

Abstract
Mutations in centrosome genes deplete neural progenitor cells (NPCs) during brain development, causing microcephaly. While NPC attrition is linked to TP53‐mediated cell death in several microcephaly models, how TP53 is activated remains unclear. In cultured cells, mitotic delays resulting from centrosome loss prevent the growth of unfit daughter cells by activating a pathway involving 53BP1, USP28, and TP53, termed the mitotic surveillance pathway. Whether this pathway is active in the developing brain is unknown. Here, we show that the depletion of centrosome proteins in NPCs prolongs mitosis and increases TP53‐mediated apoptosis. Cell death after a delayed mitosis was rescued by inactivation of the mitotic surveillance pathway. Moreover, 53BP1 or USP28 deletion restored NPC proliferation and brain size without correcting the upstream centrosome defects or extended mitosis. By contrast, microcephaly caused by the loss of the non‐centrosomal protein SMC5 is also TP53‐dependent but is not rescued by loss of 53BP1 or USP28. Thus, we propose that mutations in centrosome genes cause microcephaly by delaying mitosis and pathologically activating the mitotic surveillance pathway in the developing brain.
Keywords: centrosome, DNA damage, microcephaly, mitotic surveillance pathway, TP53 activation
Subject Categories: Cell Cycle, Neuroscience
A mechanism that delays mitosis upon centrosomal loss in cultured cells appears to also underlie the in vivo consequences for brain size and neural progenitor proliferation in developing mouse brains.
Introduction
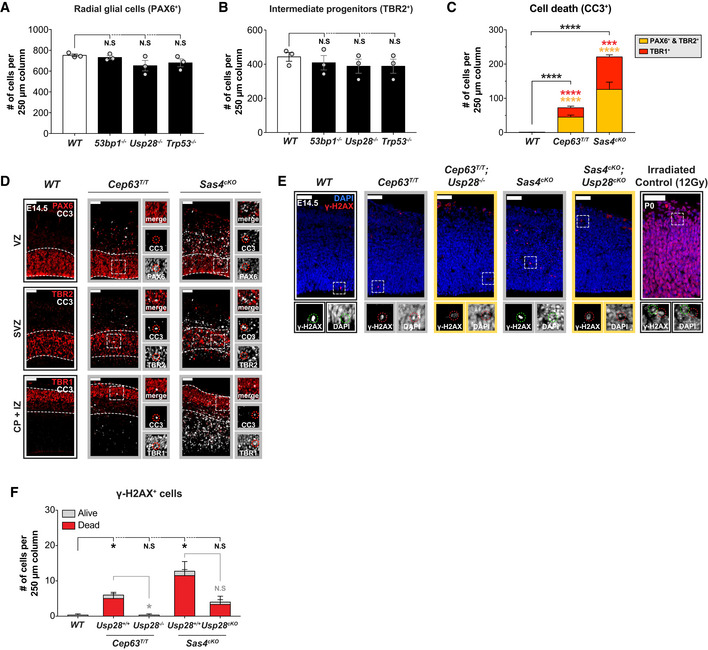
Neocortical development relies on the extensive proliferation of neural progenitor cells (NPCs) during embryonic development (Noctor et al, 2001; Florio & Huttner, 2014). Radial glial cells (RGCs) are located in the ventricular zone (VZ) and constitute the major subtype of NPC within the developing neocortex (Malatesta et al, 2000; Noctor et al, 2001; Heins et al, 2002). During cortical development, RGCs undergo proliferative symmetric divisions to replenish themselves and asymmetric divisions to produce a second type of NPC termed intermediate progenitors (IPs) (Miyata et al, 2004; Noctor et al, 2004; Franco & Muller, 2013). IPs reside in the sub‐ventricular zone (SVZ) and are thought to produce the majority of cortical neurons (Haubensak et al, 2004; Englund et al, 2005; Kowalczyk et al, 2009). Newborn neurons migrate outward toward the cortical plate giving rise to the inside‐out formation of the lamina layers of the mature neocortex (Rakic, 1972; Noctor et al, 2001). Defects in NPC divisions result in the generation of incorrect numbers of progenitors, which in turn influences brain size and function (Feng & Walsh, 2004; Lizarraga et al, 2010; McIntyre et al, 2012; Chen et al, 2014; Insolera et al, 2014; Marjanovic et al, 2015; Ding et al, 2019; Shi et al, 2019; Sheehan et al, 2020).
Autosomal recessive primary microcephaly (MCPH) is an inherited disorder of fetal brain growth characterized by a dramatically reduced cerebral cortex that is architecturally normal (Jayaraman et al, 2018). MCPH is caused by a depletion of the NPC pool and death of neurons during embryonic development, both of which lead to the generation of fewer mature neurons in the developed cortex (Faheem et al, 2015). So far, MCPH has been linked to mutations in ~ 25 genes (Jayaraman et al, 2018; Degrassi et al, 2019). Remarkably, many of these genes encode proteins required for centrosome duplication, underscoring a critical role of these organelles in the production of normal neuronal populations in the cortex (Marthiens & Basto, 2020).
Centrosomes are microtubule‐nucleating organelles with important roles in interphase and in forming the mitotic spindle. Each centrosome comprises a pair of centrioles, which assemble a surrounding protein matrix known as the pericentriolar material (PCM) that harbors proteins required for microtubule nucleation (Woodruff et al, 2014). Forming the core of centrosomes, a mature centriole can dock at the plasma membrane and form a basal body, which templates the formation of cilia (Firat‐Karalar & Stearns, 2014; Breslow & Holland, 2019). Multiple mouse models with mutations in centrosome proteins have been found to exhibit microcephaly (Lizarraga et al, 2010; Gruber et al, 2011; McIntyre et al, 2012; Chen et al, 2014; Insolera et al, 2014; Marjanovic et al, 2015; Jayaraman et al, 2016; Ding et al, 2019; Lin et al, 2020). Analysis of these animals revealed that NPCs are depleted by both apoptosis and the precocious onset of neurogenic divisions. Importantly, for several of these models, microcephaly can be rescued by deletion of TP53, highlighting a central role of this transcription factor in NPC attrition and microcephaly pathogenesis (Insolera et al, 2014; Marjanovic et al, 2015; Lin et al, 2020). Nevertheless, the pathway responsible for TP53 activation in NPCs with defective centrosomes remains unknown.
Recent work has established that in vitro, centrosome loss triggers activation of a USP28‐53BP1‐TP53 signaling axis hereafter referred to as the mitotic surveillance pathway (Fong et al, 2016; Lambrus et al, 2016; Meitinger et al, 2016). Cultured cells with defects in centrosome number or function take longer to assemble a bipolar spindle, resulting in an increased mitotic duration (Bazzi & Anderson, 2014; Lambrus et al, 2015; Wong et al, 2015). This, in turn, triggers a G1 arrest in daughter cells to prevent the expansion of potentially unfit progeny. The current evidence suggests that centrosome defects activate the mitotic surveillance pathway by prolonging mitosis (Uetake & Sluder, 2010; Fong et al, 2016; Lambrus et al, 2016; Meitinger et al, 2016). Nevertheless, it remains unclear in which context the mitotic surveillance pathway functions in vivo.
Consistent with work in cell culture, depletion of centrosome proteins in the developing mouse brain leads to an increased mitotic index in NPCs (Gruber et al, 2011; Insolera et al, 2014; Marjanovic et al, 2015; Pilaz et al, 2016; Ding et al, 2019; Shi et al, 2019). In addition, prolonging mitosis in NPCs with pharmacological agents or through depletion of the exon junction complex protein MAGOH, leads to increased death or premature differentiation of the progeny from these divisions (Pilaz et al, 2016; Mitchell‐Dick et al, 2020). The intriguing link between extended mitotic duration and NPC cell fate motivated us to ask whether activation of the mitotic surveillance pathway is responsible for the depletion of NPC in microcephalic brains with centrosome defects.
Here, we show that NPCs with centrosome defects delay in mitosis but undergo normal chromosome segregation. The increase in mitotic duration in NPCs activates the mitotic surveillance pathway and suppresses the proliferation of the progeny from these divisions. Disabling the mitotic surveillance pathway prevents TP53 activation and restores the expansion of the NPC pool and brain size in two microcephaly mouse models with centrosome defects. Finally, we show that loss of the non‐centrosome protein SMC5 leads to microcephaly that is not rescued by the inactivation of the mitotic surveillance pathway. Since mutations in centrosome and spindle proteins are the major cause of primary microcephaly, we propose that activation of the mitotic surveillance pathway is a central mechanism underlying microcephaly pathogenesis in human patients.
Results
Neural progenitors with centrosome defects delay in mitosis
To investigate the impact of centrosome defects on NPC divisions, we analyzed two published microcephaly mouse models with decreased levels of the centrosome proteins CEP63 or SAS4 (Bazzi & Anderson, 2014; Insolera et al, 2014; Marjanovic et al, 2015). CEP63 localizes at the base of the parent centriole, and mutations in Cep63 cause Seckel syndrome, which is characterized by both growth retardation and microcephaly (Sir et al, 2011). SAS4/CENPJ/CPAP is a centriole protein and mutations in Sas4 cause microcephaly in humans (Bond et al, 2005).
Mice that are homozygous for a gene‐trap insertion in the Cep63 gene (Cep63T/T) exhibit growth retardation and microcephaly (Marjanovic et al, 2015). Consistently, we observed that the telencephalic area of Cep63T/T animals was reduced by 26% and cortical thickness was reduced by 19% at P60 (Figs 1A and B, and EV1A and B). Conditional deletion of Sas4 from NPCs using Nestin‐Cre (hereafter referred to as Sas4cKO) produced a more severe microcephaly phenotype leading to a 30% reduction in telencephalic area and a 17% reduction in cortical thickness at P14 (Figs 1C and D, and EV1C and D) (Insolera et al, 2014). As previously reported, Sas4cKO mice also lack cilia and develop severe hydrocephalus and cerebellar hypoplasia (Chizhikov et al, 2007; Insolera et al, 2014). As a consequence, 33% of Sas4cKO mice failed to survive past P14 (Fig EV3D).
Figure 1. NPCs with centrosome defects delay in mitosis.
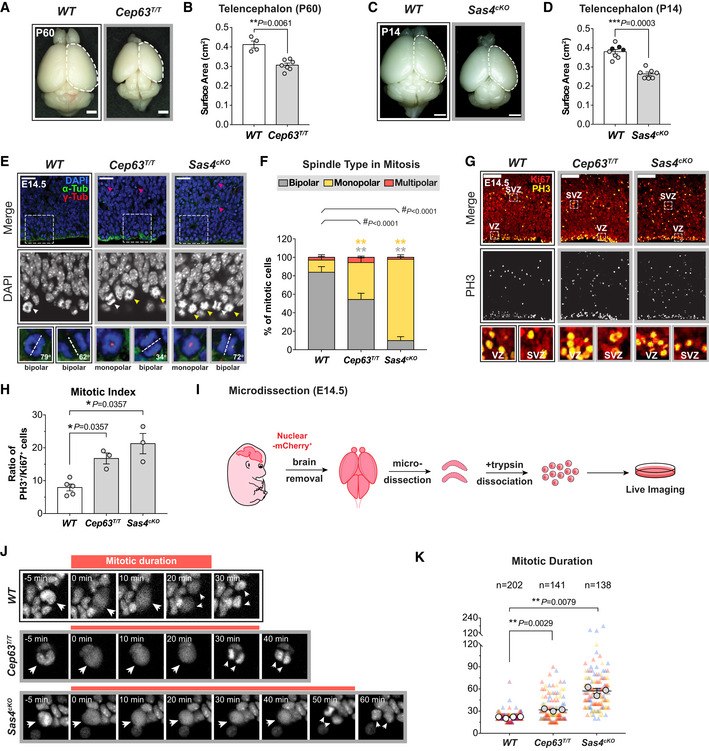
- Representative whole mount images of WT and Cep63T/T brains at P60. Scale bar = 0.2 cm.
- Telencephalon area of WT and Cep63T/T brains. WT littermates N = 4, Cep63T/T N = 7; two‐tailed Mann–Whitney t‐test.
- Representative whole mount images of WT and Sas4cKO brains at P14. Scale bar = 0.2 cm.
- Telencephalon area of WT and Sas4cKO brains. WT littermates N = 8, Sas4cKO N = 7; two‐tailed Mann–Whitney t‐test. Black circles represent Nestin‐Cre+ animals.
- Representative images of E14.5 WT, Cep63T/T, and Sas4cKO cortices stained with antibodies against α‐tubulin (green), γ‐tubulin (red), and DAPI (blue). Magenta arrows indicate dead cells; white and yellow arrows indicate cells with bipolar and monopolar spindles, respectively. Insets showing zoomed in view of DAPI and 2 representative cells. Dashed lines showing the orientation of the cleavage plane relative to the ventricular surface. Scale bar = 25 μm.
- Graph showing the percentage of bipolar, monopolar, and multipolar cells at the ventricular surface of E14.5 cortices. WT n = 201 cells, N = 3 embryos; Cep63T/T n = 312 cells, N = 3 embryos; Sas4cKO n = 296 cells, N = 3 embryos; #, chi‐square test with *, post hoc analysis, comparisons are made to WT.
- E14.5 cortices stained with antibodies against Ki67 (red) and phosphorylated‐Histone H3 (PH3, yellow). Insets showing zoomed in view of cells in the ventricular zone (VZ) and sub‐ventricular zone (SVZ). Scale bar = 100 μm.
- Ratio of mitotic (PH3+) to cycling (Ki67+) cells in E14.5 cortices. WT N = 5, Cep63T/T N = 3, Sas4cKO N = 3; two‐tailed Mann–Whitney t‐test.
- Protocol for the establishment of primary NPC cultures for live imaging.
- Representative images from live‐cell movies of Nuclear‐mCherry+ WT, Cep63T/T, and Sas4cKO NPCs. The timing of mitosis began at nuclear envelope breakdown (NEBD) and finished at nuclear envelope reformation. White arrows track the mitotic events of the mother cell from NEBD to the generation of daughter cells following cytokinesis.
- Plot showing the duration of mitosis in WT, Cep63T/T, and Sas4cKO NPCs. Triangles represent individual cells; triangles of the same color represent cells derived from the same embryo. Circles represent the average mitotic duration of cells from each embryo. WT n = 202 cells, N = 4 embryos; Cep63T/T n = 141 cells, N = 3 embryos; Sas4cKO n = 138 cells, N = 3 embryos; two‐tailed Welch’s t‐test.
