

Abstract
Objectives
Regulatory T cells (Tregs) are a vital sub‐population of CD4+ T cells with major roles in immune tolerance and homeostasis. Given such properties, the use of regulatory T cells for immunotherapies has been extensively investigated, with a focus on adoptive transfer of ex vivo expanded natural Tregs (nTregs). For immunotherapies, induced Tregs (iTregs), generated in vitro from naïve CD4+ T cells, provide an attractive alternative, given the ease of generating cell numbers required for clinical dosage. While the combination of TGF‐β, ATRA and rapamycin has been shown to generate highly suppressive iTregs, the challenge for therapeutic iTreg generation has been their instability. Here, we investigate the impact of rapamycin concentrations and α‐CD3/CD28 bead ratios on human iTreg stability.
Methods
We assess iTregs generated with various concentrations of rapamycin and differing ratios of α‐CD3/CD28 beads for their differentiation, stability, expression of Treg signature molecules and T helper effector cytokines, and Treg‐specific demethylation region (TSDR) status.
Results
iTregs generated in the presence of TGF‐β, ATRA, rapamycin and a higher ratio of α‐CD3/CD28 beads were highly suppressive and stable upon in vitro re‐stimulation. These iTregs exhibited a similar expression profile of Treg signature molecules and T helper effector cytokines to nTregs, in the absence of TSDR demethylation.
Conclusion
This work establishes a method to generate human iTregs which maintain stable phenotype and function upon in vitro re‐stimulation. Further validation in pre‐clinical models will be needed to ensure its suitability for applications in adoptive transfer.
Keywords: immunotherapy, induced regulatory t cells, rapamycin, regulatory t cells, TCR stimulation
Induced regulatory T cells are an attractive alternative for adoptive transfer in an allogeneic transplant setting. We report that rapamycin and abundant TCR stimulation are required to generate stable human induced regulatory T cells.

Introduction
Regulatory T cells (Tregs) are a sup‐population of CD4+ T cells with immune‐suppressive and immune‐modulatory properties. With such properties, Tregs form a vital part of immune homeostasis, providing tolerance to self and non‐pathogenic foreign antigens, and down‐regulating immune responses once pathogens are cleared in order to minimise tissue‐damage. 1 Currently, human natural Tregs (nTregs) are defined by the surface phenotype CD4+CD25hiCD127lo, and expression of the master regulator of Treg‐lineage, FOXP3. 2 , 3 , 4 , 5 , 6 , 7 Within the CD4+CD25hiCD127‐FOXP3+ population, there are two main subgroups of nTregs: thymic‐derived thymic Tregs (tTregs) and peripherally induced peripheral Tregs (pTregs). tTregs are generated within the thymus from self‐reactive CD4+ T cells and account for 5–10% of circulating CD4+ T cells. 8 Most self‐reactive CD4+ T cells are negatively selected and deleted by apoptosis to establish central tolerance; however, some self‐reactive CD4+ T cells with relatively high affinity for self‐antigens receive signals to differentiate into nTregs by the induction of FOXP3 expression. 9 tTregs express a T‐cell receptor (TCR) repertoire skewed towards self‐antigens allowing them to curtail autoimmune responses by self‐reactive conventional T cells that have escaped central tolerance and thus prevent autoimmunity. 10 , 11 In contrast, peripheral naïve CD4+ T cells, which normally account for 30–50% of circulating CD4+ T cells in adults aged between 18 to 70, 12 , 13 can acquire FOXP3 expression upon activation, becoming pTregs. As pTregs are differentiated from conventional T cells, they express a TCR repertoire skewed towards foreign antigens and help establish tolerance to commensal microbiota, environmental and food allergens, and foetal alloantigens during pregnancy. 11 , 14 The induction of pTregs can be mimicked in vitro to generate induced Tregs (iTregs). 14 , 15
iTregs provide an attractive alternative to Treg‐based immunotherapies in allogeneic transplantation. Currently, most Treg‐based immunotherapies employ adoptive transfer of ex vivo expanded nTregs. 16 To generate the required number of cells for clinical dosage, which requires up to 5 billion Tregs per patient, nTregs need to be ex vivo expanded for a prolonged period of time due their low frequency in peripheral blood. 17 , 18 Thus, it would be beneficial to generate large number of Tregs in a shorter time frame, by differentiating iTregs from naïve CD4+ T cells, which are at a significantly higher frequency in peripheral blood. Furthermore, the TCR repertoire of iTregs is potentially more relevant for allogeneic transplantation, as rejection of the donor tissue occurs in response to foreign antigens against which the nTreg pool has not been educated. 15 In addition, the broader TCR repertoire of iTregs compared with nTregs has advantages in the generation of antigen‐specific Tregs, potentially providing a more targeted therapy. 14 , 19
The pivotal point in the generation of iTregs has been the discovery of differentiation induction molecules converting naïve CD4+ T cells into pTregs. One particular environment in which pTregs are present in significant numbers is the gut. 20 , 21 The gut mucosal environment contains TGF‐β, all‐trans retinoic acid (ATRA), and short chain fatty acids, such as butyrate, which have been shown to promote pTreg differentiation. 22 , 23 Additionally, manipulation of ex vivo iTreg generation has validated various molecules such as IL‐2, rapamycin and progesterone as enhancer of pTreg differentiation. 15 To date, different approaches using combinations of these molecules have been explored to generate human iTregs. 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 In particular, the combination of TGF‐β, ATRA and rapamycin has been shown to generate highly suppressive iTregs that are stable upon resting 26 ; however, phenotypic instability of iTregs upon re‐stimulation remains a major challenge significantly limiting the use of iTregs for therapeutic applications.
Here, we aimed to optimise an iTreg differentiation method for the robust and reproducible production of iTregs with phenotypic and functional stability in vitro. Various concentrations of rapamycin have been utilised previously for the induction of human iTregs, ranging from 0.45 to 100 ng mL−1. 26 , 27 , 29 , 33 , 34 , 35 , 36 In addition, the effect of rapamycin concentration (1, 10 and 100 ng mL−1) on the proportion of cells that differentiate into iTregs has been studied; 29 however, the effect on iTreg stability remains unknown. Thus, we investigated which concentration of rapamycin is optimal for the generation of stable iTregs, comparing low‐dose rapamycin of 1 ng mL−1, medium‐dose rapamycin of 10 ng mL−1 and high‐dose rapamycin of 100 ng mL−1. Various TCR stimulation methods appear across the literature for the generation of human iTregs, with no clear consensus. 24 , 25 , 26 , 27 , 28 In particular, different ratios of α‐CD3/CD28 beads (3:1 and 1:10 bead to cell) have been utilised. 25 , 27 , 29 Hence, we explored the impact of TCR stimulation bead ratio by using 1:1 and 1:10 α‐CD3/CD28 beads to represent ‘high’ and ‘low’ ratios of α‐CD3/CD28 beads, respectively. We assessed phenotype and suppressive function of iTregs generated under various conditions after initial stimulation and upon re‐stimulation without iTreg‐differentiating factors. We further characterised the functionality of iTregs by measuring expression of Treg signature molecules and T helper signature cytokines. In addition, phenotypic stability and T helper signature cytokine production of iTregs upon challenge with Th17‐polarising cytokines were evaluated. Lastly, Treg‐specific demethylation region (TSDR) methylation status of iTregs was analysed.
Results
Phenotype and function of iTregs generated with varied rapamycin concentration
To investigate the impact of rapamycin concentration on human iTreg induction, naïve CD4+ T cells and naïve nTregs were isolated from human peripheral blood (Supplementary figure 1). Naïve CD4+ T cells were differentiated into iTregs over a 7‐day stimulation using TGF‐β, ATRA, IL‐2, α‐CD3/CD28 beads (1:1 bead to cell) and various concentrations of rapamycin (0, 1, 10 and 100 ng mL−1; these iTregs were termed iTreg‐0, iTreg‐1, iTreg‐10 and iTreg‐100, respectively). Naïve nTregs were stimulated using IL‐2 and α‐CD3/CD28 beads (1:1 bead to cell) as a positive control for Treg phenotype and function (nTreg), and naïve CD4+ T cells were stimulated using IL‐2 and α‐CD3/CD28 beads (1:1 bead to cell) without iTreg differentiation components as a mock stimulation control (Tconv). Following the 7‐day stimulation, all cell types were rested for 7 days in the presence of IL‐2 and were assessed after 3 days and 7 days of rest. After a 3‐day rest, cells were assessed for expression of Treg markers FOXP3 and CD25. FOXP3 and CD25 expression levels were measured via flow cytometry and expressed as a percentage (%FOXP3+CD25+) of the viable CD4+ population, and as protein expression levels by MFI (Supplementary figure 2). In each experiment, CD25 and FOXP3 MFI were normalised to the highest raw MFI and represented as nMFI (%). Tconv cells were 77% FOXP3+CD25+, iTreg‐0 cells were 87% FOXP3+CD25+, nTreg and iTreg‐1 cells were 91% FOXP3+CD25+, and iTreg‐10 and iTreg‐100 cells were 95% FOXP3+CD25+. %FOXP3+CD25+ of Tconv was significantly lower than nTreg, iTreg‐1, iTreg‐10 and iTreg‐100 (P = 0.0398, 0.0343, 0.0105 and 0.0088). FOXP3 MFI of Tconv was significantly lower than nTreg, iTreg‐10 and iTreg‐100 (P = 0.0121, 0.0017 and 0.0011) while FOXP3 MFI of iTreg‐0 was significantly lower compared with iTreg‐10 and iTreg‐100 (P = 0.0080 and 0.0049). CD25 MFI of iTreg‐10 and iTreg‐100 were significantly higher than other cell types (Figure 1a; raw MFI for CD25 and FOXP3 shown in Supplementary figure 3; nTreg: P = 0.0228 and 0.0180, Tconv: P = 0.0017 and 0.0013, iTreg‐0: P = 0.0217 and 0.0172, and iTreg‐1: P = 0.0330 and 0.0260). Generated iTregs were then assessed for their suppressive function. Cell Trace Violet (CTV) was used to track cell proliferation of responder cells (naïve CD4+ T cells) in the presence of α‐CD3/CD28 beads (1:5 bead to cell) and different ratios of Tregs (1:1 to 1:8 Treg to Tresponder). Based on the positive proliferation control (CTV‐stained and stimulated with no Tregs), suppression was calculated (Supplementary figure 4). Suppressive activities of iTreg generated in the presence of rapamycin showed no significant differences compared with nTreg, while Tconv and iTreg‐0 displayed significantly lower suppressive activities than nTreg, iTreg‐1, iTreg‐10 and iTreg‐100 at most Treg:Tresponder ratios (1:1 – Tconv: P = 0.0409, 0.0250, 0.0233 and 0.0250, 1:2 – Tconv: P < 0.0001 for all and iTreg‐0: P = 0.0688, 0.0331, 0.0226 and 0.0250, 1:4 – Tconv: P < 0.0001 for all and iTreg‐0: P = 0.0020, P < 0.0001, 0.0001 and 0.0001, 1:8 – Tconv: P < 0.0001 for all and iTreg‐0: P = 0.0038, P < 0.0001, 0.0001 and 0.0001). Additionally, suppressive activities of Tconv were significantly lower than iTreg‐0 (Figure 1b; 1:2 – P = 0.0387 and 1:4 – P = 0.0336). After a 7‐day rest, FOXP3 and CD25 expression were assessed again. nTreg, iTreg‐0, iTreg‐1, iTreg‐10 and iTreg‐100 retained their %FOXP3+CD25+ at 87%, 78%, 86%, 91% and 92%, respectively. % FOXP3+CD25+ of Tconv was significantly lower than nTreg, iTreg‐0, iTreg‐1, iTreg‐10 and iTreg‐100 at 45% (P < 0.0001, P = 0.0004, P < 0.0001, 0.0001 and 0.0001, respectively). While no significant differences in FOXP3 MFI between Tconv and other cell types were observed, CD25 MFI of Tconv was significantly lower than nTreg, iTreg‐0, iTreg‐1, iTreg‐10 and iTreg‐100 (P = 0.0116, 0.0254, 0.0006, P < 0.0001 and 0.0001). CD25 MFI of iTreg‐10 and iTreg‐100 remained significantly higher than nTreg and iTreg‐0 (Figure 1c; raw MFI for CD25 and FOXP3 shown in Supplementary figure 5; iTreg‐10: P = 0.0096 and 0.0045 and iTreg‐100: P = 0.0022 and 0.0011). Furthermore, cell growth rates at the end of the stimulation cycle were assessed, which showed iTreg‐10 and iTreg‐100 with significantly lower cell growth rate compared with nTreg, Tconv and iTreg‐0 (Supplementary figure 6; iTreg‐10: P = 0.0135, 0.0578, 0.0267 and iTreg‐100: P = 0.0064, 0.0267, 0.0021).
