

Abstract
Nucleosomes are dynamic entities with wide‐ranging compositional variations. Human histone variants H2A.B and H2A.Z.2.2 play critical roles in multiple biological processes by forming unstable nucleosomes and open chromatin structures, but how H2A.B and H2A.Z.2.2 confer these dynamic features to nucleosomes remains unclear. Here, we report cryo‐EM structures of nucleosome core particles containing human H2A.B (H2A.B‐NCP) at atomic resolution, identifying large‐scale structural rearrangements in the histone octamer in H2A.B‐NCP. H2A.B‐NCP compacts approximately 103 bp of DNA wrapping around the core histones in approximately 1.2 left‐handed superhelical turns, in sharp contrast to canonical nucleosome encompassing approximately 1.7 turns of DNA. Micrococcal nuclease digestion assay reveals that nineteen H2A.B‐specific residues, including a ROF (“regulating‐octamer‐folding”) sequence of six consecutive residues, are responsible for loosening of H2A.B‐NCPs. Unlike H2A.B‐NCP, the H2A.Z.2.2‐containing nucleosome (Z.2.2‐NCP) adopts a less‐extended structure and compacts around 125 bp of DNA. Further investigation uncovers a crucial role for the H2A.Z.2.2‐specific ROF in both H2A.Z.2.2‐NCP opening and SWR1‐dependent histone replacement. Taken together, these first high‐resolution structure of unstable nucleosomes induced by histone H2A variants elucidate specific functions of H2A.B and H2A.Z.2.2 in enhancing chromatin dynamics.
Keywords: H2A.B, H2A.Z.2.2, histone variant, nucleosome dynamics, open nucleosome
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Structural Biology
High‐resolution cryo‐EM structures of nucleosome core particles containing distinct human histone H2A variants reveal how these variants facilitate formation of unstable nucleosomes and open chromatin.
Introduction
Nucleosomes, the fundamental units of eukaryotic chromatin, are highly dynamic entities that compose variable histone contents, DNA sequences, and epigenetic markers (Zhou et al, 2019b). Nucleosomes achieve different levels of dynamics owing to the compositional and conformational alternations. They play a pivotal role in modulating chromatin structures during all vital biological processes that require access to DNA. Conformational changes of nucleosomal DNA include nucleosome opening (DNA unwrapping and breathing) and nucleosome gaping (H2A‐H2B dimer splitting and DNA gyres transition in the direction perpendicular to the DNA superhelical axis) (Falk et al, 2015; Ngo & Ha, 2015). The open nucleosome showing large‐scale conformational changes and DNA detachment has been implicated in nucleosome dynamics and chromatin structure regulation. However, these nucleosomes have only been captured in the transiently formed unwrapped states of the canonical nucleosome (Bilokapic et al, 2018) or in the reaction intermediates of nucleosomes bound to RNA polymerase (Ehara et al, 2019) or chromatin remodeling complexes (Farnung et al, 2017; Liu et al, 2017; Ayala et al, 2018; Eustermann et al, 2018; Willhoft et al, 2018). The high‐resolution structure of open nucleosome in the free form is not yet available.
Histone variants are a large variety of histones which share sequence homology with their canonical histones (Buschbeck & Hake, 2017). Histone variants play critical roles in multiple biological processes by conferring nucleosome composition variability (Zhou et al, 2019b) and regulating chromatin heterogeneity and dynamics (Andrews & Luger, 2011; Buschbeck & Hake, 2017). The structural plasticity of nucleosome conferred by histone H3 variant CENP‐A has been implicated in centromere function (Tachiwana et al, 2011; Roulland et al, 2016). The unfolded terminal DNA segments have been observed in the crystal structure of nucleosome containing CENP‐A (CENP‐A‐NCP) (Tachiwana et al, 2011; Roulland et al, 2016). In contrast, recent structural studies using the cryo‐EM approach showed that the CENP‐A‐NCP terminal DNA segments undergo dynamic wrapping/unwrapping (Chittori et al, 2018; Ali‐Ahmad et al, 2019; Zhou et al, 2019a). Notably, structures of nucleosome containing other histone variants, such as H3.3 and H2A.Z, largely resemble that of the canonical nucleosome (Suto et al, 2000). It remains unclear to what extent histone variants can alter the nucleosome structure.
H2A.B (previously termed H2A.Bbd) and H2A.Z.2.2 (termed Z.2.2) are H2A variants identified from mammals and primates. Incorporation of either H2A.B or Z.2.2 leads to the formation of the unstable, highly dynamic nucleosome (Bao et al, 2004; Bonisch et al, 2012, 2; Arimura et al, 2013). H2A.B presents the lowest similarity to canonical H2A by lacking the last 19 residues in the C‐terminal region and several acidic residues that form the nucleosome acidic patch. H2A.B is associated with gene transcription, DNA replication, RNA splicing (Ishibashi et al, 2010; Tolstorukov et al, 2012; Sansoni et al, 2014), and contributes to tumor cell proliferation (Winkler et al, 2012). Z.2.2 is the alternatively spliced histone H2A.Z variant preferentially found in brain tissue (Bonisch & Hake, 2012). The C‐terminal region of Z.2.2 is 14 residues shorter than H2A.Z, and the last six residues of Z.2.2 are distinct from their H2A.Z counterpart (Bonisch & Hake, 2012). Z.2.2 undergoes rapid chromatin exchanges, which is catalyzed by the chromatin remodeling complex SRCAP and p400/Tip60 (SWR1 in yeast) (Bonisch et al, 2012). Notably, the C‐terminal region of H2A.Z has been implicated in H2A.Z incorporation and function by an as‐yet‐unknown mechanism (Wang et al, 2011; Wratting et al, 2012). H2A.B‐NCP and Z.2.2‐NCP organize less DNA than canonical nucleosomes and adopt open nucleosome structures (Bonisch et al, 2012, 2; Arimura et al, 2013). These unique nucleosome structures are critical for H2A.B and Z.2.2 functions (Bonisch et al, 2012, 2; Arimura et al, 2013). While some specific residues of H2A.B and Z.2.2 have been implicated in open nucleosomes formation and chromatin folding (Shukla et al, 2011; Bonisch et al, 2012, 2), little is known about the structure of H2A.B‐NCP or Z.2.2‐NCP, not to mention the mechanism underlying the conformational variability.
In this study, we determined the cryo‐EM structure of human H2A.B‐NCP and Z.2.2‐NCP, which easily fall apart due to low stability. We performed biochemical analyses to identify the essential H2A.B and Z.2.2 residues and elucidate the mechanism by which H2A.B and Z.2.2 regulate nucleosome dynamics to fulfill their biological functions. Our study provides insights into the structure and function of the H2A.B‐NCP and Z.2.2‐NCP and sheds new light on the mechanism governing the open nucleosome formation.
Results
Structure determination of H2A.B‐NCP to atomic resolution
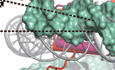
Our initial trial of nucleosome assembly using a regular H2A.B‐H2B dimer resulted in severe aggregation and material losses. To increase the reconstitution efficiency and overall yield of H2A.B‐NCP, we generated a linked H2B‐H2A.B (lnkH2B‐H2A.B) by fusing the N‐terminus of human H2A.B with the C‐terminus of human H2B. The linked H2B‐H2A adopts the same structure as H2B‐H2A dimer and can be reconstituted into histone chaperone complex and nucleosome properly (Hong et al, 2014, 1; Hu et al, 2017). We next assembled H2A.B‐NCP containing lnkH2B‐H2A.B using the 147‐bp Widom 601 sequence DNA (Lowary & Widom, 1998) (Fig EV1A and B). However, the assembly efficiency of H2A.B‐NCP is markedly lower than that of the canonical nucleosome, and the assembled H2A.B‐NCP falls apart during the process of GraFix, likely due to the dynamic nature of H2A.B‐NCP (Stark, 2010). To solve this problem, we performed the cross‐linking experiment before the gradient centrifugation, which is commonly used in the cryo‐EM sample preparation. This treatment improves the yield and homogeneity of H2A.B‐NCP samples for cryo‐EM study (Fig EV1C and D). We collected cryo‐EM data of H2A.B‐NCP and classified ~ 140,000 particles into several distinct classes (Fig EV1E and F). Further analyses revealed a dataset consisting of ~ 38,000 particles, leading to determination of the H2A.B‐NCP structure at 3.9 Å resolution (Figs 1A and B, and EV1G–I and Table 1).
Figure EV1. Biochemical characterization and data processing of the H2A.B‐NCP .
-
ANative‐PAGE analysis of the purified human H2A‐NCP and H2A.B‐NCP.
-
BSDS–PAGE analysis of the purified human H2A.B‐NCP. The linked H2B‐H2A.B dimer (lnkH2B‐H2A.B) was used for nucleosome reconstitution.
-
C, DSDS–PAGE (C) and Native‐PAGE (D) analyses of pre‐crosslinked H2A.B‐NCP subject to GraFix treatment. Peak fractions (18–19) containing intact H2A.B‐NCP were processed for further electron microscopy analysis. Experiments were repeated multiple times (n > 5).
-
DRepresentative cryo‐EM image (left) and 2D class averages (right) of H2A.B‐NCP particles.
-
EFlowchart of data processing of the H2A.B‐NCP.
-
FFSC curve of the final density maps of the H2A.B‐NCP. Reported resolutions were based on the FSC = 0.143 criteria.
-
GThe final 3D density map of H2A.B‐NCP shown in the indicated views is colored according to the local resolution estimated by the software ResMap.
-
HExamples of cryo‐EM density maps of the histones and DNA generated in Pymol.
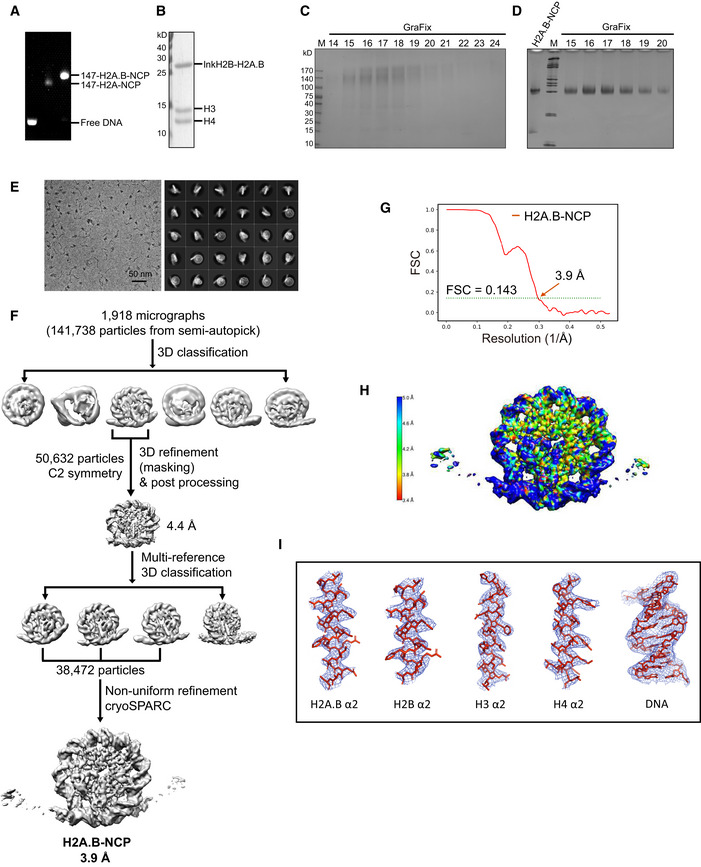
Figure 1. Overall structure of nucleosome containing human H2A.B.
- Cryo‐EM density map of the H2A.B‐NCP at 3.9 Å resolution: disc view (left) and gyre view (right).
- Atomic model of the H2A.B‐NCP in disc view (left) and gyre view (right). The nucleosome dyad is indicated with Φ. The superhelical turns of H2A.B nucleosomal DNA are designated as superhelical locations SHL ‐5 through SHL + 5.
