

The exact disease mechanism of MEGF10 deficiency, which causes a rare inherited myopathy, remains unknown. In mice, Megf10 deficiency is associated with slower proliferation and migration of satellite cells, as well as impaired skeletal muscle regeneration after skeletal muscle injury experiments. These studies demonstrate that Megf10 makes important contributions to the function of satellite cells in muscle regeneration.
Keywords: MEGF10 myopathy, satellite cells, skeletal muscle regeneration
Abstract
Biallelic loss‐of‐function MEGF10 mutations lead to MEGF10 myopathy, also known as early onset myopathy with areflexia, respiratory distress, and dysphagia (EMARDD). MEGF10 is expressed in muscle satellite cells, but the contribution of satellite cell dysfunction to MEGF10 myopathy is unclear. Myofibers and satellite cells were isolated and examined from Megf10−/− and wild‐type mice. A separate set of mice underwent repeated intramuscular barium chloride injections. Megf10−/− muscle satellite cells showed reduced proliferation and migration, while Megf10−/− mouse skeletal muscles showed impaired regeneration. Megf10 deficiency is associated with impaired muscle regeneration, due in part to defects in satellite cell function. Efforts to rescue Megf10 deficiency will have therapeutic implications for MEGF10 myopathy and other inherited muscle diseases involving impaired muscle regeneration.
Abbreviations
- AMP
adult muscle precursor
- ANOVA
analysis of variance
- bHLH
basic helix‐loop‐helix
- Drpr
Draper
- EDL
extensor digitorum longus
- EMARDD, early onset myopathy
areflexia, respiratory distress, and dysphagia
- IACUC
institutional animal care and use committee
- IF
immunofluorescence
- MEGF10 (human) or Megf10 (mouse)
multiple EGF‐like domains 10
- MRF
myogenic regulatory factor
- PBS
phosphate‐buffered saline
- SEM
standard error of the mean
- SSRI
selective serotonin reuptake inhibitor
- TA
tibialis anterior
MEGF10 is an orphan receptor that is expressed in developing myoblasts and muscle satellite cells. Biallelic loss‐of‐function mutations in the encoding gene MEGF10 cause a recessive congenital muscle disease called MEGF10 myopathy [1, 2, 3, 4, 5, 6, 7]; the classic form of this disease is also known as early onset myopathy with areflexia, respiratory distress, and dysphagia (EMARDD) [8]. Minicores have been seen on muscle biopsy in several cases of MEGF10 myopathy [2, 5], and an adult onset form of this disease has also been described [4, 5]. The molecular mechanism of disease in MEGF10 myopathy/EMARDD involves impaired tyrosine phosphorylation [9] and impaired interactions between MEGF10 and the Notch pathway [10, 11], whereas the cellular mechanism of disease appears to involve potential defects in myogenesis, particularly myoblast proliferation [10] and migration [11], consistent with the congenital onset of disease in the classic EMARDD phenotype.
Satellite cells are muscle stem cells that are located between the sarcolemma and the basal lamina of muscle fibers and were first described in frogs [12]. Embryologically, they originate from the dermomyotome cell population [13]. Satellite cells undergo asymmetric division, yielding both a self‐perpetuating cell population and cells that are destined to fuse with adjacent muscle fibers to assist with muscle growth and regeneration [14]. In the settings of muscle injury and chronic muscle disease, satellite cells help resuscitate injured and regenerating muscle fibers by fusing with them [15]. The regulation of satellite cell states, including quiescence, activation, proliferation, and differentiation/fusion, is complex, with many details described but many questions still unanswered [16]. In the context of MEGF10, prior work has shown that satellite cell dysfunction contributes to the pathogenesis of MEGF10 myopathy and that Megf10 is expressed in quiescent and activated murine satellite cells [10]. Dll1 [17], a Notch ligand, and myogenin [18] have been shown to be positive regulators of Megf10 expression, whereas the combination of an Rbpj conditional mutation in Pax3+ myogenic precursors combined with mutant MyoD downregulate Megf10 expression [17]. However, it is not yet clear whether Megf10 deficiency has an impact on key cellular processes such as satellite cell migration, along with the larger biological process of muscle regeneration. Experiments on a mouse model of MEGF10 myopathy as well as satellite cells isolated from these mice will help answer these questions.
There are several methods used to replicate muscle injury in mouse models [19]. For the current study, we selected chemical injury with barium chloride due to its reliability and low toxicity [20]. A prior study from our group showed that muscle injury induced by intramuscular barium chloride injections yielded subtle signs of delayed regeneration in Megf10−/− mice compared to wild‐type mice [11]. However, repeated intramuscular injections to deplete the satellite cell pool have not previously been performed [21].
A better understanding of the contribution of MEGF10 to satellite cell function and muscle regeneration, along with the effects of MEGF10 deficiency on these important processes, could help identify novel targets for therapeutic strategies for MEGF10 myopathy. If such therapeutic strategies specifically augment satellite cell function in skeletal muscle, they could also be applied to a range of muscle diseases associated with impaired regeneration, including the muscular dystrophies. The aims of the current study are to examine patterns of satellite cell detachment from myofibers, expression patterns of MyoD in these satellite cells, and skeletal muscle regenerative capacity in a mouse model of MEGF10 myopathy.