Figure 1.
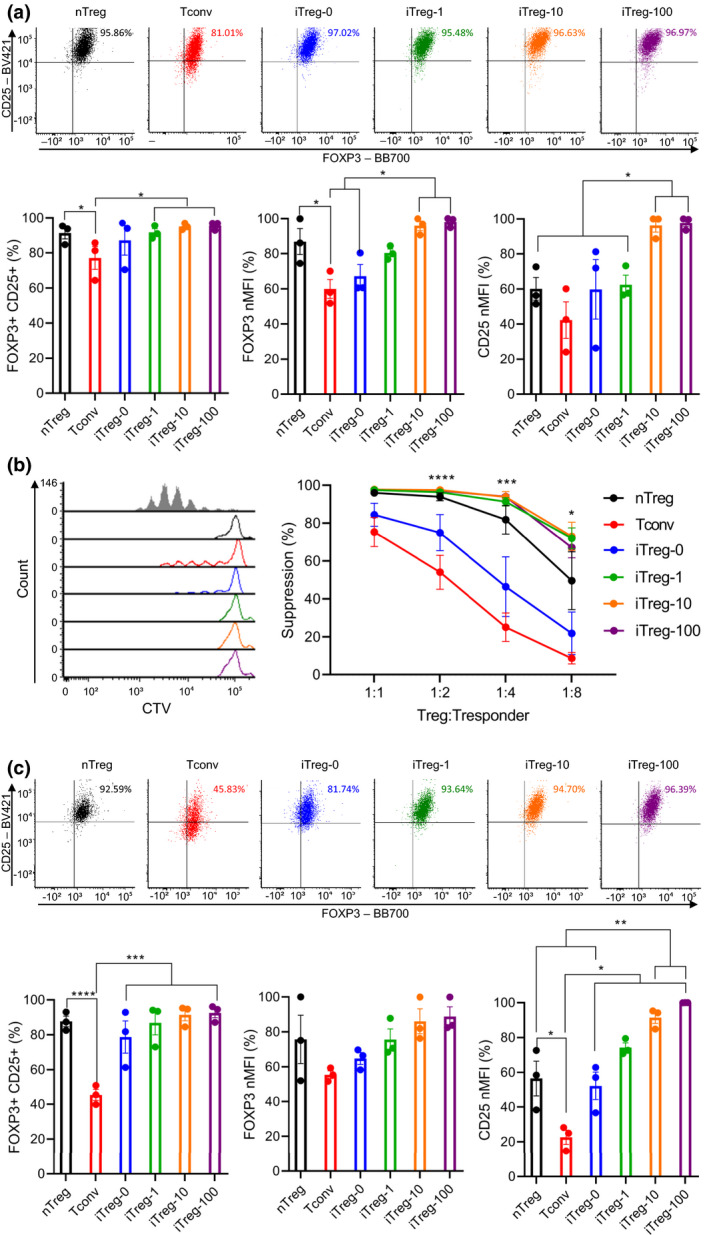
The impact of rapamycin concentrations on iTreg differentiation. Naïve nTregs were stimulated with a 1:1 α‐CD3/CD28 beads and IL‐2 (nTreg). Naïve CD4+ T cells were mock‐stimulated with a 1:1 α‐CD3/CD28 beads and IL‐2 (Tconv). iTregs were differentiated from naïve CD4+ T cells using a 1:1 α‐CD3/CD28 beads, IL‐2 TGF‐β, ATRA and various concentrations of rapamycin (0, 1, 10 and 100 ng mL−1; iTreg‐0, iTreg‐1, iTreg‐10 and iTreg‐100). Following a 7‐day stimulation, cells were rested up to 7 days with IL‐2. After 3 days of rest, (a) expression of FOXP3 and CD25 (b) suppressive activities were evaluated. After 7 days of rest, (c) expression of FOXP3 and CD25 was evaluated. MFI of FOXP3 and CD25 were normalised to highest raw MFI value in each experiment and represented as nMFI (%). Raw MFI for (a and c) Supplementary figures 3 and 5, respectively. For (b), grey‐shaded histogram represents positive control (no Treg control). Data are represented as mean ± sem, N = 3 in three independent experiments. For each donor (N), technical triplicates were utilised, and the average of technical replicates was used for each datapoint. Statistical significance identified by RM one‐way (a and c) and RM two‐way (b) ANOVA with Dunnett’s multiple comparisons test: *P < 0.05, **0.01, ***0.001, ****0.0001.
Reductions in FOXP3 expression and suppressive activities have been observed in human iTregs upon re‐stimulation without iTreg differentiation components, 25 , 26 which could occur in vivo resulting in generation of non‐functional ex‐Tregs. Thus, the impact of rapamycin concentration on human iTreg stability upon in vitro re‐stimulation was evaluated. Following a 7‐day stimulation and 3‐day rest, cells were re‐stimulated using IL‐2 and α‐CD3/CD28 beads (1:1 bead to cell) without the iTreg differentiation factors for 7 days, then extensively washed and rested for 3 days in the presence of IL‐2. After re‐stimulation, FOXP3 and CD25 expression and suppressive function of cells were assessed. nTreg, iTreg‐1, iTreg‐10 and iTreg‐100 retained their %FOXP3+CD25+ at 89%, 81%, 89% and 90%. %FOXP3+CD25+ of Tconv was significantly lower than nTreg, iTreg‐0, iTreg‐1, iTreg‐10 and iTreg‐100 at 38% (P < 0.0001, P = 0.0007, P < 0.0001, 0.0001 and 0.0001). iTreg‐0 exhibited significantly lower %FOXP3+CD25+ than nTreg, iTreg‐1 and iTreg‐10 and iTreg‐100 at 66% (P = 0.0032, 0.0127, 0.0038 and 0.0028). FOXP3 MFI of Tconv and iTreg‐0 were significantly lower than nTreg (P = 0.0349 and 0.0496). Tconv showed significantly lower CD25 MFI compared with nTreg, iTreg‐1, iTreg‐10 and iTreg‐100 (P = 0.0002, 0.0011, 0.0003 and 0.0002), and iTreg‐0 displayed significantly lower CD25 MFI than nTreg, iTreg‐10 and iTreg‐100 (Figure 2a; raw MFI for CD25 and FOXP3 shown in Supplementary figure 7; P = 0.0053, 0.0085 and 0.0051). Furthermore, iTreg‐1, iTreg‐10 and iTreg‐100 retained their suppressive function with no significant differences compared with nTreg. Tconv and iTreg‐0 exhibited significantly lower suppressive activities than nTreg, iTreg‐1, iTreg‐10 and iTreg‐100 (Figure 2b; 1:1 – Tconv: P = 0.0001, 0.0001, P < 0.0001 and 0.0001, and iTreg‐0: P = 0.0134, 0.0125, 0.0036 and 0.0055, 1:2 – Tconv: P < 0.0001, P = 0.0006, 0.0001 and 0.0003, iTreg‐0: P = 0.0012, 0.0375, 0.0098 and 0.0202, 1:4 – Tconv: P < 0.0001, P < 0.0099, 0.0046 and 0.0037, iTreg‐0: P = 0.0019, 0.1149, 0.0630 and 0.0525 and 1:8 – Tconv: P = 0.0356, 0.1588, 0.0427 and 0.0315). Notably, assessment of cell growth rates upon re‐stimulation revealed significantly higher fold expansion in iTreg‐10 and iTreg‐100 than in nTreg, Tconv and iTreg‐0 (iTreg‐10: P = 0.0303, 0.0005 and 0.0010, and iTreg‐100: P = 0.0042, 0.0001 and 0.0002). Moreover, cell growth rate of iTreg‐1 was significantly higher than Tconv and iTreg‐0 (Supplementary figure 8; P = 0.0051 and 0.0114).
Figure 2.
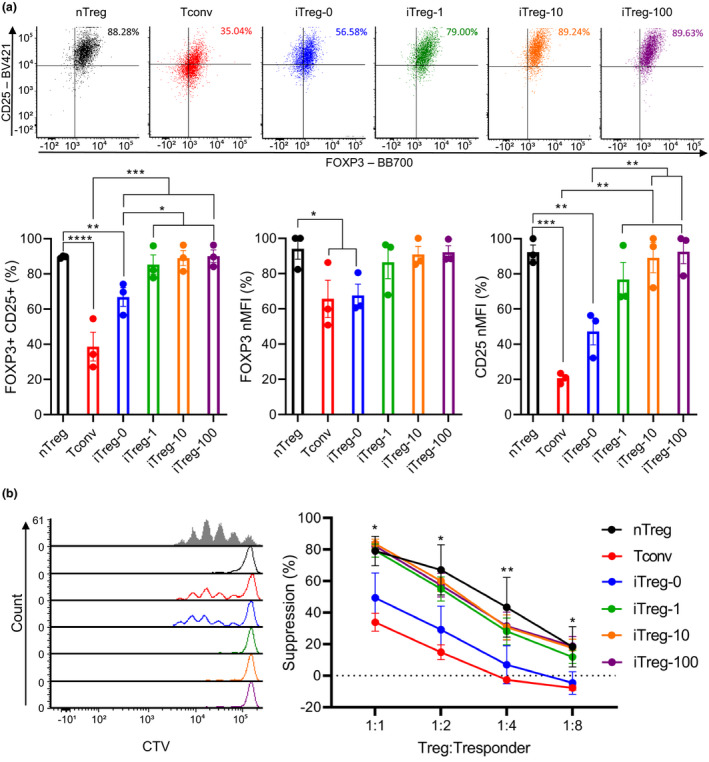
The impact of rapamycin concentrations on iTreg stability upon re‐stimulation. Following a 7‐day stimulation and a 3‐day rest, cells were re‐stimulated using a 1:1 α‐CD3/CD28 beads and IL‐2 without iTreg differentiation factors for 7 days then rested with IL‐2 for 3 days. After 3 days of rest, (a) expression of FOXP3 and CD25 (b) suppressive activities were evaluated. MFI of FOXP3 and CD25 were normalised to highest raw MFI value in each experiment and represented as nMFI (%). Raw MFI for (a) is shown in Supplementary figure 7. For (b), grey‐shaded histogram represents positive control (no Treg control). Data are represented as mean ± sem, N = 3 in three independent experiments. For each donor (N), technical triplicates were utilised, and the average of technical replicates was used for each datapoint. Statistical significance identified by RM one‐way (a) and RM two‐way (b) ANOVA with Dunnett’s multiple comparisons test: *P < 0.05, **0.01, ***0.001, ****0.0001.