- Cryo‐EM density map of the H2A.B‐NCP at 2.8 Å resolution: disc view (left) and gyre view (right).
Table 1.
Cryo‐EM data collection, refinement and validation statistics
| H2A.B‐NCP (EMD‐30078) (PDB 6M4H) | Pb‐H2A.B‐NCP C1 symmetry | Pb‐H2A.B‐NCP C2 symmetry (EMD‐30077) (PDB 6M4G) | Pb‐H2A.B‐NCP Class 2 | H2A.Z.2.2‐NCP (EMD‐30076) (PDB 6M4D) | |
|---|---|---|---|---|---|
| Data collection and processing | |||||
| Microscope | Titan Krios | Titan Krios | Titan Krios | Titan Krios | Titan Krios |
| Camera | K2 Summit | K2 Summit | K2 Summit | K2 Summit | K2 Summit |
| Magnification | 130,000 | 130,000 | 130,000 | 130,000 | 130,000 |
| Voltage (kV) | 300 | 300 | 300 | 300 | 300 |
| Pixel size (Å) | 1.04 | 1.04 | 1.04 | 1.04 | 1.04 |
| Electron exposure (e−/Å2) | 50 | 50 | 50 | 50 | 50 |
| Exposure per frame (e−/Å2) | 1.35 | 1.35 | 1.35 | 1.35 | 1.5 |
| Number of frames collected | 37 | 37 | 37 | 37 | 32 |
| Defocus range (μm) | −0.4 to −1.0 | −0.4 to −1.0 | −0.4 to −1.0 | −0.4 to −1.0 | −2.0 to −3.0 |
| Phase shift (π) | 0.2 to 0.8 | 0.2 to 0.8 | 0.2 to 0.8 | 0.2 to 0.8 | — |
| Symmetry imposed | C2 | C1 | C2 | C1 | C2 |
| Micrographs recorded/used (no.) | 1,918/1,700 | 3,130/2,086 | 3,130/2,086 | 3,130/2,086 | 1,201/1,020 |
| Initial particle images (no.) | 141,738 | 416,922 | 416,922 | 416,922 | 181,600 |
| Final particle images (no.) | 38,472 | 110,355 | 62,980 | 77,017 | 111,800 |
| Final reconstruction package | cryoSPARC | cisTEM | cisTEM | RELION‐3.0 | RELION‐3.0 |
| Map resolution (Å) | 3.9 | 3.2 | 2.8 | 6.2 | 4.4 |
| FSC threshold | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 |
| Refinement | |||||
| Initial model used (PDB) | 2CV5 | 2CV5 | 1F66 | ||
| Model composition | |||||
| Non‐hydrogen atoms | 9,420 | 9,032 | 10,303 | ||
| Protein residues | 656 | 657 | 682 | ||
| Ligands | 0.00 | 0.00 | 0.00 | ||
| Validation | |||||
| MolProbity score | 1.86 | 1.42 | 2.17 | ||
| Clashscore | 12.21 | 5.11 | 21.88 | ||
| Poor rotamers (%) | 0 | 0.71 | 0.00 | ||
| Ramachandran plot | |||||
| Favored (%) | 97.19 | 97.19 | 95.20 | ||
| Allowed (%) | 2.81 | 2.81 | 3.00 | ||
| Disallowed (%) | 0.00 | 0.00 | 1.80 | ||
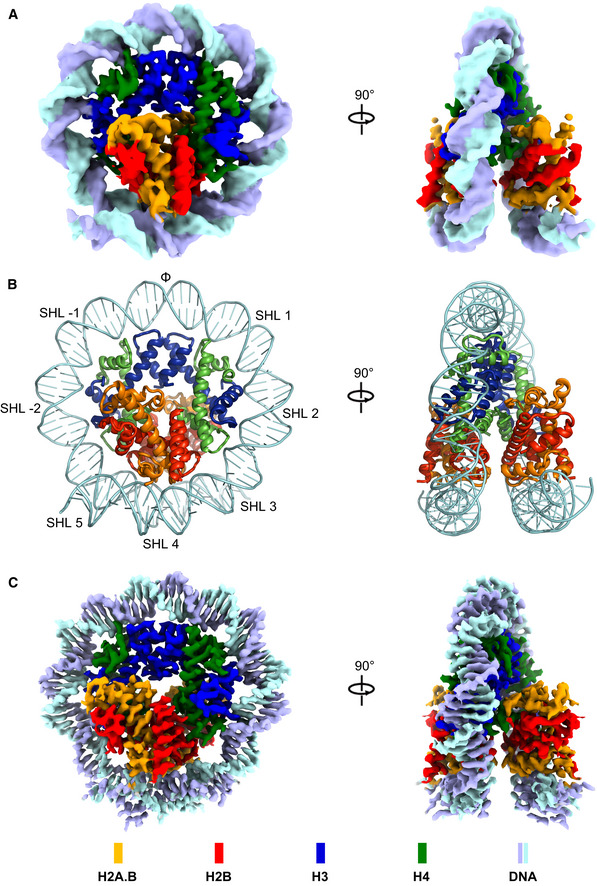
H2A.B‐NCP dissociation during cryogenic sample preparation limits the resolution of the H2A.B‐NCP structure. A previous study using a single‐chain antibody fragment (scFv) recognizing nucleosomes can successfully stabilize nucleosome and achieve the atomic resolution nucleosome cryo‐EM structures (Zhou et al, 2019a). In light of this finding, we predicted that non‐histone proteins with demonstrated nucleosome binding ability might stabilize H2A.B‐NCP by attenuating nucleosome opening. We screened chromatin‐associated proteins using EMSA assay and identified the human poly(ADP‐ribose) polymerase 1 DNA binding domain (PARP1 residues 1–373, designated as PARP1‐DBD) interacting with H2A.B‐NCP (Fig EV2A and B). Notably, PARP1 and linker histone H1 compete for nucleosome binding in vitro and exhibit a reciprocal pattern of the chromatin binding (Kim et al, 2004; Shukla et al, 2011). As PAPR1‐DBD has a demonstrated ability to interact with H2A.B‐NCP, we asked whether the linker histone can bind to H2A.B‐NCP as well. However, the globular domain of linker histone H5 (GH5 residues 22–102, designated as GH5‐GD) failed to interact H2A.B‐NCP, suggesting the binding is PAPR1‐DBD specific. Indeed, PARP1‐DBD prevents H2A.B‐NCP from falling apart during the Grafix and vitrification processes and substantially stabilizes H2A.B‐NCP (Fig EV2C and D).
Figure EV2. Biochemical characterization and data processing of the PARP1‐DBD bound H2A.B‐NCP (Pb‐H2A.B‐NCP).
-
ASDS–PAGE analysis of the purified human PARP1 DNA binding domain (hPARP1‐DBD).
-
BEMSA analyses of H2A.B‐NCP interacting with linker histone H5 and PARP1 DNA binding domain. Chicken H5 residues 22–102 (GH5‐GD) and human PARP1 DNA binding domain (hPARP1‐DBD) were added in two‐ or fourfold molar excess.
-
C, DSDS–PAGE (C) and Native‐PAGE (D) analyses of Pb‐H2A.B‐NCP subject to GraFix treatment. H2A.B‐NCP bound to PARP1 DNA binding domain (Pb‐H2A.B‐NCP) were subjected to 10–30% glycerol gradient centrifugation with 0–0.15% glutaraldehyde. Peak fractions (19–20) containing intact Pb‐H2A.B‐NCP were processed for further electron microscopy analysis. Experiments were repeated multiple times (n > 10).
-
DRepresentative cryo‐EM image (left) and 2D class averages (right) of Pb‐H2A.B‐NCP particles.
-
EFlow chart of data processing of the Pb‐H2A.B‐NCP particles. Highlighted with green color refers to the extra density corresponding to hPARP1‐DBD.
-
FThe final 3D density map of Pb‐H2A.B‐NCP shown in the indicated views is colored according to the local resolution estimated by the software ResMap.
-
GFSC curves of the final density maps of the Pb‐H2A.B‐NCP C1 (blue) and the Pb‐H2A.B‐NCP C2 (yellow). Reported resolutions were based on the FSC = 0.143 criteria.
-
HExamples of cryo‐EM density maps of the histones and DNA in Pb‐H2A.B‐NCP generated in Pymol.
The cryo‐EM sample prepared using PARP1‐DBD bound H2A.B‐NCP (Pb‐H2A.B‐NCP) presents higher yield and less heterogeneity. We collected a high‐quality dataset consisting of ~ 416,000 particles and determined a structure of Pb‐H2A.B‐NCP at 2.8 Å resolution (Figs 1C and EV2E–I). The density maps of the 3.9 Å H2A.B‐NCP and 2.8 Å H2A.B‐NCP are similar (Appendix Fig S1A). The atomic models show identical structural features, indicating the PARP1‐DBD binding does not perturb the H2A.B‐NCP structure (Appendix Fig S1B and C). Interestingly, the 2.8 Å H2A.B‐NCP lacks ~ 5 bp terminal DNA shown in the 3.9 Å H2A.B‐NCP, indicating this part of DNA is flexible (Appendix Fig S1).
Structural classifications of Pb‐H2A.B‐NCP revealed one dataset (~ 77,000 particles) that can be reconstructed to 6.2 Å resolution (Fig EV2F). The structure shows that PARP1‐DBD tethers the outer wrap DNA at SHL 6‐SHL 7 to the inner wrap DNA at SHL ‐2, suggesting that PAPR1‐DBD might stabilize H2A.B‐NCP by reducing DNA unwrapping (Fig EV2F). This observation coincides with the DNA binding ability of PAPR1‐DBD that consists of three Zinc finger domains (Ali et al, 2012). Moreover, structural classifications failed to reveal PARP1‐DBD density in other classes, suggesting a dynamic nature of PARP1‐DBD binding (Fig EV2F). The 3.9 Å H2A.B‐NCP structure is used for further analyses as it closely resembles the H2A.B‐NCP structure determined at atomic resolution.
Structure of H2A.B‐NCP is distinct from known nucleosomes
Structure analyses show that H2A.B‐NCP compacts ~ 103 bp of DNA wrapping around the histone octamer for ~ 1.2 left‐handed superhelical turns, in sharp contrast to the ~ 1.7 turns of DNA in the canonical nucleosome (Fig 1A and B). The H2A.B‐NCP structure is substantially distinct from those of all known nucleosomes, including the canonical nucleosome comprising ~ 145 bp of DNA (PDB: 3LZ0) and CENP‐A‐NCP comprising ~ 121 bp of DNA (PDB: 3AN2; Figs 2 and EV4A). The canonical nucleosome DNA contains 14 superhelical locations ranging from SHL 7 to SHL ‐7. In contrast, the H2A.B‐NCP structure displays the detachment of ~ 22 bp DNA through SHL 5 to SHL 7 and through SHL ‐5 to SHL ‐7, leaving only ~ 103 bp of DNA wrapping around the histone octamer (SHL 4 to SHL ‐4; Fig 2A). Notably, the last ~ 5 bp DNA at each end show less contact with histone octamer and form an outward tilting, suggesting that the SHL ± 5 DNA undergo conformational fluctuation (Fig 2A). In line with this observation, MNase digestion experiments show protection of ~ 103 bp DNA in H2A.B‐NCP (Fig EV3A and B). The unusual DNA detachment tends to cause H2A.B‐NCP disassembly, leading to highly unstable H2A.B‐NCP.
Figure 2. Conformational changes of H2A.B‐NCP in comparison with canonical nucleosome.
