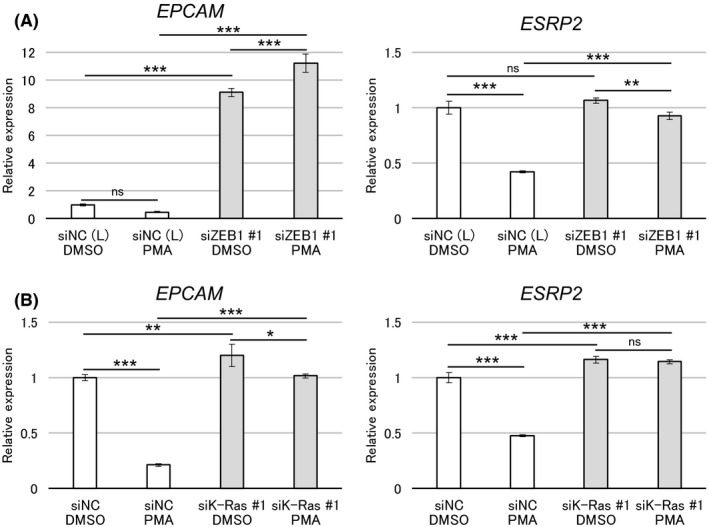
Effect of the zinc finger E‐box–binding homeobox 1 (ZEB1)‐Ras regulatory switch on other epithelial genes. A, PANC‐1 cells were transfected with siRNA against ZEB1 or nonspecific control siRNA (NC). After 24 h of transfection, cells were treated with phorbol 12‐myristate 13‐acetate (PMA) (10 nmol/L) for 48 h. B, PANC‐1 cells were transfected with siRNA against K‐Ras or nonspecific control siRNA (NC). After 72 h of transfection, cells were treated with PMA (10 nmol/L) for 48 h. A and B, Expressions of EPCAM and ESRP2 mRNA were analyzed by quantitative RT‐PCR (triplicate determination). The value in DMSO control is shown as “1.” Data are presented as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001; ns, not significant
4. DISCUSSION
E‐cadherin is a cell adhesion molecule exclusively expressed in epithelial cells. It exerts two opposite functions in invasion and metastasis of cancer cells. E‐cadherin expression is regulated in multiple ways, among which temporal and reversible regulation of E‐cadherin expression appears to be crucial during cancer progression. Cancer cells that acquire a stimulus‐dependent E‐cadherin regulatory system could have an advantage in metastasis, as they can regulate E‐cadherin level appropriately during metastasis, depending on the environmental factors in primary tumor sites or distant organs. In this study, we identified a novel mode of stimulus‐dependent repression of E‐cadherin expression at the mRNA level mediated by ZEB1 and oncogenic Ras signaling.
ZEB1 is a transcriptional repressor implicated in EMT and cancer progression. Particularly, it has been demonstrated that ZEB1 is involved in metastasis and acquisition of tumor‐initiating capacity of pancreatic ductal carcinoma cells, whereas Snail and Twist are dispensable. 49 , 50 Hence, ZEB1 plays a primary role in the malignant progression of certain types of cancers. Notably, studies have shown that E‐cadherin expression inversely correlates with the expression of ZEB1, and not Snail, in pancreatic, lung, and breast cancer cells. 36 , 51 , 52
Several modes of ZEB1‐mediated suppression of E‐cadherin expression have been reported. ZEB1 is associated with two E‐box sequences located in the proximal promoter region of E‐cadherin gene (CDH1) to downregulate its expression. 34 , 35 , 53 In this process, ZEB1 recruits HDAC1 and HDAC2 to the promoter region, 36 thus rendering the downregulation sensitive to HDAC inhibitors. This regulatory system is metastable, as it is primarily dependent on the expression level of ZEB1, which can be altered by the perturbation of a regulatory network of ZEB1 expression. ZEB1 also contributes to the epigenetic silencing of E‐cadherin by recruiting DNA methyltransferase 1 to the CDH1 promoter region and maintains its DNA methylation in the basal‐like subtype of breast cancer cells. 21 Derepression from ZEB1‐assisted epigenetic silencing requires DNA demethylation. Although it is a reversible process, it is virtually a stable regulation. In this study, we identified another regulatory system involved in the downregulation of E‐cadherin expression by ZEB1, which is dependent on K‐Ras signaling and some external cue(s). Furthermore, this regulatory switch did not mediate ZEB1 binding to the CDH1 promoter region. Hence, these three ZEB1‐dependent regulatory systems appear to have distinct properties in regulating E‐cadherin expression. The external cue–induced E‐cadherin downregulation by ZEB1 requires oncogenic Ras signaling, whereas the ZEB1‐assisted epigenetic silencing of E‐cadherin accompanies the recruitment of DNA methyltransferase 1. Therefore, the three regulatory systems described above appear to be differently regulated, depending on additional cofactors.
PMA‐induced protein downregulation of E‐cadherin in /WiDir colorectal cancer cells has been previously reported 54 ; however, the underlying mechanism was not further explored. Although HT‐29 cells do not harbor mutated KRAS, they harbor BRAF and PIK3CA mutations. 55 Therefore, these cells may have exhibited a similar phenomenon.
Previous studies have also reported E‐cadherin downregulation induced by external stimuli, such as prostaglandin E2 or hypoxic conditions, 56 , 57 both of which accompany the induction of ZEB1. In contrast, the ZEB1‐Ras regulatory switch required for E‐cadherin downregulation, identified in the present study, does not require the induction of ZEB1. In PANC‐1 cells, ZEB1 is expressed, but E‐cadherin mRNA expression is not extensively downregulated. However, after the knockdown of ZEB1, E‐cadherin expression was elevated twofold. These results indicate that endogenous ZEB1 itself can repress E‐cadherin expression moderately, but not extensively. Therefore, ZEB1 requires the assistance of external cues to extensively downregulate E‐cadherin expression, which is primarily based on the functional control of ZEB1 activity. However, the mechanism by which the ZEB1‐mediated repression of E‐cadherin expression is modulated by external cues yet remains to be elucidated (Figure S9). Such modulation may target either ZEB1 itself, other factors that interact with ZEB1, or downstream effectors of ZEB1. Further studies would unveil the intricate mechanisms underlying the regulation of ZEB1 activity.
DISCLOSURE
The authors have no conflict of interest.
Supporting information
Figures S1‐S9
Tables S1‐S3
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a Grant for Practical Research for Innovative Cancer Control (18dk030766s0303 to KM) from the Japan Agency for Medical Research Development (AMED) and KAKENHI Grant‐in‐Aid for young Scientists (B) (19K16537 to SO) from the Japan Society for the Promotion of Science (JSPS).
Otake S, Itoh Y, Omata C, Saitoh M, Miyazawa K. ZEB1 and oncogenic Ras constitute a regulatory switch for stimulus‐dependent E‐cadherin downregulation. Cancer Sci. 2021;112:205–216. 10.1111/cas.14701
REFERENCES
- 1. Takeichi M. Cadherin cell adhesion receptors as a morphogenetic regulator. Science. 1991;251:1451‐1455. [DOI] [PubMed] [Google Scholar]
- 2. Behrens J, Mareel MM, Van Roy FM, Birchmeier W. Dissecting tumor cell invasion: epithelial cells acquire invasive properties after the loss of uvomorulin‐mediated cell‐cell adhesion. J Cell Biol. 1989;108:2435‐2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Frixen UH, Behrens J, Sachs M, et al. E‐cadherin‐mediated cell‐cell adhesion prevents invasiveness of human carcinoma cells. J Cell Biol. 1991;113:173‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Perl AK, Wilgenbus P, Dahl U, et al. A causal role for E‐cadherin in the transition from adenoma to carcinoma. Nature. 1998;392:190‐193. [DOI] [PubMed] [Google Scholar]
- 5. Kim NG, Koh E, Chen X, Gumbiner BM. E‐cadherin mediates contact inhibition of proliferation through Hippo signaling‐pathway components. Proc Natl Acad Sci USA. 2011;108:11930‐11935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cepek KL, Shaw SK, Parker CM, et al. Adhesion between epithelial cells and T lymphocytes mediated by E‐cadherin and the αEβ7 integrin. Nature. 1994;372:190‐193. [DOI] [PubMed] [Google Scholar]
- 7. Witta SE, Gemmill RM, Hirsch FR, et al. Restoring E‐cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res. 2006;66:944‐950. [DOI] [PubMed] [Google Scholar]
- 8. De Leeuw WJ, Berx G, Vos CB, et al. Simultaneous loss of E‐cadherin and catenins in invasive lobular breast cancer and lobular carcinoma in situ. J Pathol. 1997;183:404‐411. [DOI] [PubMed] [Google Scholar]
- 9. Borcherding N, Cole K, Kluz P, et al. Re‐evaluating E‐Cadherin and β‐catenin: a pan‐cancer proteomic approach with an emphasis on breast cancer. Am J Pathol. 2018;188:1910‐1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yazdani J, Ghavimi MA, Jabbari Hagh E, Ahmadpour F. The role of E‐cadherin as a prognostic biomarker in head and neck squamous carcinoma: a systematic review and meta‐analysis. Mol Diagn Ther. 2018;22:523‐535. [DOI] [PubMed] [Google Scholar]
- 11. Labernadie A, Kato T, Brugués A, et al. A mechanically active heterotypic E‐cadherin/N‐cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat Cell Biol. 2017;19:224‐237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang H, Yu C, Gao X, et al. The osteogenic niche promotes early‐stage bone colonization of disseminated breast cancer cells. Cancer Cell. 2015;27:193‐210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Padmanaban V, Krol I, Suhail Y, et al. E‐cadherin is required for metastasis in multiple models of breast cancer. Nature. 2019;573:439‐444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reichert M, Bakir B, Moreira L, et al. Regulation of epithelial plasticity determines metastatic organotropism in pancreatic cancer. Dev Cell. 2018;45:696‐711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Venhuizen JH, Jacobs FJC, Span PN, Zegers MM. P120 and E‐cadherin: Double‐edged swords in tumor metastasis. Semin Cancer Biol. 2020;60:107‐120. [DOI] [PubMed] [Google Scholar]
- 16. Katsuno Y, Meyer DS, Zhang Z, et al. Chronic TGF‐β exposure drives stabilized EMT, tumor stemness, and cancer drug resistance with vulnerability to bitopic mTOR inhibition. Sci Signal. 2019;12:eaau8544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Berx G, Cleton‐Jansen AM, Nollet F, et al. E‐cadherin is a tumour/invasion suppressor gene mutated in human lobular breast cancers. EMBO J. 1995;14:6107‐6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Corso G, Carvalho J, Marrelli D, et al. Somatic mutations and deletions of the E‐cadherin gene predict poor survival of patients with gastric cancer. J Clin Oncol. 2013;31:868‐875. [DOI] [PubMed] [Google Scholar]
- 19. Dong C, Wu Y, Yao J, et al. G9a interacts with Snail and is critical for Snail‐mediated E‐cadherin repression in human breast cancer. J Clin Invest. 2012;122:1469‐1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dong C, Wu Y, Wang Y, et al. Interaction with Suv39H1 is critical for Snail‐mediated E‐cadherin repression in breast cancer. Oncogene. 2013;32:1351‐1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fukagawa A, Ishii H, Miyazawa K, Saitoh M. δEF1 associates with DNMT1 and maintains DNA methylation of the E‐cadherin promoter in breast cancer cells. Cancer Med. 2015;4:125‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166:21‐45. [DOI] [PubMed] [Google Scholar]
- 23. Bure IV, Nemtsova MV, Zaletaev DV. Roles of E‐cadherin and noncoding RNAs in the epithelial‐mesenchymal transition and progression in gastric cancer. Int J Mol Sci. 2019;20:2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ma L, Young J, Prabhala H, et al. miR‐9, a MYC/MYCN‐activated microRNA, regulates E‐cadherin and cancer metastasis. Nat Cell Biol. 2010;12:247‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang H, Wu Q, Zhang Y, Zhang HN, Wang YB, Wang W. TGF‐β1‐induced epithelial‐mesenchymal transition in lung cancer cells involves upregulation of miR‐9 and downregulation of its target, E‐cadherin. Cell Mol Biol Lett. 2017;22:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fujita Y, Krause G, Scheffner M, et al. Hakai, a c‐Cbl‐like protein, ubiquitinates and induces endocytosis of the E‐cadherin complex. Nat Cell Biol. 2002;4:222‐231. [DOI] [PubMed] [Google Scholar]
- 27. Manshouri R, Coyaud E, Kundu ST, et al. ZEB1/NuRD complex suppresses TBC1D2b to stimulate E‐cadherin internalization and promote metastasis in lung cancer. Nat Commun. 2019;10:5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Aiello NM, Maddipati R, Norgard RJ, et al. EMT subtype influences epithelial plasticity and mode of cell migration. Dev Cell. 2018;45:681‐695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Matsuyoshi N, Hamaguchi M, Taniguchi S, Nagafuchi A, Tsukita S, Takeichi M. Cadherin‐mediated cell‐cell adhesion is perturbed by v‐src tyrosine phosphorylation in metastatic fibroblasts. J Cell Biol. 1992;118:703‐714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shibamoto S, Hayakawa M, Takeuchi K, et al. Tyrosine phosphorylation of β‐catenin and plakoglobin enhanced by hepatocyte growth factor and epidermal growth factor in human carcinoma cells. Cell Adhes Commun. 1994;1:295‐305. [DOI] [PubMed] [Google Scholar]
- 31. Saitoh M. Involvement of partial EMT in cancer progression. J Biochem. 2018;164:257‐264. [DOI] [PubMed] [Google Scholar]
- 32. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial‐mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72:2457‐2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Eger A, Aigner K, Sonderegger S, et al. DeltaEF1 is a transcriptional repressor of E‐cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene. 2005;24:2375‐2385. [DOI] [PubMed] [Google Scholar]
- 35. Shirakihara T, Saitoh M, Miyazono K. Differential regulation of epithelial and mesenchymal markers by δEF1 proteins in epithelial mesenchymal transition induced by TGF‐β. Mol Biol Cell. 2007;18:3533‐3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aghdassi A, Sendler M, Guenther A, et al. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E‐cadherin expression in pancreatic cancer. Gut. 2012;61:439‐448. [DOI] [PubMed] [Google Scholar]
- 37. Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10:116‐129. [DOI] [PubMed] [Google Scholar]
- 38. Kang X, Lin Z, Xu M, et al. Deciphering role of FGFR signalling pathway in pancreatic cancer. Cell Prolif. 2019;52:e12605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Horiguchi K, Shirakihara T, Nakano A, Imamura T, Miyazono K, Saitoh M. Role of Ras signaling in the induction of snail by transforming growth factor‐β. J Biol Chem. 2009;284:245‐253. [DOI] [PubMed] [Google Scholar]
- 40. Saitoh M, Shirakihara T, Fukasawa A, et al. Basolateral BMP signaling in polarized epithelial cells. PLoS One. 2013;8:e62659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ishii H, Saitoh M, Sakamoto K, et al. Epithelial splicing regulatory proteins 1 (ESRP1) and 2 (ESRP2) suppress cancer cell motility via different mechanisms. J Biol Chem. 2014;289:27386‐27399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Takahashi K, Ehata S, Miyauchi K, et al. The role of neurotensin receptor 1 signaling in the progression of pancreatic cancer. Mol Oncol. in press. 10.1002/1878-0261.12815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Itoh Y, Koinuma D, Omata C, et al. A comparative analysis of Smad‐responsive motifs identifies multiple regulatory inputs for TGF‐β transcriptional activation. J Biol Chem. 2019;294:15466‐15479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Brose N, Rosenmund C. Move over protein kinase C, you've got company: alternative cellular effectors of diacylglycerol and phorbol esters. J Cell Sci. 2002;115:4399‐4411. [DOI] [PubMed] [Google Scholar]
- 45. Akhurst RJ, Fee F, Balmain A. Localized production of TGF‐β mRNA in tumour promoter‐stimulated mouse epidermis. Nature. 1988;331:363‐365. [DOI] [PubMed] [Google Scholar]
- 46. Takahashi K, Ehata S, Koinuma D, et al. Pancreatic tumor microenvironment confers highly malignant properties on pancreatic cancer cells. Oncogene. 2018;37:2757‐2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Terauchi R, Kitamura N. Requirement of regulated activation of Ras for response of MDCK cells to hepatocyte growth factor/scatter factor. Exp Cell Res. 2000;256:411‐422. [DOI] [PubMed] [Google Scholar]
- 48. Rosen EM, Meromsky L, Goldberg I, et al. Studies on the mechanism of scatter factor. Effects of agents that modulate intracellular signal transduction, macromolecule synthesis and cytoskeleton assembly. J Cell Sci. 1990;96:639‐649. [DOI] [PubMed] [Google Scholar]
- 49. Krebs AM, Mitschke J, Lasierra Losada M, et al. The EMT‐activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat Cell Biol. 2017;19:518‐529. [DOI] [PubMed] [Google Scholar]
- 50. Zheng X, Carstens JL, Kim J, et al. Epithelial‐to‐mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527:525‐530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ohira T, Gemmill RM, Ferguson K, et al. WNT7a induces E‐cadherin in lung cancer cells. Proc Natl Acad Sci USA. 2003;100:10429‐10434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Horiguchi K, Sakamoto K, Koinuma D, et al. TGF‐β drives epithelial‐mesenchymal transition through δEF1‐mediated downregulation of ESRP. Oncogene. 2012;31:3190‐3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Remacle JE, Kraft H, Lerchner W, et al. New mode of DNA binding of multi‐zinc finger transcription factors: δEF1 family members bind with two hands to two target sites. EMBO J. 1999;18:5073‐5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fabre M, García de Herreros A. Phorbol ester‐induced scattering of HT‐29 human intestinal cancer cells is associated with down‐modulation of E‐cadherin. J Cell Sci. 1993;106:513‐521. [DOI] [PubMed] [Google Scholar]
- 55. Ahmed D, Eide PW, Eilertsen IA, et al. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis. 2013;2:e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dohadwala M, Yang SC, Luo J, et al. Cyclooxygenase‐2‐dependent regulation of E‐cadherin: prostaglandin E(2) induces transcriptional repressors ZEB1 and snail in non‐small cell lung cancer. Cancer Res. 2006;66:5338‐5345. [DOI] [PubMed] [Google Scholar]
- 57. Krishnamachary B, Zagzag D, Nagasawa H, et al. Hypoxia‐inducible factor‐1‐dependent repression of E‐cadherin in von Hippel‐Lindau tumor suppressor‐null renal cell carcinoma mediated by TCF3, ZFHX1A, and ZFHX1B. Cancer Res. 2006;66:2725‐2731. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1‐S9
Tables S1‐S3
Supplementary Material