Phenotype and function of iTregs generated with differing ratios of α‐CD3/CD28 beads
To investigate the impact of TCR stimulation bead ratios on human iTreg induction, iTregs were generated from naïve CD4+ T cells using TGF‐β, ATRA, 100 ng mL‐1 rapamycin, IL‐2 and differing ratios of α‐CD3/CD28 beads (1:1 and 1:10 bead to cell; termed iTreg‐100 as previous and iTreg‐1:10). As above, stimulated naïve nTreg and naïve CD4+ T cells were used as controls (nTreg and Tconv), and cells were stimulated and rested for 7 days each. After 3 days of rest, cells were assessed for their FOXP3 and CD25 expression and suppressive function. iTreg‐1:10 showed significantly lower %FOXP3+CD25+ than nTreg (91%), Tconv (85%) and iTreg‐100 (95%) at 54% (P = 0.0015, 0.0036 and 0.0009). This was accompanied with a significant difference in FOXP3 MFI between iTreg‐1:10 and nTreg (P = 0.0452). Additionally, Tconv showed significantly lower FOXP3 MFI than nTreg (P = 0.0422). CD25 MFI of iTreg‐100 was significantly higher than nTreg, Tconv and iTreg‐1:10 (Figure 3a; raw MFI for CD25 and FOXP3 shown in Supplementary figure 9; P = 0.0002, 0.0003 and P < 0.0001). The suppressive activities of Tconv were significantly lower than nTreg, iTreg‐100 and iTreg‐1:10 (Figure 3b; 1:2 – P = 0.0348, 0.0009 and 0.0493, and 1:4 – P = 0.0109, 0.0002 and 0.0479, and 1:8 – P = 0.04151, 0.0316 and 0.6079). After a 7‐day rest, expression of FOXP3 and CD25 were further assessed. %FOXP3+CD25+ of Tconv and iTreg‐1:10 were significantly lower than nTreg (86%) and iTreg‐100 (93%) at 38% and 48%, respectively (Tconv: P = 0.0027 and 0.0012, and iTreg‐1:10: P = 0.0090 and 0.0037). FOXP3 MFI of iTreg‐100 were significantly higher than nTreg and Tconv (P = 0.0436 and 0.0244). CD25 MFI of iTreg‐100 remained significantly higher than nTreg, Tconv and iTreg‐1:10 (P = 0.0174, 0.0002, and 0.0002), and nTreg exhibited significantly higher CD25 MFI than Tconv and iTreg‐1:10 (Figure 3c; raw MFI for CD25 and FOXP3 shown in Supplementary figure 10 P = 0.0039 for both). Additionally, evaluation of cell growth rates showed that fold expansion of iTreg‐1:10 is significantly lower than nTreg, Tconv and iTreg‐100 (P = 0.0064, 0.0010 and 0.0391). Cell growth rate of iTreg‐100 was significantly lower than in Tconv (Supplementary figure 11; P = 0.0179).
Figure 3.
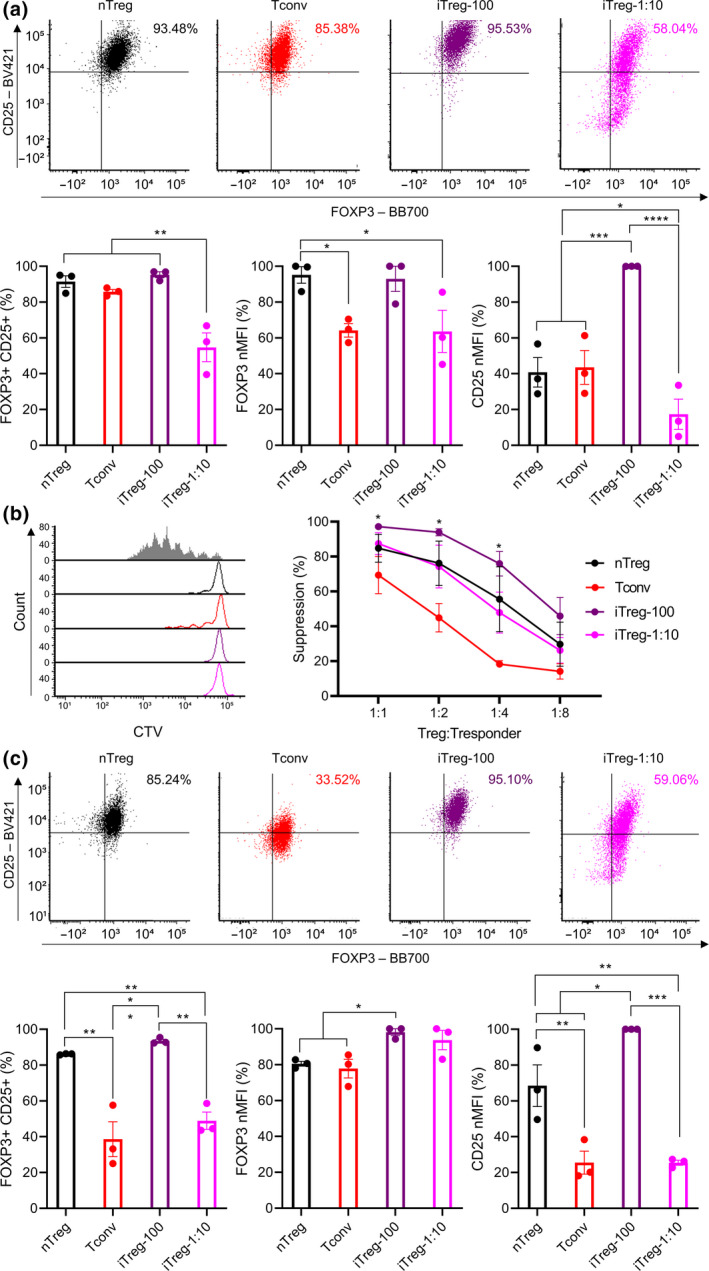
The impact of α‐CD3/C28 bead ratios on iTreg differentiation. Naïve nTregs were stimulated with a 1:1 α‐CD3/CD28 beads and IL‐2 (nTreg). Naïve CD4+ T cells were mock‐stimulated with a 1:1 α‐CD3/CD28 beads and IL‐2 (Tconv). iTregs were differentiated from naïve CD4+ T cells using IL‐2 TGF‐β, ATRA and 100 ng mL−1 rapamycin and various ratios of α‐CD3/CD28 beads (1:1 and 1:10 bead to cell; iTreg‐100 as before and iTreg‐1:10). Following a 7‐day stimulation, cells were rested up to 7 days with IL‐2. After 3 days of rest, (a) expression of FOXP3 and CD25 (b) suppressive activities were evaluated. After 7 days of rest, (c) expression of FOXP3 and CD25 was evaluated. MFI of FOXP3 and CD25 were normalised to highest raw MFI value in each experiment and represented as nMFI (%). Raw MFI for (a and c) is shown in Supplementary figures 9 and 10, respectively. For (b), grey‐shaded histogram represents positive control (no Treg control). Data are represented as mean ± sem, N = 3 in three independent experiments. For each donor (N), technical triplicates were utilised, and the average of technical replicates was used for each datapoint. Statistical significance identified by RM one‐way (a and c) and RM two‐way (b) ANOVA with Dunnett’s multiple comparisons test: *P < 0.05, **0.01, ***0.001, ****0.0001.
Following this up, the impact of TCR stimulation bead ratios on human iTreg stability upon in vitro re‐stimulation was assessed. As above, after initial stimulation and rest, cells were re‐stimulated using IL‐2 and α‐CD3/CD28 beads (1:1 bead to cell) for 7 days, then rested for 3 days in the presence of IL‐2. Upon re‐stimulation, FOXP3 and CD25 expression and suppressive activities of cells were evaluated. Significantly lower %FOXP3+CD25+ were observed in Tconv and iTreg‐1:10 than in nTreg (91%) and iTreg‐100 (91%) at 52% and 63%, respectively (Tconv: P = 0.0042 and 0.0039, and iTreg‐1:10: P = 0.0185 and 0.0168). While Tconv showed significantly lower FOXP3 MFI than nTreg and iTreg‐100 (P = 0.0078 and 0.0244), iTreg‐1:10 displayed no significant differences in FOXP3 MFI compared with nTreg and iTreg‐100. CD25 MFI showed similar trends to %FOXP3+CD25+, with Tconv and iTreg‐1:10 exhibiting significantly lower CD25 MFI than nTreg and iTreg‐100 (Figure 4a; raw MFI for CD25 and FOXP3 shown in Supplementary figure 12; Tconv: P = 0.0011 and 0.0003, and iTreg‐1:10: P = 0.0016 and 0.0004). Moreover, no significant differences were shown with suppressive activities of nTreg, iTreg‐100 and iTreg‐1:10, while Tconv displayed significantly lower suppressive activities compared with nTreg, iTreg‐100 and iTreg‐1:10 (Figure 1, 4, 1:1 – P = 0.0582, 0.0062 and 0.0167, 1:2 – P = 0.0009, 0.0002 and 0.0009, and 1:4 – P = 0.0010, 0.0030 and 0.0206). In addition, cell growth rates upon re‐stimulation were measured. Tconv exhibited significantly lower fold expansion compared with iTreg‐100 and iTreg‐1:10 (P = 0.0179 and 0.0117), and cell growth rate of nTreg was significantly lower than iTreg‐1:10 (Supplementary figure 13; P = 0.494).
Figure 4.