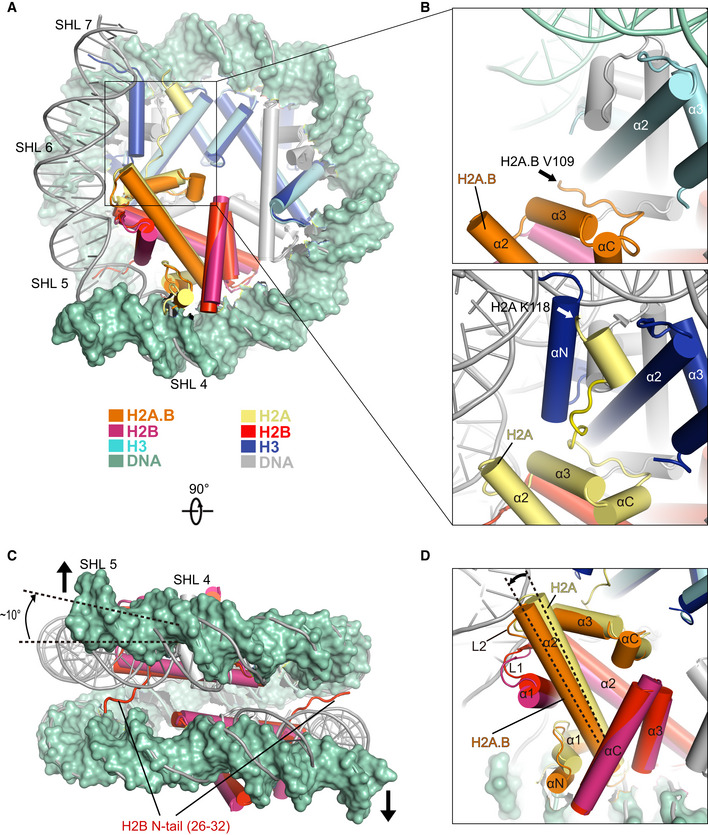
- Comparison of the cryo‐EM structure of H2A.B‐NCP with the structure of canonical nucleosome (PDB: 3LZ0) in disc view. Histone octamers containing H2A.B and canonical H2A were superimposed for comparison. The ˜ 103 bp DNA of H2A.B‐NCP in green color is shown in surface mode. The 145 bp DNA of canonical nucleosome is colored in gray. Histones H2A.B, H2B, H3, and DNA at SHL 5 through SHL 7 are highlighted to show the structural differences between two nucleosomes. Histones H2A.B, H2B, and H3 in H2A.B‐NCP are colored in orange, warmpink, and cyan. Their counterparts in canonical nucleosome are colored in yellow, red, and blue. Histone H4 is colored in gray.
- Close‐up view of the structures of H2A.B C‐terminal regions (top) and H2A C‐terminal regions (bottom). Black and white arrows indicate the last residue at the C‐terminus of H2A.B (V109) and H2A (K118) that are observable in the structures.
- Comparison of the cryo‐EM structure of H2A.B‐NCP with the structure of canonical nucleosome in gyre view. Arrows indicate the direction of H2A.B‐NCP DNA movement during nucleosome gaping transition. The dashed lines indicate the ˜ 10 degrees rotation of H2A.B DNA at SHL 5. H2B N‐tail refers to H2B residues 26–32 which are exclusively observed in the canonical nucleosome structure. Histones H2B in H2A.B‐NCP are colored in warmpink, Their counterparts in canonical nucleosome are colored in red. H2A.B‐NCP DNA in green is shown in surface mode for clear comparison.
- Close‐up view of H2A.B‐H2B dimer in the structural comparison. The secondary structure of H2A.B‐H2B dimer is indicated. The dashed lines highlight the tilt of H2A.B α2‐helix.
Figure EV4. Structural comparison of nucleosomes containing H2A.B, H2A.Z.2.2, and other histone variants.
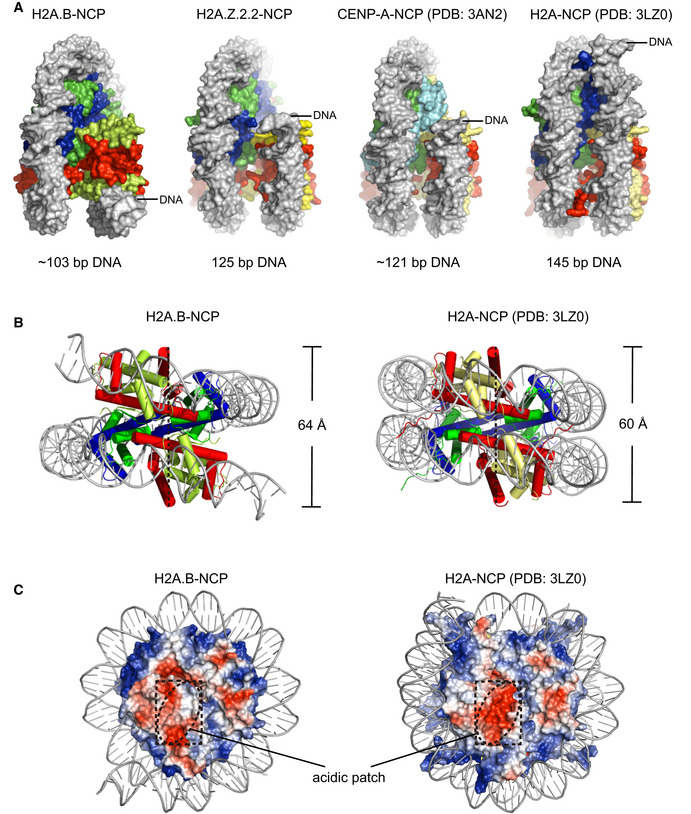
- Structural comparison of nucleosome containing H2A.B, H2A.Z.2.2, CENP‐A (PDB: 3AN2), and H2A (PDB: 3LZ0) in gyre view. The DNA visible in these structures are colored in gray for comparison. The histones H2A or variants, H2B, H3, or CENP‐A are colored in yellow, red, blue, and green, respectively. The nucleosomal DNA is colored in gray.
- Structural comparison of nucleosome containing H2A.B (left) and H2A (right, PDB: 3LZ0) in bottom view. The dashed lines indicate the distance between the two H2B S113.
- Structural comparison of nucleosome containing H2A.B (left) and H2A (right, PDB: 3LZ0) in disc view. Structures of core histones are shown in the surface mode and colored according to the electrostatic potential. The acidic patches of H2A.B‐NCP and canonical nucleosome are highlighted by dashed line for comparison.
Figure EV3. Characterization of DNA in H2A.B‐NCP .
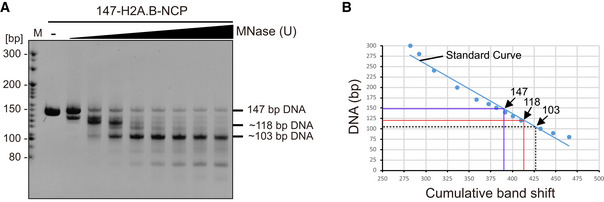
-
A, BProtection of H2A.B‐NCP DNA against MNase digestion. The H2A.B nucleosomes containing a 147‐bp Widom 601 sequence DNA were treated with increasing amounts of MNase at 30°C. After 30 min incubation, the reactions were quenched by adding EGTA and proteinase K. The protected DNA was extracted and analyzed by 10% Native‐PAGE with ethidium bromide staining (A). Length of DNA protected against MNase digestion. A standard curve is prepared by plotting the length of each DNA standard versus the mobility measured from the EMSA. The length of the 147‐bp DNA, 118‐bp DNA, and 103‐bp DNA are then calculated from the standard curve (B).
The histone octamer of H2A.B‐NCP undergoes large‐scale conformational changes and structural rearrangements (Fig 2A–D). The cryo‐EM densities corresponding to the H3 αN‐helix (residues 1–59), the H2A.B C‐terminal region (residues 109–114; Fig 2A and B), and the H2B N‐terminal region (residues 1–32; Fig 2C) are absent in the H2A.B‐NCP structure, suggesting these structural elements shown in canonical nucleosome are unfolded in the H2A.B‐NCP. Moreover, H2A.B‐H2B dimer tilts away from the histone H3–H4 tetramer, resulting in the distortion of the H2A.B‐NCP SHL ± 4 DNA (Fig 2A and D). These conformational changes increase the maximal distance between two H2A.B‐H2B dimers (64 Å) as compared to that observed in the canonical nucleosome (60 Å), indicative of the pronounced nucleosome gaping transition (Figs 2C and EV4B). Furthermore, H2A.B‐specific residues alter the surface charge of the H2A.B‐H2B dimer structure and generate a neutralized acidic patch in H2A.B‐NCP (Fig EV4C). Collectively, we concluded that H2A.B‐NCP adopts an unconventional, open nucleosome structure by compacting ~ 103 bp of DNA and undergoing both opening and gaping transition.
H2A.B presents 48% sequence identity to canonical H2A and lacks the last 19 residues in the C‐terminal region. We searched for H2A.B‐specific residues that are essential for H2A.B‐NCP structural changes by comparing structures of H2A.B‐NCP and canonical nucleosome (Fig 3A–D). In comparison with the well‐folded H2A C‐terminal region, the H2A.B C‐terminal region residues VAPGED (residues 109–114) are not observed in the H2A.B‐NCP structure (Fig 2B and Appendix Fig S3). The absence of H2A.B VAPGED sequence weakens the interactions between the H2A.B C‐terminal region, the H3 αN‐helix, and the DNA at SHL ± 6 and SHL ± 7 (Fig 2A and B). Moreover, unlike H2A residues K74, K75, which form multiple polar interactions with DNA at SHL ± 6 in the canonical nucleosome, H2A.B counterpart residues G78, E79 lose the positive charges that are critical for DNA interaction (Fig 3A). Similarly, the substitution of positively charged H2A residues R29, H31, and K36 with H2A.B counterparts Q33, E35, and E40 impair the bindings between H2A α1‐helix and DNA at SHL ± 4.5 in H2A.B‐NCP (Fig 3B). H2A.B residues reduce the association between H2A.B and DNA, leading to the unwrapping of nucleosomal DNA.
Figure 3. Close‐up view of H2A.B‐NCP structure in comparison with the canonical nucleosome structure.
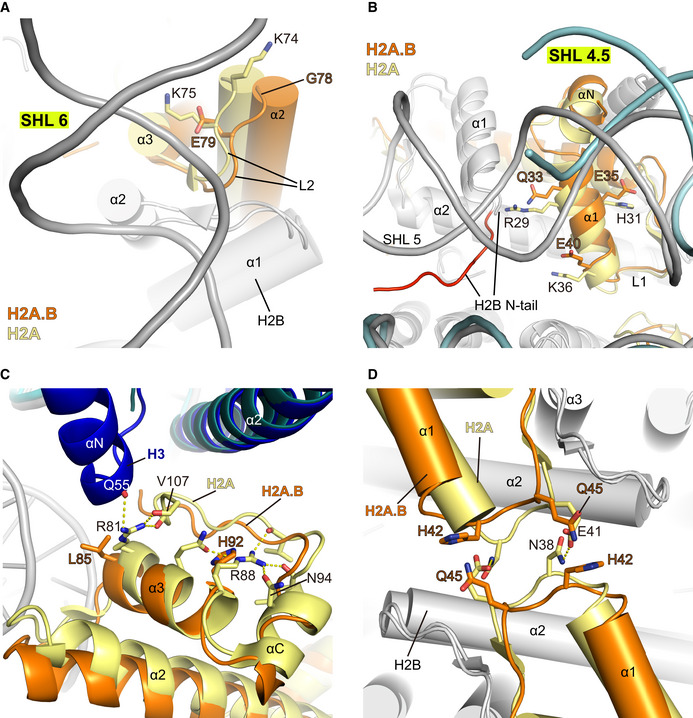
-
A–DStructures of H2A.B‐NCP and canonical nucleosome were superimposed to show local interaction differences. Displayed are H2A and H2A.B residues located close to the canonical nucleosomal DNA at super helical location SHL 6 (A) and SHL 4.5 (B), and H2A and H2A.B residues in α3–αC helices (C) and the L1 loop (D). H2A.B, H3, and DNA in H2A.B‐NCP are colored in orange, cyan, and light blue. H2A, H3, and DNA in canonical nucleosome are colored in yellow, blue, and gray, respectively. All other histones are colored in gray. H2B N‐tail highlighted in (B) refers to H2B residues 26–32 which are exclusively observed in the canonical nucleosome structure. The nucleosomal DNA is not shown in (D) for clear presentation.
The structural comparison revealed several structural changes that may affect histone–histone interactions (Fig 3C and D). First, the substitution of H2A residues R81, R88 by H2A.B counterparts L85, H92 abolish the interactions between H2A.B α3–αC helices and histone H3, due to the disruption of hydrogen bonding network formed in the canonical nucleosome (Fig 3C). Second, replacement of H2A L1‐loop residues N38, E41 with H2A.B counterparts H42, Q45 disrupt the contact between H2A residues N38, E41 (Fig 3D). Third, the H2B N‐terminal tails displayed in the canonical nucleosome are absent in H2A.B‐NCP. The absence of H2B N‐terminal tail disrupts the interactions between H2B residues 26–32 and two gyres of DNA (Fig 3B). Losses of binding between H2A.B and other core histones result in an outward tilting of H2A.B‐H2A dimers and a ~ 10 degrees kink of H2A.B‐NCP SHL ± 4 DNA, leading to H2A.B‐NCP gaping transition (Figs 2C and D, and EV4B).
H2A.B residues responsible for H2A.B‐NCP opening
To investigate the roles of the identified H2A.B residues in modulating H2A.B‐NCP structure, we interchanged H2A.B residues with their H2A counterparts and measured the resistance of nucleosomal DNA to MNase digestion (Fig 4A–E). The deletion of H2A residues 112–129 (termed H2A‐ΔC) slightly reduces the resistance of H2A‐NCP against MNase digestion (Fig 4A, B and D), suggesting a mild effect of H2A C‐terminal region residues on protecting DNA against MNase digestion. Conversely, the substitution of either the H2A N‐terminal domain (designated as H2A‐NTDH2A.B) or H2A docking domain (termed as H2A‐DDH2A.B) with their H2A.B counterparts decreases the resistance of H2A‐NCP to MNase digestion (Fig 4B and D). These observations are consistent with previous results, in which the H2A.B docking domain and the N‐terminal domain are both involved in H2A.B‐NCP destabilization (Doyen et al, 2006).
Figure 4. H2A.B residues dictate nucleosome resistance to MNase digestion.
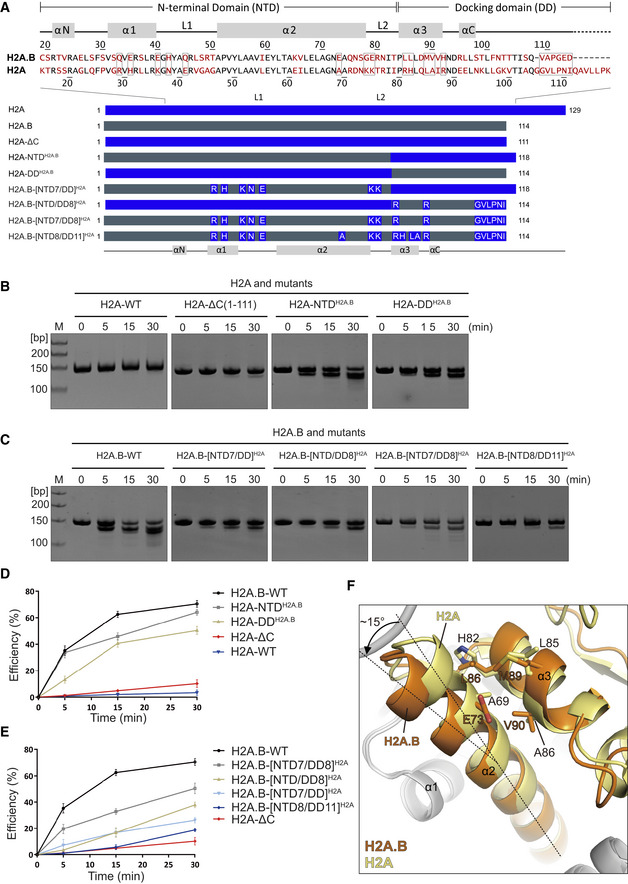
-
ATop: Sequence alignment of human H2A.B and H2A. Residues not conserved in H2A.B and H2A are colored in red. Secondary structures are indicated above. H2A.B N‐terminal domain (NTD) and the docking domain (DD) are indicated. H2A.B residues subject to mutagenesis analyses are indicated as boxes. Bottom: Schematic view of mutants used for MNase analysis. The H2A and H2A.B sequences are indicated by blue and gray, respectively.
-
B, CEffects of H2A mutations (B) and H2A.B mutations (C) on nucleosome resistance to MNase digestion. MNase digested nucleosomal DNA is analyzed by 10% Native‐PAGE. All MNase digestion experiments are repeated twice.
-
D, EGraph presentation of effects of H2A mutations (D) and H2A.B mutations (E) on nucleosome resistance to MNase digestion. MNase digestion efficiency for each sample is calculated from quantitation of the digested DNA against total DNA. All experiments are repeated twice. Data are mean ± SD, n = 2.
-
DStructural comparison of H2A.B α2‐α3 helices with their H2A counterparts. Structures of H2A‐H2B dimer in the canonical nucleosome (yellow, PDB: 3LZ0) and H2A.B‐H2B dimer in the H2A.B‐NCP (orange) were superimposed for comparison. The dashed lines indicate the ˜ 15 degrees kink of C‐terminal region of H2A.B α2 helix.
We next asked whether the diminished protection of H2A.B‐NCP could be restored to the same level as protection of H2A‐ΔC‐NCP by converse replacement of H2A.B residues with H2A counterparts on H2A.B backbones. The H2A.B‐[NTD7/DD]H2A mutant (containing the entire docking domain and the N‐terminal seven residues Q33, E35, E40, H42, Q45, G78, E79 of H2A) and H2A.B‐[NTD/DD8]H2A mutant (containing the entire NTD and the docking domain eight residues L85, H92, V109, A110, P111, G112, E113, and D114 of H2A) are generated for MNase digestion analyses (Fig 4A and C). Notably, H2A.B‐[NTD7/DD]H2A show higher MNase digestion resistance as compared to H2A‐NTDH2A.B, a H2A.B‐NCP mutant containing the entire docking domain of H2A (Fig 4B and C). Similarly, H2A.B‐NCP containing H2A NTD/DD8 is more resistant to MNase digestion than H2A.B‐NCP containing H2A NTD (i.e., H2A‐DDH2A.B; Fig 4B and C). Although these results underscore the importance of the selected H2A.B residues (NTD7/DD8) to form a less compact H2A.B‐NCP, replacement of these residues with H2A counterparts fail to restore the nucleosome resistance to MNase digestion (Fig 4C and E), suggesting that other H2A.B residues might be required to restore the H2A.B‐NCP resistance.
Further structural comparison revealed that the H2A.B α2‐helix in H2A.B‐NCP displays a ~ 15° tilt as compared to its H2A counterpart in the canonical nucleosome (Fig 4F). The H2A.B‐H2B dimer structure (PDB: 6A7U) (Dai et al, 2018) exhibits a similar tilt in H2A.B α2‐helix, suggesting the tilt is intrinsically formed in H2A.B‐H2B dimer (Appendix Fig S2A). Notably, H2A.B residues E73, L86, M89, and V90, which located at the binding interface of α2 and α3 helices, display enlarged side chains as compared to their H2A counterparts (A69, H82, L85, A86; Fig 4F). Therefore, the engagement of bulky H2A.B residues might induce the kink of H2A.B α2‐helix, leading to the outward tilt of H2A.B‐H2B dimers and the gaping transition of H2A.B‐NCP (Figs 2C and EV4B). To assess the destabilizing effect of these four H2A.B residues, we generated the H2A.B‐[NTD8/DD11]H2A mutant by combining H2A residues A69, H82, L85, and A86 with H2A.B‐[NTD7/DD8]H2A mutations (Fig 4A). Indeed, the MNase digestion resistance of H2A.B‐[NTD8/DD11]H2A mutation is increased to the same level as that of H2A‐ΔC‐NCP (Fig 4C and E). Collectively, these results suggested that H2A counterparts of all 19 H2A.B residues are required to restore the diminished DNA protection of H2A.B‐NCP.
Identification of the H2A regulating‐octamer‐folding sequence
A previous study has shown that H2A.B is unable to form histone octamer with other core histones (Bao et al, 2004). Consistent with this result, the size‐exclusion chromatography experiment results showed that lnkH2B‐H2A.B failed to form octamer with core histones H3 and H4, whereas the octamer assembly using linked H2B‐H2A (lnkH2B‐H2A) is normal (Fig 5A and B). Histone octamer assembly is not affected by the substitution of H2A NTD with H2A.B residues (H2A‐NTDH2A.B), nor deletion of H2A residues 112–129 (H2A‐ΔC; Fig 5B), but deletion of H2A residues 106–129 (termed H2A‐ΔROFΔC, see below) cause remarkable octamer folding defects (Fig 5B). These results suggested that H2A residues 106–111 play an essential role in histone octamer folding. Indeed, replacement of H2A residues 106–111 with H2A.B counterpart residues 109–114 (H2A‐ROFH2A.B, see below) is sufficient to disrupt histone octamer folding, indicating a regulatory role of the six consecutive residue in histone octamer folding. We therefore designated the six consecutive residue harbored by all H2A family members as the regulating‐octamer‐folding (ROF) sequence (Fig 5A and C).
Figure 5. Effect of H2A.B and H2A.Z.2.2 ROF on modulating histone octamer assembly.
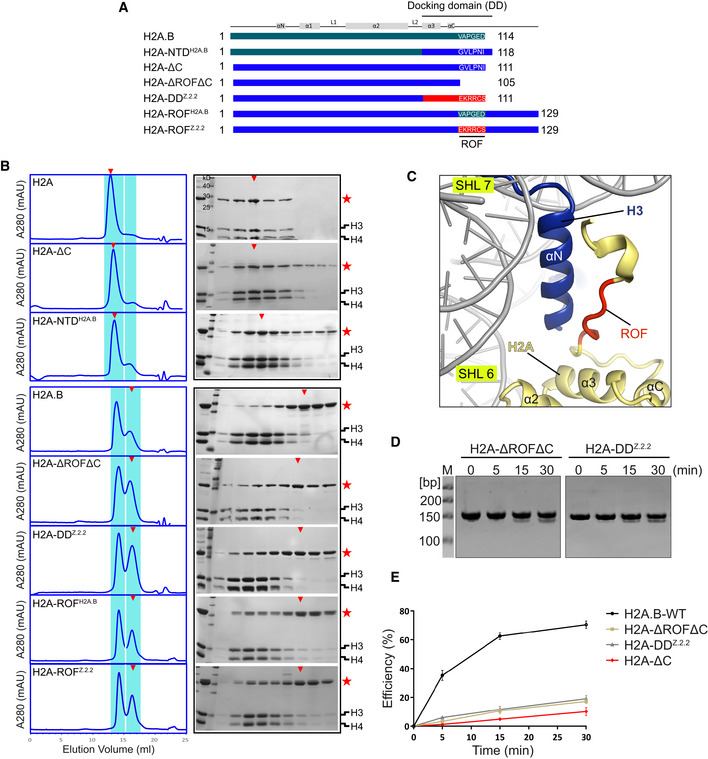
-
ASchematic view of canonical and variant H2A used for octamer assembly analysis. The H2A.B, H2A, and H2A.Z.2.2 sequences are indicated by gray, blue, and orange, respectively. The regulating‐octamer‐folding (ROF) sequences from different H2A histones are displayed for comparison.
-
BEffect of the canonical and noncanonical H2A ROF on histone octamer assembly. Left: Gel filtration chromatography profiles of octamers assembled by core histones including various H2A family members or mutants. Highlighted in blue shadow are chromatography fractions subject to SDS–PAGE analysis. Right: SDS–PAGE analysis of histone octamer integrity by visualizing histone stoichiometry. The stars indicate lnkH2B‐H2A containing different H2A family members or mutants present in chromatography fractions. The triangles indicate the chromatography fractions containing the majority of the lnkH2B‐H2A or their mutants.
-
CClose‐up view of structure of H2A C‐terminal regions shown in cartoon representation. Histones H2A, H3, and ROF region in H2A‐NCP (PDB: 3LZ0) are colored in yellow, blue, and red, respectively.
-
D, EEffects of the canonical and noncanonical H2A ROF on nucleosome resistance to MNase digestion. Displayed are Native‐PAGE results (D) and graph of MNase digestion efficiency calculated from quantitation of the digested DNA against total DNA (E). All experiments are repeated twice. Data are mean ± SD, n = 2.
Comparative analyses revealed highly conserved ROF sequences in major H2A family members but not in the variant H2A.B and Z.2.2 (Appendix Fig S3A–C). The C‐terminal region of Z.2.2 is 14 residues shorter than H2A.Z (Bonisch & Hake, 2012), and the last six residues form a noncanonical ROF (Appendix Fig S3B). Indeed, the substitution of H2A docking domain (H2A‐DDZ.2.2) or H2A ROF (H2A‐ROFZ.2.2) with Z.2.2 counterparts both disrupt histone octamer folding (Fig 5A and B). These results underscore the defective effect of H2A.B and Z.2.2 noncanonical ROFs on histone octamer assembly (Figs 5B and EV5E).
Figure EV5. Data processing of the H2A.Z.2.2‐NCP .
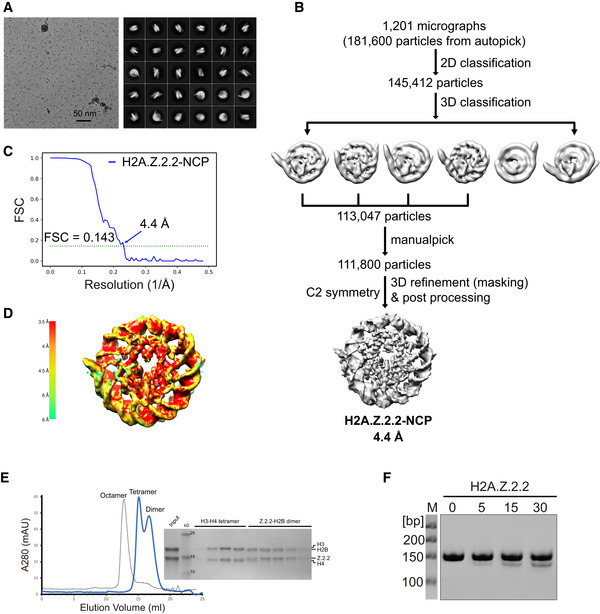
- Representative cryo‐EM image (left) and 2D class averages (right) of H2A.Z.2.2‐NCP particles.
- Flow chart of data processing of the Z.2.2‐NCP particles.
- FSC curves of the final density map of the Z.2.2‐NCP. Reported resolutions were based on the FSC = 0.143 criteria.
- The final 3D density map of Z.2.2‐NCP shown in the indicated views is colored according to the local resolution estimated by the software ResMap.
- Z.2.2 does not constitute stable histone octamers with H2B, H3, and H4 in vitro. Left: Size‐exclusion chromatography of refolding reactions using recombinant human H3, H4, and H2B proteins together with either H2A (dashed line) or Z.2.2 (blue line). Peaks corresponding to histone octamers, tetramers, or dimers are labeled, respectively. Right: Fractions corresponding to blue line were analyzed by 4–16% gradient SDS–PAGE and stained with Coomassie brilliant blue.
- Effects of the Z.2.2 on nucleosome resistance to MNase digestion. MNase digested nucleosomal DNA is analyzed by 10% Native‐PAGE.
Role of noncanonical ROF to facilitate nucleosome opening and histone replacement
Compared to the full‐length H2A.B, H2A‐ROFH2A.B displays less octamer folding defect (Fig 5B). This suggests that H2A.B residues outside the ROF region contribute to octamer unfolding and H2A.B ROF itself may not be sufficient to form the H2A.B‐NCP encompassing ~ 103 bp of DNA. Surprisingly, previous studies have shown that H2A.Z.2.2 lacking the canonical ROF and C‐terminal tail of H2A.Z indeed induces nucleosome opening (Bonisch et al, 2012, 2), indicating the Z.2.2 ROF induces the nucleosome opening, albeit to limited extents. To unveil the effect of noncanonical ROF on regulating nucleosome structure, we determine the cryo‐EM structure of nucleosome containing human H2A.Z.2.2 (Z.2.2‐NCP) at 4.4 Å resolution (Fig EV5A–D). The overall structure of Z.2.2‐NCP, which retains several features of the H2A.B‐NCP structure, is substantially distinct from that of the canonical nucleosome. Structural analyses show detachment of ~ 11 bp DNA at both entry/exit sites of Z.2.2‐NCP, leaving only ~ 125 bp of DNA wrapping around the histone octamer (Figs 6A and EV4A). Similar to H2A.B‐NCP structure, Z.2.2‐NCP structure show absence of the H3 αN‐helix and Z.2.2 ROF, which causes the disruption of interactions between the H3 αN‐helix and DNA at SHL ± 6 and SHL ± 7 (Fig 6B). Moreover, the absence of Z.2.2 ROF and H2B N‐terminal domains impair the binding of H2A.Z.2.2‐H2B dimer to H3–H4 tetramer or DNA at SHL ± 5 (Fig 6B and C). These structural changes cause the outward tilting of Z.2.2‐H2B dimers and the increases of H2B distance ranging from 60 to 63 Å, resulting in the gapping transition of Z.2.2‐NCP structure (Fig 6C). It is worth noting that Z.2.2‐NCP presents a less open structure as compared to H2A.B‐NCP. The Z.2.2‐NCP DNA at SHL ± 6 remains associated with the Z.2.2 L2‐loop (Fig 6A). In line with this observation, deletion of H2A ROF (H2A‐ΔROFΔC) and substitution with Z.2.2 ROF (H2A‐DDZ.2.2) slightly reduce the nucleosome resistance to MNase digestion, as does wild‐type Z.2.2, (Figs 5D and E, and EV5F), underscoring a subtle effect of noncanonical ROF on nucleosomal DNA protection. Together, the cryo‐EM structures of Z.2.2‐NCP and H2A.B‐NCP suggested that the noncanonical ROF induces substantial conformational changes in Z.2.2‐NCP.
Figure 6. H2A.Z.2.2 ROF modulates nucleosome dynamics and facilitates histone replacement.
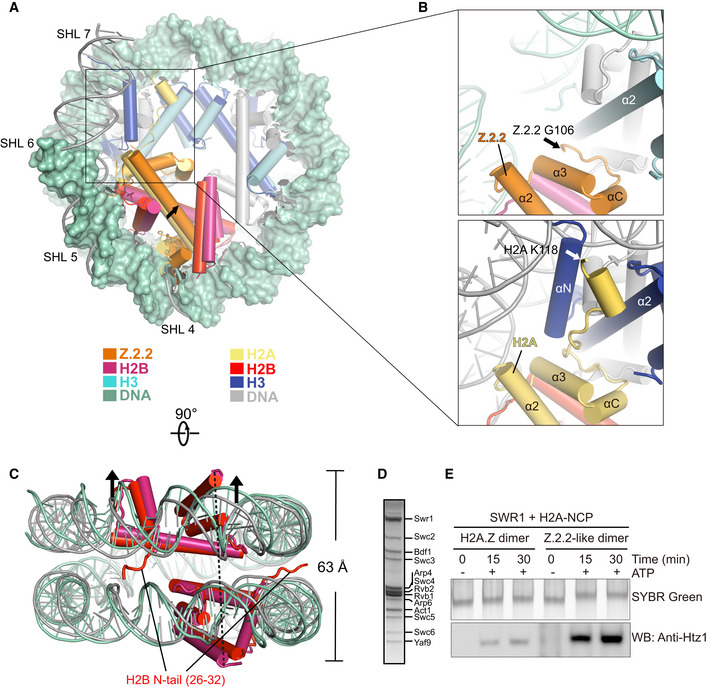
- Comparison of the cryo‐EM structure of H2A.Z.2.2‐NCP with the structure of canonical nucleosome (PDB: 3LZ0) in disc view. Histone octamers containing H2A.Z.2.2 and canonical H2A are superimposed for comparison. The 125 bp DNA of Z.2.2‐NCP in green color is shown in surface mode. The 145 bp DNA of canonical nucleosome is colored in gray. Histones H2A.B, H2B, H3, and DNA at SHL 6 through SHL 7 are highlighted to show the structural differences between two nucleosomes. The Z.2.2‐NCP histones are colored in orange, warmpink, and cyan. Their counterparts in canonical nucleosome are colored in yellow, red, and blue. Histone H4 is colored in gray.
- Close‐up view of the structures of Z.2.2 C‐terminal regions (top) and H2A C‐terminal regions (bottom). Black and white arrows indicate last residue at the C‐terminus of Z.2.2 (G106) and H2A (K118) that are observable in the structures.
- Comparison of the cryo‐EM structure of Z.2.2‐NCP with the crystal structure of canonical nucleosome (PDB: 3LZ0) in gyre view. Arrows indicate the direction of Z.2.2‐NCP DNA movement during nucleosome gaping transition. H2B N‐tail refers to H2B residues 26–32 which are exclusively observed in the canonical nucleosome structure. Histones H2B and DNA in Z.2.2‐NCP are colored in warmpink and green, their counterparts in canonical nucleosome are colored in red and gray.
- SDS–PAGE analysis of SWR1 complex purified from Saccharomyces cerevisiae.
- Effect of H2A.Z.2.2 ROF on facilitating histone replacement. Incorporations of yeast H2A.Z‐H2B dimers containing either the wild‐type H2A.Z or Z.2.2‐like H2A.Z were analyzed by SWR1‐catalyzed histone replacement assay. H2A.Z and Z.2.2‐like H2A.Z incorporated into nucleosome were resolved by 6% Native‐PAGE and detected by SYBR Green staining and Western blotting.
The chromatin incorporation of human H2A.Z and H2A.Z.2.2 are catalyzed by remodeling complexes SRCAP and p400/Tip60 (SWR1 in yeast) (Liang et al, 2016; Latrick et al, 2016, 1). Human Z.2.2 displays rapid chromatin turnover as compared to H2A.Z, suggesting that the chromatin deposition of Z.2.2 might be faster than H2A.Z (Bonisch et al, 2012, 2). Given that the H2A.Z and SRCAP or p400/Tip60 are highly conserved across eukaryotic species, we constructed a Z.2.2‐like H2A.Z by substituting yeast H2A.Z residues 113–133 with human Z.2.2 ROF residues. We next analyzed the replacement of yeast H2A by Z.2.2‐like H2A.Z using purified SWR1 complex (Fig 6D). Indeed, the exchange of Z.2.2‐like H2A.Z is more efficient than that with yeast H2A.Z, suggesting Z.2.2 ROF enhances the SWR1‐catalyzed histone variant exchange (Fig 6E and Appendix Fig S4). The exchange increase conferred by Z.2.2 ROF might be due to the structural changes of either Z.2.2‐H2B dimer or Z.2.2‐NCP. Overall, this finding provides a hint to link the effect of H2A ROF on a well‐defined biological function, given that the mechanism of H2A.Z incorporation is highly conserved in all eukaryotes. However, as H2A.Z.2.2 is a primate‐specific histone variant, histone exchange assay catalyzed by SRCAP and p400/Tip60 will be required to verify the impact of Z.2.2 ROF.