Materials and methods
Mouse strains
All animal studies were performed under the auspices of a protocol approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Florida. Megf10+/‐ mice with a C57BL/6 background were generous gifts from Jeremy Kay at Duke University and Joshua Sanes at Harvard University, and were bred to yield Megf10−/− mice. Genotypes were verified as previously described, and Megf10 deficiency has previously been confirmed in this strain [11, 22]. Wild‐type C57BL/6J mice were obtained from Animal Care Services at the University of Florida. Skeletal muscle specimens for histological analysis and cell extractions were dissected ex vivo immediately after IACUC‐approved euthanasia procedures.
Single myofiber isolation and culture, with satellite cell detachment
Individual myofibers were isolated from the extensor digitorum longus (EDL) muscles of 6 Megf10−/− and 5 wild‐type mice according to a standard protocol [23]. EDL is a standard muscle from which to isolate individual myofibers from mice for satellite cell analysis [23]. Ten to fifteen live myofibers were dissected from each EDL muscle and then cultured individually in glass chamber slides (ibidi GmbH, Martinsried, Germany) coated with Matrigel (Corning®) with serum‐rich medium (20% fetal bovine serum). The satellite cells observed to be detached from each myofiber were manually counted at 24, 48, and 72 h timepoints to capture detachment and migration prior to differentiation [24].
Immunofluorescence (IF) of myofibers and satellite cells
At 72 h, the myofibers and satellite cells in glass chamber slides were washed with phosphate‐buffered saline (PBS), fixed in prewarmed 4% paraformaldehyde, incubated in 1% glycine in PBS quenching solution to minimize background staining, then permeabilized with 0.1% Triton X‐100 in PBS. After blocking with 5–10% fetal bovine serum in PBS, the myofibers and satellite cells were incubated with primary antibodies to Pax7 (Developmental Studies Hybridoma Bank) and MyoD (Developmental Studies Hybridoma Bank and Santa Cruz), then with secondary Alexa‐568 or Alexa‐488 conjugated goat anti‐rabbit antibodies (Life Technologies) or secondary Alexa‐568 anti‐mouse antibodies (Life Technologies). The nuclei were counterstained with DAPI. Pax7+ and MyoD‐+ satellite cells were counted, and the shortest distance between the center of each satellite cell to the edge of the myofiber was measured using ImageJ [25].
IF of satellite cells
Myofibers were cultured on cover slips in culture dishes and maintained in proliferation media consisting of 20% fetal bovine serum, 5% chick embryo extract, and 2% penicillin/streptomycin in DMEM (high glucose). At 72 h, the myofibers were washed away, leaving the activated satellite cells on the culture dish. IF was performed on the satellite cells as described above for MyoD, accompanied by nuclear DAPI counterstaining.
Barium chloride injury
Muscle injury was modeled in 2‐month‐old mice of both strains via injection of 50 μL 1.2% barium chloride in sterile water into the right tibialis anterior, with injection of sterile water into the left tibialis anterior as a control. Tibialis anterior is a standard muscle used for such muscle injury experiments in mice [26]. To deplete the satellite cell pool, the injections were performed a total of three times [21] starting at age 2 months, with gaps of 21–28 days between injections to permit complete recovery between injections [27]. Twenty‐one days after the third injection, the mice were euthanized and bilateral tibialis anterior muscles were harvested. The tibialis anterior muscles were snap‐frozen for hematoxylin and eosin staining.
Hematoxylin and eosin histological analyses
Tibialis anterior muscles were dissected immediately ex vivo from the two mouse strains noted above and then snap‐frozen in 2‐methylbutane cooled with liquid nitrogen. The muscle samples were sectioned at 7µm in a cryostat and transferred to superfrost microscope slides. For hematoxylin and eosin staining, the sections were incubated for 5 minutes in hematoxylin (Leica Biosystems); rinsed with cold tap water; immersed twice in eosin Y; washed consecutively with 80, 90, and 100% ethanol; and incubated three times for 2 minutes in xylene. Sections were also stained with fibronectin using standard methods [28]; fibronectin quantification was performed using ImageJ [25]. The sections were mounted with Cytoseal 60 (Richard Allan Scientific) and visualized on an Olympus BX51 upright light microscope.
Statistical analysis
Unpaired t‐tests were used to compare Megf10−/− and wild‐type groups for the various experiments described above. Means +/‐ standard error of the mean (SEM) were calculated. One‐way analysis of variance (ANOVA) was used to analyze the fibronectin quantification. GraphPad Prism (GraphPad Software, San Diego, California) was used for statistical analyses.
Results
Diminished detachment of Megf10 −/− satellite cells from myofibers compared to wild‐type cells
Manual counts showed steadily increasing numbers of satellite cells that had visibly detached from individual myofibers at 24 h (Fig. 1A), 48 h (Fig. 1B), and 72 h (Fig. 1C) of culture. Representative images are shown at 72 hours (Fig. 1D). At each time point, fewer satellite cells were detached from Megf10−/− live myofibers than from wild‐type live myofibers, indicating that Megf10 deficiency is associated with impaired satellite cell activation and migration.
Fig. 1.