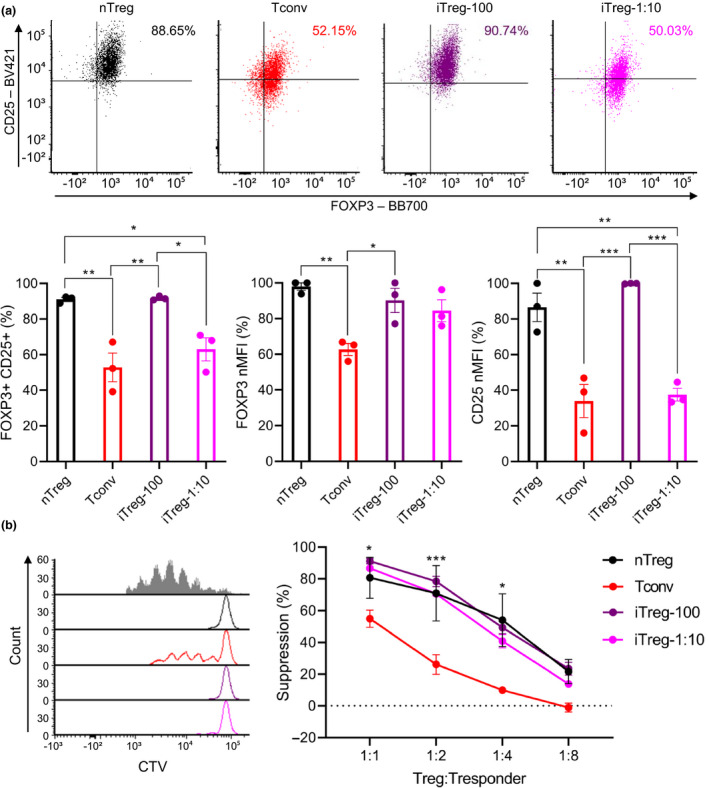
The impact of α‐CD3/C28 bead ratios on iTreg stability upon re‐stimulation. Following a 7‐day stimulation and a 3‐day rest, cells were re‐stimulated using a 1:1 α‐CD3/CD28 beads and IL‐2 without iTreg differentiation factors for 7 days then rested with IL‐2 for 3 days. After 3 days of rest, (a) expression of FOXP3 and CD25 (b) suppressive activities were evaluated. MFI of FOXP3 and CD25 were normalised to highest raw MFI value in each experiment and represented as nMFI (%). Raw MFI for (a) is shown in Supplementary figure 12. For (b), grey‐shaded histogram represents positive control (no Treg control). Data are represented as mean ± sem, N = 3 in three independent experiments. For each donor (N), technical triplicates were utilised, and the average of technical replicates was used for each datapoint. Statistical significance identified by RM one‐way (a) and RM two‐way (b) ANOVA with Dunnett’s multiple comparisons test: *P < 0.05, **0.01, ***0.001, ****0.0001.
Functional characterisation of iTregs generated with varied concentrations of rapamycin and differing ratios of α‐CD3/CD28 beads
To provide a broader understanding of iTregs generated under different conditions, their functionality was further characterised. After an initial 7‐day stimulation and 3‐day rest, expression levels of Treg signature molecules and CD4+ T cell effector cytokines were assessed by measuring CTLA4 and IL2 gene expression and production of active TGF‐β, IL‐4, IL‐6, IL‐10, TNF‐α, IFN‐γ and IL‐17A. 37 , 38 , 39 While no significant differences were observed in CTLA4 expression amongst cell types (Figure 5a), Tconv exhibited significantly higher IL2 expression compared with nTreg, iTreg‐1, iTreg‐10, iTreg‐100 and iTreg‐1:10 (Figure 5b; P = 0.0415, 0.0423, 0.0423, 0.0430 and 0.0445). No significant differences were shown in production of active TGF‐β (Figure 5c), IL‐4, IL‐6 and IL‐17A. Furthermore, IL‐10 production in nTreg was significantly higher than Tconv, iTreg‐0, iTreg‐1, iTreg‐10, iTreg‐100 and iTreg‐1:10 (P = 0.0401, 0.0489, 0.0493, 0.0400, 0.0391 and 0.0387). Tconv produced significantly more TNF‐α compared with nTreg, iTreg‐0, iTreg‐1, iTreg‐10, iTreg‐100 and iTreg‐1:10 (P < 0.0001, P = 0.0130, 0.0012, 0.0002, 0.0002 and 0.0192), and iTreg‐0 and iTreg‐1:10 produced significantly more TNF‐α compared with nTreg (P = 0.0102 and 0.0070). Lastly, IFN‐γ production in Tconv was significantly greater than nTreg, iTreg‐0, iTreg‐1, iTreg‐10, iTreg‐100 and iTreg‐1:10 (Figure 5d; P = 0.0017, 0.0056, 0.0021, 0.0017, 0.0017 and 0.0016).
Figure 5.
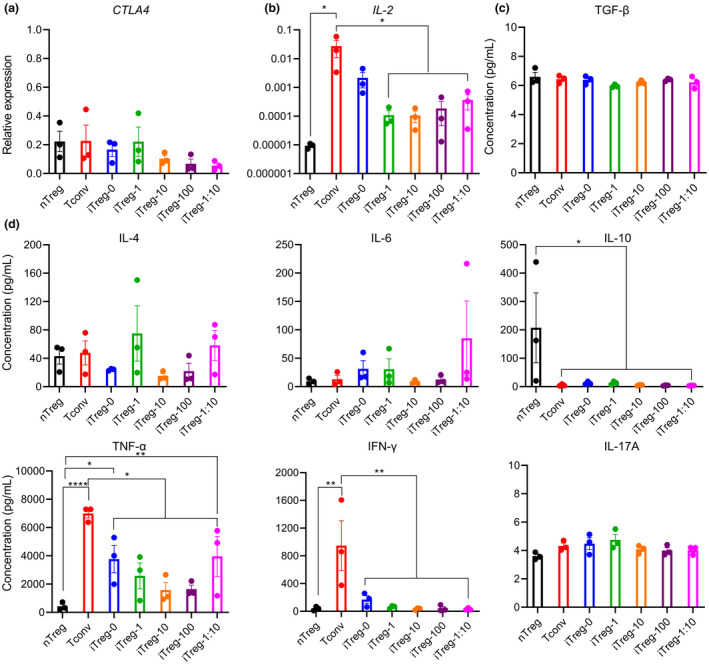
The impact of rapamycin concentrations and α‐CD3/C28 bead ratios on iTreg functionality. Following a 7‐day stimulation and a 3‐day rest, cells were stimulated using a 1:10 α‐CD3/CD28 beads and low‐dose IL‐2. After 3 days of stimulation, (a) CLTA4 gene expression, (b) IL2 gene expression, production of (c) active TGF‐β, (d) IL‐4, IL‐6, IL‐10, TNF‐α, IFN‐γ and IL‐17A were measured. For (c and d), the cells were stimulated with PMA and ionomycin for 6 hours prior to collection of cell supernatants. Cell‐free medium was used as a control to measure background cytokine levels in the medium which was supplemented with heat‐inactivated human serum. Background cytokine levels in the medium were negligible (below the lowest standards). Data are represented as mean ± sem, N = 3 in three independent experiments (a and b) or in one independent experiment (c and d). For each donor (N), technical triplicates (a and b) or technical duplicates (c and d) were utilised, and the average of technical replicates was used for each datapoint. Statistical significance identified by RM one‐way Dunnett’s multiple comparisons test: *P < 0.05, **0.01, ***0.001, ****0.0001.
Th17‐polarising challenge of iTregs generated with varied concentrations of rapamycin and differing ratios of TCR stimulation bead
It has previously been shown that iTregs can convert to pathogenic Th17 cells in a pro‐inflammatory environment, 40 , 41 and that challenging iTregs with Th17‐polarising cytokines can compromise their FOXP3 expression and protective abilities in vivo. 28 Therefore, the stability of iTregs upon Th17‐polarising challenge was investigated by culturing them for 3 days with Th17‐polarising cytokines IL‐1β, IL‐6, IL‐21 and IL‐23, 42 , 43 α‐CD3/CD28 beads (1:10 bead to cell) and low‐dose IL‐2, after initial 7‐day stimulation and 3‐day rest. Low‐dose IL‐2 was utilised to maintain cell viability with minimal stabilisation of FOXP3 expression and inhibition of Th17 polarisation during the challenge. 44 , 45 After the challenge, FOXP3 and CD25 expression, production of CD4+ T cell cytokines and gene expression of Th17 master regulator, RORC, 46 were assessed. While no significant differences were observed in %FOXP3+CD25+ and FOXP3 MFI for all cell types after the challenge, challenged nTreg, Tconv, iTreg‐0, iTreg‐1, iTreg‐10, iTreg‐100 and iTreg‐1:10 displayed significant increases in their CD25 MFI compared with ‘unchallenged’ cells (Figure 6a; raw MFI for CD25 and FOXP3 shown in Supplementary figure 14; P = 0.0439, 0.0239, 0.0318, 0.0040, 0.0370, 0.0228 and 0.0097). All cell types exhibited no significant differences in production of IL‐17A (Figure 6b). Additionally, no significant differences were shown with IL‐2, IL‐4, IL‐6, IL‐10 TNF‐α and IFN‐γ after the challenge (Supplementary figure 15). Notably, both unchallenged and challenged cells lacked RORC expression (Supplementary figure 16).
Figure 6.
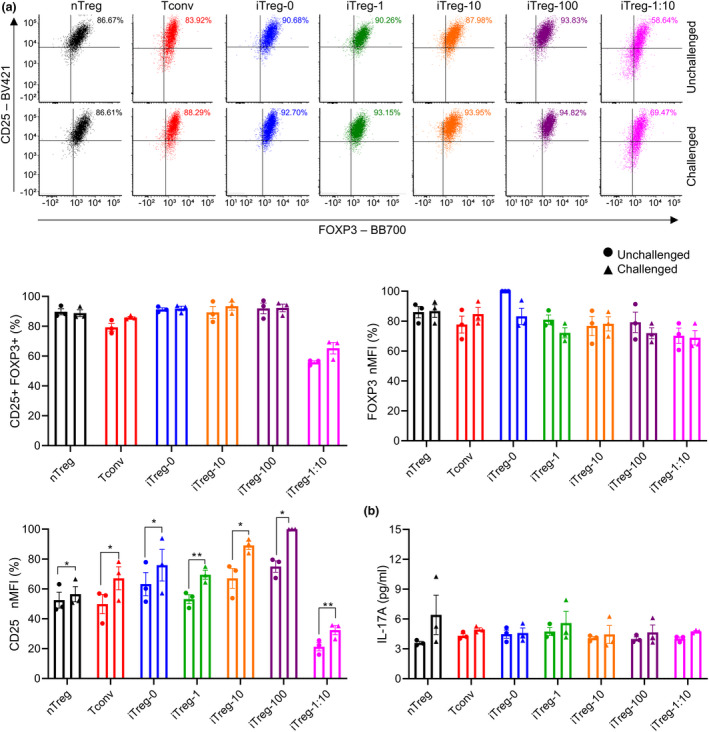
The impact of rapamycin concentrations and α‐CD3/C28 bead ratios on iTreg stability upon challenge with Th17‐polarising cytokines. Following a 7‐day stimulation and a 3‐day rest, cells were challenged using a 1:10 α‐CD3/CD28 beads, low‐dose IL‐2, IL‐1β, IL‐6, IL‐21 and IL‐23 for 3 days. Unchallenged controls were stimulated using a 1:10 α‐CD3/CD28 beads and low‐dose IL‐2 for 3 days. After 3 days of challenge, (a) expression of FOXP3 and CD25 and (b) production of IL‐17A were assessed. MFI of FOXP3 and CD25 were normalised to highest raw MFI value in each experiment and represented as nMFI (%). Raw MFI for (a) is shown in Supplementary figure 14. For (b), the cells were stimulated with PMA and ionomycin for 6 hours prior to collection of cell supernatants. Cell‐free medium was used as a control to measure background cytokine levels in the medium which was supplemented with heat inactivated human serum. Background cytokine levels in the medium were negligible (below the lowest standards). Data are represented as mean ± sem, N = 3 in three independent experiments (a) or in one independent experiment (b). For each donor (N), technical triplicates (a) or technical duplicates (b) were utilised, and the average of technical replicates was used for each datapoint. Statistical significance identified by a paired t‐test: *P < 0.05, **0.01, ***0.001, ****0.0001.