Data information: All data represent the means ± SEM. *P < 0.05; **< 0.01; ***< 0.001; ****< 0.0001 and not significant indicates P > 0.05. See also Fig EV1.
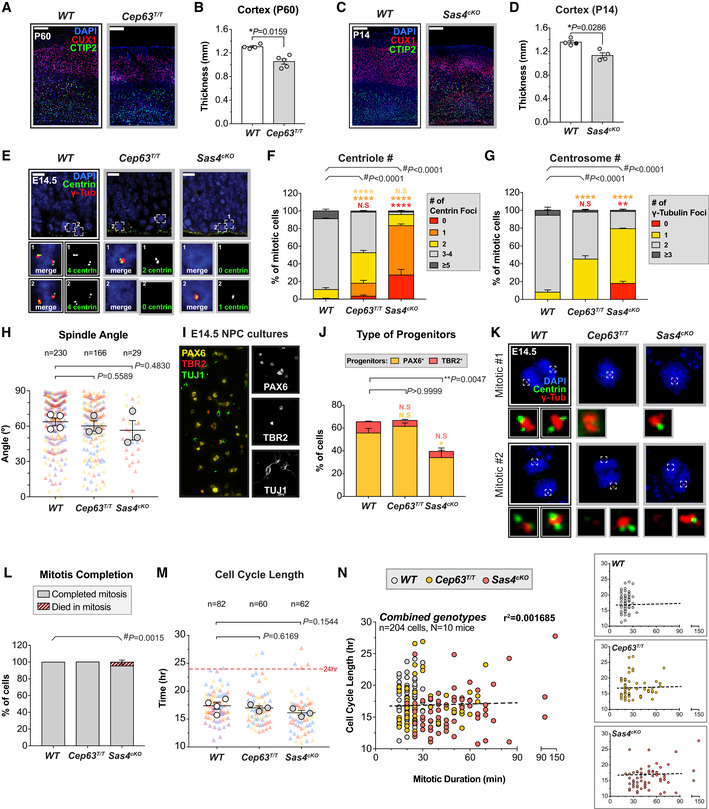
Figure EV1. Depletion of SAS4 or CEP63 leads to centrosome loss and reduced cortical size in adult mice. Related to Fig 1 .
-
AWT and Cep63T/T cortices at P60 stained with antibodies against the deep layer marker CTIP2 (green), superficial layer marker CUX1 (red) and DAPI (blue). Scale bar = 200 μm.
-
BCortical thickness of P60 brains of the indicated genotypes. WT littermates N = 4, Cep63T/T N = 5; two‐tailed Mann–Whitney test.
-
CWT and Sas4cKO cortices at P14 stained with antibodies against the deep layer marker CTIP2 (green), superficial layer marker CUX1 (red) and DAPI (blue). Scale bar = 200 μm.
-
DCortical thickness of P14 brains of the indicated genotypes. WT littermates N = 4, Sas4cKO N = 4; two‐tailed Mann–Whitney test.
-
EWT, Cep63T/T and Sas4cKO cortices at E14.5 stained with antibodies against centrin (green), γ‐tubulin (red) and DAPI (blue). Insets showing zoomed in view of 2 representative cells. Scale bar = 25 μm.
-
F, GQuantification of the number of (F) centrin foci and (G) γ‐tubulin foci in mitotic cells in the VZ of E14.5 cortices. WT n = 175 cells, N = 4 embryos; Cep63T/T n = 150 cells, N = 3 embryos; Sas4cKO n = 150 cells, N = 3 embryos; #, chi‐square test with *, post hoc analysis, comparisons are made to WT.
-
HPlot showing the spindle angle of dividing NPCs relative to the ventricular surface in WT, Cep63T/T and Sas4cKO at E14.5. Triangles represent individual cells; triangles of the same color represent cells derived from the same embryo. Circles represent the average spindle angle of mitotic cells from each embryo. WT n = 230 cells, N = 4 embryos; Cep63T/T n = 166 cells, N = 3 embryos; Sas4cKO n = 29 cells, N = 3 embryos; two‐tailed Welch’s t‐test. Note that the frequency of bipolar spindle formation is lower in Cep63T/T and Sas4cKO brains.
-
IRepresentative image from a disassociated NPC culture derived from an E14.5 WT brain stained with antibodies against the radial glial cell marker PAX6 (yellow), intermediate progenitor marker TBR2 (red) and differentiated neuron marker TUJ1 (green).
-
JGraph showing the percentage of radial glial cells (PAX6+, yellow) and intermediate progenitor cells (TBR2+, red) present in NPC cultures derived from E14.5 brains of the indicated genotypes. WT N = 6, Cep63T/T N = 3, Sas4cKO N = 5; multiple t‐tests, comparisons are made to WT.
-
KRepresentative images of mitotic cells in disassociated NPC cultures derived from E14.5 WT, Cep63T/ T and Sas4cKO brains stained with antibodies against centrin (green), γ‐tubulin (red) and DAPI (blue). Insets showing zoomed in view of the spindle poles.
-
LGraph showing the fate of mitotic NPCs of the indicated genotypes. WT n = 165 cells, N = 4 embryos; Cep63T/T n = 130 cells, N = 3 embryos; Sas4cKO n = 117 cells, N = 3 embryos. #, chi‐square test.
-
MGraph showing the total cell cycle length in WT, Cep63T/T, and Sas4cKO NPCs. The timing of the cell cycle began at NEBD of the mother cell and finished at NEBD in one of the two daughter cells. Triangles represent individual cells; triangles of the same color represent cells derived from the same embryo. Circles represent the average cell cycle length of cells from each embryo. Dashed line is set at 24 h. WT n = 82 cells, N = 4 embryos; Cep63T/T n = 60 cells, N = 3 embryos; Sas4cKO n = 62 cells, N = 3 embryos; two‐tailed Welch’s t‐test.
-
NGraphs showing cell cycle length as a function of mitotic duration in individual NPCs dissociated from WT (gray), Cep63T/T (yellow), and Sas4cKO (red) brains. The dashed line represents the best‐fit linear regression function (r 2 = 0.0017) for all genotypes combined.
Data information: All data represent the means ± SEM. *P < 0.05; **< 0.01; ****< 0.0001 and not significant indicates P > 0.05.
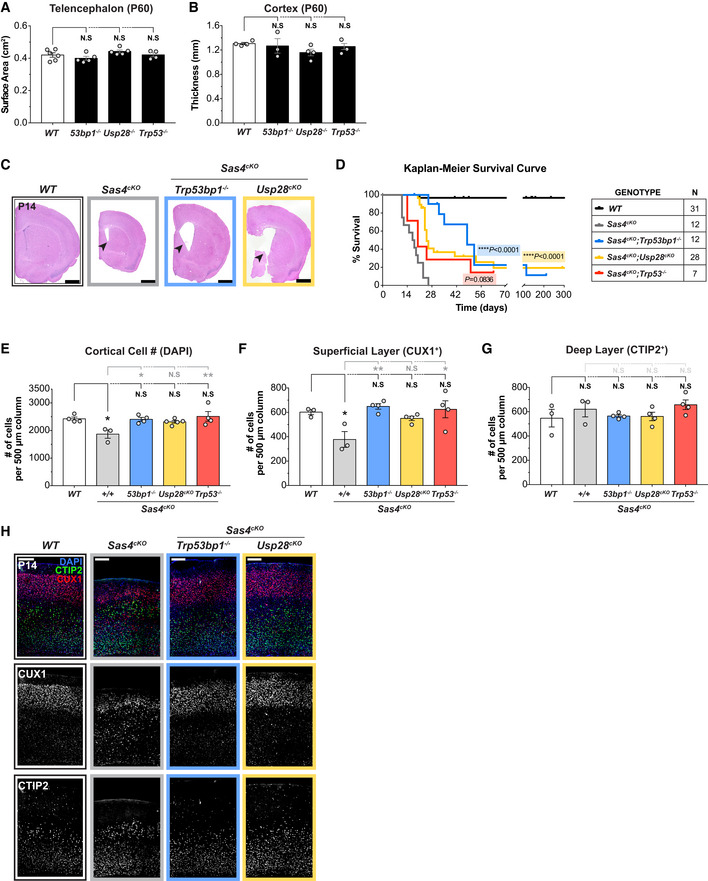
Figure EV3. Ablation of the mitotic surveillance pathway restores brain size and increases survival but does not rescue cilia defects in Sas4cKO animals. Related to Fig 3 .
-
ATelencephalon area of P60 brains of the indicated genotypes. WT littermates N = 6, Trp53bp1 −/− N = 5, Usp28 −/− N = 5, Trp53 −/− N = 4; one‐way ANOVA with post hoc analysis. 4 WT data points are from Fig 1B and shown alongside for comparison.
-
BCortical thickness of P60 brains of the indicated genotypes. WT littermates N = 4, Trp53bp1 −/− N = 3, Usp28 −/− N = 4, Trp53 −/− N = 3; one‐way ANOVA with post hoc analysis. WT data are from Fig EV1B and shown alongside for comparison.
-
CRepresentative histology images of P14 brains of the indicated genotypes. Arrows indicate the enlarged ventricles caused by the lack of motile cilia. Scale bar = 0.2 cm.
-
DKaplan–Meier survival analysis of Sas4cKO, Sas4cKO;Trp53bp1 −/−, Sas4cKO;Usp28cKO, Sas4cKO;Trp53 −/− animals compared to WT littermates. P values were calculated using the log‐rank test.
-
EQuantification of total number of cells within a 500 μm‐width column of cortices at P14. WT littermates N = 4, Sas4cKO N = 3, Sas4cKO;Trp53bp1 −/− N = 4, Sas4cKO;Usp28cKO N = 5, Sas4cKO;Trp53 −/− N = 4; one‐way ANOVA with post hoc analysis.
-
F, GQuantification of the number of cells in the (F) superficial layer (CUX1+) and (G) deep layer (CTIP2+) within a 500 μm‐width column of P14 cortices. WT littermates N = 3, Sas4cKO N = 3, Sas4cKO;Trp53bp1 −/− N = 4, Sas4cKO;Usp28cKO N = 4, Sas4cKO;Trp53 −/− N = 4; one‐way ANOVA with post hoc analysis.
-
HP14 cortices of the indicated genotypes stained with antibodies against the deep layer marker CTIP2 (green), superficial layer marker CUX1 (red) and DAPI (blue). Scale bar = 200 μm.
Data information: All data represent the means ± SEM. *P < 0.05; **< 0.01; ****< 0.0001 and not significant indicates P > 0.05.
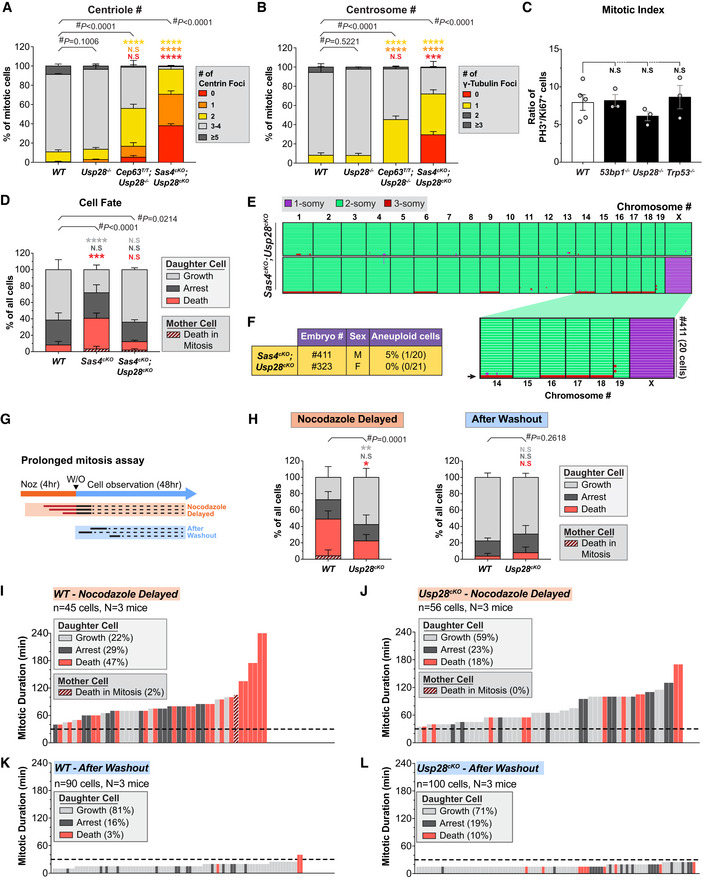
To examine how the loss of CEP63 and SAS4 disrupted centriole duplication and spindle organization in vivo, we analyzed dividing NPCs at the VZ in E14.5 cortices. Loss of CEP63 led to a decline in centriole and centrosome number and a concomitant increase in the percentage of monopolar spindles in dividing NPCs, phenotypes that were even more severe in Sas4cKO embryos (Figs 1E and F, and EV1E–G). Surprisingly, centrosome defects in Cep63T/T and Sas4cKO mice did not grossly disrupt the orientation of NPC divisions relative to the ventricular surface (Fig EV1H). To determine if centrosome loss leads to an increased mitotic duration in vivo, we calculated the fraction of phosphorylated H3‐Ser10‐positive (PH3+) mitotic cells among the Ki67+ cycling progenitors. The mitotic index of Cep63T/T and Sas4cKO developing brains were increased by 2.1‐ to 2.7‐fold compared to control brains, consistent with a delayed progression through mitosis due to spindle assembly defects (Fig 1G and H).
To establish if NPCs from Cep63T/T and Sas4cKO embryos successfully completed mitosis following a mitotic delay, we performed live imaging of dissociated Nuclear‐mCherry+ progenitors from control, Cep63T/T and Sas4cKO brains (Fig 1I). Cultures generated from embryonic brains consisted of TBR2+ intermediate progenitors (5–10%) and PAX6+ radial glial cells (55–61% in WT and Cep63T/T, 34% in Sas4cKO) (Fig EV1I and J). As expected, centrosome loss and monopolar spindle configurations were observed in cultured Cep63T/T and Sas4cKO NPCs (Fig EV1K). Nevertheless, time‐lapse imaging showed that > 95% of these progenitors successfully completed mitosis and divided into two daughter cells (Fig EV1L). However, while control NPCs had an average mitotic duration of 22 min, Cep63T/T and Sas4cKO NPCs took an average of 10 and 35 min longer to divide, respectively (Fig 1J and K). This increase in mitotic duration did not significantly alter the total cell cycle length, indicating that the duration of other cell cycle phases is not affected by the loss of CEP63 or SAS4 (Fig EV1M and N). Collectively, these data show that Cep63T/T and Sas4cKO NPCs delay in mitosis and that the length of the mitotic delay scales with the severity of centrosome loss.
Delays in NPC mitosis lead to increased frequency of cell death in the progeny
We next evaluated if the mitotic delay observed in NPCs leads to an alteration in the fate of progeny cells. To assess this, we tracked the progeny of NPCs after mitotic exit and determined if they divided, arrested, or underwent cell death (Movies EV1 and EV2). While 60% of control progeny re‐entered mitosis within the time frame of our movies, only 38% of Cep63T/T and 29% of Sas4cKO daughter cells divided again (Figs 2A–C and EV2A). In the case of control cells, the majority of the non‐proliferative progeny underwent a cell cycle arrest, while a small fraction of NPCs (9%) died in interphase (Fig 2A). By contrast, the progeny of Cep63T/T and Sas4cKO NPCs underwent much higher rates of cell death (25 and 37%, respectively) (Figs 2B and C, and EV2A).
Figure 2. Prolonged mitosis leads to an increased frequency of cell death in the progeny of NPCs.
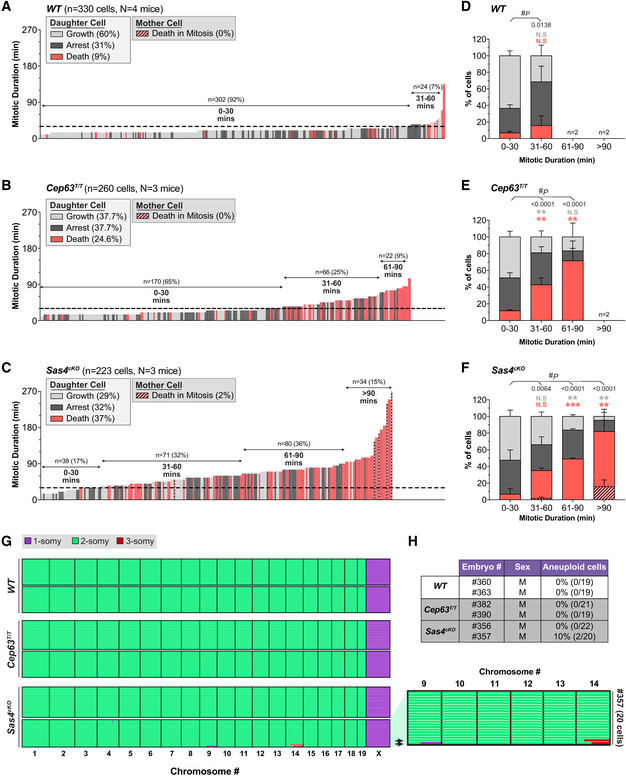
-
A–CGraph showing the mitotic duration of NPCs and the fate of their progeny in primary cultures generated from E14.5 embryos. Each bar represents one cell; its height represents the amount of time the mother cell spent in mitosis; bar color represents the fate of the progeny. Dashed line is set at 30 mins. WT n = 330 cells, N = 4 embryos; Cep63T/T n = 260 cells, N = 3 embryos; Sas4cKO n = 223 cells, N = 3 embryos.
-
D–FPercentage of proliferating, arrested and apoptotic progeny within each group of the indicated mitotic duration, calculated from data shown in (A–C); groups with n < 20 cells were excluded from this quantification; #, chi‐square test with *, post hoc analysis, within each genotype, comparisons are made to the 0–30 min group.
-
GGenome‐wide copy number plots of single cells sequenced from dissociated dorsolateral telencephalons of E14.5 embryos. Individual cells are represented in rows with copy number states indicated by colors; arrows on the zoomed in view (right) indicate aneuploid cells. WT n = 38 cells, N = 2 embryos; Cep63T/T n = 40 cells, N = 2 embryos; Sas4cKO n = 42 cells, N = 2 embryos.
-
HPercentage of aneuploid cells from the single‐cell sequencing data shown in (G).
Data information: All data represent the means ± SEM. *P < 0.05; **< 0.01; ***< 0.001; and not significant indicates P > 0.05. See also Fig EV2.
Figure EV2. Sas4cKO and Cep63T/T NPCs with centrosome loss upregulate TP53. Related to Fig 2 .
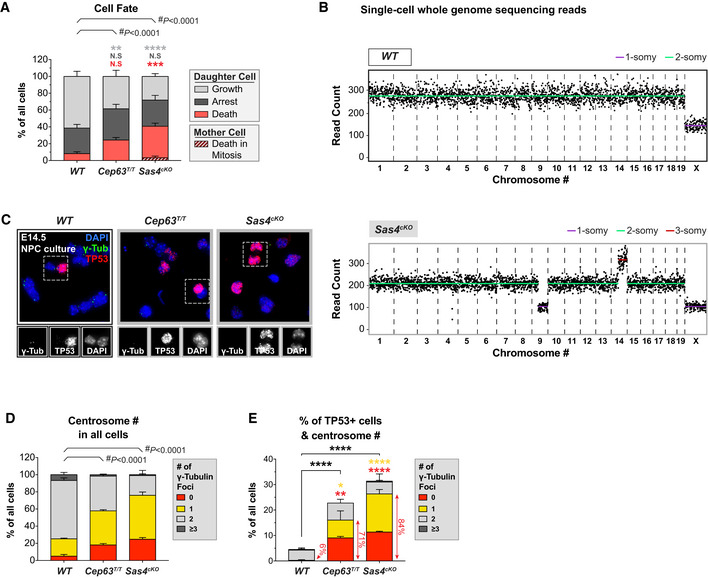
-
APercentage of proliferating, arrested and apoptotic progeny within each genotype, calculated from data shown in Fig 2A–C; #, chi‐square test with *, post hoc analysis, comparisons are made to WT.
-
BSingle‐cell whole genome sequencing data of a representative WT (top) and Sas4cKO (bottom) cell dissociated from E14.5 brains. Each black dot indicates the number of reads per 1 Mb sequence; copy number states indicated by colored lines.
-
CRepresentative images of NPCs dissociated from E14.5 WT, Cep63T/T, and Sas4cKO brains stained with antibodies against γ‐tubulin (green), TP53 (red), and DAPI (blue). Insets showing zoomed in view of TP53‐positive cells.
-
D, EQuantification of the number of γ‐tubulin foci in interphase NPCs dissociated from E14.5 cortices. Graphs show centrosome number in all cells (D) or TP53‐positive cells (E). Red arrowheads highlight the fraction of cells with centrosome loss among all TP53‐positive cells. WT n = 399 cells, N = 4 embryos; Cep63T/T n = 299 cells, N = 3 embryos; Sas4cKO n = 300 cells, N = 3 embryos; #, chi‐square test and *, multiple t‐tests, comparisons are made to WT.
Data information: All data represent the means ± SEM. *P < 0.05; **< 0.01; ***< 0.001; ****< 0.0001 and not significant indicates P > 0.05.
We hypothesized that the NPCs with mitotic delays would preferentially undergo cell death. To test this, we evaluated the fate of progeny derived from NPCs that experienced a normal or delayed mitosis. Virtually all control (92%) NPCs completed mitosis within 30 mins, and thus, we classified delayed mitosis as lasting longer than 30 mins (Fig 2A and D). Control, Cep63T/T and Sas4cKO NPCs that progressed through mitosis in ≤ 30 mins produced primarily viable progeny (Fig 2A–F). By contrast, control, Cep63T/T and Sas4cKO NPCs that delayed in mitosis produced an increased frequency of non‐viable progeny. Moreover, the frequency of cell death increased with the duration of the extended mitosis (Fig 2D–F). In the most extreme cases in which Sas4cKO NPCs spent > 90 min in mitosis, 16% of the mother cells died during cell division, while 66% of the progeny underwent cell death in the following interphase (Fig 2F). These data argue that the probability of generating non‐viable progeny increases with mitotic duration.
Since chromosome missegregation has been shown to occur following centrosome loss or delays in cell division (Lambrus et al, 2015; Wong et al, 2015), we assessed whether loss of CEP63 or SAS4 was associated with an increase in aneuploidy in the brain. We performed single‐cell DNA sequencing of dissociated NPCs from two control, Cep63T/T and Sas4cKO E14.5 cortices. While all of the control and Cep63T/T cells analyzed were diploid, 5% (2/42) of the cells from Sas4cKO brains showed gains of a large region of chromosome 14, and in one case, an accompanying loss of a large part of chromosome 9 (Figs 2G and H, and EV2B). We conclude that the mitotic delay observed in Sas4cKO NPCs is associated with only a modest increase in the level of aneuploidy.
Our findings suggest that centrosome defects prolong mitosis in NPCs, leading to an increased frequency of cell death in the progeny. Therefore, we analyzed whether TP53 was induced following centrosome loss in Sas4cKO and Cep63T/T dissociated progenitors. We observed a > 5‐fold increase in the fraction of TP53+ progenitors in dissociated cultures from Cep63T/T and Sas4cKO cortices (Fig EV2C–E). Moreover, > 70% of the TP53+ Cep63T/T and Sas4cKO NPCs had a reduction in centrosome number (Fig EV2E). Since aneuploidy is rare in the Cep63T/T and Sas4cKO brains, the upregulation of TP53 in NPCs with centrosome defects is unlikely to be a result of chromosome missegregation.
Centrosome defects activate the mitotic surveillance pathway to reduce cortical expansion
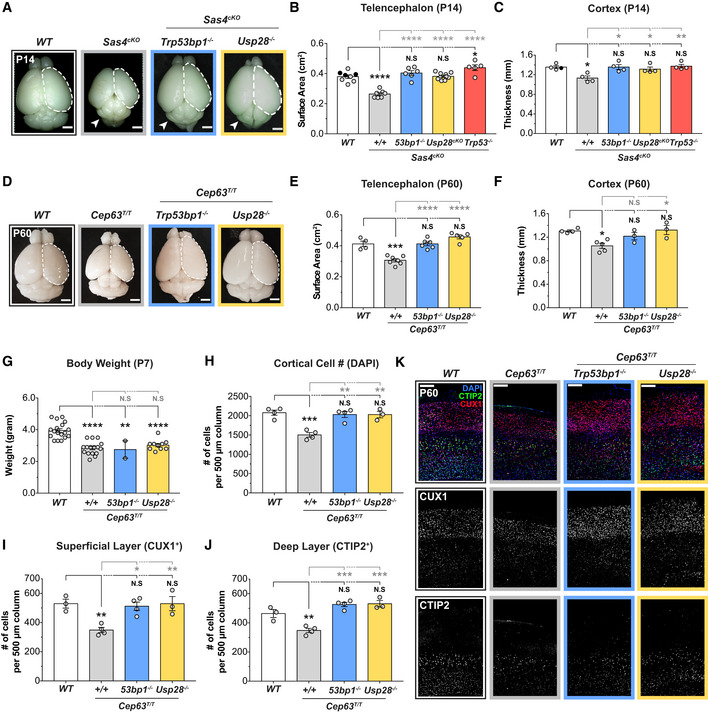
Centrosome defects have been shown to activate the mitotic surveillance pathway by delaying mitosis (Uetake & Sluder, 2010; Bazzi & Anderson, 2014; Fong et al, 2016; Lambrus et al, 2016; Meitinger et al, 2016). Thus, we set out to test whether pathological activation of the mitotic surveillance pathway causes the reduced cortical expansion in Cep63T/T and Sas4cKO animals. While knockout of Trp53, Usp28, or Trp53bp1 alone did not alter brain size (Fig EV3A and B), genetic ablation of Usp28 or Trp53bp1 largely restored the telencephalic area and cortical thickness of Cep63T/T mice at P60 and of Sas4cKO animals at P14 (hereafter referred to as double knockout animals) (Fig 3A–F). Intriguingly, knockout of Usp28 or Trp53bp1 did not rescue the reduced body weight of young Cep63T/T mice, suggesting that centrosome defects cause whole‐body growth retardation and microcephaly via distinct mechanisms (Fig 3G). As expected, the severe hydrocephalus and cerebellar hypoplasia caused by the loss of cilia in Sas4cKO brains were also not rescued by knockout of Usp28 or Trp53bp1 (Figs 3A and EV3C). Nevertheless, Sas4cKO;Usp28cKO and Sas4cKO;Trp53bp1 −/− mice had improved overall survival compared to Sas4cKO animals, showing that alleviating microcephaly extended the lifespan of these animals (Fig EV3D).
Figure 3. Centrosome defects reduce brain size through activation of the mitotic surveillance pathway.
-
ARepresentative whole mount images of WT, Sas4cKO, Sas4cKO;Trp53bp1 −/−, Sas4cKO;Usp28cKO animals at P14. Arrows indicate the cerebellar hypoplasia resulting from the lack of primary cilia. Scale bar = 0.2 cm.
-
BTelencephalon area of P14 brains of the indicated genotypes. WT littermates N = 8, Sas4cKO N = 7, Sas4cKO;Trp53bp1 −/− N = 6, Sas4cKO;Usp28cKO N = 10, Sas4cKO;Trp53 −/− N = 5; one‐way ANOVA with post hoc analysis. WT and Sas4cKO data are from Fig 1D and shown alongside for comparison. Black circles represent Nestin‐Cre+ animals.
-
CCortical thickness of P14 brains of the indicated genotypes. WT littermates N = 4, Sas4cKO N = 4, Sas4cKO;Trp53bp1 −/− N = 4, Sas4cKO;Usp28cKO N = 4, Sas4cKO;Trp53 −/− N = 4; one‐way ANOVA with post hoc analysis. WT and Sas4cKO data are from Fig EV1D and shown alongside for comparison. Black circles represent Nestin‐Cre+ animals.
-
DRepresentative whole mount images of WT, Cep63T/T, Cep63T/T;Trp53bp1 −/−, Cep63T/T;Usp28 −/− animals at P60. Scale bar = 0.2 cm.
-
ETelencephalon area of P60 brains of the indicated genotypes. WT littermates N = 4, Cep63T/T N = 7, Cep63T/T;Trp53bp1 −/− N = 6, Cep63T/T;Usp28 −/− N = 5; one‐way ANOVA with post hoc analysis. WT and Cep63T/T data are from Fig 1B and shown alongside for comparison.
-
FCortical thickness of P60 brains of the indicated genotypes. WT littermates N = 4, Cep63T/T N = 5, Cep63T/T;Trp53bp1 −/− N = 3, Cep63T/T;Usp28 −/− N = 3; one‐way ANOVA with post hoc analysis. WT and Cep63T/T data are from Fig EV1B and shown alongside for comparison.
-
GBody weight of P7 animals of the indicated genotypes. WT littermates N = 18, Cep63T/T N = 16, Cep63T/T;Trp53bp1 −/− N = 2, Cep63T/T;Usp28 −/− N = 10; one‐way ANOVA with post hoc analysis.
-
HQuantification of total number of cells within a 500 μm‐width column of cortices at P60. WT littermates N = 4, Cep63T/T N = 4, Cep63T/T;Trp53bp1 −/− N = 4, Cep63T/T;Usp28 −/− N = 3; one‐way ANOVA with post hoc analysis.
-
I, JQuantification of the number of cells in the (I) superficial layer (CUX1+) and (J) deep layer (CTIP2+) within a 500 μm‐width column of P60 cortices. WT littermates N = 3, Cep63T/T N = 4, Cep63T/T;Trp53bp1 −/− N = 4, Cep63T/T;Usp28 −/− N = 3; one‐way ANOVA with post hoc analysis.
-
KP60 cortices of indicated genotypes stained with antibodies against the deep layer marker CTIP2 (green), superficial layer marker CUX1 (red) and DAPI (blue). Scale bar = 200 μm.
Data information: All data represent the means ± SEM. *P < 0.05; **< 0.01; ***< 0.001; ****< 0.0001 and not significant indicates P > 0.05. See also Fig EV3.
To assess the organization of the cortical layers in the adult brain, we compared the number of CUX1+ cells in the superficial layers (layer II–IV) and CTIP2+ cells in the deep layers (layer V–VI) of the cortex between control, Cep63T/T, Sas4cKO, and double knockout animals. Cep63T/T cortices showed a loss of CUX1+ upper layer and CTIP2+ deep layer neurons, while Sas4cKO brains had a preferential reduction of CUX1+ neurons (Figs 3H–K and EV3E–H). By contrast, the overall cell density and the total number of CUX1+ and CTIP2+ neuronal subtypes in double knockout animals were similar to that of control mice (Figs 3H–K and EV3E–H). Collectively, these data demonstrate that genetic ablation of the mitotic surveillance pathway can restore brain size and the abundance of neurons in two microcephaly mouse models with centrosome defects.
Inactivation of the mitotic surveillance pathway prevents the attrition of NPCs in mice with centrosome defects
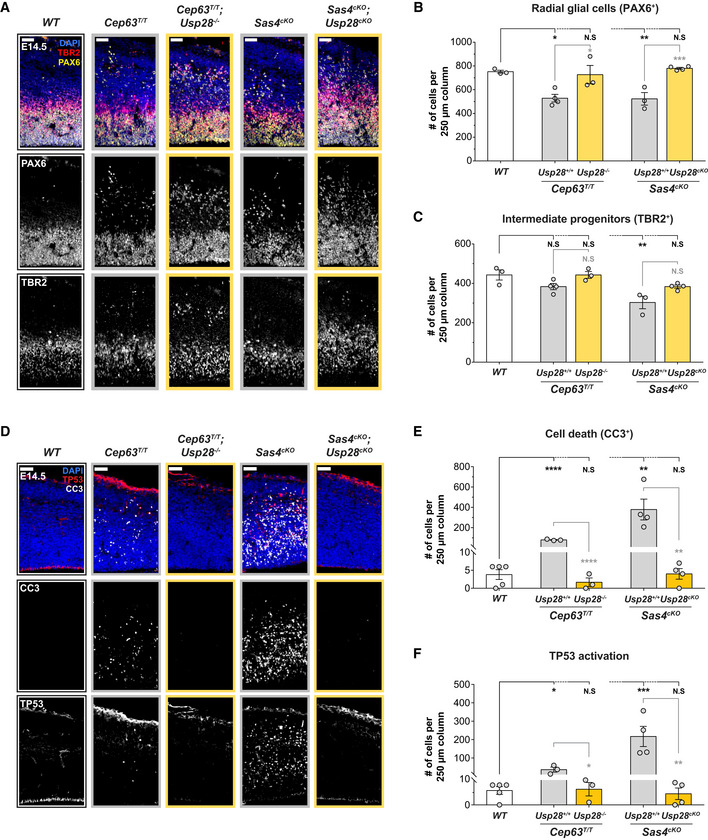
To determine if the rescue of cortical size and neuronal production in double knockout animals was associated with a restoration of the NPC pool, we analyzed the abundance of PAX6+ RGCs and TBR2+ IPs in E14.5 brains from control, Sas4cKO, Cep63T/T, and double knockout animals. Consistent with previous reports, the number of RGCs was reduced in both Cep63T/T and Sas4cKO cortices (Insolera et al, 2014; Marjanovic et al, 2015; Fig 4A and B). In addition, a portion of these progenitors were displaced from their normal position in the VZ and mislocalized to regions further away from the ventricle (Fig 4A). TBR2+ IPs were also significantly reduced in Sas4cKO but not in Cep63T/T cortices (Fig 4A and C).
Figure 4. Inactivation of the mitotic surveillance pathway prevents NPCs depletion in brains with centrosome defects.
-
ACortices from E14.5 brains stained with antibodies against the radial glial cell marker PAX6 (yellow), intermediate progenitor marker TBR2 (red) and DAPI (blue). Scale bar = 50 μm.
-
B, CQuantification of the number of (B) radial glial cells (PAX6+) and (C) intermediate progenitors (TBR2+) within a 250 μm‐width column of E14.5 cortices. WT N = 3, Cep63T/T N = 4, Cep63T/T;Usp28 −/− N = 3, Sas4cKO N = 3, Sas4cKO;Usp28cKO N = 4; one‐way ANOVA with post hoc analysis.
-
DCortices from E14.5 embryos stained with antibodies against cleaved‐caspase 3 (CC3, white), TP53 (red) and DAPI. Scale bar = 50 μm.
-
E, FQuantification of the number of CC3+ apoptotic cells (E) and cells with TP53 activation (F) within a 250 μm‐width column of E14.5 cortices. WT N = 5, Cep63T/T N = 3, Cep63T/T;Usp28 −/− N = 3, Sas4cKO N = 4, Sas4cKO;Usp28cKO N = 4; one‐way ANOVA with post hoc analysis.
Data information: All data represent the means ± SEM. *P < 0.05; **< 0.01; ***< 0.001; ****< 0.0001 and not significant indicates P > 0.05. See also Fig EV4.
While the deletion of the mitotic surveillance pathway genes alone did not alter the number of PAX6+ or TBR2+ NPCs in the developing cortex (Fig EV4A and B), knockout of Usp28 restored both populations of progenitors in Sas4cKO and Cep63T/T cortices to levels similar to that in controls (Fig 4A–C). Although the total number of progenitors in the double knockout cortices was comparable to control mice, we observed a large population of ectopic PAX6+ RGCs in the SVZ and intermediate zone (IZ) of the developing neocortex (Fig 4A). A similar phenotype has been reported in Sas4cKO;Trp53 −/−, and Cep63T/T;Trp53 −/− mice and arises due to the suppression of cell death in the misplaced RGCs (Insolera et al, 2014; Marjanovic et al, 2015). We also observed ectopic IPs in both Sas4cKO;Usp28cKO and Cep63T/T;Usp28 −/− embryonic brains that were displaced from the SVZ (Fig 4A). Altogether, these data show that inactivation of the mitotic surveillance pathway rescues the attrition of the NPC pool in mice with centrosome defects but does not prevent the NPC delamination.
Figure EV4. Sas4cKO and Cep63T/T brains show elevated cell death in NPCs and neurons without increased DNA damage. Related to Fig 4 .
-
A, BQuantification of the number of (A) radial glial cells (PAX6+) and (B) intermediate progenitors (TBR2+) within a 250 μm‐width column of E14.5 cortices. WT N = 3, Trp53bp1 −/− N = 3, Usp28 −/− N = 3, Trp53 −/− N = 3; one‐way ANOVA with post hoc analysis.
-
CQuantification of the number of apoptotic cells (CC3+) co‐stained with PAX6‐TBR2 or TBR1 within a 250 μm‐width column of E14.5 cortices. WT N = 3, Cep63T/T N = 2, Sas4cKO N = 3; multiple t‐tests, comparisons are made to WT.
-
DCortices from E14.5 embryos stained with antibodies against cleaved‐caspase 3 and markers for radial glial cells (PAX6, red), intermediate progenitors (TBR2, red), and neurons (TBR1, red). Insets showing zoomed in view of the cortical plate (CP), the intermediate zone (IZ), the sub‐ventricular zone (SVZ), and the ventricular zone (VZ), demarcated by the white dashed lines. Scale bar = 50 μm.
-
EE14.5 cortices of indicated genotypes stained with antibodies against γ‐H2AX (red) and DAPI (blue). A P0 brain section from an irradiated animal is shown alongside as a positive control for DNA damage signal. Insets showing zoomed in view of representative γ‐H2AX+ cells. Red dashed circles indicate dead cells; green dashed circles indicate live cells. Scale bar = 50 μm.
-
FQuantification of the number of cells stained positive for γ‐H2AX within a 250 μm‐width column of E14.5 cortices; cells are categorized as dead or alive based on DAPI morphology. WT N = 3, Cep63T/T N = 4, Cep63T/T;Usp28 −/− N = 3, Sas4cKO N = 4, Sas4cKO;Usp28cKO N = 3; multiple t‐tests.
Data information: All data represent the means ± SEM. *P < 0.05; ***< 0.001; ****< 0.0001 and not significant indicates P > 0.05.
To establish whether suppression of apoptosis contributed to the restoration of NPC pools, we analyzed the levels of TP53 and cleaved‐caspase 3 (CC3) at E14.5. Only rare instances of TP53 and CC3 labeling were observed in the cortex of control mice (Fig 4D–F). However, TP53+ and CC3+ cells were widespread in the cortex of Cep63T/T and Sas4cKO animals, with cell death occurring in both NPCs and TBR1+ neurons (Figs 4D–F, and EV4C and D). Strikingly, deletion of Usp28 reduced the number of TP53 and CC3‐expressing cells in the Sas4cKO and Cep63T/T cortices to levels similar to that observed in control brains (Fig 4D–F). This shows that activation of the mitotic surveillance pathway is responsible for TP53 upregulation and cell death in NPCs and neurons.
To determine whether the loss of SAS4 or CEP63 leads to DNA damage that can activate TP53, we stained cortices with the DNA double‐strand break marker γ‐H2AX. Although there was a minor increase in γ‐H2AX+ cells in both Cep63T/T and Sas4cKO animals, the staining occurred in apoptotic cells with either a condensed DNA morphology or no DNA staining (Fig EV4E and F). DNA damage occurs in dying cells as a result of caspase‐activated DNase activity (Enari et al, 1998). In accordance with this, suppression of cell death by knockout of Usp28 dramatically reduced the number of γ‐H2AX+ cells in Cep63T/T and Sas4cKO cortices (Fig EV4E and F). We conclude that centrosome defects induced by loss of SAS4 or CEP63 do not lead to an increase in DNA damage outside of apoptotic cells.
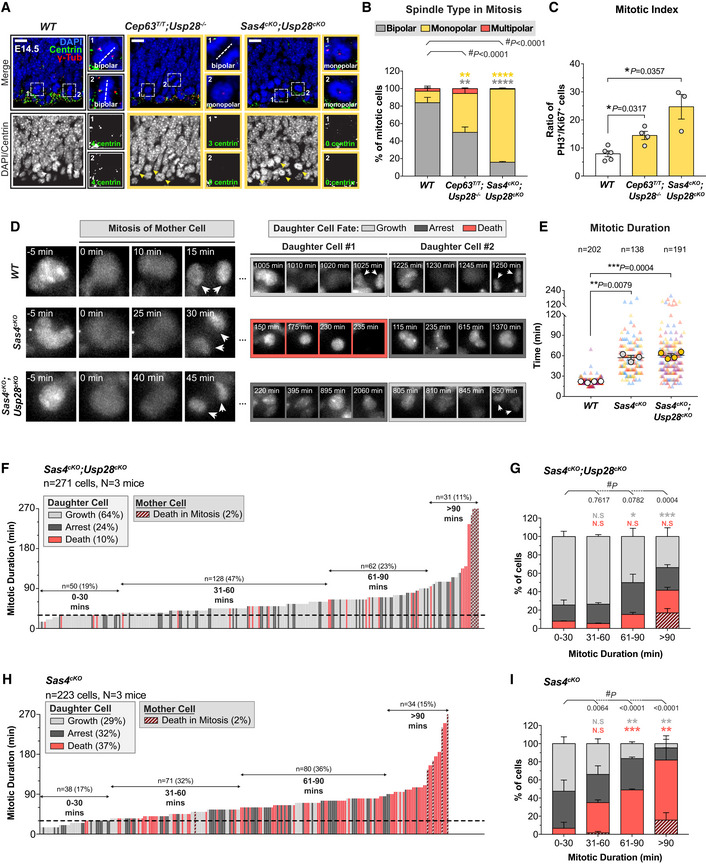
Inactivation of mitotic surveillance does not rescue centrosome function or alleviate mitotic delays in Sas4cKO and Cep63T/T NPCs
To determine if the primary deficits persist following inactivation of the mitotic surveillance pathway, we analyzed centrosome number and mitotic duration in Sas4cKO and Cep63T/T NPCs that lacked Usp28. Similar to what we observed in the cortex of Cep63T/T and Sas4cKO mice, mitotic progenitors in Cep63T/T;Usp28 −/− and Sas4cKO;Usp28cKO animals had reduced numbers of centrioles and an increased frequency of monopolar spindles (Figs 5A and B, and EV5A and B). To examine if mitotic delays persisted in Sas4cKO and Cep63T/T NPC after inactivation of the mitotic surveillance pathway, we stained E14.5 brains with PH3 and Ki67. As expected, the deletion of Trp53, Usp28, or Trp53bp1 alone did not significantly change the mitotic index in the developing cortex (Fig EV5C). By contrast, the mitotic index in Cep63T/T;Usp28 −/− and Sas4cKO;Usp28cKO cortices was increased to a level similar to that observed in the Sas4cKO and Cep63T/T mice (Fig 5C). To directly measure the mitotic duration in these NPCs, we performed live imaging of dissociated Sas4cKO;Usp28cKO NPCs. This confirmed that the inactivation of the mitotic surveillance pathway did not alter the extended mitotic duration in Sas4cKO progenitors (Fig 5D and E).
Figure 5. Inactivation of the mitotic surveillance pathway rescues NPC proliferation without alleviating the centrosome defects or mitotic delay.
-
ARepresentative images of E14.5 cortices stained with antibodies against centrin (green), γ‐tubulin (red) and DAPI (blue). White and yellow arrows indicate cells with bipolar and monopolar spindles, respectively. Insets showing zoomed in view of 2 representative cells. Dashed lines showing the orientation of the cleavage plane relative to the ventricular surface. Scale bar = 10 μm.
-
BPercentage of bipolar, monopolar and multipolar cells at the ventricular surface of E14.5 cortices. WT n = 201 cells, N = 3 embryos; Cep63T/T;Usp28 −/− n = 205 cells, N = 3 embryos; Sas4cKO;Usp28cKO n = 308 cells, N = 3 embryos; #, chi‐square test with *, post hoc analysis, comparisons are made to WT. WT data are from Fig 1F and shown alongside for comparison.
-
CRatio of mitotic (PH3+) to cycling (Ki67+) cells in E14.5 cortices. WT N = 5, Cep63T/T;Usp28 −/− N = 4, Sas4cKO;Usp28cKO N = 3; two‐tailed Mann–Whitney t‐test. 3 WT data points are from Fig 1H and shown alongside for comparison.
-
DRepresentative images from live‐cell movies of Nuclear‐mCherry+ WT, Sas4cKO, and Sas4cKO;Usp28cKO NPCs. White arrows showing the nuclei of daughter cells following cytokinesis.
-
EPlot showing quantification of mitotic duration of WT, Sas4cKO, and Sas4cKO;Usp28cKO NPCs. Triangles represent individual cells; triangles of the same color represent cells derived from the same embryo. Circles represent the average mitotic duration of cells from each embryo. WT n = 202 cells, N = 4 embryos; Sas4cKO n = 138 cells, N = 3 embryos; Sas4cKO;Usp28cKO n = 191 cells, N = 4 embryos; two‐tailed Welch’s t‐test. WT and Sas4cKO data are from Fig 1K and shown alongside for comparison.
-
FGraph showing the mitotic duration of NPCs and the fate of their progeny in primary cultures generated from Sas4cKO;Usp28cKO E14.5 embryos. Each bar represents one cell; its height represents the amount of time the mother cell spent in mitosis; bar color represents the fate of the progeny. Dashed line is set at 30 min. Sas4cKO;Usp28cKO n = 271 cells, N = 3 embryos.
-
GPercentage of proliferating, arrested, and apoptotic progeny within each group of the indicated mitotic duration, calculated from data shown in (F); #, chi‐square test with *, post hoc analysis, comparisons are made to the 0–30 min group.
-
H, IData from Fig 2C and F shown alongside for comparison with (F and G).
Data information: All data represent the means ± SEM. *P < 0.05; **< 0.01; ***< 0.001; ****< 0.0001 and not significant indicates P > 0.05. See also Fig EV5.
Figure EV5. Prolonging mitosis results in USP28‐dependent cell death in the progeny of NPC divisions. Related to Fig 5 .
-
A, BQuantification of the number of (A) centrin foci and (B) γ‐tubulin foci in mitotic cells in the VZ of E14.5 cortices. WT n = 175 cells, N = 4 embryos; Usp28 −/− n = 137 cells, N = 3 embryos; Cep63T/T;Usp28 −/− n = 150 cells, N = 3 embryos; Sas4cKO;Usp28cKO n = 150 cells, N = 3 embryos. #, chi‐square test, with * post hoc analysis, comparisons are made to WT. WT data are from Fig EV1F and G and shown alongside for comparison.
-
CRatio of mitotic (PH3+) to cycling (Ki67+) cells in E14.5 cortices. WT N = 5, Trp53bp1 −/− N = 3, Usp28 −/− N = 3, Trp53 −/− N = 3; one‐way ANOVA with post hoc analysis. WT data are from Fig 1H and shown alongside for comparison.
- D
-
EGenome‐wide copy number plots of single cells sequenced from NPCs dissociated from Sas4cKO;Usp28cKO E14.5 brains. Individual cells are represented in rows with copy number states indicated by colors; arrow on the zoomed in view (bottom) indicates an aneuploid cell. Sas4cKO;Usp28cKO n = 41 cells, N = 2 embryos.
-
FPercentage of aneuploid cells from the single‐cell sequencing data shown in E.
-
GSchematic of the prolonged mitosis assay.
-
HPercentage of proliferating, arrested and apoptotic progeny in WT and Usp28cKO dissociated NPCs with delayed mitosis (nocodazole delayed; WT n = 45 cells, N = 3 embryos; Usp28cKO n = 56 cells, N = 3 embryos) and normal mitosis (After Washout; WT n = 90 cells, N = 3 embryos; Usp28cKO n = 100 cells, N = 3 embryos) calculated from data shown in Fig EV5I–L; #, chi‐square test with *, post hoc analysis, comparisons are made to WT.
-
I, JGraph showing the mitotic duration and fate of (I) WT and (J) Usp28cKO E14.5 NPCs that were delayed in mitosis using nocodazole (0.08 μM). Each bar represents one cell; its height represents the amount of time the mother cell spent in mitosis; bar color represents the fate of the progeny. WT n = 45 cells, N = 3 embryos; Usp28cKO n = 56 cells, N = 3 embryos.
-
K, LGraph showing the mitotic duration and fate of (K) WT and (L) Usp28cKO NPCs from the same experiment in I and J that entered mitosis after nocodazole washout. WT n = 90 cells, N = 3 embryos; Usp28cKO n = 100 cells, N = 3 embryos.
Data information: All data represent the means ± SEM. *P < 0.05; **< 0.01; ***< 0.001; **** < 0.0001 and not significant indicates P > 0.05.
We next proceeded to track the fate of Sas4cKO;Usp28cKO progeny after mitotic exit. While only 29% of Sas4cKO progeny re‐entered mitosis within the time frame of our movies, 64% of Sas4cKO;Usp28cKO daughter cells divided again, which is similar to what was observed in control NPCs (Figs 5F and G, and EV5D). In the case of Sas4cKO cells, 37% of progeny underwent cell death in interphase, and this was reduced by > 3.5‐fold in Sas4cKO;Usp28cKO progenitors to a level similar to that of control NPCs (Figs 5F–I, and EV5D). Importantly, knockout of Usp28 dramatically reduced the frequency of apoptosis in the progeny that resulted from an extended mitosis (Fig 5G and I). Finally, single‐cell DNA sequencing of dissociated progenitors showed that similar to Sas4cKO mice, cells from Sas4cKO;Usp28cKO brains had only modest levels of aneuploidy (2.4%, 1/41) (Fig EV5E and F). Note that this level of aneuploidy is similar to that of Sas4cKO (5%) (Fig 2G and H). Thus, the continued proliferation of NPCs with centrosome defects does not lead to a clear increase in chromosome segregation errors. We conclude that inactivation of the mitotic surveillance pathway rescues the proliferation of NPCs downstream of the centrosome defects and extended mitoses.
To directly test if the mitotic surveillance pathway is responsible for triggering apoptosis in NPCs with extended mitotic duration, we adopted an approach to reversibly delay NPCs in mitosis using the spindle poison nocodazole (Uetake & Sluder, 2010; Pilaz et al, 2016). Dissociated control and Usp28cKO NPCs were treated with nocodazole for 4 h to delay cells in mitosis and subsequently released into drug‐free media for an additional 48 h (Fig EV5G). This approach allowed us to track the fate of progeny from cells with different mitotic durations. Control and Usp28cKO NPCs that entered mitosis following nocodazole washout divided with a normal mitotic duration, showing that the drug treatment was fully reversible (Fig EV5H, K and L). 47% of control cells that were delayed in mitosis (> 30 min) produced non‐viable progeny (Fig EV5H and I). By contrast, cell death was reduced by 2.6‐fold in Usp28cKO NPCs that experienced similarly prolonged mitoses (Fig EV5H and J). Altogether, these data confirm that USP28 is required to initiate cell death in the progeny of cells that delay in mitosis.
Distinct pathways lead to TP53 activation in microcephaly
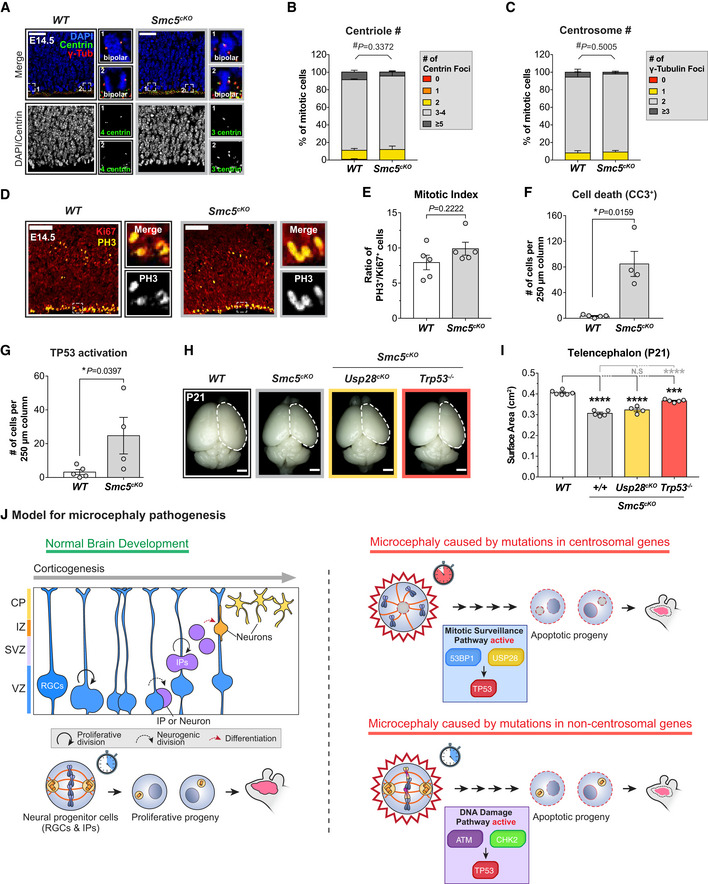
Our data suggest that activation of the mitotic surveillance pathway is a central cause of microcephaly in multiple mouse models with centrosome dysfunction. To examine whether defects in non‐centrosomal genes also cause microcephaly via activation of the mitotic surveillance pathway, we examined a microcephaly mouse model caused by loss of the structural maintenance of chromosome (SMC) complex protein SMC5. The SMC5/6 complex is best characterized for its function in DNA repair and maintenance of genome stability (Fousteri & Lehmann, 2000; Sergeant et al, 2005; Zhao & Blobel, 2005), and mutations in the SMC5/6 complex component NSMCE2 cause primordial dwarfism and microcephaly in humans (Payne et al, 2014). Conditional deletion of Smc5 using Nestin‐Cre (hereafter referred to as Smc5cKO) did not alter centriole or centrosome number in dividing NPCs (Fig 6A–C) or increase the mitotic index in the developing neocortex (Fig 6D and E). However, loss of Smc5cKO resulted in increased numbers of TP53+ and CC3+ cells in the cortex at E14.5 (Figs 6F and G, and EV6A), leading to a 24% reduction in telencephalic area at P21 (Fig 6H and I).
Figure 6. TP53 is activated through a distinct pathway in a microcephaly model caused by the loss of Smc5 .
-
AWT and Smc5cKO cortices at E14.5 stained with antibodies against centrin (green), γ‐tubulin (red) and DAPI (blue). Insets showing zoomed in view of 2 representative cells. Scale bar = 25 μm.
-
B, CQuantification of the number of (B) centrin foci and (C) γ‐tubulin foci in mitotic cells in the VZ of E14.5 cortices. WT n = 175 cells, N = 4 embryos; Smc5cKO n = 134 cells, N = 3 embryos. #, chi‐square test. WT data are from Fig EV1F and G and shown alongside for comparison.
-
DE14.5 cortices stained with antibodies against Ki67 (red) and phosphorylated‐Histone H3 (PH3, yellow). Insets showing zoomed in view of cells in the VZ and SVZ. Scale bar = 100 μm.
-
ERatio of mitotic (PH3+) to cycling (Ki67+) cells in E14.5 cortices. WT N = 5, Smc5cKO N = 5; two‐tailed Mann–Whitney t‐test. WT data are from Fig 1H and shown alongside for comparison.
-
F, GQuantification of the number of (F) apoptotic cells (CC3+) and (G) cells with TP53 activation within a 250 μm‐width column of E14.5 cortices. WT N = 5, Smc5cKO N = 4; two‐tailed Mann–Whitney t‐test. WT data are from Fig 4E and F and shown alongside for comparison.
-
HRepresentative whole mount images of WT, Smc5cKO, Smc5cKO;Usp28cKO, Smc5cKO;Trp53 −/− brains at P21. Scale bar = 0.2 cm.
-
ITelencephalon area of P21 brains of the indicated genotypes. WT littermates N = 6, Smc5cKO N = 5, Smc5cKO;Usp28cKO N = 4, Smc5cKO;Trp53 −/− N = 5; one‐way ANOVA with post hoc analysis.
-
JModel for two TP53‐dependent pathways underlying primary microcephaly. In the first model, NPCs with mutations in centrosome genes take longer to assemble their spindle in mitosis. This delay triggers the activation of the mitotic surveillance pathway, which activates TP53 through 53BP1 and USP28. By contrast, in microcephaly caused by mutations in genes required for DNA repair or genome stability, centrosome function and mitotic duration are unaffected. Instead, accumulated DNA lesions trigger DNA damage signaling in NPCs to activate TP53. In both models, increased TP53 activity leads to apoptosis, thus depleting the NPC pool and decreasing the final number of neurons.
Data information: All data represent the means ± SEM. *P < 0.05; ***< 0.001; ****< 0.0001 and not significant indicates P > 0.05. See also Fig EV6.
Figure EV6. Smc5cKO embryonic brains show TP53 upregulation. Related to Fig 6 .
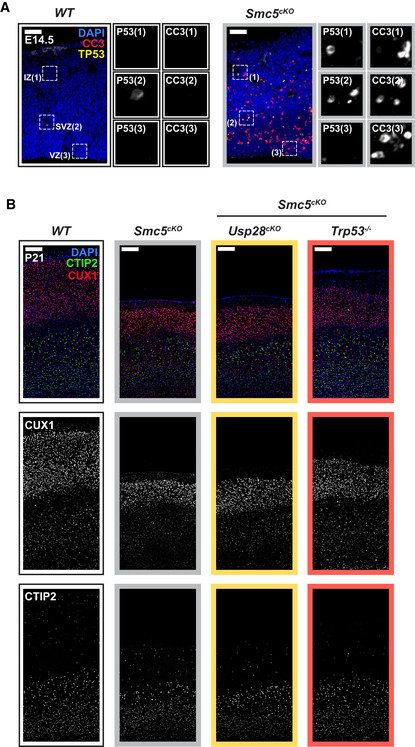
- Cortices from E14.5 embryos stained with antibodies against cleaved‐caspase 3 (CC3, red), TP53 (yellow), and DAPI (blue). Insets showing zoomed in view of the intermediate zone (IZ), the sub‐ventricular zone (SVZ), and ventricular zone (VZ). Scale bar = 50 μm.
- P21 cortices of the indicated genotypes stained with antibodies against the deep layer marker CTIP2 (green), superficial layer marker CUX1 (red) and DAPI (blue). Scale bar = 200 μm.
Prior work has shown that knockout of TP53 partially rescues progenitor numbers and the microcephaly phenotype in Smc5cKO animals (Atkins et al, 2020). We, therefore, tested if the knockout of the mitotic surveillance pathway component Usp28 can also rescue brain size in Smc5cKO mice. Genetic ablation of Usp28 failed to provide any rescue of brain size in Smc5cKO mice (Figs 6H and I, and EV6B). Thus, these data show that genetic deletion of Usp28/Trp53bp1 and Trp53 are not functionally equivalent in vivo, and that deficiency in the SMC5/6 complex leads to activation of TP53 independently of the mitotic surveillance pathway.
Discussion
Previous work in cultured cells showed that centrosome loss activates the mitotic surveillance pathway by prolonging mitosis (Fong et al, 2016; Lambrus et al, 2016; Meitinger et al, 2016). However, the consequence of activating this pathway in vivo remain unexplored. In this manuscript, we provide several lines of evidence to support the proposal that pathological activation of the mitotic surveillance pathway leads to NPC depletion and reduced brain growth in microcephaly mouse models with centrosome defects. First, NPCs with centrosome defects delay in mitosis, and the probability of producing non‐viable cells increased with mitotic duration. Second, inactivation of the mitotic surveillance pathway suppresses apoptosis and restores NPC pools and brain size in two microcephaly models. Third, this restoration of the NPC number occurs without correction of the upstream defect in centrosome function or the increase in mitotic duration. Altogether, these data argue that activation of the mitotic surveillance pathway is a central cause of microcephaly in mice, and potentially human patients, with centrosome dysfunction.
Our findings are consistent with previous work showing that extending mitosis in NPCs leads to increased apoptotic progeny and premature differentiation (Pilaz et al, 2016; Mitchell‐Dick et al, 2020). We show that depletion of centrosome proteins that are mutated in human patients with microcephaly delays bipolar spindle assembly and prolongs mitosis in NPCs. Delayed NPCs complete mitosis with minimal chromosome segregation errors, but the extended mitotic duration increases the frequency of death in the resulting progeny. This apoptotic phenotype was suppressed by the deletion of USP28, leading to a full rescue of telencephalon size and cell number in the adult brains of Sas4cKO and Cep63T/T animals. Although Cep63T/T;Usp28 −/− and Sas4cKO;Usp28cKO embryonic brains have restored NPC pools, some progenitors are located outside of the VZ and SVZ. This is consistent with prior work showing that centrosome loss leads to the delamination of RGCs from the VZ (Insolera et al, 2014; Marjanovic et al, 2015). Knockout of USP28 suppresses apoptosis in these ectopic RGCs, allowing them to continue to proliferate and produce the correct number of IPs and neurons. Thus, while our analysis of cortical layer markers showed that the gross organization of adult Cep63T/T;Usp28 −/− and Sas4cKO;Usp28cKO cortices are normal, we do not rule out specific defects in lamina organization or brain function. Nevertheless, our data clearly demonstrate that genetic ablation of the mitotic surveillance pathway is sufficient to restore the proliferation of NPCs with centrosome defects and rescue neuronal loss and microcephaly in adult animals.
Although multiple microcephaly genes have established functions at the centrosome, several of these genes have reported roles in DNA damage signaling and defects in many DNA repair and DNA damage signaling factors also result in microcephaly (Jayaraman et al, 2018). Several lines of evidence argue that the depletion of NPCs in Sas4cKO and Cep63T/T animals is a result of activating the mitotic surveillance pathway and not a consequence of DNA damage signaling. First, while long mitotic delays of > 8 h can lead to the initiation of apoptosis and subsequent DNA damage (Orth et al, 2012), the mitotic delay caused by loss of CEP63 or SAS4 in NPCs was well below this threshold, with the majority of Cep63T/T and Sas4cKO progenitors completing mitosis in less than 90 min. Second, we, and others, did not observe an increase in DNA breaks outside of the apoptotic cells in the cortices of Cep63T/T and Sas4cKO animals (Insolera et al, 2014; Marjanovic et al, 2015). Third, despite not being critical for the DNA damage response in vitro and in vivo (Knobel et al, 2014), USP28 removal can dramatically rescue NPC abundance and suppress cell death in both Cep63T/T and Sas4cKO embryonic brains. Finally, prior work showed that unlike removing TP53, loss of ATM or CHK2 does not rescue the microcephaly phenotype in Cep63T/T mice (Marjanovic et al, 2015). Taken together with previous data in cultured cells demonstrating that the mitotic surveillance pathway is mechanistically distinct from the DNA damage response (Fong et al, 2016; Lambrus et al, 2016; Meitinger et al, 2016), our data strongly indicate that activation of the mitotic surveillance pathway is the major mechanism leading to NPC depletion in the developing neocortex with centrosome defects.
Nevertheless, our work highlights that multiple mechanisms can promote TP53 activation and the attrition of NPCs in the neocortex. Defects in DNA double‐strand break (DSB) repair factors, including LIG4, NBS1 and RAD50, have been shown to result in apoptosis in the developing brain and underlie multiple human syndromes characterized by microcephaly (McKinnon, 2017). Loss of ATM, CHK2, or TP53 rescued viability and suppressed neuronal apoptosis in LIG4‐deficient mice and Trp53 deletion suppressed cell death in mice lacking NBS1 (Carney et al, 1998; Varon et al, 1998; Frank et al, 2000; Lee et al, 2000; O'Driscoll et al, 2001; Frappart et al, 2005; Waltes et al, 2009; Foster et al, 2012), indicating that DSBs potently activate TP53 through the ATM‐CHK2 pathway during neurodevelopment. Consistent with these previous findings, our data show that while knockout of TP53 largely rescued brain size in Smc5cKO animals, the loss of USP28 had no effect. In addition, loss of SMC5 in cultured cells activates CHK2 (Venegas et al, 2020) and knockout of Chk2 also rescues microcephaly in Smc5cKO animals (Atkins et al, 2020), further establishing that the role of USP28 in rescuing NPCs is uncoupled from DSB signaling. Thus, we propose that centrosome defects activate TP53 via the USP28‐dependent mitotic surveillance pathway, while DNA damage signaling in response to DSBs can independently upregulate TP53 through ATM and CHK2 (Fig 6J). The pathways that feed into TP53 activation in NPCs are therefore likely to vary depending on the particular function of the microcephaly gene that is mutated. In this regard, it is also worth noting that microcephaly caused by hypomorphic mutations in ATR, an ATM‐related kinase that responds to replication stress and is mutated in Seckel Syndrome, is exacerbated by Trp53 deletion (O'Driscoll et al, 2003; Murga et al, 2009). This indicates that while TP53 activation is a prominent cause of microcephaly, there are also TP53‐independent pathways relevant to microcephaly that are caused by non‐centrosomal defects.
While mutations in some centrosome proteins cause both primary microcephaly and dwarfism (Chavali et al, 2014), several microcephaly genes only affect brain size (Mahmood et al, 2011). This leads to an outstanding conundrum of why mutations in ubiquitously expressed centrosome proteins selectively impact brain development. One explanation could be that NPCs have a lower threshold for activating the mitotic surveillance pathway compared to other cell types. Our data from dissociated cultures suggest that this threshold is set at around 30 min for NPCs, with cell death steadily increasing with additional time spent in mitosis. Whether this threshold is indeed higher for other proliferating cells in the body remains to be tested. Another possible explanation is that activation of the mitotic surveillance pathway promotes different cellular responses between NPCs and other cell types. In retinal pigment epithelial (RPE1) cells, TP53 targets CDKN1A (P21) and triggers a G1 arrest instead of cell death following an extended mitosis (Fong et al, 2016; Lambrus et al, 2016; Meitinger et al, 2016). This suggests that TP53 signaling occurs through different downstream factors to induce apoptosis in NPCs. Last but not least, our data reveal that although inactivation of the mitotic surveillance pathway restores brain size in Cep63T/T mice, these double knockout animals still display body growth retardation. This argues that mutations in centrosome genes can cause brain and body size reduction through distinct pathological mechanisms.
In conclusion, our data provide evidence to support the pathological role of the mitotic surveillance pathway in neurodevelopmental disorders. Inhibitors of USP28 are under development and may have therapeutic utility in disorders of brain growth (Wrigley et al, 2017). Understanding the biochemical “timing” mechanism that allows cells to measure mitotic duration and what sets the sensitivity of this response are important questions for the future.
Materials and Methods
Animals
Cep63T/T mice were obtained from the laboratory of T. Stracker (Institute for Research in Biomedicine, Spain; Marjanovic et al, 2015). The following primers were used for genotyping: Cep63‐5P2, 5′‐GTAGGACCAGGCCTTAGCGTTAG‐3′; Cep63‐3P1a, 5′‐TAAGTGTAAAAGCCGGGCGTGGT‐3′ and MutR (B32), 5′‐Caaggcgattaagttgggtaacg‐3′.
Sas4cKO mice were obtained from the laboratory of K. Anderson (Memorial Sloan Kettering Cancer Center, USA; Bazzi & Anderson, 2014). The following primers were used for genotyping: Cenpj LoxP2 F, 5′‐TGCTTGCTTGTCTCTCCTGA‐3′; Cenpj LoxP2 R 5′‐GCTGACACCAAGTGGGAAAT‐3′; Cenpj FRT1F 5′‐GGGGAGCAGACTTCAACACT‐3′.
Smc5cKO mice were generated in the laboratory of P. Jordan (Johns Hopkins Bloomberg School of Public Health, USA; Atkins et al, 2020). The following primers were used for genotyping: Smc5F (WT/Flox) Primer1 Fwd 5′‐ACTCAGTCTCACACGGCAAG‐3′; Smc5F (WT/Flox) Primer2 Rev 5′‐ATCCTTCCCACCTTGGAAAC‐3′.
Nestin‐Cre mice were obtained from The Jackson Laboratory (stock #003771). The following primers were used for genotyping: Nestin‐Cre Fwd 5′‐ATTGCTGTCACTTGGTCGTGGC‐3′; Nestin‐Cre Rev 5′‐GGAAAATGCTTCTGTCCGTTTGC‐3′.
Trp53bp1 −/− mice were obtained from the laboratory of S. Franco at (Johns Hopkins University School of Medicine, USA; Rybanska‐Spaeder et al, 2013). The following primers were used for genotyping: 53bp1 Common 10694 5′‐GAACTTGGCTCACACCCATT‐3′; 53bp1 WT 10693 5′‐CTCCAGAGAGAACCCAGCAG‐3′; 53bp1 Mut oIMR5316 5′‐CTAAAGCGCATGCTCCAGAC‐3′.
Usp28F /‐ mice were obtained from the laboratory M. Eilers (University of Wuerzburg, Germany; Diefenbacher et al, 2014). The following primers were used for genotyping: Usp28 Common Fwd 1718 27 5′‐GAGGCTTGAGTTATGACTGG‐3′; Usp28 WT Rev 1718 m28 5′‐AGAACACCTGCTGCTTAAGC‐3′; Usp28 Del Rev 5′‐TCCCCCAAGAGTGTTTTCAC‐3′.
Trp53 −/− mice were obtained from The Jackson Laboratory (stock #02101). The following primers were used for genotyping: TP53 COMMON 01 MR 0336 5′‐ATAGGTCGGCGGTTCAT‐3′; TP53 WT 01MR 0337 5′‐CCCGAGTATCTGGAAGACAG‐3′; TP53 MUT 01MR 0013 5′‐CTTGGGTGGAGAGGCTATTC‐3′.
Nuclear‐mCherryFl / Fl mice were obtained from The Jackson Laboratory (stock #023139). The following primers were used for genotyping: Nuclear‐mCherry COM Fwd oIMR8545 5′‐AAAGTCGCTCTGAGTTGTTAT‐3′; Nuclear‐mCherry WT Rev oIMR8546 5′‐GGAGCGGGAGAAATGGATATG‐3′; Nuclear‐mCherry Flox Rev 10507 5′‐TTATGTAACGCGGAACTCCA‐3′.
Analysis of Smc5cKO and Cep63T / T mice and associated genotypes was performed on a congenic C57BL6 background, while analysis of Sas4cKO mice and associated genotypes was performed on a mixed FVB/NJ and C57BL6 background. Genotyping was carried out using standard PCR protocols. Embryos and adults from both genders were included in our analysis. Mice were housed and cared for in an AAALAC‐accredited facility, and all animal experiments were conducted in accordance with Institute Animal Care and Use Committee approved protocols.
Post‐natal brain dissection, whole‐brain imaging, and histology
Post‐natal day 14 (P14), P21 and P60 animals were anesthetized with Avertin before being perfused with PBS and 1% paraformaldehyde. The brains were removed from the skull and post‐fixed in 4% paraformaldehyde overnight at 4°C. The next day, the brains were rinsed three times with PBS before being weighed and imaged using a MU1003 digital camera attached to a dissecting microscope (Amscope). The two hemispheres were separated down the midline; one hemisphere was processed for cryosectioning (details below), while the other hemisphere was sent for histological analysis. The Johns Hopkins University, School of Medicine, phenotyping core performed the tissue processing, paraffin embedding and hematoxylin & eosin staining.
Brain section, immunohistochemistry, and confocal imaging
For analysis of embryonic brain sections, timed pregnant females carrying the respective transgenes were anesthetized with isoflurane before cervical dislocation. The heads of the embryos were removed and rinsed with ice‐cold PBS, followed by incubation in 4% paraformaldehyde for 4 h (for TBR1 and Mouse‐Pax6 antibody specifically) or overnight (for all other antibodies) at 4°C. The next day, the brains were dissected out of the skulls, rinsed three times in PBS before being incubated in 30% sucrose at 4°C for 24 h. Brains were embedded in O.C.T medium and frozen for cryosectioning. 20 µm brain sections were cut using a Leica CM1950 or CM3050 cryostat and collected on Superfrost microscope slides (Thermo Fisher Scientific). For immunohistochemistry staining, the brain sections were rehydrated with PBS, blocked and permeabilized with blocking solution (10% donkey serum (Sigma Aldrich), 0.1‐0.5% Triton X‐100 in PBS), and incubated with the respective primary antibodies diluted in blocking solution for 12 h at 4°C or 1.5 h at RT (Table EV3). The following primary antibodies were used: Rabbit‐PAX6 (Covance, PRB‐278P, 1:500), Chicken‐TBR2 (EMD Millipore, AB15894, 1:250), Rat‐α‐tubulin (Pierce Antibodies, MA1‐80017, 1:500), Goat‐γ‐tubulin (homemade, polyclonal, raised against the peptide CDEYHAATRPDYISWGTQEQ, 1:500), Rabbit‐Ki67 (D3B5) (Cell Signaling, 9129, 1:500), Mouse‐PH3 (Cell Signaling, 9701, 1:100), Rabbit‐CC3 (Asp175) (Cell Signaling, 9661, 1:500), Rabbit‐centrin (in house, raised against human centrin2 (a.a. 1–172); 1:500), Rabbit‐γ‐H2AX (p‐Ser139) (Cell Signaling, 2577, 1:250), Mouse‐TP53 (1C12) (Cell Signaling, 2524S, 1:250), Mouse‐TBR1 (ProteinTech, 66564‐1‐Ig, 1:250) and Mouse‐PAX6 (Thermo Fisher Scientific, MA1‐109, 1:250). Following primary antibody staining, the tissue slides were washed 3 times with PBST (0.1–0.5% Triton X‐100) and incubated with secondary antibodies and DAPI diluted in blocking solution for 1 h at room temperature. Following secondary antibody staining, the tissue slides were washed three times with PBST before mounting. Secondary antibodies were conjugated to Alexa Fluor 488, 555, or 647 (Thermo Fisher Scientific, 1:500).
For analysis of P14, P21 and P60 brain sections, the brains were dissected and post‐fixed in 4% paraformaldehyde at 4°C overnight as described above. The next day, brains were rinsed with PBS, incubated in 30% sucrose for at least 48 h and embedded in O.C.T medium. 20 µm brain sections were collected onto Superfrosted microscope slides. For antigen retrieval, slides containing the brain sections were rehydrated with PBS and boiled in citrate buffer (10 mM citric acid, 0.1% Triton X‐100, pH 6.0) at 95°C for 15 min. Slides were then rinsed three times in PBS before being incubated with blocking solution and stained with respective primary and secondary antibodies, as described above. The following primary antibodies were used: Rat‐CTIP2 (Abcam, ab18465, 1:1,000) and Rabbit‐CUX1 (ProteinTech, 11733‐1‐AP, 1:1,000). Secondary antibodies were conjugated to Alexa Fluor 488, 555, or 647 (Thermo Fisher Scientific, 1:500).
Stained tissue sections were mounted in ProLong Gold Antifade Mountant (Invitrogen). Images of stained sections were obtained using an LSM700 (Zeiss) or SP8 (Leica Microsystems) confocal microscope. For centrosome and centriole staining, images were collected using a Leica 63× 1.40 NA oil objective at 0.25 μm z‐sections. For all other staining, images were collected using a Zeiss 20× 0.8 NA air objective at 1 μm z‐sections.
Neural progenitor primary cultures
Embryos at embryonic day 14.5 (E14.5) were removed from pregnant females following cervical dislocation. The brain was removed from the skull and the dorsolateral telencephalon isolated as previously described (Landeira et al, 2017). Following dissection, the dorsolateral telencephalon tissues were incubated with Trypsin‐EDTA (0.05%) for 10 min at 37°C. Proliferation medium (10% FBS, 0.4% glucose, 1% penicillin/streptomycin in DMEM) was added to stop Trypsin activity, and the cells were mechanically dissociated by gentle pipetting. Cells were then washed twice with proliferation medium to remove residual Trypsin before being counted. For time‐lapse imaging, 500,000 cells were seeded into each well of a 48‐well glass‐bottom plate (MatTek Corporation) or 8‐chamber glass‐bottom dish (ibidi) coated with Poly‐d‐Lysine. For immunofluorescence staining, 1,000,000 cells were seeded onto 12‐mm glass coverslips (Thermo Fisher Scientific) coated with Poly‐d‐Lysine. Seeded cells were maintained at 37°C, 5% CO2 for 2–5 h before being used for fixed or live‐cell imaging.
Time‐lapse Imaging of neural progenitor culture
Cells were maintained in proliferation media at 37°C, 5% CO2 with an environmental control station throughout the entire imaging session. 24 to 52‐h time‐lapse movies were captured using a SP8 microscope (Leica microsystems) controlling a Leica DFC9000 GTC sCMOS camera. Images were acquired with a 40× 0.8 NA air objective. Every 5 min, 6 × 3‐μm z‐sections were acquired in the Texas‐Red channel to observe the Nuclear‐mCherry signal. Note that Nuclear‐mCherry corresponds to a transgenic H2B‐mCherry mouse obtained from Jax (stock #023139). While the H2B‐mCherry fusion protein fails to localize to the chromosomes, it provides a nuclear signal that disperses at the beginning of mitosis and reappears in late anaphase when nuclear envelope reformation occurs. Thus, we were able to make use of the H2B‐mCherry protein to monitor mitotic duration by determining the time taken from NEBD to nuclear envelope reformation.
Prolonged mitosis assay
500,000 cells were seeded into each well of a 48‐well glass‐bottom plate as described above. After 2 h of incubation at 37°C, media with nocodazole (0.08 µM final concentration) was added, and fields of cells were continuously followed by video time‐lapse microscopy at 37°C for 4 h. After 4 h, media with nocodazole was removed and cells washed with fresh medium several times. The same fields of view were continuously followed for an additional 48 h.
Immunofluorescence staining of neural progenitor culture
Cells were grown on glass coverslips and fixed for 10 mins in 4% paraformaldehyde at room temperature. Cells were blocked in blocking solution (2.5% FBS, 200 mM glycine, and 0.1% Triton X‐100 in PBS) for 1 h at room temperature or overnight at 4°C. Cells were then incubated in primary antibody diluted in the blocking solution for 1 h, rinsed with PBST (0.1% Triton X‐100 in PBS), followed by secondary antibody staining prepared in the same blocking solution. DNA was stained with DAPI, and cells were mounted in ProLong Gold Antifade Mountant (Invitrogen). The following primary antibodies were used: Rabbit‐PAX6 (Covance, PRB‐278P, 1:1,000), Chicken‐TBR2 (EMD Millipore, AB15894, 1:1,000), Mouse‐TUJ1 (Covance, MMS‐435P, 1:1,000), Goat‐γ‐tubulin (homemade, polyclonal, raised against the peptide CDEYHAATRPDYISWGTQEQ, 1:1,000) and Mouse‐TP53 (1C12) (Cell Signaling, 2524S, 1:1,000). Secondary antibodies were conjugated to Alexa Fluor 488, 555, or 647 (Thermo Fisher Scientific, 1:1,000).
Single‐cell sequencing
Single cells were isolated from embryonic brains at E14.5, as described above, and immediately stored in proliferation medium with 10% DMSO at −80°C until sorted. Single‐cell karyotype analysis was performed and analyzed as previously described (Bakker et al, 2016; Levine et al, 2017).
Quantification and statistical analysis
Image and movie analysis
Imaris software version 9.2.1 (Bitplane) was used to quantify the total number of nuclei per field of view in the tissues stained with Ki67, PH3, PAX6, TBR2, CC3, TP53, CTIP2, CUX1, γ‐H2AX, and DAPI. ImageJ software (US National Institutes of Health) was used to quantify the telencephalon area from whole‐brain images and cortical thickness from stained brain sections. The same software was also used to quantify the fraction of dissociated neural progenitor cells stained with PAX6, TBR2, and TP53.
LAX software (Leica) was used to measure mitotic duration (from NEBD to nuclear envelope reformation), cell cycle length (from NEBD of the mother cell to the NEBD of one of the two daughter cells), and track cell fate following cell division in Nuclear‐mCherry+ dissociated neural progenitor cultures. For cell cycle length and cell fate analysis, dissociated NPC were filmed for 48 h and only cells that enter mitosis within the first 24 h period were selected for fate tracing. The same software was also used to quantify spindle angle or the number of centrioles and centrosomes per NPC in embryonic sections.
Statistical analysis
Statistical analysis was performed using GraphPad Prism software. Differences between samples were analyzed using a two‐tailed unpaired Student t‐tests (Mann–Whitney t‐test or Welch’s t‐test), chi‐square test with post hoc analysis, one‐way ANOVA with post hoc analysis, or a log‐rank test for survival analysis as indicated in the figure legends. Error bars represent SEM unless otherwise indicated. Figure legends state the number of cells or animals used per experiment (Tables EV1 and EV2).
Author contributions
TPP performed and analyzed the majority of the experiments and prepared the figures. ALM assisted with experiments using dissociated NPCs and characterization of the adult Smc5cKO mice. CAB and JL assisted with animal husbandry and brain dissections. AA performed the initial characterization of the Smc5cKO mice and associated double knockout animals. AT, DCJS, and FF performed the single‐cell sequencing experiments. THS provided the Cep63T/T mice, HB provided the Sas4F/F mice, and PWJ provided the Smc5F/F mice. AJH and TPP wrote the manuscript. All authors edited the manuscript. AJH conceived and supervised the study.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Movie EV1
Movie EV2
Review Process File
Acknowledgements
The research grant supporting this project was terminated by March of Dimes one year after the project began (FY17‐698, https://www.nature.com/articles/d41586‐018‐05875‐7). We are grateful to Jeremy Nathans, Antony Rosen, and the Johns Hopkins Institute for Basic Biomedical Sciences for providing bridge support to allow the completion of this work. We also thank the members of the Nathans and Mueller laboratory for their valuable insights and technical support. This work also was supported by the National Institutes of Health grants R01GM114119 and R01GM133897, an American Cancer Society Scholar grant RSG‐16‐156‐01‐CCG, and an American Cancer Society Mission Boost Grant MBG‐19‐173‐01‐MBG (to A.J.H). T.P was funded by the National Institutes of Health training grant (T32GM007445).
The EMBO Journal (2021) 40: e106118.
Data availability
This study includes no data deposited in external repositories.
References
- Atkins A, Xu MJ, Li M, Rogers NP, Pryzhkova MV, Jordan PW (2020) Neurogenesis requires SMC5/6 to avoid catastrophic replicative DNA damage in early progenitors. Elife, in revision. [Google Scholar]
- Bakker B, Taudt A, Belderbos ME, Porubsky D, Spierings DC, de Jong TV, Halsema N, Kazemier HG, Hoekstra‐Wakker K, Bradley A et al (2016) Single‐cell sequencing reveals karyotype heterogeneity in murine and human malignancies. Genome Biol 17: 115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzi H, Anderson KV (2014) Acentriolar mitosis activates a p53‐dependent apoptosis pathway in the mouse embryo. Proc Natl Acad Sci USA 111: E1491–E1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond J, Roberts E, Springell K, Lizarraga SB, Scott S, Higgins J, Hampshire DJ, Morrison EE, Leal GF, Silva EO et al (2005) A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat Genet 37: 353–355 [DOI] [PubMed] [Google Scholar]
- Breslow DK, Holland AJ (2019) Mechanism and regulation of centriole and cilium biogenesis. Annu Rev Biochem 88: 691–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney JP, Maser RS, Olivares H, Davis EM, Le Beau M, Yates JR 3rd, Hays L, Morgan WF, Petrini JH (1998) The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double‐strand break repair to the cellular DNA damage response. Cell 93: 477–486 [DOI] [PubMed] [Google Scholar]
- Chavali PL, Putz M, Gergely F (2014) Small organelle, big responsibility: the role of centrosomes in development and disease. Philos Trans R Soc Lond B Biol Sci 369: 20130468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CT, Hehnly H, Yu Q, Farkas D, Zheng G, Redick SD, Hung HF, Samtani R, Jurczyk A, Akbarian S et al (2014) A unique set of centrosome proteins requires pericentrin for spindle‐pole localization and spindle orientation. Curr Biol 24: 2327–2334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chizhikov VV, Davenport J, Zhang Q, Shih EK, Cabello OA, Fuchs JL, Yoder BK, Millen KJ (2007) Cilia proteins control cerebellar morphogenesis by promoting expansion of the granule progenitor pool. J Neurosci 27: 9780–9789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degrassi F, Damizia M, Lavia P (2019) The mitotic apparatus and kinetochores in microcephaly and neurodevelopmental diseases. Cells 9: 49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diefenbacher ME, Popov N, Blake SM, Schulein‐Volk C, Nye E, Spencer‐Dene B, Jaenicke LA, Eilers M, Behrens A (2014) The deubiquitinase USP28 controls intestinal homeostasis and promotes colorectal cancer. J Clin Invest 124: 3407–3418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding W, Wu Q, Sun L, Pan NC, Wang X (2019) Cenpj regulates cilia disassembly and neurogenesis in the developing mouse cortex. J Neurosci 39: 1994–2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S (1998) A caspase‐activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 391: 43–50 [DOI] [PubMed] [Google Scholar]
- Englund C, Fink A, Lau C, Pham D, Daza RA, Bulfone A, Kowalczyk T, Hevner RF (2005) Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci 25: 247–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faheem M, Naseer MI, Rasool M, Chaudhary AG, Kumosani TA, Ilyas AM, Pushparaj P, Ahmed F, Algahtani HA, Al‐Qahtani MH et al (2015) Molecular genetics of human primary microcephaly: an overview. BMC Med Genomics 8(Suppl 1): S4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Walsh CA (2004) Mitotic spindle regulation by Nde1 controls cerebral cortical size. Neuron 44: 279–293 [DOI] [PubMed] [Google Scholar]
- Firat‐Karalar EN, Stearns T (2014) The centriole duplication cycle. Philos Trans R Soc Lond B Biol Sci 369: 20130460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florio M, Huttner WB (2014) Neural progenitors, neurogenesis and the evolution of the neocortex. Development 141: 2182–2194 [DOI] [PubMed] [Google Scholar]
- Fong CS, Mazo G, Das T, Goodman J, Kim M, O'Rourke BP, Izquierdo D, Tsou MF (2016) 53BP1 and USP28 mediate p53‐dependent cell cycle arrest in response to centrosome loss and prolonged mitosis. Elife 5: e16270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster SS, De S, Johnson LK, Petrini JH, Stracker TH (2012) Cell cycle‐ and DNA repair pathway‐specific effects of apoptosis on tumor suppression. Proc Natl Acad Sci USA 109: 9953–9958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fousteri MI, Lehmann AR (2000) A novel SMC protein complex in Schizosaccharomyces pombe contains the Rad18 DNA repair protein. EMBO J 19: 1691–1702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco SJ, Muller U (2013) Shaping our minds: stem and progenitor cell diversity in the mammalian neocortex. Neuron 77: 19–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank KM, Sharpless NE, Gao Y, Sekiguchi JM, Ferguson DO, Zhu C, Manis JP, Horner J, DePinho RA, Alt FW (2000) DNA ligase IV deficiency in mice leads to defective neurogenesis and embryonic lethality via the p53 pathway. Mol Cell 5: 993–1002 [DOI] [PubMed] [Google Scholar]
- Frappart PO, Tong WM, Demuth I, Radovanovic I, Herceg Z, Aguzzi A, Digweed M, Wang ZQ (2005) An essential function for NBS1 in the prevention of ataxia and cerebellar defects. Nat Med 11: 538–544 [DOI] [PubMed] [Google Scholar]
- Gruber R, Zhou Z, Sukchev M, Joerss T, Frappart PO, Wang ZQ (2011) MCPH1 regulates the neuroprogenitor division mode by coupling the centrosomal cycle with mitotic entry through the Chk1‐Cdc25 pathway. Nat Cell Biol 13: 1325–1334 [DOI] [PubMed] [Google Scholar]
- Haubensak W, Attardo A, Denk W, Huttner WB (2004) Neurons arise in the basal neuroepithelium of the early mammalian telencephalon: a major site of neurogenesis. Proc Natl Acad Sci USA 101: 3196–3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heins N, Malatesta P, Cecconi F, Nakafuku M, Tucker KL, Hack MA, Chapouton P, Barde YA, Gotz M (2002) Glial cells generate neurons: the role of the transcription factor Pax6. Nat Neurosci 5: 308–315 [DOI] [PubMed] [Google Scholar]
- Insolera R, Bazzi H, Shao W, Anderson KV, Shi SH (2014) Cortical neurogenesis in the absence of centrioles. Nat Neurosci 17: 1528–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman D, Kodani A, Gonzalez DM, Mancias JD, Mochida GH, Vagnoni C, Johnson J, Krogan N, Harper JW, Reiter JF et al (2016) Microcephaly proteins Wdr62 and Aspm define a mother centriole complex regulating centriole biogenesis, apical complex, and cell fate. Neuron 92: 813–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman D, Bae BI, Walsh CA (2018) The genetics of primary microcephaly. Annu Rev Genomics Hum Genet 19: 177–200 [DOI] [PubMed] [Google Scholar]
- Knobel PA, Belotserkovskaya R, Galanty Y, Schmidt CK, Jackson SP, Stracker TH (2014) USP28 is recruited to sites of DNA damage by the tandem BRCT domains of 53BP1 but plays a minor role in double‐strand break metabolism. Mol Cell Biol 34: 2062–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczyk T, Pontious A, Englund C, Daza RA, Bedogni F, Hodge R, Attardo A, Bell C, Huttner WB, Hevner RF (2009) Intermediate neuronal progenitors (basal progenitors) produce pyramidal‐projection neurons for all layers of cerebral cortex. Cereb Cortex 19: 2439–2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrus BG, Uetake Y, Clutario KM, Daggubati V, Snyder M, Sluder G, Holland AJ (2015) p53 protects against genome instability following centriole duplication failure. J Cell Biol 210: 63–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrus BG, Daggubati V, Uetake Y, Scott PM, Clutario KM, Sluder G, Holland AJ (2016) A USP28‐53BP1‐p53‐p21 signaling axis arrests growth after centrosome loss or prolonged mitosis. J Cell Biol 214: 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landeira BS, Araujo JAM, Schroeder T, Muller U, Costa MR (2017) Live imaging of primary cerebral cortex cells using a 2D culture system. J Vis Exp 126: 56063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Barnes DE, Lindahl T, McKinnon PJ (2000) Defective neurogenesis resulting from DNA ligase IV deficiency requires Atm. Genes Dev 14: 2576–2580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine MS, Bakker B, Boeckx B, Moyett J, Lu J, Vitre B, Spierings DC, Lansdorp PM, Cleveland DW, Lambrechts D et al (2017) Centrosome amplification is sufficient to promote spontaneous tumorigenesis in mammals. Dev Cell 40: 313–322.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YN, Lee YS, Li SK, Tang TK (2020) Loss of CPAP in developing mouse brain and its functional implication in human primary microcephaly. J Cell Sci 133: jcs243592 [DOI] [PubMed] [Google Scholar]
- Lizarraga SB, Margossian SP, Harris MH, Campagna DR, Han AP, Blevins S, Mudbhary R, Barker JE, Walsh CA, Fleming MD (2010) Cdk5rap2 regulates centrosome function and chromosome segregation in neuronal progenitors. Development 137: 1907–1917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmood S, Ahmad W, Hassan MJ (2011) Autosomal Recessive Primary Microcephaly (MCPH): clinical manifestations, genetic heterogeneity and mutation continuum. Orphanet J Rare Dis 6: 39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malatesta P, Hartfuss E, Gotz M (2000) Isolation of radial glial cells by fluorescent‐activated cell sorting reveals a neuronal lineage. Development 127: 5253–5263 [DOI] [PubMed] [Google Scholar]
- Marjanovic M, Sanchez‐Huertas C, Terre B, Gomez R, Scheel JF, Pacheco S, Knobel PA, Martinez‐Marchal A, Aivio S, Palenzuela L et al (2015) CEP63 deficiency promotes p53‐dependent microcephaly and reveals a role for the centrosome in meiotic recombination. Nat Commun 6: 7676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marthiens V, Basto R (2020) Centrosomes: The good and the bad for brain development. Biol Cell 112: 153–172 [DOI] [PubMed] [Google Scholar]
- McIntyre RE, Lakshminarasimhan Chavali P, Ismail O, Carragher DM, Sanchez‐Andrade G, Forment JV, Fu B, Del Castillo V‐H, Edwards A, van der Weyden L et al (2012) Disruption of mouse Cenpj, a regulator of centriole biogenesis, phenocopies Seckel syndrome. PLoS Genet 8: e1003022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon PJ (2017) Genome integrity and disease prevention in the nervous system. Genes Dev 31: 1180–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meitinger F, Anzola JV, Kaulich M, Richardson A, Stender JD, Benner C, Glass CK, Dowdy SF, Desai A, Shiau AK et al (2016) 53BP1 and USP28 mediate p53 activation and G1 arrest after centrosome loss or extended mitotic duration. J Cell Biol 214: 155–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell‐Dick A, Chalem A, Pilaz LJ, Silver DL (2020) Acute lengthening of progenitor mitosis influences progeny fate during cortical development in vivo . Dev Neurosci 41: 300–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata T, Kawaguchi A, Saito K, Kawano M, Muto T, Ogawa M (2004) Asymmetric production of surface‐dividing and non‐surface‐dividing cortical progenitor cells. Development 131: 3133–3145 [DOI] [PubMed] [Google Scholar]
- Murga M, Bunting S, Montana MF, Soria R, Mulero F, Canamero M, Lee Y, McKinnon PJ, Nussenzweig A, Fernandez‐Capetillo O (2009) A mouse model of ATR‐Seckel shows embryonic replicative stress and accelerated aging. Nat Genet 41: 891–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noctor SC, Flint AC, Weissman TA, Dammerman RS, Kriegstein AR (2001) Neurons derived from radial glial cells establish radial units in neocortex. Nature 409: 714–720 [DOI] [PubMed] [Google Scholar]
- Noctor SC, Martinez‐Cerdeno V, Ivic L, Kriegstein AR (2004) Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci 7: 136–144 [DOI] [PubMed] [Google Scholar]
- O'Driscoll M, Ruiz‐Perez VL, Woods CG, Jeggo PA, Goodship JA (2003) A splicing mutation affecting expression of ataxia‐telangiectasia and Rad3‐related protein (ATR) results in Seckel syndrome. Nat Genet 33: 497–501 [DOI] [PubMed] [Google Scholar]
- O'Driscoll M, Cerosaletti KM, Girard PM, Dai Y, Stumm M, Kysela B, Hirsch B, Gennery A, Palmer SE, Seidel J et al (2001) DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency. Mol Cell 8: 1175–1185 [DOI] [PubMed] [Google Scholar]
- Orth JD, Loewer A, Lahav G, Mitchison TJ (2012) Prolonged mitotic arrest triggers partial activation of apoptosis, resulting in DNA damage and p53 induction. Mol Biol Cell 23: 567–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne F, Colnaghi R, Rocha N, Seth A, Harris J, Carpenter G, Bottomley WE, Wheeler E, Wong S, Saudek V et al (2014) Hypomorphism in human NSMCE2 linked to primordial dwarfism and insulin resistance. J Clin Invest 124: 4028–4038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilaz LJ, McMahon JJ, Miller EE, Lennox AL, Suzuki A, Salmon E, Silver DL (2016) Prolonged mitosis of neural progenitors alters cell fate in the developing brain. Neuron 89: 83–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P (1972) Mode of cell migration to the superficial layers of fetal monkey neocortex. J Comp Neurol 145: 61–83 [DOI] [PubMed] [Google Scholar]
- Rybanska‐Spaeder I, Reynolds TL, Chou J, Prakash M, Jefferson T, Huso DL, Desiderio S, Franco S (2013) 53BP1 is limiting for NHEJ repair in ATM‐deficient model systems that are subjected to oncogenic stress or radiation. Mol Cancer Res 11: 1223–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeant J, Taylor E, Palecek J, Fousteri M, Andrews EA, Sweeney S, Shinagawa H, Watts FZ, Lehmann AR (2005) Composition and architecture of the Schizosaccharomyces pombe Rad18 (Smc5‐6) complex. Mol Cell Biol 25: 172–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehan CJ, McMahon JJ, Serdar LD, Silver DL (2020) Dosage‐dependent requirements of Magoh for cortical interneuron generation and survival. Development 147: dev182295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Qalieh A, Lam MM, Keil JM, Kwan KY (2019) Robust elimination of genome‐damaged cells safeguards against brain somatic aneuploidy following Knl1 deletion. Nat Commun 10: 2588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sir JH, Barr AR, Nicholas AK, Carvalho OP, Khurshid M, Sossick A, Reichelt S, D'Santos C, Woods CG, Gergely F (2011) A primary microcephaly protein complex forms a ring around parental centrioles. Nat Genet 43: 1147–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetake Y, Sluder G (2010) Prolonged prometaphase blocks daughter cell proliferation despite normal completion of mitosis. Curr Biol 20: 1666–1671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varon R, Vissinga C, Platzer M, Cerosaletti KM, Chrzanowska KH, Saar K, Beckmann G, Seemanova E, Cooper PR, Nowak NJ et al (1998) Nibrin, a novel DNA double‐strand break repair protein, is mutated in Nijmegen breakage syndrome. Cell 93: 467–476 [DOI] [PubMed] [Google Scholar]
- Venegas AB, Natsume T, Kanemaki M, Hickson ID (2020) Inducible degradation of the human SMC5/6 complex reveals an essential role only during interphase. Cell Rep 31: 107533 [DOI] [PubMed] [Google Scholar]
- Waltes R, Kalb R, Gatei M, Kijas AW, Stumm M, Sobeck A, Wieland B, Varon R, Lerenthal Y, Lavin MF et al (2009) Human RAD50 deficiency in a Nijmegen breakage syndrome‐like disorder. Am J Hum Genet 84: 605–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YL, Anzola JV, Davis RL, Yoon M, Motamedi A, Kroll A, Seo CP, Hsia JE, Kim SK, Mitchell JW et al (2015) Reversible centriole depletion with an inhibitor of Polo‐like kinase 4. Science 348: 1155–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff JB, Wueseke O, Hyman AA (2014) Pericentriolar material structure and dynamics. Philos Trans R Soc Lond B Biol Sci 369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrigley JD, Gavory G, Simpson I, Preston M, Plant H, Bradley J, Goeppert AU, Rozycka E, Davies G, Walsh J et al (2017) Identification and characterization of dual inhibitors of the USP25/28 deubiquitinating enzyme subfamily. ACS Chem Biol 12: 3113–3125 [DOI] [PubMed] [Google Scholar]
- Zhao X, Blobel G (2005) A SUMO ligase is part of a nuclear multiprotein complex that affects DNA repair and chromosomal organization. Proc Natl Acad Sci USA 102: 4777–4782 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Movie EV1
Movie EV2
Review Process File
Data Availability Statement
This study includes no data deposited in external repositories.