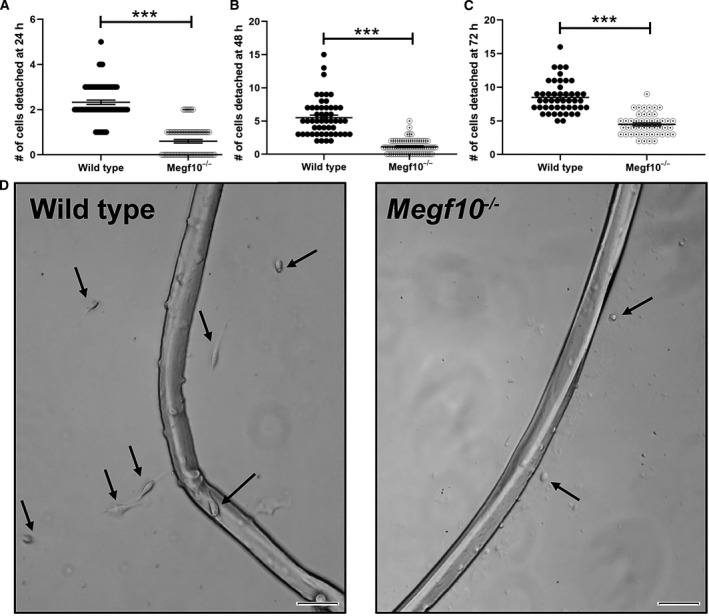
(A) Single intact live myofibers were isolated from wild‐type and Megf10−/− mice and cultured in serum‐rich medium in glass chamber slides. Detached satellite cells were counted for each myofiber at (A) 24, (B) 48, and (C) 72 h timepoints. Each dot represents the satellite cell count for one myofiber. The number of myofibers evaluated were: (A) wild‐type, n = 70 myofibers; Megf10−/−, n = 70 myofibers; (B) wild‐type, n = 52 myofibers; Megf10−/−, n = 70 myofibers; (C) wild‐type, n = 47 myofibers; Megf10−/−, n = 47 myofibers. Comparisons between wild‐type and Megf10−/− myofiber counts were performed at each time point by unpaired t‐tests; ***, P < 0.001. (D) Representative images are shown at 72 h. Arrows indicate satellite cells that have detached and migrated from the isolated myofibers. Scale bar, 50 μm.
Deficiency in MyoD expression of Megf10 −/− satellite cells
To quantify satellite cell activation and migration patterns in greater detail, IF was performed to identify Pax7265947092+/MyoD619236543+ cells associated with individual myofibers, including those attached to and detached from the myofibers, at 72 h of culture (Fig. 2A). Manual counts of attached and detached Pax7+/MyoD+ cells showed that these cells were less abundant for Megf10−/− myofibers compared to wild‐type myofibers (Fig. 2B). Measurements of distances migrated by the Pax7+/MyoD+ cells at the same timepoint showed less robust migration for cells associated with Megf10−/− myofibers compared to wild‐type myofibers (Fig. 2C). Satellite cells detached from myofibers were cultured separately, showing less abundant MyoD expression among DAPI‐positive nuclei of Megf10−/− satellite cells compared to wild‐type satellite cells (Fig. 2D,E). These results provide additional evidence that Megf10 deficiency is associated with reduced satellite cell activation and migration.
Fig. 2.
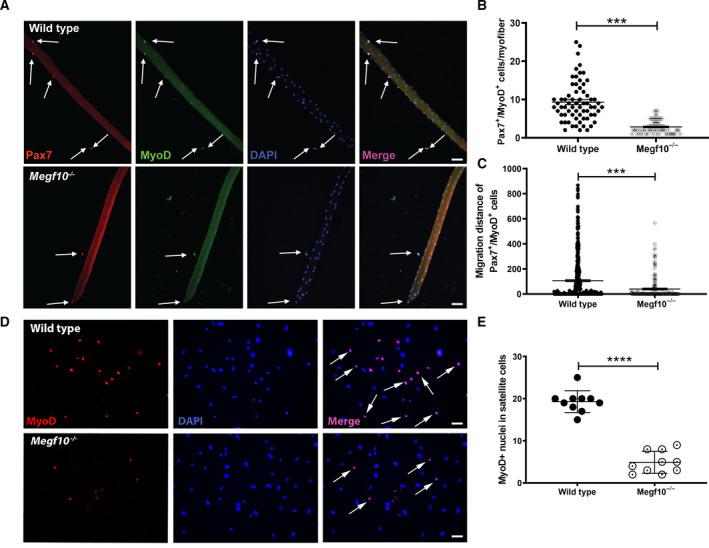
(A) Representative individual and merged images of satellite cells stained with anti‐Pax7 (red), anti‐MyoD (green), and DAPI at 72 h of myofiber culture. Arrows indicate satellite cells expressing both Pax7 and MyoD. Scale bar = 50 μm. (B) Scatter plot showing numbers of Pax7+/MyoD+ satellite cells associated with individual myofibers at 72 h. Each dot represents the cell count from one myofiber: wild‐type, n = 67; Megf10−/−, n = 71. The comparison of the cell counts between the wild‐type and Meg10−/− groups was performed by an unpaired t‐test; ***, P < 0.001. (C) The distances between Pax7169005038+/MyoD865006223+ satellite cells and their associated myofiber were measured at 72 h of culture. Each dot represents one Pax7+/MyoD+ cell. Arbitrary units noted on y‐axis. Wild‐type: n = 471 cells counted from 67 fibers; Megf10−/−: n = 165 cells counted from 71 fibers. The comparison of the migration distances between the wild‐type and Megf10−/− groups was performed by an unpaired t‐test; ***, P < 0.001. (D) MyoD+ nuclei are less abundant in Megf10−/− detached satellite cells compared to wild‐type cells. (E) Quantification of MyoD+ nuclei in proliferating satellite cells. Megf10−/− cells have significantly reduced MyoD+ nuclei in plated satellite cells after 96 h of single myofiber culture. Scatter plots represent counts from n = 10 images; mean ± SEM shown; ****, P < 0.0001.