TSDR methylation status of iTreg generated with varied concentrations of rapamycin and differing ratios of α‐CD3/CD28 beads
The demethylation status of Treg‐specific demethylation region (TSDR) is an important indicator of stability of FOXP3 expression. 47 , 48 Methylation levels of 11 CpG motifs in the TSDR were measured via targeted bisulphite pyrosequencing. DNA was collected after an initial 7‐day stimulation and 3‐day rest. TSDR methylation levels of Tconv, iTreg‐0, iTreg‐1, iTreg‐10, iTreg‐100 and iTreg‐1:10 were above 90% in average with each CpG motifs showing various levels of methylation between 78% and 100%, while nTreg showed TSDR methylation level of 45% in average with each CpG motifs showing various levels of methylation between 40% and 67% (Figure 7).
Figure 7.
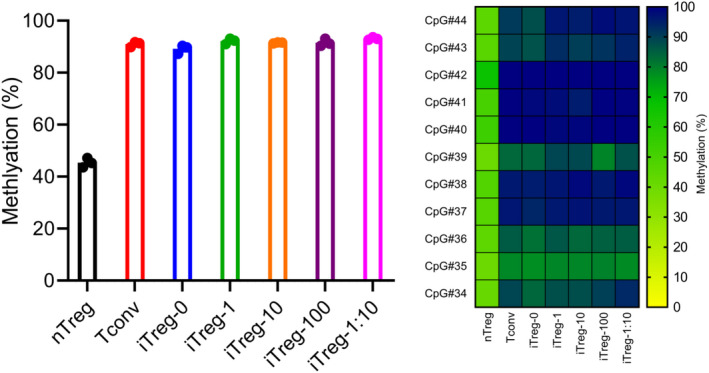
The impact of rapamycin concentrations and α‐CD3/C28 bead ratios on TSDR methylation status. Following a 7‐day stimulation and a 3‐day rest, cells were harvested for TSDR methylation assay. TSDR methylation levels are represented as average methylation level of 11 CpG locations for each donor and average methylation levels of individual CpG locations across the donors (heatmap). Data are represented as mean ± sem or mean, N = 3 in one independent experiment. No technical replicates were utilised. Each datapoint represent different donor.
Discussion
In this study, we optimised an iTreg differentiation method utilising TGF‐β, ATRA, rapamycin, IL‐2 and α‐CD3/CD28 beads to differentiate and expand human iTregs from naïve CD4+ T cells, which are 7‐ and 20‐fold more frequent in human peripheral blood compared with nTregs and naïve nTregs. (Supplementary figure 17). These molecules have previously been shown to induce or enhance iTreg generation. TGF‐β secreted by CD103+ intestinal dendritic cells (DCs) plays an essential role in the generation of pTregs in the gut by inducing binding of the transcription factors Smad2 and Smad3 to conserved non‐coding DNA sequence 1 (CNS1) region of FOXP3 locus, 14 , 15 which is crucial for FOXP3 induction in pTregs but not in nTregs. 49 Additionally, ATRA secreted by CD103+ intestinal DCs reinforces binding of Smad3 to CNS1 region through histone acetylation of Smad3‐binding region, preventing potential Th17 polarisation. 14 , 15 , 50 Furthermore, the mTOR (mammalian target of rapamycin)‐targeting drug, rapamycin, has been shown to induce FOXP3 expression, promote expansion and stabilise FOXP3 expression via inhibition of mTORC1 complex. 51 , 52 Rapamycin also enhances purity of generated iTregs by selectively inhibiting the activation of conventional T cells, 8 as Tregs utilise an IL‐2R‐dependent STAT5 pathway for activation while conventional T cells require the mTOR pathway for activation. 53 , 54 Lastly, IL‐2 promotes FOXP3 expression and Treg expansion, and inhibits Th17 polarisation. 15 , 55 The combination of TGF‐β, ATRA, rapamycin and IL‐2 has previously been used to successfully generate iTregs with superior in vitro suppressive activities. 26 , 27 However, these iTregs lost FOXP3 expression upon re‐stimulation and were then not able to prevent the onset of xenogeneic graft‐versus‐host disease in a humanised mouse model. 26
We demonstrated that any concentration of rapamycin in conjunction with 1:1 beads robustly generate highly suppressive iTregs that are phenotypically and functionally stable upon in vitro re‐stimulation, without compromising cell viability (Supplementary figure 18). While all three concentrations of rapamycin‐induced high expression of FOXP3 and CD25, medium‐ and high‐dose rapamycin resulted in elevated FOXP3 and CD25 expression after initial stimulation, compared with low‐dose rapamycin. Conversely, iTregs generated without rapamycin were not as suppressive as nTregs or rapamycin‐induced iTregs despite having comparable FOXP3 and CD25 expression to nTregs. These iTregs lost FOXP3 and CD25 expression upon re‐stimulation. This demonstrated the importance of rapamycin for suppressor function and stability upon in vitro re‐stimulation. Interestingly, with 1:10 beads, FOXP3 and CD25 expression of rapamycin‐induced iTregs were not comparable to nTregs and yet were highly suppressive after initial stimulation and upon re‐stimulation. This was in line with a previously published protocol utilising the combination of TGF‐β, ATRA, high‐dose rapamycin and 1:10 beads, even though the authors re‐sorted CD25+FOXP3+ iTregs prior to their functional analysis. 27 Moreover, with 1:10 beads, extremely limited cell growth was observed, which together indicated cells have limited accessibility to beads when 1:10 beads are utilised. iTregs generated with 1:10 beads showed stabilisation of FOXP3 expression similar to nTregs after a 7‐day rest and upon re‐stimulation, while their CD25 expression remained low compared with nTregs and rapamycin‐induced iTregs with 1:1 beads. Together, these suggested that using a high ratio of α‐CD3/CD28 beads is beneficial for generation of iTregs. In addition, while we compared different ratios of α‐CD3/CD28 beads, direct comparison to other forms of TCR stimulation, such as plate‐bound α‐CD3 and soluble α‐CD28, 24 , 26 would provide valuable insights, as different forms would present the cells with varying TCR stimulation intensity and density. Indeed, the importance of TCR signal intensity on iTreg generation has been extensively investigated. While some have demonstrated that sub‐optimal TCR stimulation, which results in low AKT activation, favors FOXP3 induction and iTreg generation, 56 , 57 , 58 it has also been shown that low doses of strong TCR stimulation induce stable and persistent iTregs. 59 Given this controversy, it has been proposed that the ‘Goldilocks principle’, which has been associated with development of tTregs, 60 could apply to iTregs, requiring a fine balance in TCR stimulation with the stimulation being ‘just right’. 61 , 62 Furthermore, while CD28 co‐stimulation is necessary for the development of tTreg, it has been shown to promote mTOR activation and inhibit differentiation of naïve T cells into iTregs. 62 , 63 , 64 Thus, it is possible that rapamycin counteracts activation of AKT/mTOR pathways driven by α‐CD3 and α‐CD28 on the beads, 26 ultimately modulating TCR stimulation intensity to favor iTreg generation, which could explain why iTregs generated with medium‐ and high‐dose rapamycin displayed reduced cell growth.
Notably, with 1:1 α‐CD3/CD28 beads, even mock‐stimulated Tconvs highly expressed FOXP3 and CD25, which were only lost upon in vitro re‐stimulation. In contrast, previously published protocols utilising 1:10 α‐CD3/CD28 beads and plate‐bound α‐CD3 and soluble α‐CD28 showed moderate expression of FOXP3 in mock‐stimulated Tconvs. Evidently, it has been shown that stimulation‐induced FOXP3 expression in Tconvs is dependent on the amount of stimulation used. In particular, almost 100% of Tconvs exhibited FOXP3 expression similar to nTregs under 1:1 α‐CD3/CD28 beads, while less than 40% of Tconvs expressed FOXP3 under 1:16 α‐CD3/CD28 beads or plate‐bound α‐CD3 and soluble α‐CD28. 65 In addition, mock‐stimulated Tconvs exhibited moderate suppressive activities, which was in line with murine Tconvs acquiring potent suppressor function upon activation. 66 Dose‐dependent suppression shown with mock‐stimulated Tconvs was comparable to iTregs generated without rapamycin at some ratios, which was shown with a previously published protocol. 26 Of note, in this protocol, mock‐stimulated Tconvs and iTregs generated without rapamycin displayed substantial differences in their FOXP3 expression, which suggests that suppressive capacity in these cells may be unspecific suppression instead of being induced by FOXP3 expression. 26 This highlights the need for assessment of phenotype and function upon in vitro re‐stimulation for therapeutic applications of Treg products, given the used stimulation can induce non‐Treg‐like and non‐stable FOXP3. Furthermore, mock‐stimulated Tconvs and ‘no rapamycin’ iTregs displayed limited cell growth upon re‐stimulation. This suggests that perhaps 1:1 α‐CD3/CD28 beads over‐stimulated these cells, inducing T‐cell exhaustion, 67 while rapamycin‐induced iTregs were able to expand upon re‐stimulation, potentially as a result of rapamycin dampening TCR‐driven exhaustion. 26
Tregs have versatile modes of suppression via expression of various suppressive molecules 37 and also are able to produce T helper effector cytokines. 68 Interestingly, expression levels of canonical Treg suppressive molecules in cell types – no differences observed in active TGF‐β production, higher IL‐10 production in nTregs, and differences shown in CD25 expression – did not correlate with their suppressive activities. Of note, while no differences were shown in CTLA4 gene expression, CTLA4 functionality is largely controlled by externalisation of CTLA4 proteins to the cell surface, 69 , 70 thus this did not indicate much. In addition, while it has been previously shown that expression levels of FOXP3 can be directly correlated with suppressive activity, 71 , 72 FOXP3 expression levels after initial stimulation were not indicative of their suppressive activities. Furthermore, rapamycin‐induced iTregs with 1:1 α‐CD3/CD28 beads were most comparable to nTregs in terms of their T helper effector cytokine production profile. Expectedly, mock‐stimulated Tconvs displayed most discrepancies, showing high expression of IL2 and producing more pro‐inflammatory cytokines such as TNF‐α and IFN‐γ. While nTregs and rapamycin‐induced iTregs exhibited repressed IL2 expression in line with nTregs not being able to produce IL‐2 for autocrine signalling, 73 iTregs generated without rapamycin displayed comparable IL2 expression to mock‐stimulated Tconvs. In addition, TNF‐α production by iTregs generated without rapamycin and rapamycin‐induced iTregs with 1:10 α‐CD3/CD28 beads were comparable to mock‐stimulated Tconvs. These reinforced the importance of rapamycin and high ratio of α‐CD3/CD28 beads for iTreg differentiation. Notably, while TNF‐α production in rapamycin‐induced iTregs with 1:1 α‐CD3/CD28 beads was substantially lower than mock‐stimulated Tconvs, they still produced a considerable amount of TNF‐α. Considering the pro‐inflammatory characteristics of TNF‐α, 74 this could affect in vivo functionality of these cells, despite showing no differences in suppression of responder T‐cell proliferation compared with nTregs in vitro. Indeed, suppression of cytokine production in responder T cell is another aspect of Treg‐mediated suppression, 75 , 76 , 77 which may be altered by TNF‐α. Overall, in‐depth analysis of transcriptomes and proteomes may be required to fully understand which factors and genes are attributing to discrepancies in the suppressor function and the phenotype of these cells. Indeed, recent transcriptomic and proteomic profiling of iTregs generated by multiple differentiation protocols revealed iTreg‐specific molecular pathways and molecules, 78 even though these protocols were not able to generate stable iTregs.