Discussion
It has been speculated for a long time that nucleosomes containing either H2A.B or Z.2.2 form the least stable nucleosomes with flexible DNA segments. The underlying mechanism by which H2A.B or Z.2.2 confers the flexibility to nucleosome remains elusive. In this study, we determine the cryo‐EM structures of H2A.B‐NCP and Z.2.2‐NCP at high resolution. Comparative analyses reveal nineteen H2A.B residues playing roles in forming the extended nucleosome structure. The noncanonical regulating‐octamer‐folding (ROF) sequence in H2A.B and Z.2.2 abrogate histone octamer assembly and contribute to destabilizing nucleosome. Moreover, The Z.2.2 ROF induces the formation of Z.2.2‐NCP, which contains ~ 125‐bp of DNA, and enhances the SWR1‐catalyzed H2A.Z exchange. Our study reveals the mechanism of how H2A.B and H2A.Z.2.2 confer dynamics to open nucleosomes and shed light on the structure and function of H2A.B‐NCP and Z.2.2‐NCP.
The H2A.B‐NCP structure displays several features not shown in the Z.2.2‐NCP structure. For example, H2A.B‐specific residues induce a kink of the α2 helix, which enhances the outward tilting of H2A.B‐H2B dimer. The kinked α2 helix is exclusively dictated by changes of intramolecular interactions within H2A.B‐H2B dimer, underscoring the role of internal plasticity of H2A.B‐H2B dimer in modulating H2A.B‐NCP dynamics. Conformational rearrangement of H2A‐H2B dimer was also described in the recent study (Bilokapic et al, 2018). Comparisons of H2A.B‐NCP to the unwrapped states of the canonical nucleosome reveals similar conformational changes of H2A.B and H2A (Fig EV4B and C), even though the tilted canonical H2A‐H2B only observed in the transiently formed unwrapped state nucleosome. Moreover, H2A.B‐specific residues form a neutralized acidic patch in H2A.B‐NCP, providing direct evidence to corroborate the speculation that H2A.B‐NCP's acidic patch fails to interact with the histone tails from neighboring nucleosomes (Fig EV4C). This observation is in coincidence with previous studies in which H2A.B naturally lacking an intact acidic patch inhibits the formation of the higher‐order structure of chromatin (Zhou et al, 2007).
The incorporation of histone variants in chromatin leads to specific histone residue displacements, which subsequently change the nucleosome structure and dynamics to fulfill their physiological function. Our findings reveal that H2A variants display impaired interactions with H3–H4 tetramer and nucleosomal DNA. Comparison of H2A.B‐NCP and Z.2.2‐NCP structures elucidate two distinct mechanisms by which histone variants H2A.B and Z.2.2 residues dictate the nucleosomes instability and flexibility. On the one hand, H2A.B ROF and Z.2.2 ROF abolish histone octamer assembly and enhance nucleosome local structure fluctuation (Appendix Fig S3C). On the other hand, H2A.B residues outside the ROF sequence disrupt multiple interactions between H2A.B‐H2B dimer and DNA, leading to large‐scale DNA detachment and a drastic decrease of nucleosome resistance to MNase digestion. Unlike Z.2.2 residues, H2A.B residues prevent the H2A.B‐H2B dimer from interacting with both H3–H4 tetramer and DNA, leading to a nucleosome with even more open structure. These results underscore the critical roles of H2A.B residues outside the ROF sequence in dynamic modulation.
H2A C‐terminal tail has been implicated in nucleosome stability regulation. A recent study showed that CENP‐C binds and destabilizes the H2A C‐terminal tail, which increases DNA unwrapping (Ali‐Ahmad et al, 2019). Post‐translational modifications at H2A.Z C‐terminus, such as mono‐ubiquitination, might contribute to the regulatory process as well (Henikoff & Smith, 2015). These data indicate the H2A C‐terminal tail regulates nucleosome stability. In this study, the change of ROF sequence and absence of the C‐terminal tail in H2A.B and H2A.Z.2.2 show effects on nucleosome stability. Importantly, our observation of the facilitating effect of yeast Z.2.2‐like H2A.Z on the SWR1‐catalyzed H2A.Z exchange rate is consistent with the results of previous studies that show the function of H2A.Z C‐terminal tail in regulating the association of H2A.Z with nucleosomes (Wang et al, 2011; Wratting et al, 2012). However, it remains unclear whether the increase of histone exchange rate is due to the dynamic nature of Z.2.2‐NCP or recognition changes of Z.2.2‐like histone dimer.
PARP1 is a highly abundant and ubiquitous chromatin‐associated enzyme in nucleus. PARP1‐DBD contains three Zinc finger domains (Zn1, Zn2, and Zn3) that can all bind to DNA (Kim et al, 2004; Muthurajan et al, 2014) and confer PARP1 the ability to recognize a diverse range of DNA breaks and atypical DNA secondary structures (Alemasova & Lavrik, 2019). Structural classifications of Pb‐H2A.B‐NCP reveal one dataset that can be reconstructed to 6.2 Å resolution, wherein the PARP1‐DBD was found to interact with both the out wrap and inner wrap of DNA (Fig EV2F). The low resolution precludes the determination of binding details for PARP1‐DBD. Notably, whether this binding mode is adopted by full‐length PARP1 remains unknown. Further studies are needed to answer this question. Nonetheless, the feature of PAPR1‐DBD tethering out wrap and inner wrap of DNA provides a rationale for H2A.B‐NCP stabilization by PARP1‐DBD, which prevents unstable H2A.B‐NCP from falling apart. This strategy may be applicable to the cryo‐EM structural study of other nucleosomes or nucleosome complexes.
Materials and Methods
Cloning, expression, and purification of recombinant proteins
To improve the stability of H2A.B‐H2B dimer, the human H2A.B and H2B were covalently linked into one expression cassette to make a linked H2B‐H2A.B (lnkH2B‐H2A.B), as reported in earlier structural studies, which showed this construct has no impact on the structures of histone dimers (Liang et al, 2016, 1; Hu et al, 2017; Dai et al, 2018). The lnkH2B‐H2A.B, lnkH2B‐H2A, and their mutant expression cassettes were cloned into bacterial expression plasmids (pET17b, Novagen) for protein production in Escherichia coli. In brief, BL21(DE3)‐Codon Plus‐RIPL cells were transfected with expression plasmids and grown in LB medium at 37°C until reaching an OD600 of 0.8. Protein expressions were induced by the addition of 0.5 mM IPTG for 4 h at 37°C. Cells were harvested by centrifugation at 4,670 g for 15 min at 4°C, and cell pellets were washed with 10 ml of cold PBS/l of culture. Cells were resuspended and lysed in 10 ml of lysis buffer (20 mM Tris–HCl pH 8.0, 0.5 M NaCl, 5 mM β‐ME) per liter of culture by passage through an EmulsiFlex‐C3 homogenizer (Avestin). The lysates were clarified by centrifugation, and the supernatants were bound to 4 ml SP sepharose fast flow beads (GE Healthcare). The SP beads were transferred to an Econo column (Bio‐Rad) and washed with lysis buffer. The target proteins were eluted using elution buffer (20 mM Tris–HCl pH 8.0, 1 M NaCl, 5 mM β‐ME) and further purified over HiTrap SP cation‐exchange column (GE Healthcare) equilibrated with lysis buffer using an ÄKTA FPLC system (GE Healthcare). Fractions were pooled and stored at −80°C for further use.
Human H2A.Z.2.2, H2B, H3.1, H4, and their mutants were produced as recombinant protein in E. coli BL21(DE3)‐Codon Plus‐RIPL (Novagen). Expressed histones were purified form the inclusion body using HiTrap SP column with ÄKTA FPLC (GE Healthcare) under denaturing condition (Luger et al, 1999).
Human PARP1‐DBD (1–373) with an N‐terminal His6‐SUMO tag was subcloned into bacterial expression plasmids (pET28a, Novagen) for protein production in E. coli BL21(DE3)‐Codon Plus‐RIPL (Novagen). Briefly, E.coli cells were transfected with expression plasmids and grown in LB medium at 37°C until reaching an OD600 of 0.6. Protein expression was induced by addition of 0.3 mM IPTG and 0.1 mM ZnCl2 overnight at 16°C. Cells were harvested by centrifugation at 4,000 RPM for 15 min at 4°C, and cell pellets were washed with 10 ml of cold PBS/l of culture. Cell was lysed in 15 ml of lysis buffer (20 mM Tris–HCl pH 8.0, 2 M NaCl, 25 mM imidazole and 5 mM β‐ME) per liter of culture and initially purified by Ni‐NTA affinity chromatography (Qiagen). The His6‐SUMO tag cleaved from the hPARP1‐DBD construct by Ulp1 protease and removed by reverse Ni‐NTA affinity chromatography. The protein was further purified over HiTrap SP cation‐exchange column (GE Healthcare) equilibrated with SP buffer (20 mM Tris–HCl pH 8.0, 150 mM NaCl, and 5 mM β‐ME) using an ÄKTA FPLC system (GE Healthcare). Fractions containing hPARP1‐DBD were pooled and dialyzed against buffer containing 20 mM Tris–HCl pH 8.0, 150 mM NaCl, and 5 mM β‐ME. The purified target proteins were stored at −80°C for further use.
Preparation of nucleosomal DNAs
Large‐scale quantities of 147‐bp Widom 601 DNA were purified as described previously from the pUC19 12 × 147 bp 601‐sequence using EcoRV restriction enzyme to digest the DNA into fragments (Luger et al, 1999). The backbone DNA was removed by polyethylene glycol (PEG) precipitation. The sequence of 147‐bp Widom 601 DNA is as follows:
ATCGAGAATCCCGGTGCCGAGGCCGCTCAATTGGTCGTAGACAGCTCTAGCACCGCTTAAACGCACGTACGCGCTGTCCCCCGCGTTTTAACCGCCAAGGGGATTACTCCCTAGTCTCCAGGCACGTGTCAGATATATACATCCGAT
Reconstitution of nucleosome
Nucleosomes were assembled by mixing DNA, lnkH2B‐H2A.B (or human H2A.Z.2.2‐H2B dimer), and human H3–H4 tetramer at 1:2.2:2.2 molar ratio in 2TEN buffer (20 mM Tris–HCl pH7.5, 2 M NaCl, 1 mM EDTA and 5 mM β‐ME), followed by dialysis in the same buffer with a gradual decrease in the NaCl concentration from 2 to 0.25 M over 18 h. The H2A.B nucleosome was heat re‐positioned by incubating the reconstituted nucleosome at 37°C for 2 h. All nucleosomes were purified from extra free DNA using DEAE‐5PW column with ÄKTA FPLC system (GE Healthcare). The reconstitution of nucleosomes containing histone mutants was identical to those of their wide type.
GraFix
To avoid the problems arising from dissociation of H2A.B nucleosome during ultracentrifugation, 200 μl of 10 μM 147‐H2A.B‐NCP was crosslinked in buffer containing 10 mM HEPES‐NaOH pH 7.5, 1 mM EDTA, and 0.15% glutaraldehyde for 1 h at 4°C. Subsequently, the NCP was applied onto the top of the gradient solution (10–28% glycerol gradient with 0–0.15% glutaraldehyde, in 10 mM HEPES‐NaOH pH 7.5 and 1 mM EDTA) and was centrifuged at 171,500 g (MLS‐50 Beckman) for 18 h at 4°C. Immediately, fractions were collected and checked by SDS–PAGE, Native‐PAGE, and electronic microscope in negative stain, followed by a one‐step buffer exchange to remove the glycerol. In contrast, regular grafix process was used for the treatment of 147‐Z.2.2‐NCP.
The PARP1‐DBD‐H2A.B‐NCP complex sample was prepared by adding 0.2 ml of 30 μM hPARP1‐DBD dropwise into 5 ml of 0.4 μM 147‐H2A.B‐NCP, followed by incubation for 0.5 h at 4°C. The sample was then concentrated to 200 μl at ~ 10 μM concentration, and applied onto the top of the gradient solution (10–30% glycerol gradient with 0–0.15% glutaraldehyde, in 10 mM HEPES‐NaOH pH 7.5 and 1 mM EDTA), and was centrifuged at 40,000 RPM (MLS‐50 Beckman) at 4°C for 18 h. The next steps were performed as described above.
Micrococcal nuclease digestion assay
To evaluate nucleosome stability, the 147‐H2A.B‐NCP, 147‐H2A‐NCP, and their mutants were digested with 0.08 U MNase in 400 μl reaction buffer (10 mM HEPES‐NaOH pH 7.5, 25 mM NaCl, and 1 mM CaCl2) at 30°C for 0, 5, 15, and 30 min, respectively. Each reaction was stopped and deproteinized by adding 8 μl of 0.2 M EGTA and 5 μl proteinase K (5 mg/ml), with incubating at 37°C for 30 min. DNA fragments were extracted, precipitated, and analyzed on 10% non‐denaturing polyacrylamide gels (in 0.5× TBE). The gels were stained with ethidium bromide. The band size of DNA fragments was quantitated with Gel Image System ver.4.2 (Tanon). The band intensity of DNA fragments was quantitated with Quantity One (Bio‐Rad).
To define the length of DNA that is actually protected by H2A.B‐NCP, H2A.B nucleosome (2 μg NCP per reaction) assembled with 147 bp Widom 601 was digested with increasing amounts of MNase (Sigma) in 400 μl reaction buffer (10 mM HEPES‐NaOH pH 7.5, 25 mM NaCl, and 1 mM CaCl2) for 30 min at 30°C. The next steps were identical to those described above.
Histone exchange assay
The SWR1 complex was purified with Swr1–3 × FLAG affinity purification, as described previously (Ranjan et al, 2013). The yeast histone purification and nucleosome reconstitution for this exchange assay were identical to human histones mentioned above. In vitro histone exchange assays were performed as previously described (Liang et al, 2016, 1). Briefly, 5 nM yeast nucleosomes, 2 nM SWR1 complex, 5 nM yeast H2A.Z‐H2B dimer (or Z.2.2‐like‐H2A.Z‐H2B dimer), and 2 mM ATP were dissolved into exchange buffer (25 mM HEPES‐KOH, pH 7.6, 0.37 mM EDTA, 0.35 mM EGTA, 10% glycerol, 0.017% NP‐40, 1 mM DTT, 70 mM KCl, 3.6 mM MgCl2, 100 μg/ml BSA, and protease inhibitors) and used in 20 μl reaction mixes. The reactions were quenched at the indicated time points with excess (100 ng) λ DNA. The reactions were subjected to 5% Native‐PAGE with SYBR Green staining. SYBR Green was directly detected in gels with a Typhoon 7000 (GE Healthcare). The gels were soaked in SDS buffer (25 mM Tris–HCl pH 7.5, 192 mM glycine and 0.1% SDS), followed by transferring onto PVDF membranes for Western blotting. Western blotting was performed using commercially available monoclonal antibody raised against the yeast H2A.Z (Htz1) epitope (Abcam, ab4626).
Cryo‐EM grid preparation and data collection
A drop of 3 μl sample was deposited onto a freshly glow discharged holey carbon copper grid (GIG R2/2 or Quantifoil R1.2/1.3, 300 mesh). Grids were blotted for 4–5 s (waiting time 5 s) under 100% humidity at 4°C before rapidly plunge frozen into a liquid ethane bath cooled by liquid nitrogen using a Mark IV Vitrobot (Thermo Fisher Sci). The datasets of both H2A.B‐NCP and Pb‐H2A.B‐NCP were collected on the Titan Krios microscope at 300 kV, equipped with a GIF quantum energy filter (slit width 20 eV), a K2 summit (Gatan) direct electron detector, and a Volta phase plate (Thermo Fisher Sci). Images were recorded in super‐resolution counting mode using Serial EM (Mastronarde, 2005) for semi‐automatic acquisition. The data collection with phase plate refers to the approach as previously described (Danev et al, 2017). The dataset of H2A.Z.2.2 was also collected on the Titan Krios microscope at 300 kV, but without using Volta phase plate. The magnification, pixel size, dose rate, and other Krios microscope settings are listed in Table 1.
Image processing
Image stacks of H2A.B‐NCP and Pb‐H2A.B‐NCP were recorded with a Volta phase plate. A total of 1,918 and 3,130 stacks were processed for motion correction with MotionCor2 (Zheng et al, 2017), and the CTF parameters for each micrograph were then calculated with Gctf (Zhang, 2016) and CTFFIND4 (Rohou & Grigorieff, 2015). According to the defocus and phase parameters, 1,700 and 2,086 micrographs were selected, respectively, for the following processing. Particle picking, 2D, and 3D classification were performed with twofold binned datasets, while in 3D reconstruction, the original images were used.
For data processing of H2A.B‐NCP, particles were selected automatically with Gautomatch (http://www.mrclmb.cam.ac.uk/kzhang) before more accurate manual picking, yielding a total of 141,738 particles. Subsequent analysis processes were performed with RELION 2.1.0 (Scheres, 2012). After several rounds of 3D classification, the classes with poor features were discarded and 50,632 particles were obtained. C2 symmetry was imposed on the best particle images, with a mask that encompasses the main part of nucleosome. H2A.B‐NCP was further refined to a density map at a resolution of 4.4 Å. After several further rounds of 3D classification with these particles in RELION 2.1.0, 38,472 of them were selected and a non‐uniform refinement was performed in cryoSPARC v2.14.2(Punjani et al, 2017), which resulted in a final density map at a resolution of 3.9 Å, with C2 symmetry imposed.
For data processing of Pb‐H2A.B‐NCP, the H2A.B‐NCP were stabilized by hPARP1‐DBD and more data were collected. The initial processing procedures were similar to the dataset of H2A.B‐NCP, resulting in 416,922 semi‐automated selected particles for further processing. After iterative rounds of 3D classification using RELION 2.1.0, the best three classes (239,144 particles) were combined and subjected to further 3D classification with a mask that only contains the NCP part. 110,355 good particles were selected for refinement in cisTEM, with C1 symmetry and C2 symmetry applied to improve the resolution to 3.2 and 3.0 Å, respectively. Through re‐importing the cisTEM particles into RELION for 3D classification, 62,980 particles were obtained for the final refinement in cisTEM (Grant et al, 2018), resulting in a resolution at 2.8 Å, with C2 symmetry imposed.
Meanwhile, a subset of particles containing clear density of hPARP1‐DBD (class 2, 77,017 particles) was subjected to final 3D reconstruction in RELION, resulting in a final density map at a resolution of 6.2 Å.
For data processing of 147‐H2A.Z.2.2‐NCP, the collection and processing steps refer to the dataset of H2A.B‐NCP, except for the usage of VPP in data collection, which resulted in a final density map at a resolution of 4.4 Å, with C2 symmetry imposed.
The global resolution calculations of all structures were based on the gold standard Fourier Shell Correlation (FSC) 0.143 criterion, with automatic B factor determined in RELION (Scheres, 2012). The local resolution estimation of the maps was calculated by ResMap (Kucukelbir et al, 2014).
Model building and refinement
The model building was first performed on H2A.B‐NCP using the map of Pb‐H2A.B‐NCP C2 (2.8 Å). The human nucleosome structure (PDB: 2CV5) (Tsunaka et al, 2005) was used as the initial template for the model building. The template was manually fitted into the density map in Chimera (Pettersen et al, 2004), followed by manual rebuilding using Coot (Emsley & Cowtan, 2004). Importantly, the densities of either nucleotides or histones are sufficient for the de novo building of the atomic model (Fig EV2I). The model was refined using Phenix.real_space_refine (Adams et al, 2002) with secondary structure restraints, geometry restraints, and DNA‐specific restraints applied. After iteratively rebuilding and refinement by COOT (Emsley & Cowtan, 2004) and PHENIX (Adams et al, 2002), the final structural model was obtained, and statistics are presented in Table 1.
For the 3.9 Å map of H2A.B‐NCP, model building was performed using the similar procedures, and the 2.8 Å model was used as the initial template. The atomic model for Z.2.2‐NCP is built using the similar steps described above. A crystal structure of nucleosome containing H2A.Z was taken as the initial model (PDB: 1F66) (Suto et al, 2000), and Pymol were used for figure preparation.
Author contributions
LD performed biochemical experiments. LS and YH assisted with LD prepared samples for structural study. LD and FW performed the histone exchange assay. MZ and ZG prepared cryo‐grids and collected data. CL, MZ, and ZG processed data and determined the structure. ZZ, PZ, and LD conceived the study and wrote the manuscript. All authors contributed to experimental design and commented on the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank Drs. Yawen Bai and David Tremethick for discussion and comments. Gang Ji, Xiaojun Huang and Boling Zhu from the Center for Biological Imaging (CBI), Institute of Biophysics, CAS for the help in cryo‐EM data collection. The study was supported by grants from the Ministry of Science and Technology of China (2019YFA0508902, 2017YFA0504700, 2015CB856200), grants from the National Natural Science Foundation of China (31521002, 31671344, 31425007, 31730023), and the Strategic Priority Research Program from Chinese Academy of Sciences (XDB08010100, XDB37010100).
The EMBO Journal (2021) 40: e105907
Contributor Information
Ping Zhu, Email: zhup@ibp.ac.cn.
Zheng Zhou, Email: zhouzh@ibp.ac.cn.
Data availability
The cryo‐EM density maps have been deposited in the Electron Microscopy Data Bank (https://www.ebi.ac.uk/pdbe/emdb): accession number EMD‐30078 for the H2A.B‐NCP, EMD‐30077 for the Pb‐H2A.B‐NCP and EMD‐30076 for the H2A.Z.2.2‐NCP. Atomic coordinates have been deposited in the Protein Data Bank: accession number PDB ID 6M4H (https://www.rcsb.org/structure/6M4H) for the H2A.B‐NCP, PDB ID 6M4G (https://www.rcsb.org/structure/6M4G) for the Pb‐H2A.B‐NCP and PDB ID 6M4D (https://www.rcsb.org/structure/6M4D) for the H2A.Z.2.2‐NCP.