Reduced regeneration potential and increased fibrosis in Megf10 −/− mice after muscle injury
Three sequential barium chloride injections into mouse tibialis anterior were performed for both wild‐type and Megf10−/− mice, with saline intramuscular injections serving as controls (Fig. 3A). Hematoxylin and eosin staining of representative sections from the injected muscles revealed significantly reduced regeneration potential with pronounced pockets of atrophic fibers and greater overall fiber size variability in Megf10−/− tibialis anterior muscles injected with barium chloride compared to wild‐type tibialis anterior muscles (Fig. 3B). Fibronectin staining was performed on muscle sections to demonstrate the degree of fibrosis in the injected tibialis anterior muscles. Greater densities of fibronectin were measured via ImageJ quantification in Megf10−/− muscles compared to wild‐type muscles (Fig. 3C). A representative IF image demonstrates the thicker bands of fibronectin seen in Meg10−/− muscle sections compared to wild‐type muscle sections (Fig. 3D).
Fig. 3.
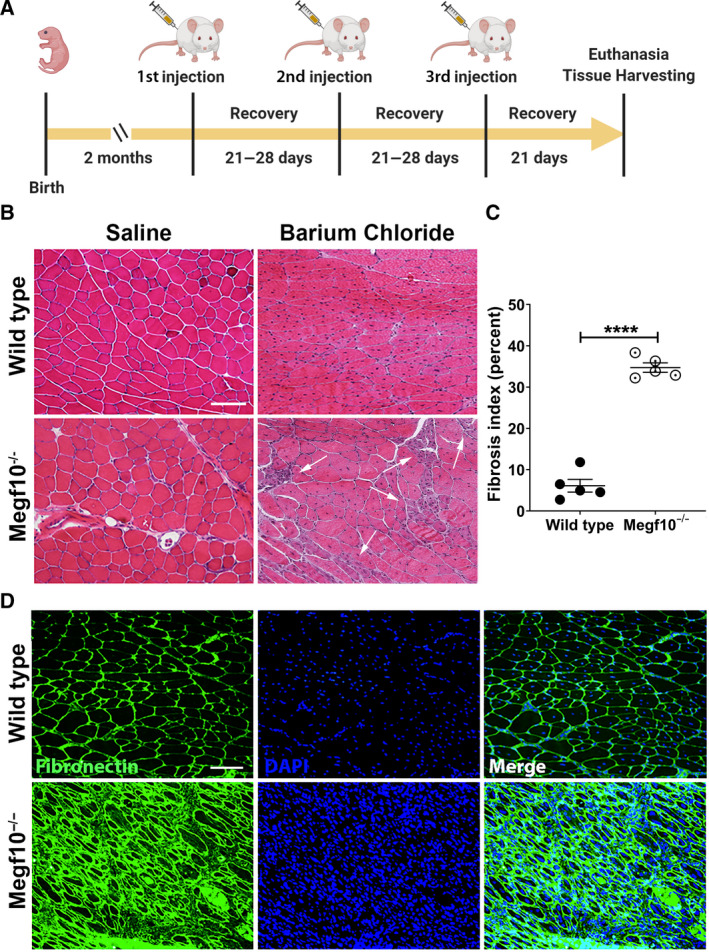
(A) Timeline of mouse injury experiments, showing intervals between birth, 3 sequential barium chloride and saline (control) injections into tibialis anterior, and euthanasia followed by tissue harvesting. (B) Hematoxylin and eosin staining of sections from tibialis anterior muscles that were injected with either barium chloride or saline in wild‐type and Megf10−/− mice. Arrows in the barium chloride Megf10−/− image indicate clusters of small myofibers with internalized nuclei; such clusters are not present in the barium chloride wild‐type image. Scale bar, 100μm. (C) Fibrosis quantification was performed on 5 images of fibronectin stained sections of tibialis anterior muscles injected with barium chloride in wild‐type and Megf10−/− mice. The percent fibrosis index was calculated as fibrotic area/total area × 100, indicated as percentages. An unpaired t‐test and one‐way ANOVA both yielded P < 0.0001 between wild‐type and Megf10−/− groups. Scatter plots represent fibrosis indices from n = 5 images; mean ± SEM shown; ****, P < 0.0001. (D) Representative IF images of muscle sections show fibronectin (green) with DAPI (blue) counterstaining in barium chloride injected tibialis anterior muscles from wild‐type and Megf10−/− mice. Scale bar, 100 μm.
Discussion
In the decade since the gene MEGF10 was first associated with a distinct human muscle disease, it has become apparent that the protein product MEGF10 plays a key role in satellite cell function. Over time, various aspects of MEGF10’s biochemical and cellular functions have been elucidated. The current study fills in another piece to the puzzle and provides further details on how MEGF10 deficiency affects satellite cell behavior and offers novel information regarding the impact of such a deficiency on skeletal muscle regenerative capacity.
Satellite cells are mononuclear progenitor stem cells in skeletal muscle that make key contributions to the process of muscle repair after injury. The name ‘satellite cell’ was derived from the original observation that these cells are found in the niche between the basal lamina and sarcolemma of a myofiber [12]. The satellite cell cycle includes symmetric and asymmetric components, the former consisting of self‐renewal within the quiescent state [29] and the latter leading to a terminal exit from that state. Quiescent and self‐renewing satellite cells can be identified by the detection of several distinct protein markers, most commonly the paired box 7 (Pax7) transcription factor in their nuclei [30]. Of particular relevance to Megf10, which interacts with Notch1 [10, 11], extrinsic Notch signaling is critical for the maintenance of the quiescent state [31, 32] and Notch1 activation promotes proliferation of satellite cells [14]. The classic stages of an exit from the quiescent state are demarcated by the expression patterns of key myogenic regulatory factors (MRFs) that are all basic helix‐loop‐helix (bHLH) transcription factors [33]: Myf5 [34], MyoD [35], myogenenin [36], and Mrf4 [37]. All four of these MRFs are induced during various stages of satellite cell activation [38, 39]. For our studies, we focused on Pax7+/MyoD+ cells as the co‐expression of these two markers is generally accepted as indicating the presence of activated satellite cells that proliferate and migrate [40], and our prior work indicated that Megf10 deficiency is associated with impaired proliferation and migration in C2C12 myoblasts and primary mouse myoblasts [11], suggesting a role for Megf10 in this early stage of the satellite cell myogenic pathway.
In this context, what role might MEGF10 play in the regulation of the early stages of the satellite cell myogenic pathway? It is an orphan receptor that is expressed at the cell surface with a single transmembrane domain and thus is not an MRF. It interacts with Notch1 and thus likely performs an extrinsic regulatory task [10], though the interaction occurs at both proteins’ intracellular domains [11]. Our current data show that Megf10 deficiency impairs migration of Pax7+/MyoD+ activated satellite cells and is associated with reduced numbers of MyoD + cells, providing further evidence that Megf10 contributes to an early stage of satellite cell activation during which the cells migrate to target myofibers. This information could help explain the persistent nature of the clinical manifestations of MEGF10 myopathy, as muscle function generally does not improve in human patients affected by this disease [1, 2].
In previous studies, we have shown that downregulation of draper (drpr), the Drosophila homolog of Megf10, in quiescent adult muscle precursors (satellite cell‐like AMPs) leads to decreased motor activity in corresponding mutant flies [41]. These cells, however, are insensitive to drpr/Megf10 overexpression, which is deleterious later in myogenesis, that is, at the migration/differentiation stages (suggesting that Drpr levels are finely tuned during these steps) [42]. It is well established that drpr plays a crucial role in mediating the migration of glia toward injured neurons [43, 44], as well as the migration of immune cells toward tissue wound [45]. Whether a parallel drpr/Megf10‐dependent molecular mechanism participates in regulating the migration of satellite cells toward developing, or injured, muscle fibers remains to be elucidated. Regeneration of injured muscle has been reported in Drosophila [46]. The fly may provide a useful model in which to further elucidate the conserved pathways downstream of drpr/Megf10 that contribute to muscle development and potentially repair. Future Drosophila studies could address the question of satellite cell migration by examining the effects of drpr deficiency on the migration patterns of AMPs, which behave much as muscle satellite cells do in mammals [47].
Regardless of the exact cellular role of Megf10 in the satellite cell cycle, our data show a clear impact on skeletal muscle regeneration. Impaired muscle regeneration has significant chronic consequences for muscle homeostasis. Thus, MEGF10 bears further investigation as a therapeutic target, not only for MEGF10 deficiency itself, but also for other inherited muscle diseases, including various forms of muscular dystrophy. Recently, we published a study that identified selective serotonin reuptake inhibitors, notably sertraline, as having a beneficial effect on cellular and in vivo (i.e., Drosophila and zebrafish) models of Megf10 deficiency [48]. Serotonin did not replicate this effect in Drosophila, whereas sertraline behaved as a Notch pathway agonist in these cellular models, suggesting another link between MEGF10 and the Notch pathway [48] in addition to evidence from various selective serotonin reuptake inhibitors (SSRIs) in other contexts [49, 50, 51, 52]. Further investigations of the effects of sertraline on Megf10 deficiency should include detailed mouse experiments that could both confirm the therapeutic effect and also elucidate the interactions between Megf10 and the satellite cell cycle. These studies could then be supplemented by examinations of the effects of sertraline on mouse models for other inherited muscle diseases, to explore potential broader applications of manipulating this therapeutic target.
Overall, more precise information regarding the relationship between MEGF10 and the Notch pathway could be drawn out by exploring potential interactions with other Notch pathway components, as well as with protein O‐glucosyltransferase 1 (encoded by POGLUT1 and previously known as Rumi); the latter is associated with muscular dystrophy and is also known to be a regulator of the Notch pathway [53, 54]. POGLUT1 mutations have been associated with a reduction in muscle satellite cells [55]. Detailed fluorescence activated cell sort (FACS) experiments could also elucidate how MEGF10 deficiency could alter early stages of the satellite cell cycle. Such knowledge could then lead to the development of standardized in vitro markers to gauge potential therapeutic effects of sertraline or other candidate therapies for MEGF10 myopathy. MEGF10 could provide a therapeutic target for manipulation of Notch signaling to augment skeletal muscle regenerative capacity via satellite cell enhancement, which could lead to new treatments not only for MEGF10 myopathy, but also for other inherited muscle diseases accompanied by impaired muscle regeneration, including a broad range of muscular dystrophies.
Conflict of interest
The authors declare no conflict of interest.
Author contribution
PBK conceived and supervised the study and designed experiments. CL, MS, RMD, and KAM performed experiments. CL, DV, MS, CAP, and PBK analyzed data. CL, DV, MS, ID, CAP, and PBK interpreted data and wrote the manuscript.
Acknowledgements
This work was supported by NIH R01 NS080929 and the Ferlita Family Fund.
Data Availability Statement
The corresponding author will share data upon reasonable request.
References
- 1. Logan CV, Lucke B, Pottinger C, Abdelhamed ZA, Parry DA, Szymanska K, Diggle CP, van Riesen A, Morgan JE, Markham G et al (2011) Mutations in MEGF10, a regulator of satellite cell myogenesis, cause early onset myopathy, areflexia, respiratory distress and dysphagia (EMARDD). Nat Genet 43, 1189–1192. [DOI] [PubMed] [Google Scholar]
- 2. Boyden SE, Mahoney LJ, Kawahara G, Myers JA, Mitsuhashi S, Estrella EA, Duncan AR, Dey F, DeChene ET, Blasko‐Goehringer JM et al (2012) Mutations in the satellite cell gene MEGF10 cause a recessive congenital myopathy with minicores. Neurogenetics 13, 115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alabdullatif MA, Al Dhaibani MA, Khassawneh MY and El‐Hattab AW (2017) Chromosomal microarray in a highly consanguineous population: diagnostic yield, utility of regions of homozygosity, and novel mutations. Clin Genet 91, 616–622. [DOI] [PubMed] [Google Scholar]
- 4. Harris E, Marini‐Bettolo C, Töpf A, Barresi R, Polvikoski T, Bailey G, Charlton R, Tellez J, MacArthur D, Guglieri M et al (2018) MEGF10 related myopathies: a new case with adult onset disease with prominent respiratory failure and review of reported phenotypes. Neuromuscul Disord 28, 48–53. [DOI] [PubMed] [Google Scholar]
- 5. Liewluck T, Milone M, Tian X, Engel AG, Staff NP and Wong LJ (2016) Adult‐onset respiratory insufficiency, scoliosis, and distal joint hyperlaxity in patients with multiminicore disease due to novel Megf10 mutations. Muscle Nerve 53, 984–988. [DOI] [PubMed] [Google Scholar]
- 6. Pierson TM, Markello T, Accardi J, Wolfe L, Adams D, Sincan M, Tarazi NM, Fajardo KF, Cherukuri PF, Bajraktari I et al (2013) Novel SNP array analysis and exome sequencing detect a homozygous exon 7 deletion of MEGF10 causing early onset myopathy, areflexia, respiratory distress and dysphagia (EMARDD). Neuromuscul Disord 23, 483–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Takayama K, Mitsuhashi S, Shin J‐Y, Tanaka R, Fujii T, Tsuburaya R, Mukaida S, Noguchi S, Nonaka I and Nishino I (2016) Japanese multiple epidermal growth factor 10 (MEGF10) myopathy with novel mutations: a phenotype‐genotype correlation. Neuromuscul Disord 26, 604–609. [DOI] [PubMed] [Google Scholar]
- 8. Hartley L, Kinali M, Knight R, Mercuri E, Hubner C, Bertini E, Manzur Ay, Jimenez‐Mallebrera C, Sewry Ca and Muntoni F (2007) A congenital myopathy with diaphragmatic weakness not linked to the SMARD1 locus. Neuromuscul Disord 17, 174–179. [DOI] [PubMed] [Google Scholar]
- 9. Mitsuhashi S, Mitsuhashi H, Alexander MS, Sugimoto H and Kang PB (2013) Cysteine mutations cause defective tyrosine phosphorylation in MEGF10 myopathy. FEBS Lett 587, 2952–2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Holterman CE, Le Grand F, Kuang S, Seale P and Rudnicki MA (2007) Megf10 regulates the progression of the satellite cell myogenic program. J Cell Biol 179, 911–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Saha M, Mitsuhashi S, Jones MD, Manko K, Reddy HM, Bruels CC, Cho K‐A, Pacak CA, Draper I and Kang PB (2017) Consequences of MEGF10 deficiency on myoblast function and Notch1 interactions. Hum Mol Genet 26, 2984–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mauro A (1961) Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol 9, 493–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gros J, Manceau M, Thome V and Marcelle C (2005) A common somitic origin for embryonic muscle progenitors and satellite cells. Nature 435, 954–958. [DOI] [PubMed] [Google Scholar]