The tendency of iTregs to switch to a pro‐inflammatory phenotype, such as Th17, in a pro‐inflammatory microenvironment, needed to be assessed, as conversion of iTregs into pathogenic pro‐inflammatory T cells in vivo is the major challenge to the use of iTregs for therapeutic purposes. 40 , 41 , 79 In addition, Tregs can be polarised to Th17‐like Tregs, which express Th17 signature markers while retaining regulatory phenotype and function, under pro‐inflammatory conditions. 80 , 81 Given that IL‐1β, IL‐6, IL‐21 and IL‐23 are known promoters of Th17 polarisation, 42 , 43 mock‐stimulated Tconvs were expected to display Th17 phenotypes upon challenge. Unexpectedly, all cell types including mock‐stimulated Tconvs exhibited no differences in their FOXP3 expression, T helper effector cytokine production and RORC gene expression upon challenge with Th17‐polarising cytokines, while showing elevated CD25 expression. In particular, negligible amounts of IL‐17A were produced, and RORC gene expression was undetectable in all cell types with and without challenge, possibly as a result of high expression of FOXP3 in these cells. While, this is in line with TGF‐β/ATRA‐induced human iTregs showing increased expression of Treg suppressive molecules, PD‐1 and GITR, upon treatment with IL‐1β and IL‐6 28 it is difficult to confirm iTreg stability in a pro‐inflammatory environment without a positive control which expresses Th17 markers upon challenge. Thus, a more physiological environment may be required to validate iTreg stability under pro‐inflammatory conditions. Indeed, humanised graft‐versus‐host disease (GVHD), skin transplantation and islet transplantation models which present in vivo reactivation and pro‐inflammatory milieu have been utilised to assess Tregs. 26 , 28 , 29 , 32 , 82 , 83 In conjunction, the safety and effectiveness of iTregs can be corroborated through these humanised models as well, to ensure clinical relevance of this iTreg differentiation protocol.
Currently, demethylation of Treg‐specific demethylation region (TSDR), the CpG‐rich CNS2 region of FOXP3 locus, is thought to be the hallmark of stable FOXP3 expression, thus stable Treg phenotype. 14 , 84 , 85 In humans, it has been shown that nTregs exhibit full demethylation of TSDR, while Tconvs present fully methylated TSDR. 48 Even though rapamycin‐induced iTregs with 1:1 α‐CD3/CD28 beads demonstrated phenotypic and functional stability upon in vitro re‐stimulation, demethylation of TSDR in these cells was not observed. Mock‐stimulated Tconvs, ‘no rapamycin’ iTregs and rapamycin‐induced iTregs with 1:10 α‐CD3/CD28 beads also showed no demethylation of TSDR. Thus, this iTreg differentiation protocol could be modified to induce TSDR demethylation. Indeed, hypoxia and vitamin C were shown to enhance expression and activity of TET (ten eleven translocation) enzymes and facilitate demethylation of the TSDR in mice and human. 86 , 87 , 88 , 89 Interestingly, nTregs analysed for methylation status only displayed partial demethylation. As a result of the anonymity of donated buffy coats, we were not able to confirm the gender of the donors, which is crucial for evaluation of TSDR data, as FOXP3 is located on the X chromosome resulting in partial demethylation of TSDR in female caused by X chromosome inactivation. 90 , 91 Indeed, given the low inter‐donor variation in TSDR demethylation rates of nTregs and similarity of these rates to published data, 90 it is possible that all donors were females.
In summary, we demonstrated the importance of rapamycin and high ratio of α‐CD3/CD28 beads for iTreg differentiation and stability. The combination of TGF‐β, ATRA, any concentration of rapamycin, IL‐2 and 1:1 α‐CD3/CD28 beads generated iTregs which are superior to iTregs generated without rapamycin and iTregs generated with 1:10 α‐CD3/CD28 beads. Notably, medium‐dose and high‐dose rapamycin‐induced superior expression of CD25 compared with low‐dose rapamycin, without differences in stability and function. These iTregs were highly suppressive, and stable upon in vitro re‐stimulation. Expression of Treg signature molecules and T helper effector cytokines in these iTregs was largely comparable to nTregs. Furthermore, these iTregs were stable in the presence of Th17‐polarising cytokines with no differences in FOXP3 expression, T helper effector cytokine production and RORC expression; however, further validation in humanised mice models will be required as these results were also observed in mock‐stimulated Tconvs. Despite their stability upon in vitro re‐stimulation, demethylation of TSDR was not shown for these iTregs. While TSDR demethylation has been thought to be a surrogate for Treg stability, there might be further factors involved in stabilisation of Treg phenotype. Indeed, it has been shown that demethylation of other Treg signature genes such as TNFRSF18, CTLA‐4, IKZF4 and IL2RA, dubbed as Treg‐specific demethylation patterns (‘TSDP’), is crucial for Treg development and stable FOXP3 expression, as well as TSDR demethylation. 92
Methods
Cell isolation and stimulation
Human buffy coat (Australian Red Cross) was treated with a RossetteSep Human CD4+ T‐cell enrichment cocktail (STEMCELL Technologies, Vancouver, Canada) for 20 min on a platform mixer at 80 rpm. Treated buffy coat was diluted with PBS (+2% foetal calf serum (FCS), Bovogen, Keilor East, Victoria, Australia) prior to isolation of CD4+ T cells by density‐gradient centrifugation over Lymphoprep (STEMCELL Technologies). Enriched CD4+ T cells were surface‐stained for CD4‐APC‐H7 (SK3), CD25‐PE‐Cy7 (M‐A251), CD127‐FITC (HIL‐7R‐M21) and CD45RA‐PE (HI100; all BD Biosciences, San Jose, CA, USA). CD4+ CD25+ CD127‐ CD45RA+ T cells (naïve natural regulatory T cells or naïve nTregs) and CD4+ CD25‐ CD45RA+ T cells (naïve CD4+ T cells) were sorted by fluorescence‐activated cell sorting (FACS; BD FACSAria Fusion, BD Biosciences). Sorted naïve nTregs and naïve CD4+ T cells were rested overnight in a complete X‐vivo medium at 1 × 106 cells mL−1 (X‐vivo: serum‐free with gentamycin and phenol red, Lonza, Basel, Switzerland). Complete X‐vivo medium (cX‐vivo) was always supplemented with 2% HEPES (Gibco, Carlsbad, CA, USA), 1% L‐glutamine (Hyclone, Logan, UT, USA) and 5% human serum (Heat inactivated; Sigma‐Aldrich, St. Louis, MO, USA). 500 U mL−1 of IL‐2 (Novartis Vaccines and Diagnostics, Cambridge, MA, USA) was added to complete X‐vivo medium unless otherwise stated. After overnight resting, nTregs were stimulated with 500 U mL−1 of IL‐2 and a 1:1 ratio of Human T‐expander CD3/CD28 Dynabeads™ (Gibco) in cX‐Vivo at 1 × 106 cells mL−1 (nTreg). Naïve CD4+ T cells were stimulated with 500 U mL−1 of IL‐2 and a 1:1 ratio of expander beads in cX‐Vivo containing (1) no other factors (mock stimulation; Tconv) or (2) 5 ng mL−1 of human TGF‐β (eBioscience, San Diego, CA, USA), 10 nM of all‐trans retinoic acid (ATRA; Sigma‐Aldrich), and varying concentrations of rapamycin (0, 1, 10 and 100 ng mL−1; iTreg‐0, iTreg‐1, iTreg‐10 and iTreg‐100, respectively; LC Laboratories, Woburn, MA, USA) at 1 × 106 cells mL−1. Naïve CD4+ T cells were also stimulated with 500 U mL−1 of IL‐2 and a 1:10 ratio of expander beads in cX‐Vivo containing 5 ng mL−1 of human TGF‐β, 10 nM of ATRA and 100 ng mL−1 of rapamycin (iTreg‐1:10). Stimulation was carried out for 7 days. Media was replenished on day 3 and 5 to keep the cell density at 1 × 106 cells mL−1. On day 7 of stimulation, cells were washed three times with PBS (+2% FCS). After washing, expander beads were magnetically removed. Cells were re‐suspended in cX‐Vivo with 500 U mL−1 of IL‐2 at 2 × 106 cells mL−1 and rested up to 7 days. On day 3 of rest, cells were used for flow cytometric analysis, in vitro suppression assay and TSDR methylation assay. On day 7 of rest, cells were used for flow cytometric analysis. Cell cultures were carried out using flat bottom well plates (6, 12, 24 and 48 wells depending on total cell numbers) and culture flasks (T25, T75 and T175 depending on total cell numbers). Within an experiment, culturing formats were usually equivalent.
iTreg stability evaluation
For evaluation of iTreg stability, cells were (1) re‐stimulated without iTreg differentiation components and (2) challenged with Th17‐polarising cytokines. For (1), cells were re‐stimulated using 500 U mL−1 of IL‐2 and a 1:1 expander beads without addition of TGF‐β, ATRA and rapamycin, for 7 days at 1 × 106 cells mL−1 then rested with 500 U mL−1 of IL‐2 for 3 days at 2 × 106 cells mL−1, after an initial 7‐day stimulation and 3‐day rest. On day 3 of rest, cells were used for flow cytometric analysis and in vitro suppression assay. For (2), cells were challenged for 3 days with Th17‐polarising cytokines, IL‐1β, IL‐6, IL‐21 and IL‐23 (10 ng mL−1 for all, Biolegend, San Diego, CA, USA), in the presence of 25 U mL−1 of IL‐2 and a 1:10 expander beads at 1 × 106 cells mL‐1, after an initial 7‐day stimulation and 3‐day rest. Cells were stimulated with 25 U mL−1 of IL‐2 and a 1:10 expander beads at 1 × 106 cells mL−1 as unchallenged controls. On day 3 of challenge, cells were washed three times with PBS (+2% FCS). After washing, expander beads were magnetically removed. Cells were used for flow cytometric analysis, cytokine production assays and reverse transcription quantitative polymerase chain reaction.
Flow cytometric analysis for CD25, FOXP3 and viability
1 × 105 cells were stained for viability (Fixable Viability Stain 780, FVS780; BD Biosciences) and CD4‐FITC (OKT4, BD Biosciences) and CD25‐BV421 (M‐A251, BD Biosciences). Cells were fixed and permeabilised (Foxp3/Transcription factor staining buffer set, eBioscience) then intracellularly stained for FOXP3‐BB700 (236A/E7, BD Biosciences). Compensation controls were utilised and freshly isolated naïve CD4+ T cells were used for as a control. All samples were analysed on a BD FACS Canto II flow cytometer (BD Biosciences), and the data analysed with FCS Express 6 (De Novo Software, Pasadena, CA, USA).
