

Abstract
We present a study to evaluate the feasibility and clinical utility of amplicon‐based Oncomine Pan‐Cancer cell‐free assay to detect circulating tumor DNA (ctDNA) in patients with early or advanced breast cancer. In this study, 109 early and metastatic breast cancer patients were recruited before the initiation of treatment. ctDNA mutation profiles were assessed through unique molecular tagging (UMT) and ultradeep next generation sequencing (NGS). For patients with mutations, DNA from corresponding white blood cells (WBC) was sequenced to exclude variants of clonal‐hematopoietic (CH) origin. UMT targeted sequencing from plasma of 109 patients achieved a median total coverage of 55 498X and a median molecular coverage of 4187X. Among 53 ctDNA positive samples, 38% were mutation positive by WBC sequencing, indicating potentially false‐positive results contributed by CH origin. Prevalence of CH‐related mutations was associated with age (P = 7.51 × 10−4). After exclusion of CH mutations, ctDNA detection rates were 37% for local or locally advanced breast cancer (stage I‐III) and 81% for metastatic or recurrent breast cancer. The ctDNA detection rate correlated with disease stage (P = 2.60 × 10−4), nodal spread (P = 6.49 × 10−3) and the status of distant metastases (P = 5.00 × 10−4). ctDNA variants were detected mostly in TP53, PIK3CA and AKT1 genes, with variants showing therapeutic relevance. This pilot study endorses the use of targeted NGS for non‐invasive molecular profiling of breast cancer. Paired sequencing of plasma ctDNA and WBC should be implemented to improve accurate interpretation of liquid biopsy.
Keywords: amplicon‐based cell‐free assay, breast cancer, circulating tumor DNA, clonal hematopoiesis, liquid biopsy
We present a study to evaluate the feasibility and clinical utility of amplicon‐based liquid biopsy assay to detect circulating tumor DNA (ctDNA) in patients with early or advanced breast cancer. After excluding CH mutations, the ctDNA detection rate correlated with disease stage, nodal spread and the status of distant metastases. Paired sequencing of plasma ctDNA and WBC should be implemented to improve accurate interpretation of liquid biopsy.

Abbreviations
- CA15‐3
cancer antigen 15‐3
- CEA
carcinoembryonic antigen
- cfDNA
cell‐free DNA
- cfTNA
cell‐free total nucleic acid
- CH
clonal hematopoiesis
- CNV
copy number variant
- ctDNA
circulating tumor DNA
- ER
estrogen receptor
- HER2
human epidermal growth factor receptor 2
- IDC
invasive ductal carcinoma
- IRB
institutional review board
- NGS
next generation sequencing
- PR
progesterone receptor
- SNV
single nucleotide variant
- TARDIS
tagged digital sequencing
- UMT
unique molecular tagging
- VAF
variant allele frequency
- WBC
white blood cells
1. INTRODUCTION
Breast cancer is the most common form of cancer affecting women. 1 The 5‐year survival rate decreases when the disease is diagnosed at an advanced stage. 2 Serum‐based protein biomarkers such as CEA and CA15‐3 are commonly used in monitoring of breast cancer patients but are not suitable for diagnostic purposes because of their low sensitivity and specificity. 3 Tissue‐based biomarkers such as ER, PR and HER2 have been useful in guiding the treatment of breast cancer, 4 but these biomarkers are generally unable to capture evolutionary changes in cancer cells.
Tumor tissue biopsies remain the “gold standard” for clinical genomic testing. Given its invasive nature, growing interest has developed in the field of non‐invasive blood liquid biopsy based on mutation detection of ctDNA. In breast cancer, liquid biopsy has shown great promise for clinical applications such as residual disease monitoring, 5 therapeutic response monitoring 6 , 7 and mutation profiling. 8 , 9 , 10 , 11 , 12
Although NGS‐based liquid biopsy has reported encouraging results, there are some limitations to be considered. For example, the ligation‐based methods are only clinically applicable when higher amounts of cfDNA are available due to its low conversion rate in the library construction process. 9 , 13 Alternatively, amplicon‐based methods achieve higher conversion rates in library construction 8 , 14 at the expense of investigating a more limited number of genes. In a recent study, McDonald and colleagues developed a hybrid ligation‐amplicon‐based approach called TARDIS that aims to leverage the advantages of both methods. 5
NGS results in average error rates of 0.1%‐1% in a single read. 15 These errors acquired during PCR amplification or NGS processes can confound the detection of low‐frequency variants when carrying out ultradeep sequencing for liquid biopsy. In order to distinguish PCR/sequencing artifacts from actual variants, an error‐correction approach was developed by incorporating short oligonucleotides called UMT or molecular barcodes. 16 These UMT were attached to the DNA fragments prior to amplification and carried through the entire NGS workflow. Through this method of DNA barcoding, PCR/sequencing artifacts/errors can be eliminated based on the consensus sequence of reads with the same UMT. 17 , 18
Detection of CH mutations in cfDNA is a critical issue that needs to be addressed in liquid biopsy interpretation and forms a key narrative in the clinical application of liquid biopsy. Clonal hematopoiesis mutations, which are derived from clonal expansion of hematopoietic stem cells, may cause false‐positive results and complicate the interpretation of liquid biopsy data. 19 Most CH‐related mutations are observed in hematological cancers. For example, some CH mutations have been reported in the TP53 gene. 13 , 20 As TP53 is the most commonly mutated gene in human cancers, further clarification will be needed to define the origin of TP53 variants when detected by liquid biopsy. In order to mitigate false‐positive results and improve the accuracy of liquid biopsy in the clinical setting, paired cfDNA‐WBC sequencing is of utmost importance. 13 , 20 To date, there are limited breast cancer liquid biopsy studies 10 , 12 , 21 that carry out paired sequencing of plasma cfDNA and WBC DNA to evaluate the status of CH. Therefore, the possibility of misinterpretation due to CH‐associated variants in the previous studies cannot be excluded. Inaccurate interpretation of liquid biopsy results can lead to inappropriate clinical management of breast cancer.
Here we investigate the feasibility of the Oncomine Pan‐Cancer cell‐free assay (Ion Torrent™), an amplicon‐based panel that uses UMT to carry out targeted‐gene NGS on cfDNA. Paired cfDNA‐WBC sequencing was performed for all cfDNA mutation‐positive cases to exclude CH‐related mutations and ensure accuracy of the liquid biopsy analyses. Through this single‐center pilot study, we aim to evaluate the utility of the Oncomine Pan‐Cancer liquid biopsy panel to profile the tumor mutational landscape, interrogate tumor heterogeneity and assess association of tumor variants with clinicopathological parameters of breast cancer.
2. MATERIALS AND METHODS
2.1. Patient recruitment
For this study, we recruited 109 breast cancer patients from the Cancer Institute Hospital of Japanese Foundation for Cancer Research from April 2018 to September 2019. Among the recruited patients, 12 patients were at stage 0 (ductal carcinoma in situ), 45 patients were at stage I, 29 patients were at stage II, 7 patients were at stage III and 16 patients had metastatic or recurrent disease. The timing of blood collection for all patients is presented in Table S1. In all 93 patients with stage 0‐III, blood was collected from treatment‐naive patients before surgery or before the neoadjuvant treatment regimen. We recruited 16 patients with metastatic or recurrent breast cancer. In 5 of the 16 patients, initial diagnosis was metastatic breast cancer and patients were treatment naive. Blood was collected before any intervention. In the remaining 11 of the 16 patients, blood was collected prior to treatment for metastatic or recurrent disease, albeit with exposure to previous lines of treatment. All patients provided written informed consent for cfDNA and WBC sequencing. Approval for collection and genomic profiling of patient samples was granted by the ethical committee of the Japanese Foundation for Cancer Research (IRB No. 2015‐1056). Clinical information was obtained to evaluate the association of clinicopathological parameters with plasma cell‐free DNA. Collection and processing of whole blood has been described in a previous publication. 22 Briefly, 14 mL of whole blood was collected using EDTA‐2Na tubes (Terumo). Whole blood was centrifuged at 2000 × g at 4°C for 10 minutes to separate plasma from white blood cells and red blood cells. The plasma layer was further centrifuged at 16 000 × g at 4°C for 10 minutes to remove cell debris. Both plasma and white blood cells were stored at −80°C until nucleic acid extraction.
2.2. Cell‐free total nucleic acid (cfTNA) extraction
A total of 109 plasma samples from 109 patients were collected and cfTNA was extracted using MagMAX Cell‐Free Total Nucleic Acid Isolation kit (Applied Biosystems) according to the manufacturer’s protocol. Genomic DNA from white blood cells was extracted using FlexiGene DNA Kit (Qiagen). Amount of cfTNA and genomic DNA from each patient was quantified using Qubit DNA HS Assay Kit and Qubit DNA Broad range assay kit (Life Technologies), respectively. The TapeStation 2200 automated electrophoresis system (Agilent) was used to assess fragment sizes of the extracted cfTNA with High Sensitivity D5000 ScreenTape (Agilent). Fragment sizes of genomic DNA were assessed using Genomic DNA ScreenTape (Agilent).
2.3. Library preparation and targeted NGS
Somatic variants from plasma cfTNA were assessed by Oncomine Pan‐Cancer Cell‐Free Assay following the manufacturer’s protocol (Ion Torrent™). Library construction and subsequent NGS of cfTNA and genomic DNA were carried out as previously described. 22 Total of 7.5‐25 ng of cfTNA was used for library construction. Templating and sequencing were carried out using either Ion 540 or Ion 550 kit on the Ion Chef and Ion GeneStudio S5 Prime System (Ion Torrent, Life Technologies). Sequencing of genomic DNA was done using the same methods. Total of 30 ng of genomic DNA from WBC was used for the library construction. Further details are provided in Appendix S1.
2.4. Sequencing data analysis
As previously described, 22 alignment to hg19 and variant calling was carried out using Torrent Suite 5.10.1 and Ion Reporter 5.10.3.0 software, respectively. Raw sequence files were aligned to hg19 using the torrent mapping alignment program (TMAP) with default analysis parameters. Variant calling was performed followed by annotation using Oncomine Pan‐Cancer Annotations v1, a proprietary list of databases (Appendix S1). Library conversion rate was evaluated using the theoretical assumption that 10 ng of cfDNA would be equivalent to ~3000 haploid genomes. To evaluate performance of individual amplicons, the molecular coverage of each UMT corresponding to individual amplicons was normalized by median molecular coverage of all amplicons per sample, giving a relative molecular coverage (RMC) level. An amplicon is deemed to show poor amplification if it shows RMC of <0.5 in at least 25% of 109 patients.
2.5. Statistical analyses
We used the Mann‐Whitney U test to test the difference between average cfTNA concentrations stratified by binary clinical variables. We used the Spearman rank correlation to test the significance of correlation of cfTNA concentration with disease stage or patients’ age. To test association between clinical variables and ctDNA detection rates, we used χ2 test of independence. All statistical analyses were carried out in R version 3.6.3.
3. RESULTS
3.1. Patient characteristics
Patient characteristics at the time of blood collection are presented in Table 1. Total of 109 breast cancer patients were recruited for this study. Median age was 56 years with a range from 25 to 84 years. Of the 109 patients recruited for this study, 11% (12 patients) were at stage 0, 41% (45 patients) were at stage I, 27% (29 patients) were at stage II, 6% (7 patients) were at stage III and 15% (16 patients) had metastatic or recurrent breast cancer. Majority of the patients harbored T1 (56%) tumor status followed by T2 (22%). Lymph node metastases were observed in 32% of patients and 13% of the patients had distant metastases. The majority of the patients (84%) had invasive ductal carcinoma (IDC) histological subtype. Most patients (83%) had tumor subtype of ER+/HER2‐, whereas 6% had ER+/HER2+ and the remaining 11% had triple‐negative breast cancer. Details of patient characteristics are shown in Table S1.
Table 1.
Patient and tumor characteristics
| Characteristics | |
|---|---|
| No. of patients | 109 |
| Median age, years (range) | 56 (25‐84) |
| Disease stage, n = 109 (%) a | |
| Stage 0 | 12 (11%) |
| Stage I | 45 (41%) |
| Stage II | 29 (27%) |
| Stage III | 7 (6%) |
| Metastatic or recurrent disease | 16 (15%) |
| Subtype, n = 109 (%) | |
| ER+/PR+‐/HER2‐ | 90 (83%) |
| ER+‐/PR+‐/HER2+ | 7 (6%) |
| ER‐/PR‐/HER2‐ | 12 (11%) |
| Histology, n = 107 (%) b | |
| Invasive ductal carcinoma | 90 (84%) |
| Ductal in situ carcinoma | 12 (11%) |
| Others | 5 (5%) |
| Lymph node status, n = 98 (%) c | |
| Node positive | 31 (32%) |
| Node negative | 67 (68%) |
| Tumor size, n = 98 (%) c | |
| Tis | 12 (12%) |
| T1 | 55 (56%) |
| T2 | 22.4 (23%) |
| T3 | 3 (3%) |
| T4 | 6 (6%) |
| Metastases status, n = 107 (%)d | |
| M0 | 93 (87%) |
| M1 | 14 (13%) |
In patients with bilateral breast cancer, cancer with a higher stage was used for the analyses.
Tumor histological subtype not counted for 2 patients (P147, P174; refer Table S1) with bilateral breast cancer presenting different histological subtypes.
Tumor and lymph node stage data unavailable for some metastatic breast cancer patients.
Metastases status evaluation excludes recurrent disease patients (P49, P278; refer Table S1).
3.2. Analyses of cfTNA sequencing from plasma of patients
We successfully extracted cfTNA from all patients (median amount of 27 ng from 14 mL of blood) (Table S2). Greater amount of plasma cfTNA concentration correlated with more advanced disease stage (P unadj = 4.47 × 10−3, ρunadj = 0.27) (Figure S1A). Greater amount of plasma cfTNA concentration was also observed with increasing patient age (P = 9.65 × 10−3, ρ = 0.25) (Figure S1B). Correlation between the plasma cfTNA concentration and disease stage was not confounded by patients’ age (P adj = 3.95 × 10−3, ρadj = 0.28) (Figure S1A).
Cell‐free total nucleic acid from all 109 samples were successfully sequenced with a median total coverage of 55 498X (27 148X to 85 414X) and a median molecular coverage of 4187X (1740X to 6133X) (Table S2). Median conversion rate of cfTNA molecules to libraries was 81% (58%‐98%) with the assumption that 10 ng cfDNA is equivalent to ~3000 haploid genomes (Figure S2; Table S3). The majority of the amplicons were well amplified (Figure S3) with the exception of 6 amplicons in FGFR3, FGFR4, ESR1, FGFR1, RET and MAP2K2 genes (Figure S4). ESR1 is an important gene as activating mutations in ESR1 play a key role in resistance towards hormonal therapy in estrogen receptor‐positive breast cancer. One amplicon, ESR1_chr6_152332832, which covers ESR1 E380Q and V392I, was consistently poorly amplified (Figure S5).
Mutation analyses of plasma cfTNA identified 74 SNV and 7 CNV (Figure S6A). Overall ctDNA detection rate was 49% with 53 of the 109 patients carrying at least a single alteration in cfTNA. Of the 74 SNV detected, the majority of the variants were found in 5 genes, namely TP53 (37%), PIK3CA (16%), GNAS (7%), EGFR (5%) and AKT1 (5%) (Figure S6B).
3.3. Clonal hematopoiesis affects interpretation of ctDNA variants
Sequencing of genomic DNA from WBC was done for patients who were mutation positive in cfDNA. We successfully extracted genomic DNA (gDNA) with a median of 50.9 µg genomic DNA per 14 mL blood (Table S4). We achieved similar coverage to cfTNA sequencing with a median total coverage of 57 137X (42 589X to 80 008X) and median molecular coverage of 5142X (1934X to 7891X) (Table S4).
Somatic variants in cfTNA may derive from tumor (DNA from tumors that shed into the circulation) or from CH. In order to mitigate false‐positive results in the cfTNA, we compared the variants obtained from matched cfTNA‐WBC sequencing to ensure accurate variant interpretation. In total, 26 variants were found in WBC sequencing with 17 of them also detected in the cfTNA (Figure 1A). These 17 cfTNA‐WBC‐matched variants accounted for 23% of the 74 variants detected in plasma cfTNA. The variants were distributed across TP53, GNAS, EGFR, IDH2, IDH1 and PTEN genes. Most variants were in TP53 (7 of 17 variants, 41%) followed by GNAS (4 of 17 variants, 24%) (Figure 1B), with some variants being clinically relevant (Table S5). Before excluding CH‐related mutations, overall ctDNA detection rate was 49%. After excluding CH‐related mutations, detection rate was 40% (Figure 1C). ctDNA detection rates grouped according to breast cancer disease stages are as follows: Stage I (42% with‐, 38% without CH mutations); Stage II‐III (53% with‐, 36% without CH mutations); metastatic or recurrent disease (86% with‐, 81% without CH mutations) (Figure 1D). Details of the matched cfTNA‐WBC sequencing variants are shown in Table S6.
Figure 1.
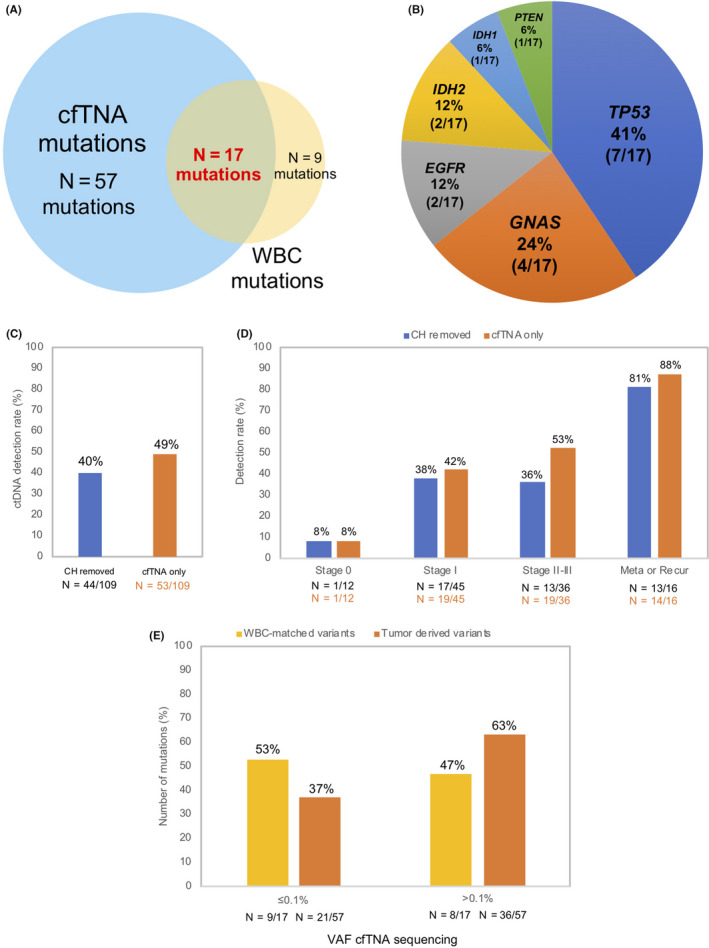
Clonal hematopoiesis (CH) variants and circulating tumor DNA (ctDNA) detection rates. A, Number of variants detected from cfTNA and white blood cell (WBC) sequencing. B, Genomic landscape of cell‐free total nucleic acid (cfTNA)‐WBC‐matched variants. C, Overall ctDNA detection rate with and without excluding CH‐associated variants. D, ctDNA detection rates across breast cancer disease stages with and without excluding CH‐associated variants. Number of patients shown below each corresponding bar. E, Comparison of WBC‐matched with tumor‐derived variants variant allele frequency (VAF) solely from cfTNA sequencing. Tumor‐derived variants (n = 57), WBC‐matched variants (n = 17). Number of variants shown below each corresponding bar
Our results show an overlap of VAF between tumor‐derived (57 variants) and CH‐related mutations (17 variants) from the cfTNA sequencing (Figure 1E). From the 57 tumor‐derived variants, 37% of variants (21 of 57 variants) showed VAF ≤ 0.1% whereas the remaining 63% of variants had VAF > 0.1%. In the 17 CH‐related mutations (cfTNA‐WBC‐matched variants), 53% of variants (9 of 17 variants) showed VAF ≤ 0.1% whereas the remaining 47% of variants had VAF > 0.1%. These results highlight the challenges faced in distinguishing tumor‐derived variants from CH origin in cfTNA in the absence of paired sequencing (Figure 1E).
We observed that patients having detectable clonal hematopoiesis were significantly older (P = 7.51 × 10−4) (Figure 2A) with increased detection of WBC somatic variants associated with advancing years (31‐50 years, 12%; 51‐60 years, 38%; 61‐70 years, 50%; ≥71 years, 75%; P = .03) (Figure 2B, Table S7). We did not observe any correlation between the detection rates of CH and clinical stages (P = .36) (Table S7). VAF detected in cfTNA were correlated with the CH‐related VAF in WBC DNAs (P = 2.97 × 10−3; ρ = 0.67), making it unlikely that these variants were due to sequencing error or background noise of the assay (Figure 2C).
Figure 2.
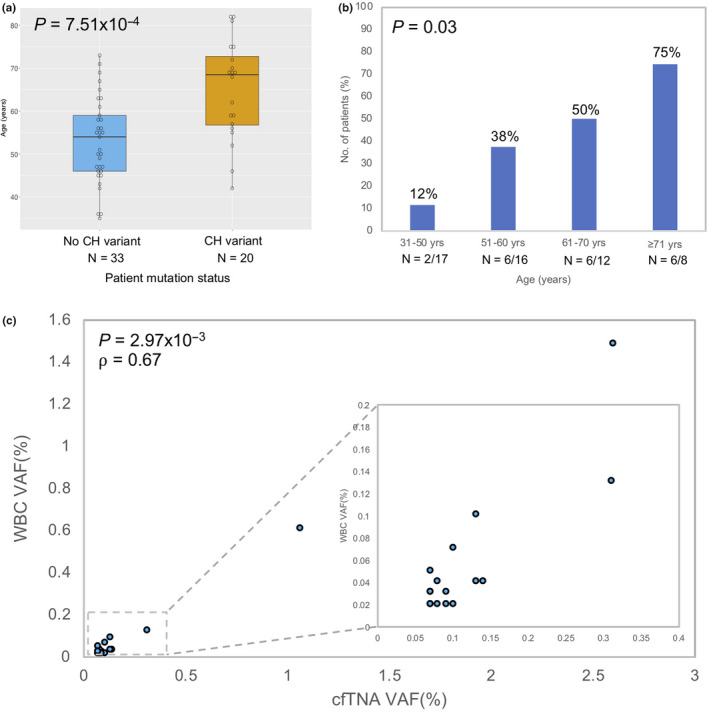
Clonal hematopoiesis (CH) variants characteristics. A, Association between CH variant occurrence and age in breast cancer patients, total samples, n = 53. P‐value was calculated using the Mann‐Whitney U test. Horizontal line represents median, box represents IQR and the whiskers denote 1.5 × IQR on either side. B, CH variant detection rates in different age groups, total samples, n = 53. P‐value was calculated using χ2 2‐sided test of independence. Number of patients shown below each corresponding bar. C, Correlation between cell‐free total nucleic acid (cfTNA) variant allele frequency (VAF) and white blood cell (WBC) VAF in variants found in both cfTNA and WBC sequencing. P‐value and correlation coefficient (⍴) were calculated using Spearman’s rank correlation test
3.4. Genomic landscape of breast cancer ctDNA after excluding CH‐related variants
After excluding CH‐related variants, we achieved an overall detection rate of 40% with 44 ctDNA‐positive samples from the 109 breast cancer plasma samples. The detection rates of ctDNA increased according to disease stage (Stage 0, 8%; Stage I, 38%; Stage II‐III, 36%; metastatic or recurrent disease, 81%) (P = 2.60 × 10−4) (Figure 3A). Patients with metastatic or recurrent disease (16 patients) had varied clinical history (Table S1). In 5 of 16 patients, initial diagnosis was metastatic breast cancer and patients were treatment naive. ctDNA was detected in 4 of the 5 patients (80%) (Table S8). In 11 of 16 patients, plasma was collected prior to metastatic or recurrence treatment. Majority of these patients (10 of 11 patients) were exposed to previous lines of treatment prior to recurrence. ctDNA was detected in 9 of the 11 patients (81%). There was no difference in the ctDNA detection rates between treatment‐naive metastatic breast cancer patients compared to patients with prior exposure to previous lines of treatment (Table S8). As expected, we found a higher proportion of multiple variants in patients with metastatic or recurrent disease (44%) compared to stage 0‐III (6%) (P = 4.02 × 10−5) (Figure 3B). The detection rate of ctDNA was higher in patients with lymph node metastases (P = 6.49 × 10−3) and in those with distant metastases (P = 5.00 × 10−4), but correlation with tumor size (P = .14) was not statistically significant (Figure 3C).
Figure 3.
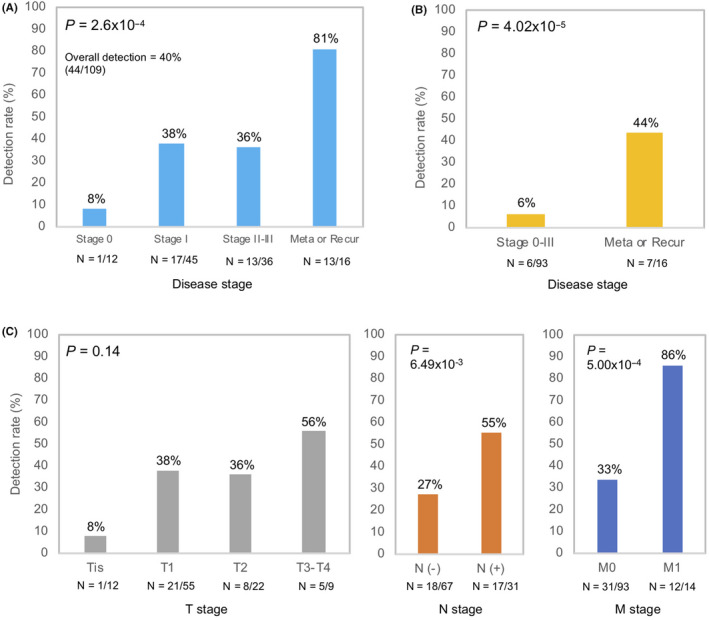
Circulating tumor DNA (ctDNA) detection rates in tumor‐related parameters of breast cancer patients after excluding clonal hematopoiesis (CH)‐associated variants. A, Breast cancer disease stage. B, Multiple mutations detection rate. C, Tumor size, nodal status and metastases status. P‐values were calculated using χ2 2‐sided test of independence. Disease staging follows the American Joint Committee on Cancer (AJCC) TNM staging criteria for non‐metastatic disease patients. ctDNA detection rates shown after excluding CH‐associated variants. Number of patients shown below each corresponding bar. Stage II and stage III breast cancer patients were combined into a single group due to small sample size
We evaluated detection rates across different metastatic lesions, namely visceral or non‐visceral lesions. Of the 14 metastatic breast cancer patients, 6 patients developed visceral (liver, pleura, lung) and 8 patients developed non‐visceral (bone and lymph node) metastatic lesions. The ctDNA detection rate in patients showing visceral metastasis was 83% (5 of 6 patients) whereas in patients showing non‐visceral metastasis, detection rate was 88% (7 of 8 patients) (Table S8). Based on the results of our study, we did not observe any distinctive difference in ctDNA detection rates across the different types of metastatic site in metastatic or recurrent disease patients (Table S8).
We detected 57 SNV (median VAF = 0.11%; range VAF 0.07%‐24.14%) and 7 CNV in FGFR1, CCND1, MYC, MET and FGFR2 (median CN = 3.88; range CN 2.57‐9.92) (Figure 4A, Tables S9 and S10) after excluding CH‐related mutations. Of the 57 SNV detected, the majority of variants were found in five genes including TP53 (37%), PIK3CA (21%), AKT1 (7%), EGFR (5%) and KRAS (5%) (Figure 4B). TP53, PIK3CA and AKT1 are well known driver genes for breast cancer. CNV were detected only in metastatic breast cancer patients. Among the frequently mutated genes in breast cancer, ctDNA detection rate of variants in this study were relatively lower compared to the results of tissue DNA analysis from The Cancer Genome Atlas (TCGA) database (Figure 4C). cfTNA sequencing was able to capture multiple variants (Table S11) in some patients from different disease stages. Some patients (P69, P137, P216, P304) harbored variants with distinct mutation frequencies, highlighting the ability of cfDNA to capture tumor heterogeneity (Table S11).
Figure 4.
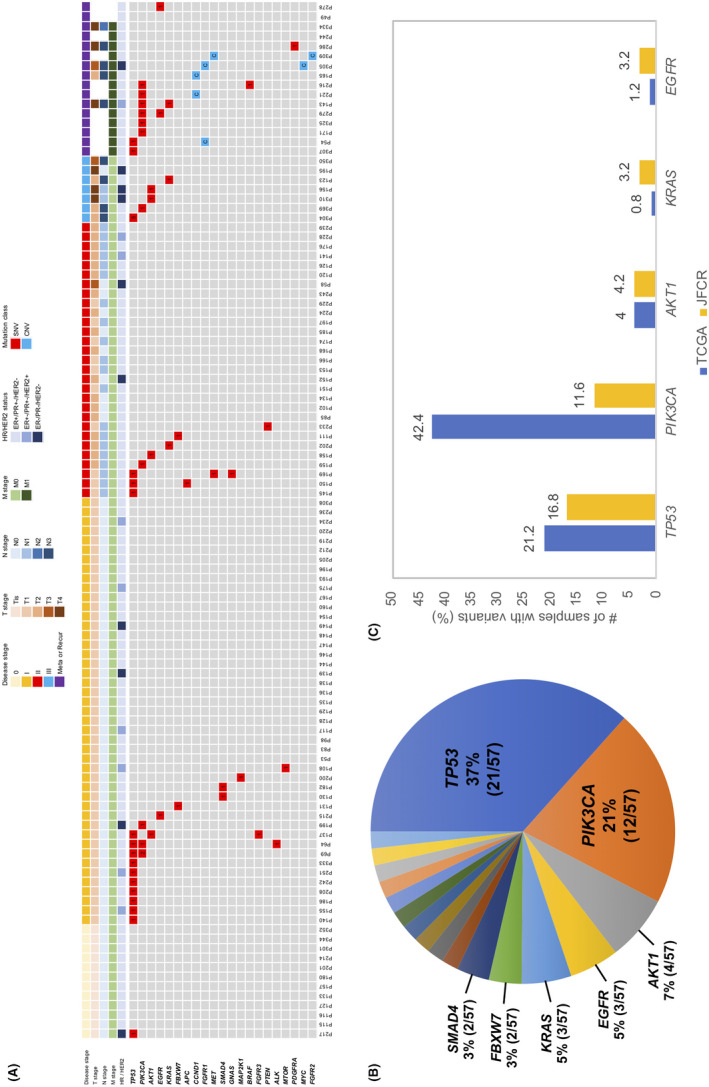
Genomic landscape of circulating tumor DNA (ctDNA) variants from breast cancer after excluding clonal hematopoiesis (CH)‐associated variants. A, Heatmap showing all ctDNA SNV and CNV detected from plasma. The clinical characteristics of patients are represented by the tiles at the top of the heatmap with details stated in the legend. Type of variant (SNV or CNV) is denoted by color in the heatmap whereas numbers signify the number of SNV for a gene per patient. Variants listed in heatmap after removing variants from WBC‐matched sequencing. B, Overall distribution of SNV calculated as number of SNV per gene over the total 57 SNV detected. C, Comparison of gene mutation rates in ctDNA against The Cancer Genome Atlas tissue sequencing data. Only the top 5 mutated genes are shown. Mutation rates for ctDNA genes from the Japanese Foundation for Cancer Research (JFCR) cohort calculated over total sample size = 95 (Stage I, II, III and metastatic disease)
4. DISCUSSION
We present a single‐center study for molecular profiling of breast cancer with blood liquid biopsy. To our knowledge, this is one of a few studies 22 , 23 demonstrating the feasibility of a high‐coverage NGS through an amplicon‐based panel with UMT, and the first in breast cancer. Blood from stage 0‐III patients was sampled prior to primary treatment (surgery or neoadjuvant therapy), while blood from metastatic or recurrence patients were sampled prior to treatment or intervention for recurrence or metastatic disease. The ctDNA detection rates reported in our study are reflective of the disease state of the patients and were not influenced by treatment intervention or the type of metastatic lesion. The detection rate of ctDNA, after excluding CH‐related mutations, were high in patients with metastatic or recurrent breast cancer (81%), but more modest for those with local or locally advanced breast cancer (stage I 38%; stage II‐III 36%). Through cfDNA sequencing, studies have detected variants related to CH, with a higher prevalence in older populations. 13 , 24 As the majority of CH‐related variants are individual‐specific, it is imperative that paired sequencing of plasma cfDNA and WBC DNA be carried out to exclude CH variants and avoid inaccurate interpretation of liquid biopsy analyses.
ctDNA detection in patients with early‐stage breast cancer is generally challenging due to lower shedding of ctDNA into circulation. 9 , 25 , 26 In addition, among different cancer types, Phallen and colleagues observed that breast cancer had the lowest amount of ctDNA. 9 These factors, to some extent, possibly contribute to the lower ctDNA detection rates. Previous studies have reported a shorter DNA fragment size for ctDNA in the blood. 27 , 28 Adopting size selection methods specific to ctDNA fragments might improve detection rates, but early data 29 , 30 require further validation. The big caveat is that improvements in the detection rates would be very challenging if the biological features of breast tumors as well as their microenvironment inherently cause low shedding of ctDNA into the blood.
As more therapeutic options for breast cancer become available, there is a need for companion diagnostic tests to detect therapeutic relevant variants and assist patients’ treatment selection. From our results, a substantial number of alterations fall into the PI3K/AKT/mTOR pathway (Figure 4A; Table S9). Several therapies that target PIK3CA (alpelisib, buparlisib) and MTOR (everolimus) are available at present. Previous literature 31 has confirmed the high mutation rate of PIK3CA in ER+/HER2‐ breast cancer, making it an attractive target for treatment. In fact, it has been reported that PIK3CA mutation status in ctDNA is correlated with efficacy of PI3K inhibitors such as buparlisib 32 and alpelisib. 33
In our study, cfTNA sequencing was able to capture multiple variants in some patients with different VAF, possibly reflecting intra‐ and/or inter‐tumor heterogeneity. Although the genetic profile of tumor biopsies is constrained by spatial limitations, liquid biopsy may have the potential to effectively interrogate tumor heterogeneity, detecting both clonal and subclonal variants. Previous studies on tumor evolution have proven that clonal variants (truncal variants) exist in almost all tumor cells and tend to show higher VAF in ctDNA compared with subclonal variants. 34 , 35
The ctDNA detection rates using Oncomine Pan‐Cancer cell‐free assay were comparable to or better than several reported studies focusing on amplicon‐based breast cancer liquid biopsy. 8 , 10 , 21 Detection rate of the current study is lower compared with the amplicon‐based custom panel reported by Zhang and colleagues 21 and the ligation‐based TEC‐Seq approach by Phallen and colleagues 9 (Table S12). Nevertheless, the study by Zhang and colleagues used a larger panel (68 genes) and, more importantly, matched WBC DNA sequencing was not carried out. The detection rates were closer when CH‐associated variants were not excluded (53% Zhang and colleagues vs 47% in the current study). Despite better overall detection rates in the study by Phallen and colleagues (56% TEC‐Seq vs 37% in the current study), input cfDNA for TEC‐Seq was much higher (average 115 ng, range 19‐998 ng) and the conversion rate was lower (average 31%, range 0.44%‐68%). To achieve the same input cfDNA as TEC‐Seq (average 115 ng), we would require approximately 48 mL blood (7 tubes of 7 mL blood) assuming an average cfDNA concentration of 33.4 ng per 14 mL of blood found in this study. The excessive sampling of blood contradicts the non‐invasive aspect of using liquid biopsy for cancer management, making this approach clinically inapplicable. In our study, we observed a high conversion rate of cfTNA molecules to libraries (median 80%), enabling high molecular depth of cfTNA sequencing. When compared with other liquid biopsy methods, the conversion rate of the Oncomine Pan‐Cancer approach is similar to the Digital Sequencing™ method, 36 but is approximately 1.5‐fold better than another amplicon‐based method TAm‐Seq 14 and nearly four times more efficient than ligation‐based methods 9 , 13 for cfDNA library construction.
Despite the advantages of cfDNA profiling, there are still some limitations to our study. In particular, due to the lack of tumor tissue samples, we were unable to evaluate the concordance of variants in cfTNA sequencing compared to tumor tissues.
Our results show that ctDNA can be effective in profiling the mutational landscape of breast cancer, with higher ctDNA detection rates observed in metastatic breast cancer compared to local or locally advanced breast cancer. The results of this pilot study are comparable to other liquid biopsy approaches and represent an endorsement for the implementation of liquid biopsy in breast cancer management. Detection of tumor‐derived variants through plasma cfDNA sequencing can be a powerful tool for minimally invasive monitoring and clinical management of breast cancer. This study also emphasizes the importance of paired plasma cfTNA and WBC DNA sequencing to exclude CH‐related variants and reduce false‐positive interpretation in liquid biopsy analyses. Our findings provide a basis for further development and eventual adoption of liquid biopsy as a routine molecular screening program.
DISCLOSURE
YMC is an employee of Cancer Precision Medicine, Inc. Japan. YN serves as a consultant and advisor to OncoTherapy Science, Inc. Japan. SKL serves as a consultant and advisor to Cancer Precision Medicine, Inc. Japan.
AUTHORS CONTRIBUTION
SKL conceived and designed the experiments. YT, MO, MF, TS, YM and TU were actively involved in recruiting patients and interpreting clinical data. MO and HTC performed the experiments. YMC, HTC and SKL performed the data analyses and interpretation of the results with assistance from YT, YN and TU. YMC and SKL wrote the initial draft of the manuscript with assistance from YT, HTC, TU and YN. The authors read and approved the final manuscript.
Supporting information
Fig S1‐S6
Table S1‐S12
App S1
ACKNOWLEDGMENTS
We thank the patients and their families for their participation in the study. We thank the staff of the Breast Oncology Center, Cancer Institute Hospital, Japanese Foundation for Cancer Research, Dr Jae‐Hyun Park, Cancer Precision Medicine, Inc. and OncoTherapy Science, Inc. for supporting this study. We also thank Ms Rie Hayashi and Dr Hideaki Matsuki for providing technical assistance. This work was supported by the Council for Science, Technology and Innovation (CSTI), cross‐ministerial Strategic Innovation Promotion Program (SIP), “Innovative AI Hospital System.” Funding Agency: National Institute of Biomedical Innovation, Health and Nutrition (NIBIOHN).
Chin YM, Takahashi Y, Chan HT, et al. Ultradeep targeted sequencing of circulating tumor DNA in plasma of early and advanced breast cancer. Cancer Sci. 2021;112:454–464. 10.1111/cas.14697
REFERENCES
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394‐424. [DOI] [PubMed] [Google Scholar]
- 2. Yoshimura A, Ito H, Nishino Y, et al. Recent improvement in the long‐term survival of breast cancer patients by age and stage in Japan. J Epidemiol. 2018;28:420‐427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Safi F, Kohler I, Rottinger E, Beger H. The value of the tumor marker CA 15–3 in diagnosing and monitoring breast cancer. A comparative study with carcinoembryonic antigen. Cancer. 1991;68:574‐582. [DOI] [PubMed] [Google Scholar]
- 4. Harris L, Fritsche H, Mennel R, et al. American Society of Clinical Oncology 2007 update of recommendations for the use of tumor markers in breast cancer. J Clin Oncol. 2007;25:5287‐5312. [DOI] [PubMed] [Google Scholar]
- 5. McDonald BR, Contente‐Cuomo T, Sammut SJ, et al. Personalized circulating tumor DNA analysis to detect residual disease after neoadjuvant therapy in breast cancer. Sci Transl Med. 2019;11(504):eaax7392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. O'Leary B, Cutts RJ, Liu Y, et al. The genetic landscape and clonal evolution of breast cancer resistance to palbociclib plus fulvestrant in the PALOMA‐3 Trial. Cancer Discov. 2018;8:1390‐1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. O’Leary B, Hrebien S, Morden JP, et al. Early circulating tumor DNA dynamics and clonal selection with palbociclib and fulvestrant for breast cancer. Nat Commun. 2018;9:896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rothe F, Laes JF, Lambrechts D, et al. Plasma circulating tumor DNA as an alternative to metastatic biopsies for mutational analysis in breast cancer. Ann Oncol. 2014;25:1959‐1965. [DOI] [PubMed] [Google Scholar]
- 9. Phallen J, Sausen M, Adleff V, et al. Direct detection of early‐stage cancers using circulating tumor DNA. Sci Transl Med. 2017;9(403):eaan2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cohen JD, Li L, Wang Y, et al. Detection and localization of surgically resectable cancers with a multi‐analyte blood test. Science. 2018;359:926‐930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhou Y, Xu Y, Gong Y, et al. Clinical factors associated with circulating tumor DNA (ctDNA) in primary breast cancer. Mol Oncol. 2019;13(5):1033‐1046 screens. [Cited May 2019.] Available from URL: https://www.ncbi.nlm.nih.gov/pubmed/30672098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early‐ and late‐stage human malignancies. Sci Transl Med. 2014;6(224):224ra224 screens. [Cited Feb 19 2014.] Available from URL: https://www.ncbi.nlm.nih.gov/pubmed/24553385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Razavi P, Li BT, Brown DN, et al. High‐intensity sequencing reveals the sources of plasma circulating cell‐free DNA variants. Nat Med. 2019;25:1928‐1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Forshew T, Murtaza M, Parkinson C, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med. 2012;4:136ra168. [DOI] [PubMed] [Google Scholar]
- 15. Fox EJ, Reid‐Bayliss KS, Emond MJ, Loeb LA. Accuracy of next generation sequencing platforms. Next Gener Seq Appl. 2014;1:1000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Salk JJ, Schmitt MW, Loeb LA. Enhancing the accuracy of next‐generation sequencing for detecting rare and subclonal mutations. Nat Rev Genet. 2018;19:269‐285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hiatt JB, Patwardhan RP, Turner EH, Lee C, Shendure J. Parallel, tag‐directed assembly of locally derived short sequence reads. Nat Methods. 2010;7:119‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci USA. 2011;108:9530‐9535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bauml J, Levy B. Clonal hematopoiesis: a new layer in the liquid biopsy story in lung cancer. Clin Cancer Res. 2018;24(18):4352‐4354. [Cited Sep 15 2018.] Available from URL: https://www.ncbi.nlm.nih.gov/pubmed/29748181 [DOI] [PubMed] [Google Scholar]
- 20. Ptashkin RN, Mandelker DL, Coombs CC, et al. Prevalence of clonal hematopoiesis mutations in tumor‐only clinical genomic profiling of solid tumors. JAMA Oncol. 2018;4:1589‐1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang X, Zhao W, Wei W, et al. Parallel analyses of somatic mutations in plasma circulating tumor DNA (ctDNA) and matched tumor tissues in early‐stage breast cancer. Clin Cancer Res. 2019;25:6546‐6553. [DOI] [PubMed] [Google Scholar]
- 22. Chan HT, Nagayama S, Chin YM, et al. Clinical significance of clonal hematopoiesis in the interpretation of blood liquid biopsy. Mol Oncol. 2020;14(8):1719‐1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Alborelli I, Generali D, Jermann P, et al. Cell‐free DNA analysis in healthy individuals by next‐generation sequencing: a proof of concept and technical validation study. Cell Death Dis. 2019;10(7):534 screens. [Cited Jul 11 2019.] Available from URL: https://www.ncbi.nlm.nih.gov/pubmed/31296838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood‐cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477‐2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Riva F, Bidard FC, Houy A, et al. Patient‐specific circulating tumor DNA detection during neoadjuvant chemotherapy in triple‐negative breast cancer. Clin Chem. 2017;63:691‐699. [DOI] [PubMed] [Google Scholar]
- 26. Stover DG, Parsons HA, Ha G, et al. Association of cell‐free DNA tumor fraction and somatic copy number alterations with survival in metastatic triple‐negative breast cancer. J Clin Oncol. 2018;36:543‐553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jiang P, Chan CW, Chan KC, et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc Natl Acad Sci USA. 2015;112:E1317‐E1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Underhill HR, Kitzman JO, Hellwig S, et al. Fragment length of circulating tumor DNA. PLoS Genet. 2016;12:e1006162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mouliere F, Chandrananda D, Piskorz AM, et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci Transl Med. 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu X, Liu L, Ji Y, et al. Enrichment of short mutant cell‐free DNA fragments enhanced detection of pancreatic cancer. EBioMed. 2019;41:345‐356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ellis MJ, Perou CM. The genomic landscape of breast cancer as a therapeutic roadmap. Cancer Discov. 2013;3:27‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Baselga J, Im SA, Iwata H, et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor‐positive, HER2‐negative, advanced breast cancer (BELLE‐2): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet Oncol. 2017;18(7):904‐916 screens. [Cited Jul 2017.] Available from URL: https://www.ncbi.nlm.nih.gov/pubmed/28576675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Razavi P, Dickler MN, Shah PD, et al. Alterations in PTEN and ESR1 promote clinical resistance to alpelisib plus aromatase inhibitors. Nature Cancer. 2020;1:382‐393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McGranahan N, Swanton C. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017;168:613‐628. [DOI] [PubMed] [Google Scholar]
- 35. Burrell RA, Swanton C. Re‐evaluating clonal dominance in cancer evolution. Trends Cancer. 2016;2(5):263‐276. [Cited May 2016.] Available from URL: https://www.ncbi.nlm.nih.gov/pubmed/28741512 [DOI] [PubMed] [Google Scholar]
- 36. Lanman RB, Mortimer SA, Zill OA, et al. Analytical and clinical validation of a digital sequencing panel for quantitative, highly accurate evaluation of cell‐free circulating tumor DNA. PLoS One. 2015;10:e0140712. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S6
Table S1‐S12
App S1