- 14. Conboy IM and Rando TA (2002) The regulation of Notch signaling controls satellite cell activation and cell fate determination in postnatal myogenesis. Dev Cell 3, 397–409. [DOI] [PubMed] [Google Scholar]
- 15. Chou SM and Nonaka I (1977) Satellite cells and muscle regeneration in diseased human skeletal muscles. J Neurol Sci 34, 131–145. [DOI] [PubMed] [Google Scholar]
- 16. Dumont NA, Bentzinger CF, Sincennes MC and Rudnicki MA (2015) Satellite cells and skeletal muscle regeneration. Compr Physiol 5, 1027–1059. [DOI] [PubMed] [Google Scholar]
- 17. Brohl D, , Vasyutina E, Czajkowski MT, Griger J, Rassek C, Rahn H‐P, Purfürst B, Wende H and Birchmeier C (2012) Colonization of the satellite cell niche by skeletal muscle progenitor cells depends on Notch signals. Dev Cell 23, 469–481. [DOI] [PubMed] [Google Scholar]
- 18. Park SY, Yun Y, Kim MJ and Kim IS (2014) Myogenin is a positive regulator of MEGF10 expression in skeletal muscle. Biochem Biophys Res Commun 450, 1631–1637. [DOI] [PubMed] [Google Scholar]
- 19. Hardy D, Besnard A, Latil M, Jouvion G, Briand D, Thépenier C, Pascal Q, Guguin A, Gayraud‐Morel B, Cavaillon J‐M et al (2016) Comparative study of injury models for studying muscle regeneration in mice. PLoS One 11, e0147198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Morton AB, Norton CE, Jacobsen NL, Fernando CA, Cornelison DDW and Segal SS (2019) Barium chloride injures myofibers through calcium‐induced proteolysis with fragmentation of motor nerves and microvessels. Skelet Muscle 9, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schaaf GJ, van Gestel TJM, in ‘t Groen SLM, de Jong B, Boomaars B, Tarallo A, Cardone M, Parenti G, van der Ploeg AT and Pijnappel WWMP (2018) Satellite cells maintain regenerative capacity but fail to repair disease‐associated muscle damage in mice with Pompe disease. Acta Neuropathol Commun 6, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kay JN, Chu MW and Sanes JR (2012) MEGF10 and MEGF11 mediate homotypic interactions required for mosaic spacing of retinal neurons. Nature 483, 465–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pasut A, Jones AE and Rudnicki MA (2013) Isolation and culture of individual myofibers and their satellite cells from adult skeletal muscle. J Vis Exp e50074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Keire P, Shearer A, Shefer G and Yablonka‐Reuveni Z (2013) Isolation and culture of skeletal muscle myofibers as a means to analyze satellite cells. Methods Mol Biol 946, 431–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schneider CA, Rasband WS and Eliceiri KW (2012) NIH image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cornelison DD, Wilcox‐Adelman SA, Goetinck PF, Rauvala H, Rapraeger AC and Olwin BB (2004) Essential and separable roles for Syndecan‐3 and Syndecan‐4 in skeletal muscle development and regeneration. Genes Dev 18, 2231–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tierney MT and Sacco A (2016) Inducing and evaluating skeletal muscle injury by notexin and barium chloride. Methods Mol Biol 1460, 53–60. [DOI] [PubMed] [Google Scholar]
- 28. Hadi AM, Mouchaers KT, Schalij I, Grunberg K, Meijer GA, Vonk‐Noordegraaf A, van der Laarse WJ and Belien JA (2010) Rapid quantification of myocardial fibrosis: a new macro‐based automated analysis. Anal Cell Pathol (Amst) 33, 257–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kuang S, Kuroda K, Le Grand F and Rudnicki MA (2007) Asymmetric self‐renewal and commitment of satellite stem cells in muscle. Cell 129, 999–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Seale P, Sabourin LA, Girgis‐Gabardo A, Mansouri A, Gruss P and Rudnicki MA (2000) Pax7 is required for the specification of myogenic satellite cells. Cell 102, 777–786. [DOI] [PubMed] [Google Scholar]
- 31. Mourikis P, Sambasivan R, Castel D, Rocheteau P, Bizzarro V and Tajbakhsh S (2012) A critical requirement for notch signaling in maintenance of the quiescent skeletal muscle stem cell state. Stem Cells 30, 243–252. [DOI] [PubMed] [Google Scholar]
- 32. Bjornson CR, Cheung TH, Liu L, Tripathi PV, Steeper KM and Rando TA (2012) Notch signaling is necessary to maintain quiescence in adult muscle stem cells. Stem Cells 30, 232–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zammit PS (2017) Function of the myogenic regulatory factors Myf5, MyoD, Myogenin and MRF4 in skeletal muscle, satellite cells and regenerative myogenesis. Semin Cell Dev Biol 72, 19–32. [DOI] [PubMed] [Google Scholar]
- 34. Braun T, Buschhausen‐Denker G, Bober E, Tannich E and Arnold HH (1989) A novel human muscle factor related to but distinct from MyoD1 induces myogenic conversion in 10T1/2 fibroblasts. EMBO J 8, 701–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Davis RL, Weintraub H and Lassar AB (1987) Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell 51, 987–1000. [DOI] [PubMed] [Google Scholar]
- 36. Wright WE, Sassoon DA and Lin VK (1989) Myogenin, a factor regulating myogenesis, has a domain homologous to MyoD. Cell 56, 607–617. [DOI] [PubMed] [Google Scholar]
- 37. Rhodes SJ and Konieczny SF (1989) Identification of MRF4: a new member of the muscle regulatory factor gene family. Genes Dev 3, 2050–2061. [DOI] [PubMed] [Google Scholar]
- 38. Maley MA, Davies MJ and Grounds MD (1995) Extracellular matrix, growth factors, genetics: their influence on cell proliferation and myotube formation in primary cultures of adult mouse skeletal muscle. Exp Cell Res 219, 169–179. [DOI] [PubMed] [Google Scholar]
- 39. Smith CK 2nd, Janney MJ and Allen RE (1994) Temporal expression of myogenic regulatory genes during activation, proliferation, and differentiation of rat skeletal muscle satellite cells. J Cell Physiol 159, 379–385. [DOI] [PubMed] [Google Scholar]
- 40. Zammit PS, Golding JP, Nagata Y, Hudon V, Partridge TA and Beauchamp JR (2004) Muscle satellite cells adopt divergent fates: a mechanism for self‐renewal? J Cell Biol 166, 347–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Draper I, Mahoney LJ, Mitsuhashi S, Pacak CA, Salomon RN and Kang PB (2014) Silencing of drpr leads to muscle and brain degeneration in adult Drosophila. Am J Pathol 184, 2653–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Draper I, Saha M, Stonebreaker H, Salomon RN, Matin B and Kang PB (2019) The impact of Megf10/Drpr gain‐of‐function on muscle development in Drosophila. FEBS Lett 593, 680–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Doherty J, Sheehan AE, Bradshaw R, Fox AN, Lu TY and Freeman MR (2014) PI3K signaling and Stat92E converge to modulate glial responsiveness to axonal injury. PLoS Biol 12, e1001985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ray A, Speese SD and Logan MA (2017) Glial draper rescues abeta toxicity in a Drosophila model of Alzheimer's disease. J Neurosci 37, 11881–11893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Evans IR, Rodrigues FS, Armitage EL and Wood W (2015) Draper/CED‐1 mediates an ancient damage response to control inflammatory blood cell migration in vivo. Curr Biol 25, 1606–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chaturvedi D, Reichert H, Gunage RD and VijayRaghavan K (2017) Identification and functional characterization of muscle satellite cells in Drosophila. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Figeac N, Daczewska M, Marcelle C and Jagla K (2007) Muscle stem cells and model systems for their investigation. Dev Dyn 236, 3332–3342. [DOI] [PubMed] [Google Scholar]
- 48. Saha M, Rizzo SA, Ramanathan M, Hightower RM, Santostefano KE, Terada N, Finkel RS, Berg JS, Chahin N, Pacak CA et al (2019) Selective serotonin reuptake inhibitors ameliorate MEGF10 myopathy. Hum Mol Genet 28, 2365–2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cray JJ Jr, Weinberg SM, Parsons TE, Howie RN, Elsalanty M and Yu JC (2014) Selective serotonin reuptake inhibitor exposure alters osteoblast gene expression and craniofacial development in mice. Birth Defects Res A Clin Mol Teratol 100, 912–923. [DOI] [PubMed] [Google Scholar]
- 50. Chen HF, Pan XL, Wang JW, Kong HM and Fu YM (2014) Protein‐drug interactome analysis of SSRI‐mediated neurorecovery following stroke. Biosystems 120, 1–9. [DOI] [PubMed] [Google Scholar]
- 51. Sui Y, Zhang Z, Guo Y, Sun Y, Zhang X, Xie C, Li Y and Xi G (2009) The function of Notch1 signaling was increased in parallel with neurogenesis in rat hippocampus after chronic fluoxetine administration. Biol Pharm Bull 32, 1776–1782. [DOI] [PubMed] [Google Scholar]
- 52. Ghareghani M, Zibara K, Sadeghi H, Dokoohaki S, Sadeghi H, Aryanpour R and Ghanbari A (2017) Fluvoxamine stimulates oligodendrogenesis of cultured neural stem cells and attenuates inflammation and demyelination in an animal model of multiple sclerosis. Sci Rep 7, 4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Acar M, Jafar‐Nejad H, Takeuchi H, Rajan A, Ibrani D, Rana NA, Pan H, Haltiwanger RS and Bellen HJ (2008) Rumi is a CAP10 domain glycosyltransferase that modifies Notch and is required for Notch signaling. Cell 132, 247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fernandez‐Valdivia R, Takeuchi H, Samarghandi A, Lopez M, Leonardi J, Haltiwanger RS and Jafar‐Nejad H (2011) Regulation of mammalian Notch signaling and embryonic development by the protein O‐glucosyltransferase Rumi. Development 138, 1925–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Servian‐Morilla E, Cabrera‐Serrano M, Johnson K, Pandey A, Ito A, Rivas E, Chamova T, Muelas N, Mongini T, Nafissi S et al (2020) POGLUT1 biallelic mutations cause myopathy with reduced satellite cells, alpha‐dystroglycan hypoglycosylation and a distinctive radiological pattern. Acta Neuropathol 139, 565–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The corresponding author will share data upon reasonable request.