In vitro suppression assay
Human peripheral blood mononuclear cells (PBMCs) cells were isolated from a fresh buffy coat by density‐gradient centrifugation as described above. Naïve CD4+ T cells were isolated using EasySep™ Human Naïve CD4 + T Cell Isolation kit (STEMCELL Technologies). Isolated naïve CD4+ T cells were allogeneic to the Tregs. Naïve CD4+ T cells were labelled with Cell Trace Violet (CTV; Thermo Fisher Scientific, Waltham, MA, USA) as per manufacture’s protocol. 96‐well round bottom plates were seeded with CTV‐labelled naïve CD4+ T cells (5 × 104 cells per well; ‘Tresponder’), and Tregs were added to the wells at various ratios of Treg:Tresponder (1:1, 1:2, 1:4 and 1:8). Human T‐expander CD3/CD28 Dynabeads™ (bead:Tresponder ratio of 1:5) was added to each well. CTV‐labelled naïve CD4+ T cells with and without beads were used as positive and negative controls, respectively. Naïve CD4+ T cells without CTV labelling were used as unstained control. Cell density control was prepared by mixing positive and negative controls at 1:1 ratio. The plate was incubated at 37 ˚C in 5% CO2 for 5 days. IL‐2 free cX‐vivo medium was used for suppression assay. After a 5‐day incubation, cells were stained with FVS780 then analysed by flow cytometry (FACS Canto II). Percentage proliferation was measured by CTV dilutions. Percentage suppression was calculated as follows: 100 × [1 − (percentage proliferation in the experiment sample divided by percentage proliferation in the positive control)].
Cytokine production assays
Cells were washed three times then re‐suspended in cX‐vivo supplemented with cell stimulation cocktail (2 µL mL−1 final concentration; PMA and ionomycin; eBioscience). Cells were incubated at 37 ˚C in 5% CO2 for 6 h. After 6‐h incubation, cells were spun down and supernatants were collected. Supernatants were stored at − 80 ˚C until assays. Cytokine production was analysed using Human Th1/Th2/Th17 cytometric bead array (CBA) kit (BD Biosciences) and LEGENDplex™ Human Free Active/Total TGF‐β1 Assay kit (Biolegend, San Diego, California, USA), as per manufacture’s protocols. Cell‐free cX‐vivo medium was used as a control to measure background cytokine levels as a result of human serum used to supplement the medium. Samples were analysed by flow cytometry (FACS Canto II), and the data were analysed by FCAP Array software (BD Biosciences) and LEGENDplex™ Data Analysis software (Biolegend). Human Th1/Th2/Th17 CBA kit measured IL‐2; however, these data were excluded as a result of carry‐over from recombinant IL‐2 added to the medium.
Reverse transcription quantitative polymerase chain reaction
5 × 105 cells were pelleted, and supernatants removed. Cell pellets were lysed, and RNA was isolated using RNAqueous™ Total RNA Isolation Kit (Ambion, Austin, TX, USA) as per manufacture’s protocol. Purified RNA was converted to cDNA using iScript Reverse Transcription Supermix (Bio‐Rad, Hercules, CA, USA) as per manufacture’s protocol. cDNA was then used for measurement of gene expression via quantitative polymerase chain reaction on a CFX Connect Real‐Time PCR Detection System (Bio‐Rad) using TaqMan™ Gene Expression Master Mix (Applied Biosystems, Foster City, CA, USA) and TaqMan primers for RORC (Hs01076112_m1), CTLA4 (Hs00175480_m1), IL2 (Hs00174114_m1) and RPL13A (Hs04194366_g1; Housekeeping gene; Applied Biosystems for all), as per manufacture’s protocol.
TSDR methylation assay
1 × 106 cells were spun down, and all supernatants were discarded, leaving cell pellets. Cell pellets were stored at − 80 ˚C until assay. Assay was conducted by EpigenDx (Hopkinton, MA, USA) using the assay ADS783‐FS1 and ADS783‐FS2 (Ensembl Transcript ID: ENST00000376207) which assessed the methylation status of 11 CpG sites in the TSDR region of FOXP3 CNS2 by targeted bisulphite pyrosequencing of genomic DNA isolated from the cell pellets. This region covered CpG sites −2376 (CpG#44), −2371 (CpG#43), −2330 (CpG#42), −2322 (CpG#41), −2312 (CpG#40), −2309 (CpG#39), −2303 (CpG#38), −2299 (CpG#37), −2291 (CpG#36), −2282 (CpG#35) and −2263 (CpG#34) relative to the FOXP3 ATG start codon. Internal low (3.2%), medium (63.3%) and high (91.7%) methylation controls were utilised.
Statistics
Statistical significance (P < 0.05) was analysed using the GraphPad Prism 8 (San Diego, CA, USA). RM one‐way ANOVA with Dunnett’s multiple comparisons test, RM two‐way ANOVA with Dunnuett’s multiple comparisons test and the paired two‐tailed t‐test were used to identify statistical significance. N numbers indicate biological replicates (separate buffy coats). Within each biological replicate, technical triplicates were utilised, except for CBA and Legendplex which used duplicates and TSDR methylation assay which used no technical replicates. For each data point, average value of technical replicates was used.
Author Contributions
Juewan Kim: Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Project administration; Visualization; Writing‐original draft. Christopher M Hope: Conceptualization; Methodology; Supervision; Writing‐review & editing. Griffith Perkins: Investigation; Writing‐review & editing. Sebastian Stead: Investigation; Visualization; Writing‐review & editing. Jacqueline Scaffidi: Investigation. Francis Kette: Investigation. Robert Carroll: Supervision; Writing‐review & editing. Simon C Barry: Conceptualization; Supervision; Writing‐review & editing. Patrick Toby Coates: Conceptualization; Funding acquisition; Supervision; Writing‐review & editing.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
Acknowledgments
The authors acknowledge funding from The Hospital Research Foundation, Adelaide Australia, support of Australian Red Cross Blood Services for providing human buffy coat for cell isolation. JK acknowledges Dr Randall Grose of South Australian Health and Medical Research Institute for operating fluorescence‐activated cell sorter.
REFERENCES
- 1. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell 2008; 133: 775–787. [DOI] [PubMed] [Google Scholar]
- 2. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self‐tolerance is maintained by activated T cells expressing IL‐2 Receptor Alpha‐Chains (CD25). Breakdown of a single mechanism of self‐tolerance causes various autoimmune diseases. J Immunol 1995; 155: 1151–1164. [PubMed] [Google Scholar]
- 3. Baecher‐Allan C, Brown JA, Freeman GJ, Hafler DA. CD4+CD25high regulatory cells in human peripheral blood. J Immunol 2001; 167: 1245–1253. [DOI] [PubMed] [Google Scholar]
- 4. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003; 299: 1057–1061. [PubMed] [Google Scholar]
- 5. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol 2003; 4: 330–336. [DOI] [PubMed] [Google Scholar]
- 6. Liu W, Putnam AL, Xu‐Yu Z et al CD127 expression inversely correlates with FoxP3 and suppressive function of human CD4+ T reg cells. J Exp Med 2006; 203: 1701–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Seddiki N, Santner‐Nanan B, Martinson J et al Expression of interleukin (IL)‐2 and IL‐7 receptors discriminates between human regulatory and activated T cells. J Exp Med 2006; 203: 1693–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Singer BD, King LS, Alessio FRD. Regulatory T cells as immunotherapy. Front Immunol 2014; 5: 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bettini ML, Vignali DAA. Development of thymically derived natural regulatory T cells. Ann N Y Acad Sci 2010; 1183: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pohar J, Simon Q, Fillatreau S. Antigen‐specificity in the thymic development and peripheral activity of CD4+FOXP3+ T regulatory cells. Front Immunol 2018; 9: 1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yadav M, Stephan S, Bluestone JA. Peripherally induced Tregs‐role in immune homeostasis and autoimmunity. Front Immunol 2013; 4: 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nokta MA, Li X, Nichols J, Pou A, Asmuth D, Pollard RB. Homeostasis of naive and memory T cell subpopulations in peripheral blood and lymphoid tissues in the context of human immunodeficiency virus infection. J Infect Dis 2002; 183: 1336–1342. [DOI] [PubMed] [Google Scholar]
- 13. Hannet I, Erkeller‐Yuksel F, Lydyard P, Deneys V, DeBruyère M. Developmental and maturational changes in human blood lymphocyte subpopulations. Immunol Today 1992; 13: 215–218. [DOI] [PubMed] [Google Scholar]
- 14. Kanamori M, Nakatsukasa H, Okada M, Lu Q, Yoshimura A. Induced regulatory T cells : their development, stability, and applications. Trends Immunol 2016; 37: 803–811. [DOI] [PubMed] [Google Scholar]
- 15. Schmitt EG, Williams CB. Generation and function of induced regulatory T cells. Front Immunol 2013; 4: 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fuchs A, Gliwinski M, Grageda N et al Minimum information about T regulatory cells: a step toward reproducibility and standardization. Front Immunol 2018; 8: 1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Romano M, Fanelli G, Albany CJ, Giganti G, Lombardi G. Past, present, and future of regulatory T cell therapy in transplantation and autoimmunity. Front Immunol 2019; 10: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Safinia N, Lombardi G. Regulatory T cells:serious contenders in the promise for immunological tolerance in transplantation. Front Immunol 2015; 6: 438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Relland LM, Williams JB, Relland GN et al The TCR repertoires of regulatory and conventional T cells specific for the same foreign antigen are distinct. J Immunol 2012; 189: 3566–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Russler‐Germain EV, Rengarajan S, Hsieh CS. Antigen‐specific regulatory T‐cell responses to intestinal microbiota. Mucosal Immunol 2017; 10: 1375–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Luu M, Steinhoff U, Visekruna A. Functional heterogeneity of gut‐resident regulatory T cells. Clin Transl Immunol 2017; 6: e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Furusawa Y, Obata Y, Fukuda S et al Commensal microbe‐derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013; 504: 446–450. [DOI] [PubMed] [Google Scholar]
- 23. Coombes JL, Siddiqui KRR, Arancibia‐Cárcamo CV et al A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF‐β ‐and retinoic acid‐dependent mechanism. J Exp Med 2007; 204: 1757–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang J, Huizinga TWJ, Toes REM. De Novo generation and enhanced suppression of human CD4+ CD25+ regulatory T cells by retinoic acid. J Immunol 2009; 183: 4119–4126. [DOI] [PubMed] [Google Scholar]
- 25. Rossetti M, Spreafico R, Saidin S et al Ex Vivo‐expanded but not in vitro‐induced human regulatory T cells are candidates for cell therapy in autoimmune diseases thanks to stable demethylation of the FOXP3 regulatory T cell‐specific demethylated region. J Immunol 2015; 194: 113–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schmidt A, Eriksson M, Shang MM, Weyd H, Tegnér J. Comparative analysis of protocols to induce human CD4+Foxp3+ regulatory T cells by combinations of IL‐2, TGF‐beta, retinoic acid, rapamycin and butyrate. PLoS One 2016; 11: 1–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Candia E, Reyes P, Covian C et al Single and combined effect of retinoic acid and rapamycin modulate the generation, activity and homing potential of induced human regulatory T cells. PLoS One 2017; 12: e0182009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lu L, Zhou X, Wang J, Zheng SG, Horwitz DA. Characterization of protective human CD4+CD25+FOXP3+ regulatory T cells generated with IL‐2. TGF‐β and retinoic acid. PLoS One 2010; 5: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Qian X, Wang K, Wang X, Zheng SG, Lu L. Generation of human regulatory T cells de novo with suppressive function prevent xenogeneic graft versus host disease. Int Immunopharmacol 2011; 11: 630–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zanin‐Zhorov A, Kumari S, Hippen KL et al Human in vitro‐induced regulatory T cells display Dlgh1dependent and PKC‐θ restrained suppressive activity. Sci Rep 2017; 7: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Riquelme P, Haarer J, Kammler A et al TIGIT+ iTregs elicited by human regulatory macrophages control T cell immunity. Nat Commun 2018; 9: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hippen KL, Merkel SC, Schirm DK et al Generation and large‐scale expansion of human inducible regulatory T cells that suppress graft‐versus‐host disease. Am J Transplant 2011; 11: 1148–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lu Y, Wang J, Gu J et al Rapamycin regulates iTreg function through CD39 and Runx1 pathways. J Immunol Res 2014; 2014: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Asanuma S, Tanaka J, Sugita J et al Expansion of CD4+CD25+ regulatory T cells from cord blood CD4+ cells using the common γ‐chain cytokines (IL‐2 and IL‐15) and rapamycin. Ann Hematol 2011; 90: 617–624. [DOI] [PubMed] [Google Scholar]