References
- Adams PD, Grosse‐Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC et al (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D 58: 1948–1954 [DOI] [PubMed] [Google Scholar]
- Alemasova EE, Lavrik OI (2019) Poly(ADP‐ribosyl)ation by PARP1: reaction mechanism and regulatory proteins. Nucleic Acids Res 47: 3811–3827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali AAE, Timinszky G, Arribas‐Bosacoma R, Kozlowski M, Hassa PO, Hassler M, Ladurner AG, Pearl LH, Oliver AW (2012) The zinc‐finger domains of PARP1 cooperate to recognize DNA strand breaks. Nat Struct Mol Biol 19: 685–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali‐Ahmad A, Bilokapić S, Schäfer IB, Halić M, Sekulić N (2019) CENP‐C unwraps the human CENP‐A nucleosome through the H2A C‐terminal tail. EMBO Rep 20: e48913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews AJ, Luger K (2011) Nucleosome structure(s) and stability: variations on a theme. Annu Rev Biophys 40: 99–117 [DOI] [PubMed] [Google Scholar]
- Arimura Y, Kimura H, Oda T, Sato K, Osakabe A, Tachiwana H, Sato Y, Kinugasa Y, Ikura T, Sugiyama M et al (2013) Structural basis of a nucleosome containing histone H2A.B/H2A.Bbd that transiently associates with reorganized chromatin. Sci Rep 3: 3510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala R, Willhoft O, Aramayo RJ, Wilkinson M, McCormack EA, Ocloo L, Wigley DB, Zhang X (2018) Structure and regulation of the human INO80‐nucleosome complex. Nature 556: 391–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Y, Konesky K, Park YJ, Rosu S, Dyer PN, Rangasamy D, Tremethick DJ, Laybourn PJ, Luger K (2004) Nucleosomes containing the histone variant H2A.Bbd organize only 118 base pairs of DNA. EMBO J 23: 3314–3324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilokapic S, Strauss M, Halic M (2018) Histone octamer rearranges to adapt to DNA unwrapping. Nat Struct Mol Biol 25: 101–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonisch C, Hake SB (2012) Histone H2A variants in nucleosomes and chromatin: more or less stable? Nucleic Acids Res 40: 10719–10741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonisch C, Schneider K, Punzeler S, Wiedemann SM, Bielmeier C, Bocola M, Eberl HC, Kuegel W, Neumann J, Kremmer E et al (2012) H2A.Z.2.2 is an alternatively spliced histone H2A.Z variant that causes severe nucleosome destabilization. Nucleic Acids Res 40: 5951–5964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschbeck M, Hake SB (2017) Variants of core histones and their roles in cell fate decisions, development and cancer. Nat Rev Mol Cell Biol 18: 299–314 [DOI] [PubMed] [Google Scholar]
- Chittori S, Hong JJ, Saunders H, Feng HQ, Ghirlando R, Kelly AE, Bai YW, Subramaniam S (2018) Structural mechanisms of centromeric nucleosome recognition by the kinetochore protein CENP‐N. Science 359: 339–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai L, Xie X, Zhou Z (2018) Crystal structure of the histone heterodimer containing histone variant H2A.Bbd. Biochem Biophys Res Comm 503: 1786–1791 [DOI] [PubMed] [Google Scholar]
- Danev R, Tegunov D, Baumeister W (2017) Using the Volta phase plate with defocus for cryo‐EM single particle analysis. Elife 6: e23006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyen CM, Montel F, Gautier T, Menoni H, Claudet C, Delacour‐Larose M, Angelov D, Hamiche A, Bednar J, Faivre‐Moskalenko C et al (2006) Dissection of the unusual structural and functional properties of the variant H2A.Bbd nucleosome. EMBO J 25: 4234–4244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehara H, Kujirai T, Fujino Y, Shirouzu M, Kurumizaka H, Sekine S‐I (2019) Structural insight into nucleosome transcription by RNA polymerase II with elongation factors. Science 363: 744–747 [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model‐building tools for molecular graphics. Acta Crystallogr D 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Eustermann S, Schall K, Kostrewa D, Lakomek K, Strauss M, Moldt M, Hopfner K‐P (2018) Structural basis for ATP‐dependent chromatin remodelling by the INO80 complex. Nature 556: 386–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk SJ, Guo LY, Sekulic N, Smoak EM, Mani T, Logsdon GA, Gupta K, Jansen LET, Van Duyne GD, Vinogradov SA et al (2015) Chromosomes. CENP‐C reshapes and stabilizes CENP‐A nucleosomes at the centromere. Science 348: 699–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnung L, Vos SM, Wigge C, Cramer P (2017) Nucleosome‐Chd1 structure and implications for chromatin remodelling. Nature 550: 539–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant T, Rohou A, Grigorieff N (2018) cisTEM, user‐friendly software for single‐particle image processing. Elife 7: e35383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Smith MM (2015) Histone variants and epigenetics. Cold Spring Harb Perspect Biol 7: a019364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong J, Feng H, Wang F, Ranjan A, Chen J, Jiang J, Ghirlando R, Xiao TS, Wu C, Bai Y (2014) The catalytic subunit of the SWR1 remodeler is a histone chaperone for the H2A.Z‐H2B dimer. Mol Cell 53: 498–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q, Botuyan MV, Cui G, Zhao D, Mer G (2017) Mechanisms of ubiquitin‐nucleosome recognition and regulation of 53BP1 chromatin recruitment by RNF168/169 and RAD18. Mol Cell 66: 473–487.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi T, Li A, Eirin‐Lopez JM, Zhao M, Missiaen K, Abbott DW, Meistrich M, Hendzel MJ, Ausio J (2010) H2A.Bbd: an X‐chromosome‐encoded histone involved in mammalian spermiogenesis. Nucleic Acids Res 38: 1780–1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MY, Mauro S, Gévry N, Lis JT, Kraus WL (2004) NAD+‐dependent modulation of chromatin structure and transcription by nucleosome binding properties of PARP‐1. Cell 119: 803–814 [DOI] [PubMed] [Google Scholar]
- Kucukelbir A, Sigworth FJ, Tagare HD (2014) Quantifying the local resolution of cryo‐EM density maps. Nat Methods 11: 63–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latrick CM, Marek M, Ouararhni K, Papin C, Stoll I, Ignatyeva M, Obri A, Ennifar E, Dimitrov S, Romier C et al (2016) Molecular basis and specificity of H2A.Z‐H2B recognition and deposition by the histone chaperone YL1. Nat Struct Mol Biol 23: 309–316 [DOI] [PubMed] [Google Scholar]
- Liang XP, Shan S, Pan L, Zhao JC, Ranjan A, Wang F, Zhang ZQ, Huang YZ, Feng HQ, Wei D et al (2016) Structural basis of H2A.Z recognition by SRCAP chromatin‐remodeling subunit YL1. Nat Struct Mol Biol 23: 317–323 [DOI] [PubMed] [Google Scholar]
- Liu X, Li M, Xia X, Li X, Chen Z (2017) Mechanism of chromatin remodelling revealed by the Snf2‐nucleosome structure. Nature 544: 440–445 [DOI] [PubMed] [Google Scholar]
- Lowary PT, Widom J (1998) New DNA sequence rules for high affinity binding to histone octamer and sequence‐directed nucleosome positioning. J Mol Biol 276: 19–42 [DOI] [PubMed] [Google Scholar]
- Luger K, Rechsteiner TJ, Richmond TJ (1999) Expression and purification of recombinant histones and nucleosome reconstitution. Methods Mol Biol 119: 1–16 [DOI] [PubMed] [Google Scholar]
- Mastronarde DN (2005) Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol 152: 36–51 [DOI] [PubMed] [Google Scholar]
- Muthurajan UM, Hepler MRD, Hieb AR, Clark NJ, Kramer M, Yao T, Luger K (2014) Automodification switches PARP‐1 function from chromatin architectural protein to histone chaperone. Proc Natl Acad Sci USA 111: 12752–12757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo TTM, Ha T (2015) Nucleosomes undergo slow spontaneous gaping. Nucleic Acids Res 43: 3964–3971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612 [DOI] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA (2017) cryoSPARC: algorithms for rapid unsupervised cryo‐EM structure determination. Nat Methods 14: 290–296 [DOI] [PubMed] [Google Scholar]
- Ranjan A, Mizuguchi G, FitzGerald PC, Wei D, Wang F, Huang Y, Luk E, Woodcock CL, Wu C (2013) Nucleosome‐free region dominates histone acetylation in targeting SWR1 to promoters for H2A.Z replacement. Cell 154: 1232–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohou A, Grigorieff N (2015) CTFFIND4: fast and accurate defocus estimation from electron micrographs. J Struct Biol 192: 216–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulland Y, Ouararhni K, Naidenov M, Ramos L, Shuaib M, Syed SH, Lone IN, Boopathi R, Fontaine E, Papai G et al (2016) The flexible ends of CENP‐A nucleosome are required for mitotic fidelity. Mol Cell 63: 674–685 [DOI] [PubMed] [Google Scholar]
- Sansoni V, Casas‐Delucchi CS, Rajan M, Schmidt A, Bonisch C, Thomae AW, Staege MS, Hake SB, Cardoso MC, Imhof A (2014) The histone variant H2A.Bbd is enriched at sites of DNA synthesis. Nucleic Acids Res 42: 6405–6420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SHW (2012) RELION: implementation of a Bayesian approach to cryo‐EM structure determination. J Struct Biol 180: 519–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla MS, Syed SH, Goutte‐Gattat D, Richard JLC, Montel F, Hamiche A, Travers A, Faivre‐Moskalenko C, Bednar J, Hayes JJ et al (2011) The docking domain of histone H2A is required for H1 binding and RSC‐mediated nucleosome remodeling. Nucleic Acids Res 39: 2559–2570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark H (2010) GraFix: stabilization of fragile macromolecular complexes for single particle Cryo‐EM. Methods Enzymol 481: 109–126 [DOI] [PubMed] [Google Scholar]
- Suto RK, Clarkson MJ, Tremethick DJ, Luger K (2000) Crystal structure of a nucleosome core particle containing the variant histone H2A.Z. Nat Struct Biol 7: 1121–1124 [DOI] [PubMed] [Google Scholar]
- Tachiwana H, Kagawa W, Shiga T, Osakabe A, Miya Y, Saito K, Hayashi‐Takanaka Y, Oda T, Sato M, Park SY et al (2011) Crystal structure of the human centromeric nucleosome containing CENP‐A. Nature 476: 232–235 [DOI] [PubMed] [Google Scholar]
- Tolstorukov MY, Goldman JA, Gilbert C, Ogryzko V, Kingston RE, Park PJ (2012) Histone variant H2A.Bbd is associated with active transcription and mRNA processing in human cells. Mol Cell 47: 596–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunaka Y, Kajimura N, Tate S, Morikawa K (2005) Alteration of the nucleosomal DNA path in the crystal structure of a human nucleosome core particle. Nucleic Acids Res 33: 3424–3434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AY, Aristizabal MJ, Ryan C, Krogan NJ, Kobor MS (2011) Key functional regions in the histone variant H2A.Z C‐terminal docking domain. Mol Cell Biol 31: 3871–3884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willhoft O, Ghoneim M, Lin C‐L, Chua EYD, Wilkinson M, Chaban Y, Ayala R, McCormack EA, Ocloo L, Rueda DS et al (2018) Structure and dynamics of the yeast SWR1‐nucleosome complex. Science 362: eaat7716 [DOI] [PubMed] [Google Scholar]
- Winkler C, Steingrube DS, Altermann W, Schlaf G, Max D, Kewitz S, Emmer A, Kornhuber M, Banning‐Eichenseer U, Staege MS (2012) Hodgkin's lymphoma RNA‐transfected dendritic cells induce cancer/testis antigen‐specific immune responses. Cancer Immunol Immunother 61: 1769–1779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wratting D, Thistlethwaite A, Harris M, Zeef LAH, Millar CB (2012) A conserved function for the H2A.Z C terminus. J Biol Chem 287: 19148–19157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K (2016) Gctf: real‐time CTF determination and correction. J Struct Biol 193: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache J‐P, Verba KA, Cheng Y, Agard DA (2017) MotionCor2: anisotropic correction of beam‐induced motion for improved cryo‐electron microscopy. Nat Methods 14: 331–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou JS, Fan JY, Rangasamy D, Tremethick DJ (2007) The nucleosome surface regulates chromatin compaction and couples it with transcriptional repression. Nat Struct Mol Biol 14: 1070–1076 [DOI] [PubMed] [Google Scholar]
- Zhou B‐R, Yadav KNS, Borgnia M, Hong J, Cao B, Olins AL, Olins DE, Bai Y, Zhang P (2019a) Atomic resolution cryo‐EM structure of a native‐like CENP‐A nucleosome aided by an antibody fragment. Nat Commun 10: 2301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou K, Gaullier G, Luger K (2019b) Nucleosome structure and dynamics are coming of age. Nat Struct Mol Biol 26: 3–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Data Availability Statement
The cryo‐EM density maps have been deposited in the Electron Microscopy Data Bank (https://www.ebi.ac.uk/pdbe/emdb): accession number EMD‐30078 for the H2A.B‐NCP, EMD‐30077 for the Pb‐H2A.B‐NCP and EMD‐30076 for the H2A.Z.2.2‐NCP. Atomic coordinates have been deposited in the Protein Data Bank: accession number PDB ID 6M4H (https://www.rcsb.org/structure/6M4H) for the H2A.B‐NCP, PDB ID 6M4G (https://www.rcsb.org/structure/6M4G) for the Pb‐H2A.B‐NCP and PDB ID 6M4D (https://www.rcsb.org/structure/6M4D) for the H2A.Z.2.2‐NCP.