- 35. Shan J, Feng L, Li Y, Sun G, Chen X, Chen P. The effects of rapamycin on regulatory T cells: Its potential time‐dependent role in inducing transplant tolerance. Immunol Lett 2015; 162: 74–86. [DOI] [PubMed] [Google Scholar]
- 36. Long SA, Buckner JH. Combination of rapamycin and IL‐2 increases de novo induction of human CD4+CD25+FOXP3+ T cells. J Autoimmun 2008; 30: 293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vignali DAA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol 2008; 8: 523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zubiaga AM, Munoz E, Merrow M, Huber BT. Regulation of interleukin 6 production in T helper cells. Int Immunol 1990; 2: 1047–1054. [DOI] [PubMed] [Google Scholar]
- 39. Raphael I, Nalawade S, Eagar TN, Forsthuber TG. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015; 74: 5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Beres A, Komorowski R, Mihara M, Drobyski W. Induced regulatory T cells to mitigate graft. Clin Cancer Res 2011; 17: 3969–3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ghali JR, Alikhan MA, Holdsworth SR, Kitching AR. Induced regulatory T cells are phenotypically unstable and do not protect mice from rapidly progressive glomerulonephritis. Immunology 2017; 150: 100–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Muranski P, Restifo NP. Essentials of Th17 cell commitment and plasticity. Blood 2013; 121: 2402–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Martinez GJ, Nurieva RI, Yang XO, Dong C. Regulation and function of proinflammatory TH17 cells. Ann N Y Acad Sci 2008; 1143: 188–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Elias KM, Laurence A, Davidson TS et al Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat‐3/Stat‐5 independent signaling pathway. Blood 2008; 111: 1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Laurence A, Tato CM, Davidson TS et al Interleukin‐2 signaling via STAT5 constrains T helper 17 cell generation. Immunity 2007; 26: 371–381. [DOI] [PubMed] [Google Scholar]
- 46. Castro G, Liu X, Ngo K et al RORγt and RORα signature genes in human Th17 cells. PLoS One 2017; 12: 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Floess S, Freyer J, Siewert C et al Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol 2007; 5: 0169–0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Baron U, Floess S, Wieczorek G et al DNA demethylation in the human FOXP3 locus discriminates regulatory T cells from activated FOXP3+ conventional T cells. Eur J Immunol 2007; 37: 2378–2389. [DOI] [PubMed] [Google Scholar]
- 49. Samstein R, Josefowicz S, Arvey A, Treuting P, Rudensky AY. Extrathymic generation of regulatory T cells in placental mammals mitigates maternal‐fetal conflict. Cell 2012; 150: 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Oh SA, Li MO. TGF‐β: guardian of T cell function. J Immunol 2013; 191: 3973–3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang P, Tey S‐K, Koyama M et al Induced regulatory T cells promote tolerance when stabilized by rapamycin and IL‐2 in vivo . J Immunol 2013; 191: 5291–5303. [DOI] [PubMed] [Google Scholar]
- 52. Chapman NM, Chi H. MTOR signaling, tregs and immune modulation. Immunotherapy 2014; 6: 1295–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL‐2 receptor β‐dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. J Immunol 2007; 178: 280–290. [DOI] [PubMed] [Google Scholar]
- 54. Delgoffe GM, Kole TP, Zheng Y et al The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 2009; 30: 832–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kryczek I, Wei S, Vatan L et al Cutting edge: opposite effects of IL‐1 and IL‐2 on the regulation of IL‐17+ T cell pool IL‐1 subverts IL‐2‐mediated suppression. J Immunol 2007; 179: 1423–1426. [DOI] [PubMed] [Google Scholar]
- 56. Kretschmer K, Apostolou I, Hawiger D, Khazaie K, Nussenzweig MC, von Boehmer H. Inducing and expanding regulatory T cell populations by foreign antigen. Nat Immunol 2005; 6: 1219–1227. [DOI] [PubMed] [Google Scholar]
- 57. Haxhinasto S, Mathis D, Benoist C. The AKT‐mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J Exp Med 2008; 205: 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Oliveira VG, Caridade M, Paiva RS, Demengeot J, Graca L. Sub‐optimal CD4+ T‐cell activation triggers autonomous TGF‐β‐dependent conversion to Foxp3+ regulatory T cells. Eur J Immunol 2011; 41: 1249–1255. [DOI] [PubMed] [Google Scholar]
- 59. Gottschalk RA, Corse E, Allison JP. TCR ligand density and affinity determine peripheral induction of Foxp3 in vivo . J Exp Med 2010; 207: 1701–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yun TJ, Bevan MJ. The Goldilocks conditions applied to T cell development. Nat Immunol 2001; 2: 13–14. [DOI] [PubMed] [Google Scholar]
- 61. Li MO, Rudensky AY. T cell receptor signalling in the control of regulatory T cell differentiation and function. Nat Rev Immunol 2016; 16: 220–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Geiger TL, Tauro S. Nature and nurture in Foxp3+ regulatory T cell development, stability, and function. Hum Immunol 2012; 73: 232–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mikami N, Kawakami R, Chen KY, Sugimoto A, Ohkura N, Sakaguchi S. Epigenetic conversion of conventional T cells into regulatory T cells by CD28 signal deprivation. Proc Natl Acad Sci USA 2020; 117: 12258–12268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Semple K, Nguyen A, Yu Y, Wang H, Anasetti C, Yu XZ. Strong CD28 costimulation suppresses induction of regulatory T cells from naive precursors through Lck signaling. Blood 2011; 117: 3096–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Allan SE, Crome SQ, Crellin NK et al Activation‐induced FOXP3 in human T effector cells does not suppress proliferation or cytokine production. Int Immunol 2007; 19: 345–354. [DOI] [PubMed] [Google Scholar]
- 66. Tai X, Van Laethem F, Pobezinsky L et al Basis of CTLA‐4 function in regulatory and conventional CD4+ T cells. Blood 2012; 119: 5155–5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 2015; 15: 486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Duhen T, Duhen R, Lanzavecchia A, Sallusto F, Campbell DJ. Functionally distinct subsets of human FOXP3+ Treg cells that phenotypically mirror effector Th cells. Blood 2012; 119: 4430–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Schildberg FA, Klein SR, Freeman GJ, Sharpe AH. Coinhibitory pathways in the B7‐CD28 ligand‐receptor family. Immunity 2016; 44: 955–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Valk E, Rudd CE, Schneider H. CTLA‐4 trafficking and surface expression. Trends Immunol 2008; 29: 272–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chauhan SK, Saban DR, Lee HK, Dana R. Levels of Foxp3 in regulatory T cells reflect their functional status in transplantation. J Immunol 2009; 182: e1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Barsheshet Y, Wildbaum G, Levy E et al CCR8+FOXp3+ Treg cells as master drivers of immune regulation. Proc Natl Acad Sci USA 2017; 114: 6086–6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chinen T, Kannan AK, Levine AG et al An essential role for IL‐2 receptor in regulatory T cell function. Nat Immunol 2016; 17: 1322–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Umare V, Pradhan V, Nadkar M et al Effect of proinflammatory cytokines (IL‐6, TNF, and IL‐1 β) on clinical manifestations in indian SLE patients. Mediators Inflamm 2014; 2014: 385297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Sojka DK, Fowell DJ. Regulatory T cells inhibit acute IFN‐γ synthesis without blocking T‐helper cell type 1 (Th1) differentiation via a compartmentalized requirement for IL‐10. Proc Natl Acad Sci USA 2011; 108: 18336–18341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Joshi RN, Fernandes SJ, Shang MM et al Phosphatase inhibitor PPP1R11 modulates resistance of human T cells toward Treg‐mediated suppression of cytokine expression. J Leukoc Biol 2019; 106: 413–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Schmidt A, Oberle N, Krammer PH. Molecular mechanisms oftreg‐mediatedt cell suppression. Front Immunol 2012; 3: 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Schmidt A, Marabita F, Kiani NA et al Time‐resolved transcriptome and proteome landscape of human regulatory T cell (Treg) differentiation reveals novel regulators of FOXP3. BMC Biol 2018; 16: 1–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Dons EM, Raimondi G, Cooper DKC, Thomson AW. Induced regulatory T cells: mechanisms of conversion and suppressive potential. Hum Immunol 2011; 73: 328–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Jung MK, Kwak J‐E, Shin E‐C. IL‐17A‐producing Foxp3+ regulatory T cells and human diseases. Immune Netw 2017; 17: 276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kim BS, Lu H, Ichiyama K et al Generation of RORγt+ antigen‐specific T regulatory 17 cells from Foxp3+ precursors in autoimmunity. Cell Rep 2017; 21: 195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Issa F, Hester J, Goto R, Nadig S, Goodacre TE. Ex vivo‐expanded human regulatory T cells prevent the rejection of skin allografts in a humanised mouse model. Transplantation 2010; 90: 1321–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wu DC, Hester J, Nadig SN et al Ex vivo expanded human regulatory T cells can prolong survival of a human islet allograft in a humanized mouse model. Transplantation 2013; 96: 707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Schreiber L, Pietzsch B, Floess S et al The Treg‐specific demethylated region stabilizes Foxp3 expression independently of NF‐κB signaling. PLoS One 2014; 9: e88318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Polansky JK, Kretschmer K, Freyer J et al DNA methylation controls Foxp3 gene expression. Eur J Immunol 2008; 38: 1654–1663. [DOI] [PubMed] [Google Scholar]
- 86. Ma H, Gao W, Sun X, Wang W. STAT5 and TET2 cooperate to regulate FOXP3‐TSDR demethylation in CD4+ T cells of patients with colorectal cancer. J Immunol Res 2018; 2018: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Sasidharan Nair V, Song MH, Oh KI. Vitamin C facilitates demethylation of the Foxp3 enhancer in a Tet‐dependent manner. J Immunol 2016; 196: 2119–2131. [DOI] [PubMed] [Google Scholar]
- 88. Someya K, Nakatsukasa H, Ito M et al Improvement of Foxp3 stability through CNS2 demethylation by TET enzyme induction and activation. Int Immunol 2017; 29: 365–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Yue X, Trifari S, Äijö T et al Control of Foxp3 stability through modulation of TET activity. J Exp Med 2016; 213: 377–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Rainbow DB, Yang X, Burren O et al Epigenetic analysis of regulatory T cells using multiplex bisulfite sequencing. Eur J Immunol 2015; 45: 3200–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Minskaia E, Saraiva BC, Soares MMV et al Molecular markers distinguishing T cell subtypes with TSDR strand‐bias methylation. Front Immunol 2018; 9: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ohkura N, Hamaguchi M, Morikawa H et al T cell receptor stimulation‐induced epigenetic changes and Foxp3 expression are independent and complementary events required for treg cell development. Immunity 2012; 37: 785–799. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials