

Abstract
The generation of induced pluripotent stem cells (iPSCs) from somatic cells provides an excellent model to study mechanisms of transcription factor‐induced global alterations of the epigenome and genome function. Here, we have investigated the early transcriptional events of cellular reprogramming triggered by the co‐expression of Oct4, Sox2, Klf4, and c‐Myc (OSKM) in mouse embryonic fibroblasts (MEFs) and mouse hepatocytes (mHeps). In this analysis, we identified a gene regulatory network composed of nine transcriptional regulators (9TR; Cbfa2t3, Gli2, Irf6, Nanog, Ovol1, Rcan1, Taf1c, Tead4, and Tfap4), which are directly targeted by OSKM, in vivo. Functional studies using single and double shRNA knockdowns of any of these factors caused disruption of the network and dramatic reductions in reprogramming efficiency, indicating that this network is essential for the induction and establishment of pluripotency. We demonstrate that the stochastic co‐expression of 9TR network components occurs in a remarkably small number of cells, approximating the percentage of terminally reprogrammed cells as a result of dynamic molecular events. Thus, the early DNA‐binding patterns of OSKM and the subsequent probabilistic co‐expression of essential 9TR components in subpopulations of cells undergoing reprogramming steer the reconstruction of a gene regulatory network marking the transition to pluripotency.
Keywords: cellular reprogramming, gene regulatory networks, transcriptional regulators
Subject Categories: Development & Differentiation, Transcription
Nine transcriptional regulators, which are required for induction and establishment of pluripotency, are stochastically expressed in a small number of cells correlating to the percentage of eventually reprogrammed cells.

Introduction
The cooperative and synchronized actions of signaling pathways, and transcriptional and epigenetic regulators define unique cellular functions and fates by determining specific gene expression programs (Buganim et al, 2013; Hussein et al, 2014). Transcription factors play the central role in defining gene expression programs by functioning as the end points of various molecular pathways and their synergistic and combinatorial action sets the epigenetic landscape required for appropriate regulation of gene transcription (Lambert et al, 2018). Transcription factors do not act on their own, but instead, they build enhanceosomes composed of distinct members of transcription factor families at DNA regulatory elements, thus responding to various cell signaling pathways (Merika & Thanos, 2001). In addition, they form autoregulatory networks, which maintain their expression levels and process signal integration to target the correct set of genes for ensuring cell identity. The numerous cross‐regulatory interactions within and between transcription factor networks along with the plasticity of the connections provide the necessary flexibility for adaptation and evolvement of novel gene expression programs (Li & Belmonte, 2017; Niwa, 2018).
The conversion of somatic cells to induced pluripotent stem cells (iPSCs) is achieved by the ectopic co‐expression of four transcription factors, Oct4, Sox2, Klf4, and c‐Myc (OSKM), generating cellular populations that closely resemble to embryonic stem cells (ESCs) (Takahashi & Yamanaka, 2006; Takahashi et al, 2007). Previous studies have provided evidence for the action of core transcriptional gene regulatory networks in pluripotency, where Oct4, Sox2, and Nanog recruit additional pluripotency‐associated regulatory factors to establish and maintain steady levels of expression of unique sets of genes, all of which define the stem cell phenotype (Loh et al, 2006; Chen et al, 2008; Kim et al, 2008). Cellular reprogramming is initiated with the genome‐wide DNA binding of OSKM (Koche et al, 2011; Soufi et al, 2012; Chen et al, 2016), which triggers massive transcriptional changes driven by gradual and hierarchical chromatin alterations at multiple topological genome organization and architectural levels (Polo et al, 2012; Stadhouders et al, 2018). These early transcriptional events affect the expression levels of hundreds of genes, some of which are relevant to the acquisition of the stemness identity. Induced pluripotency is characterized by a stepwise cellular de‐differentiation of the starting cell type and progressive establishment of a new pluripotent transcriptome beginning with the gradual abolishment of cell type‐specific transcriptional profiles (Polo et al, 2012; Stadtfeld et al, 2008; Buganim et al, 2012; Chronis et al, 2017). During this process, in addition to iPSC generation, many diverse developmental programs are also being produced with unknown fates (Schiebinger et al, 2019). Contrary to the opposite process, that is, the differentiation of ESCs to specialized cell types, cellular reprogramming of somatic cells is remarkably inefficient occurring stochastically in an asynchronous manner with variable latency (Hanna et al, 2009). It is highly improbable that OSKM can revert by themselves the pre‐existing epigenetic barriers and directly induce the massive dynamic transcriptional changes required for the acquisition of pluripotency without help from additional regulators. Indeed, additional factors that have been also implicated in acquisition of pluripotency are dispensable for pluripotency maintenance (Schwarz et al, 2018). Despite the seminal discoveries of the last decade, our current view regarding the fundamental aspects of the mechanisms that drive cellular reprogramming to pluripotency still remains elusive, illustrating the complexity of the molecular mechanisms underlying this process (Apostolou & Stadtfeld, 2018). Previous studies have proposed that the assembly of transcriptional regulatory networks could play a significant role in cellular reprogramming, as it is the case for the core pluripotent network active in stem cells (Chen et al, 2008; Kim et al, 2008; Niwa, 2014). However, these initial suggestions still remain unexplored.
Transcription factor networks not only maintain their own expression but they also ensure the subsequent robust expression of downstream genes often encoding for additional transcriptional regulators and other critical cellular components. These networks are stabilized through the balanced maintenance of their expression by forming interconnected autoregulatory loops receiving multiple inputs from extracellular signals (Chen et al, 2008) and operate as a whole in order to integrate the individual functions of each of the participating transcription factors to confer robustness and phenotypic reproducibility. Thus, it may not be surprising that although cellular reprogramming is a highly stochastic process (Buganim et al, 2012; Yamanaka, 2009), the fraction of the cells being reprogrammed is mainly determined by the characteristics of the starting cells (Chronis et al, 2017). While previous studies have examined the role of individual transcription factors in reprogramming, we lack essential knowledge about their dynamics and temporal hierarchy or for the involvement of transcription factor networks in reprogramming. For example, how known and unknown non‐OSKM transcriptional regulators are placed within the context of putative reprogramming networks to replace the cell‐specific networks of the starting cells? What is the relationship of putative reprogramming network(s) with core pluripotent networks known to be established at the end of the reprogramming process? What is the mechanism of assembly of putative regulatory networks active in reprogramming? Furthermore, it is still unknown whether common molecular trajectories are shared between distinct cell types during their conversion to iPSCs.
In this study, we derived the spatiotemporal dynamics of a gene regulatory network (GRN) by integrating dynamic transcriptional cascades to shed light to the transcriptional logic of cellular reprogramming. We showed that OSKM trigger the activation of a set of transcription factors common in at least two distinct cell types undergoing reprogramming, a subgroup of which constructs a gene regulatory network required for the gradual establishment of the stemness phenotype. Overall, our data provide a reasonable mechanistic explanation of how the functions of multiple transcription factors integrate to build an additional layer of coordinated regulatory pathways in order to control cellular reprogramming. Our resulting network provides the basis for transcription factor perturbations aimed at improving reprogramming efficiency, an important issue for personalized cell therapies and precision medicine.
Results
Delineating dynamic changes in gene expression during cellular reprogramming
Oct4, Sox2, Klf4, and c‐Myc (OSKM)‐induced cellular reprogramming triggers dynamic responses in mammalian gene transcription (Polo et al, 2012; Hussein et al, 2014). Herein, we carried out a detailed kinetic analysis of the transcriptional responses imposed to MEFs during cellular reprogramming following lentivirus‐based OSKM overexpression. As seen in Fig EV1A, the gene expression profile of our iPSC‐generated cell lines is very similar to that of ESCs, an observation that validates our reprogramming platform. We found that 4,083 genes (~20% of all mouse genes) changed their expression at least once, and of these, 2,540 genes were altered transiently, whereas 1,543 genes changed their expression levels permanently (Fig EV1B). Approximately equal numbers of genes were either activated or repressed during reprogramming (Fig EV1C). Figure EV1D and E demonstrate the dynamic transcriptional changes between any two sequential time points depicting the clustering into four distinct groups, thus suggesting the existence of dynamic alternate cell fates and a clear molecular discontinuity between specific time points. Our data also revealed an orchestrated activation and repression of gene expression occurring in two separate waves (Fig EV1F) (Polo et al, 2012). The first wave peaks at day 1 and involves the activation of genes characteristic to the ESC phenotype (e.g., cell cycle) and the simultaneous repression of genes marking the MEF phenotype (e.g., developmental processes). The second wave is marked by the activation of genes related to the epithelial phenotype (MET transition), followed by the continuation of repression of genes related to the MEF phenotype and the constant expression of genes required for the acquisition of pluripotency (Fig EV1F). These data further suggest that OSKM‐induced cellular reprogramming involves a complex orchestration of both early and late gene expression programs.
Figure EV1. Analysis of genes whose expression is altered during cellular reprogramming.
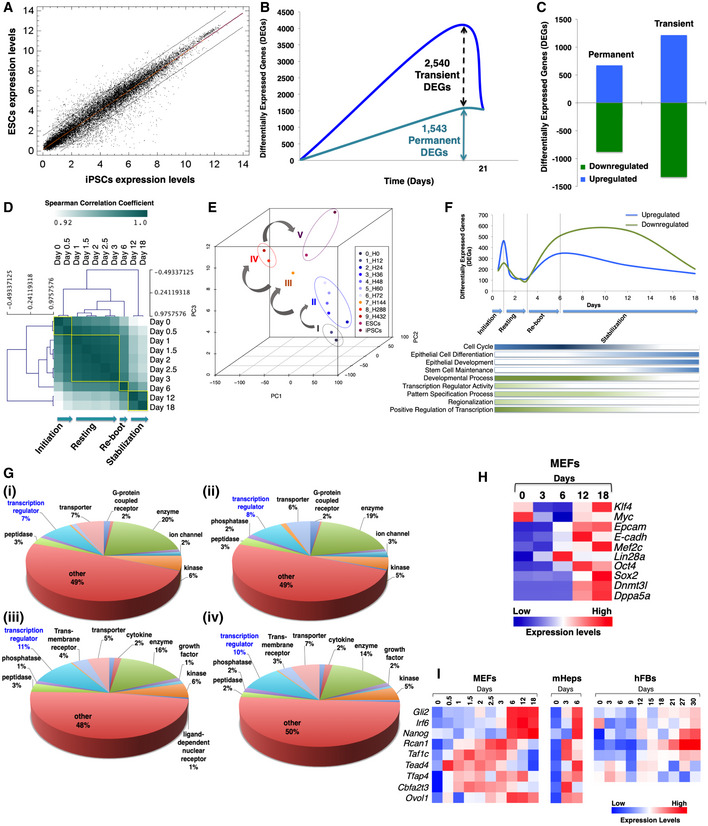
- Comparison of the transcriptomic profiles between mESCs and miPSCs. The two black diagonal lines enclose 95% of expressed genes with high correlation, that is, with similar expression profiles in both cell types. Spearman's correlation coefficient = 0.97; normalization with RMA.
- Line plot depicting the number of differentially expressed genes (DEGs), which either change their expression transiently or permanently at least once during reprogramming.
- Bar graph showing the number of up‐ and downregulated DEGs, transiently or permanently, during reprogramming.
- Hierarchical clustering of the gene expression profiles of MEFs undergoing reprogramming from day 0 to day 18. Clustering revealed four distinct phases, of which day 6 is characterized by Nanog activation.
- PCA of the gene expression profiles described in (D).
- Line plot depicting the number of upregulated (blue line) and downregulated DEGs (green line) during MEF reprogramming. The bars shown below the line plot represent the enrichment of selected gene ontology categories at each time point.
- Gene Ontology (GO) analysis categorization of up‐ and downregulated DEGs until day 6 of reprogramming (i, iii for MEFs and ii, iv for mHEPs). Transcriptional regulators are highlighted in blue color.
- Heat map depicting the pattern of expression of well‐characterized mesenchymal, epithelial, and pluripotent markers during MEF reprogramming (data derived from qPCR assays).
- Heat map showing the expression pattern of the nine TRs at selected time points during reprogramming of MEFs, mHeps (data derived from DNA microarray assay), and hFBs (data derived from qPCR assays).
Source data are available online for this figure.
Identification of transcriptional regulators required for cellular reprogramming
We hypothesized that the reprogramming of somatic cells of different developmental origins to pluripotency may utilize common transcriptional regulatory routes, that is, a shared toolbox of transcription factors acting similarly in different cell types. To test this idea, we carried out side‐by‐side reprogramming experiments followed by gene expression analyses using mouse embryonic fibroblasts (MEFs) and mouse hepatocytes (mHeps) (Fig 1A, top).
Figure 1. Identification of nine TRs required for the generation of iPSCs from cells of different origins.
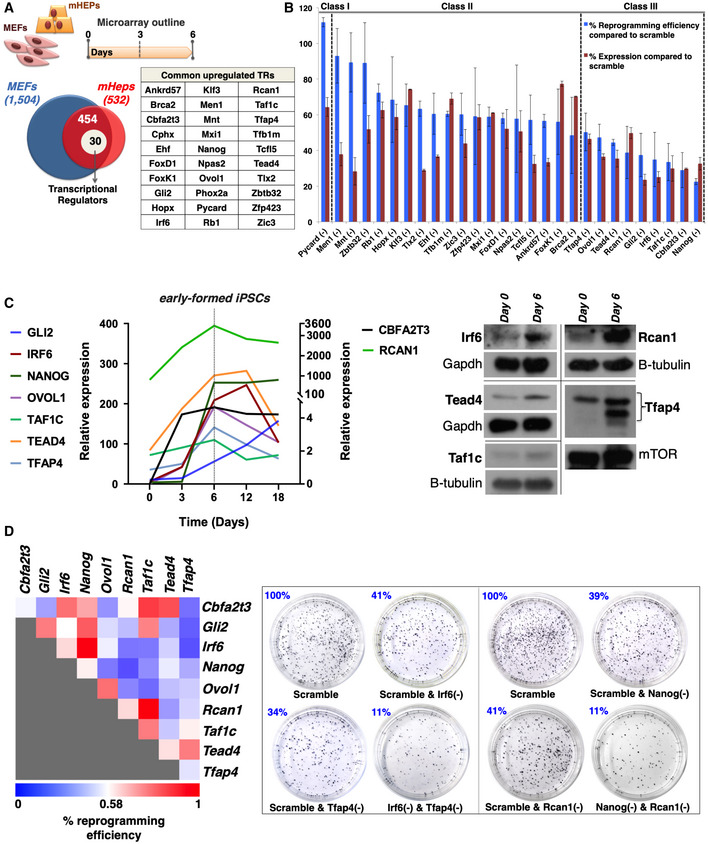
- Venn diagram depicting the total number of genes upregulated in MEFs (1504) or mHEPs (532) undergoing reprogramming and the 454 common genes upregulated in both MEFs and mHeps at day 6 of reprogramming. Of these, 30 genes, listed in the adjacent table, encode for transcriptional regulators (TRs). The top diagram indicates the experimental outline of the DNA microarrays expression studies performed in MEFs and mHEPs undergoing reprogramming.
- Bar graph summarizing the efficiency of single RNAi knockdowns for 28 out of the 30 TRs, and their effects in the reprogramming efficiency in comparison to control cells expressing scramble shRNA. The efficiency of each TR's knockdown (KD) does not correlate with its effect in reprogramming efficiency. The knockdown of 9 (class III) out of the 28 TRs decreased the efficiency of the reprogramming in a statistically significant manner (unpaired two‐tailed Student's t‐test, P‐value < 0.05). Data are shown as mean ± SEM of at least two independent experiments. Class I corresponds to one TR inhibiting reprogramming, whereas class II corresponds to 18 TRs having a weak or no effect in reprogramming. Shown are the effects of 28 out of 30 TRs, because we did not succeed in knocking down Cphx and Phox2a.
- Left: Shown is a line graph depicting normalized mRNA expression levels of the nine TRs at the indicated time points during the reprogramming of MEFs. The data were plotted after normalization with the endogenous Gapdh using the “ΔCt method”. The gray vertical dashed line depicts day 6 in which the expression levels of nearly all TR peaks. Notice that the y‐axis is broken to accommodate the large spread of expression levels. Right: Shown are Western blots using antibodies specific for Irf6, Tead4, Taf1c, Rcan1, and Tfap4. Whole‐cell extracts were prepared from MEFs at day 6 of reprogramming and run side‐by‐side with extracts from control samples (day 0). Gapdh, mTOR, and b‐tubulin were used as loading controls.
- Same as in (B) except MEFs were co‐transduced with lentiviruses expressing all possible pairwise combinations of shRNAs for the nine TRs. Left: Heat map depicting the effects of paired KD combinations in the reprogramming efficiency (RE%). Shown are the average RE values from at least two independent experiments. The REs of the double KDs were evaluated against the RE of the corresponding single KDs. Right: Alkaline phosphatase (AP)‐stained cultures of terminal reprogrammed MEFs upon representative single and double KDs of our shRNA‐based screens.
Source data are available online for this figure.
We focused on the early activation of gene expression and compared the transcriptomes of MEFs and mHeps at different time points within the first 6 days of reprogramming. During this reprogramming period, 1,504 and 532 genes were upregulated in MEFs and mHeps, respectively (Figs 1A and EV1G for gene ontologies). The number of upregulated genes in MEFs is significantly higher than in mHeps, a result consistent with the fact that the latter exhibit epithelial characteristics (Choi & Diehl, 2009), and thus do not have to undergo MET to reach pluripotency. Subsequently, we identified 454 common upregulated genes including 30 transcriptional regulators (TRs) (Fig 1A, table), some of which have been previously implicated to pluripotency and/or ESC functions, such as Nanog, Cbfa2t3, Gli2, Ovol1, and Tfap4 (Chambers et al, 2003; Tu et al, 2016; Li et al, 2013, 2; O'Malley et al, 2013; Nishiyama et al, 2013), whereas Rcan1, Taf1c, Tead4, and Irf6 had no previous involvement in ESC regulation. The validity of our assay is underscored by the confirmation of induced expression of known pluripotency markers such as E‐cadherin and Lin28A (Fig EV1H). It is important to note that apart from the 30 TRs shared between MEFs and mHeps, there are 79 TRs upregulated in MEFs only and 28 TRs upregulated specifically in mHeps (unpublished data).
To test the role of the 30 common upregulated TRs in cellular reprogramming, we carried out lentiviral‐based single or pairwise shRNA knockdown assays in at least two biological replicates and determined the reprogramming efficiencies of the knockdown cells as compared to scramble shRNA by alkaline phosphatase (AP) staining. The efficiency of each shRNA knockdown was determined by RT–qPCR and was plotted next to the corresponding reprogramming efficiency (Fig 1B). The TRs are grouped into three classes with a distinct impact in the reprogramming efficiency (Fig 1B). Class I includes one TR (PYCARD), which appears to function as inhibitor of reprogramming, class II contains 18 TRs that have a weak or no effect in reprogramming, and class III containing nine TRs, which function as positive regulators of cellular reprogramming (Fig 1B). Class III includes Cbfa2t3, Gli2, Irf6, Ovol1, Rcan1, Taf1c, Tead4, Tfap4, and the master regulator of pluripotency Nanog (see Table EV1 for their known biological properties; Heix et al, 1997; Rothermel et al, 2000; Qi et al, 2003; Richardson et al, 2006; Yagi et al, 2007; Moore et al, 2008; Rahimov et al, 2008; Cai et al, 2009; Po et al, 2010; Jackstadt et al, 2013; Wu et al, 2013; Shin et al, 2014). Figure 1C represents the quantified RNA and protein expression pattern of the class III TRs during the course of reprogramming, indicating that their induced expression peaks at day 6 of reprogramming, a time point at which the first iPSC colonies appear in the cultures (early‐iPSC colonies, see below). As a control, we showed that none of these knockdowns had an effect on cell viability or cell proliferation of either naïve or MEFs undergoing reprogramming (unpublished data). We have also carried out knockdown experiments for the nine class III TRs in mHeps undergoing reprogramming and have verified their critical role in reprogramming (unpublished data). In addition, we found that human homologs of the mouse nine TRs were also expressed in a similar manner during the reprogramming of human fibroblasts, a result consistent with the notion that human and mouse, and presumably mammalian cell reprogramming, are characterized by universally conserved transcriptional regulatory mechanisms (Fig EV1I). Taken together, our data underscore a broad role for the nine TRs in cellular reprogramming.
Next, we tested whether the nine TR function independently of each other or synergize to promote reprogramming. To do so, we evaluated the reprogramming efficiency of cells bearing all pairwise combinations of the nine TR knockdowns. Figure 1D illustrates unique modes of functional synergies between specific pairs of the nine TRs. For example, although the individual knockdowns of Cbfa2t3 and Tfap4 have a relatively weak effect, their simultaneous knockdown strongly decreased the reprogramming efficiency, suggesting that these two TRs synergize and may participate in common transcriptional regulatory pathways required for reprogramming. Similar strong synergistic effects were also observed for the knockdown pairs of Nanog‐Ovol1, Gli2‐Tfap4, Irf6‐Tfap4, and Nanog‐Rcan1 (Fig 1D). Of note, we have also detected strong anti‐synergistic effects. That is, although the single knockdowns of Rcan1 and Taf1c or Nanog and Irf6 reduced reprogramming efficiency, their double knockdowns had practically no effect. These observations underscore the complex interplay between specific TRs, giving rise to highly nonlinear processes that facilitate different spatiotemporal synergistic and antagonistic interactions. Taken together, these experiments led to the identification of nine TRs required for cellular reprogramming by participating and cooperating in common synergistic and/or antagonistic transcription regulatory routes. These observations also suggest that the nine TRs could construct a transcription factor regulatory network.
To test whether any of the nine TRs can substitute for Oct4, Sox2, Klf4, or c‐Myc in inducing reprogramming, we replaced c‐Myc with each of the nine TRs in separate lentivirus‐based transduction experiments and evaluated their ability to complement OSK in reprogramming. We chose to substitute c‐Myc, since Oct4, Sox2, and Klf4 (OSK) have well‐defined genomic targets critical for both inducing reprogramming and the maintenance of the pluripotent phenotype. While the absence of c‐Myc causes a significant delay in the kinetics of reprogramming and a decrease in reprogramming efficiency (Wernig et al, 2008; Nakagawa et al, 2008), we found that the co‐expression of Cbfa2t3, Ovol1, or Gli2 together with OSK re‐establishes the kinetics of the process and restores the reprogramming efficiency (Fig EV2A). None of the other six TRs, including Nanog, had any effect compared to the control OSK samples. Thus, we speculate that Cbfa2t3, Ovol1, and Gli2 might share common targets with c‐Myc and/or participate in interconnected regulatory networks (see below).
Figure EV2. Substitution of c‐Myc by nine TRs.
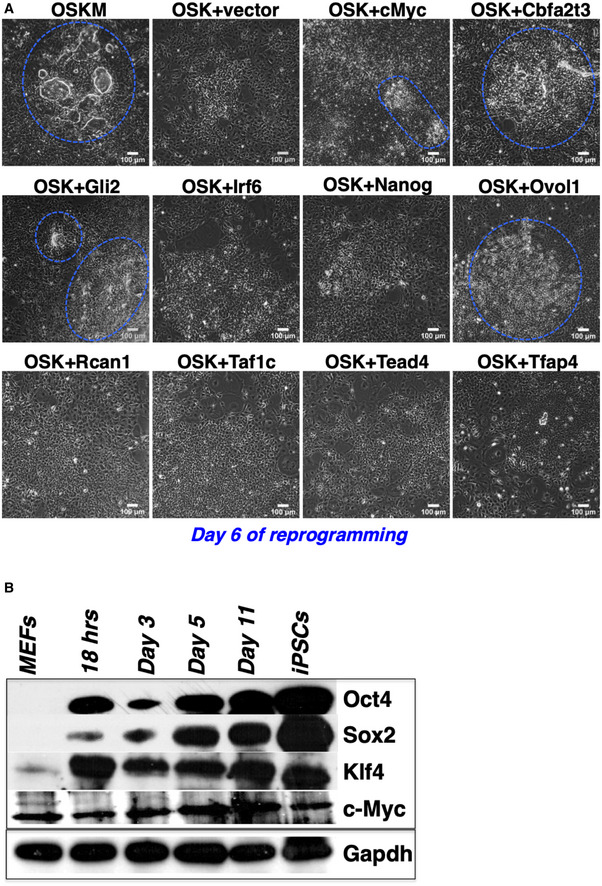
- MEFs expressing OSK were transduced with lentiviruses overexpressing each of the nine TRs or c‐Myc as a control. The transduced cultures were examined on day 6 for the formation of early‐iPSC colonies. The boundaries of the early‐iPSC formations are indicated by a blue dashed line.
- Western blot depicting the expression of O/S/K/M during MEF reprogramming. Gapdh was used as loading control. O: Oct4, S: Sox2, K: Klf4, and M: c‐Myc.
Source data are available online for this figure.
The nine TRs are co‐expressed within early‐iPSC colonies
Cellular reprogramming is an asynchronous and inefficient process that routinely produces heterogeneous intermediate cellular populations with a variable potential to become iPSCs (Hanna et al, 2009). To analyze the spatiotemporal expression pattern of the nine TRs in individual cells in the context of the dynamic cell population undergoing reprogramming, we performed RNA in situ hybridization (ISH) experiments at days 3 and 6 of reprogramming (Fig 2A). As controls, we used naïve MEFs and mESCs as well as RNA probes corresponding to the sense RNA strand (Fig EV3A–C). Remarkably, we discovered that the expression of Nanog, Cbfa2t3, Gli2, Ovol1, and Irf6 was significantly enriched in a specifically defined dynamic population of cells undergoing reprogramming lying within the early rising iPSC colonies at days 3 and 6 (Fig 2A). In contrast, a rare sporadic expression pattern for each of these factors was detected in the cells lying outside of the early‐iPSC colonies (Fig 2A). The above early‐iPSC colony‐restricted expression pattern is progressively enhanced from days 3 to 6 (Fig 2A), in agreement with the increased expression levels of these TRs (Fig 1C) and the high potential of these cells to become iPSCs. On the other hand, Rcan1, Taf1c, Tead4, and Tfap4 are broadly expressed in cells localized both within and outside the early‐iPSC colonies (Fig 2A).
Figure 2. Stochastic expression of the nine TRs in MEFs undergoing reprogramming.
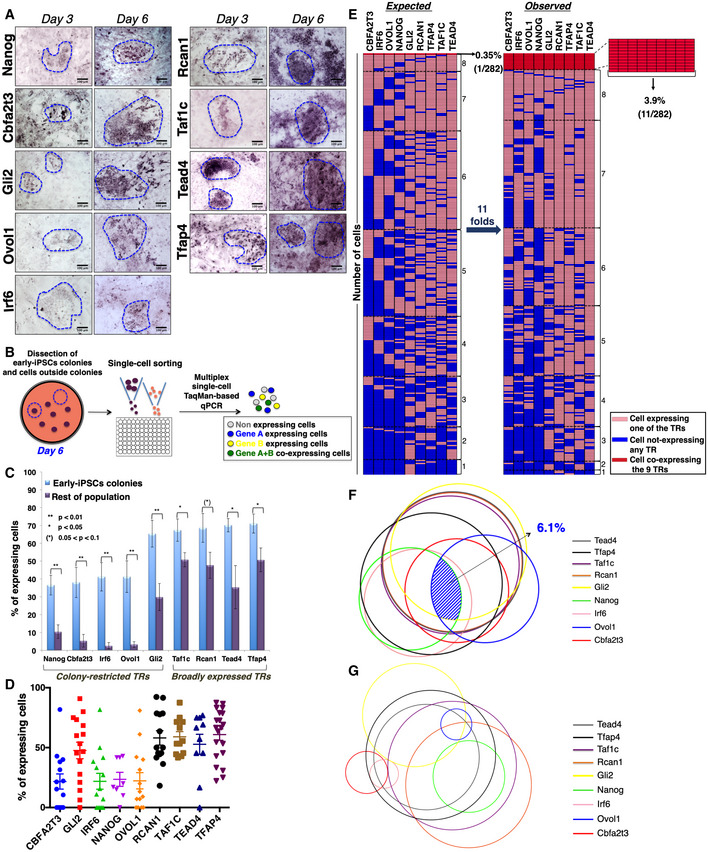
- RNA in situ hybridization depicting the expression patterns of the nine TRs in days 3 and 6 of MEF reprogramming. The boundaries of the early‐iPSC formations are indicated by a blue dashed line. Scale bars: 100 μm.
- Illustration of the experimental setup for the single‐cell TaqMan‐based qPCR assay.
- Bar graph showing the percentage of cells isolated from early‐iPSC colonies or outside of the colonies (rest of population) expressing each of the nine TRs. Data are shown as mean ± SEM of at least two independent experiments. 0.05 < (*)P < 0.1, *P < 0.05, **P < 0.01 by unpaired Student's t‐test or Welch's t‐test, when appropriate.
- Scatter plot depicting a visual unbiased representation of the percentile of cells expressing each of the nine TRs. Each dot depicts the percentage of cells expressing the indicated TRs in individual experiments. Data are shown as mean ± SEM of at least two independent experiments. ANOVA test P < 0.0001.
- Schematic representation of the statistical analysis estimating the probability of co‐expression of the nine TRs per cell within the early‐iPSC colonies. Each row consists of 9 tiles representing a single cell, and each column shows the expression status of each of the nine TRs (shown with different colors) in 282 cells (right side legend). The co‐expression of gradually increasing numbers of TRs is grouped from the bottom to the top of the figure. The left part has been generated by estimating the percentages of expression of any single TR per cell as derived from single gene expression in single cells, while the right part depicts the co‐expression of combinations of the nine TRs based on double and triple single‐cell experiments.
- Venn diagram model depicting the co‐expression of the nine TRs in cells lying within the early‐iPSC colonies on day 6 of reprogramming. The area of each circle represents the percentage of cells expressing each TR, as determined from the single single‐cell experiments, and the overlapping areas between circles represent the percentage of co‐expression of two or three TRs, as revealed from our double and triple TR single‐cell RNA expression experiments. The shaded blue area highlights the percentage of cells co‐expressing all the nine TRs.
- Same as of (F) except depicting the co‐expression of the nine TRs in cells lying outside the early‐iPSC colonies.
Source data are available online for this figure.
Figure EV3. The spatiotemporal (co)‐expression pattern of nine TRs.
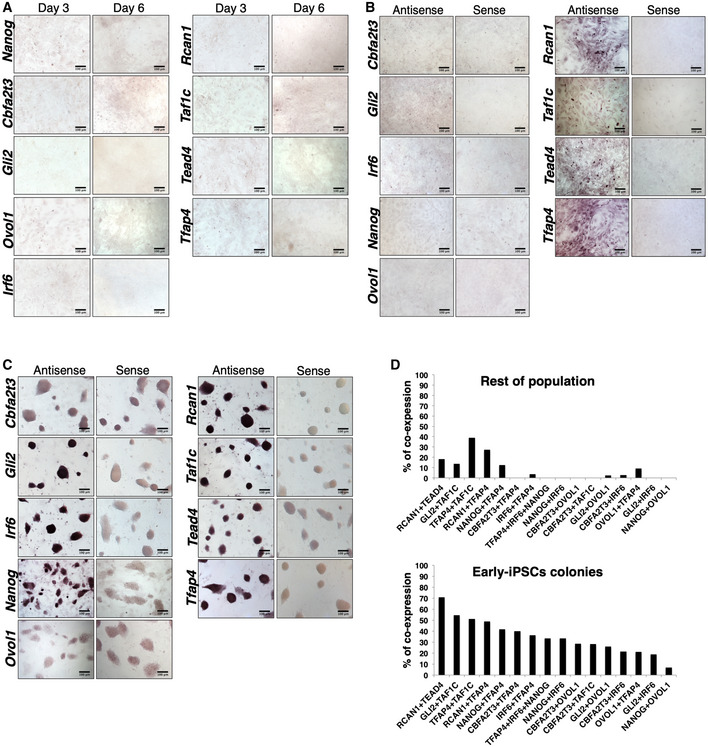
- RNA in situ hybridization (ISH) experiments in MEFs undergoing reprogramming (day 3 and day 6), depicting the hybridization pattern using the sense RNA strand of each of the nine TRs as probes. Scale bars, 100 μm.
- RNA in situ hybridization experiments using naïve MEFs hybridized with the sense or the antisense RNA strand of each of the nine TRs, as indicated at the top and at the left side of each panel, respectively. Scale bars, 100 μm.
- Same as in (B) except that mESCs were used.
- Bar graph depicting the co‐expression frequencies of the indicated combinations of nine TRs within the early‐iPSC colonies and in the rest of the population of MEFs, based on double and triple single‐cell experiments.
Source data are available online for this figure.
We next quantified the expression of the nine TRs in single cells isolated either from early‐iPSC colonies or from the rest of the culture upon cell sorting (Fig 2B). Cells were sorted from multiple experiments at day 6 of reprogramming, and RNA was isolated and analyzed by single‐cell qPCR, using TaqMan probes labeled with different fluorophores (Fig 2B). The data of Fig 2C show that Taf1c, Rcan1, Tead4, and Tfap4 are widely expressed in cells obtained from both within and outside the early‐iPSC colonies, a result consistent with the RNA in situ experiments. On the contrary, the expression of Nanog, Cbfa2t3, Irf6, Ovol1, and Gli2 is significantly enriched in cells isolated from the early‐iPSC colonies (Fig 2C). The scatter plot of Fig 2D represents the percentile distribution of cells expressing each of the corresponding nine TRs in individual experiments within the culture. Thus, for example, Cbfa2t3, Irf6, Nanog, Ovol1, and to a lesser extent Gli2 are expressed in a lower percentage of cells (~30%) when compared to Rcan1, Taf1c, Tead4, and Tfap4, which are expressed in a higher one (~65%). These results further support the data of Fig 2A and C by using an unbiased approach, that is, without any previous knowledge regarding the origin of the cells analyzed (inside or outside the early‐iPSC colonies).
The above results led us to suggest that the nine TRs could be co‐expressed in cells with higher potential to reach pluripotency, that is, in cells residing within the early‐iPSC colonies. To test this hypothesis, we examined the probability of co‐expression of the nine TRs by carrying out double and triple single‐cell qPCR assays. We note that if the co‐expression of the nine TRs within the early‐iPSC colonies was a purely random (stochastic) phenomenon, that is, the expression of any one of the nine TRs is not affected by the expression of any of the other(s), then the theoretically expected probability for their co‐expression would be:
Remarkably, however, the analysis of our multiplex single‐cell qPCR RNA expression experiments revealed that the probability of the nine TRs being co‐expressed within each of the cells lying in the early‐iPSC colonies is P(observed) = 3.9% (Fig 2E right panel), a value that is ~11‐fold higher than the one expected if the TRs were expressed independently of each other (Fig 2E left panel).
Next, the single‐cell RNA expression data were visualized as circles, a model representing the percentage of cells expressing each TR, the area of which is proportional and representative to their extent of expression in the population of cells undergoing reprogramming. The pattern and the percentage of co‐expression of the nine TRs are represented as overlapping areas between circles. Interestingly, Fig 2F demonstrates that the probability of co‐expression of all nine TRs within cells lying in the early‐iPSC colonies is 6.1%. In contrast, the corresponding probability for cells lying outside these early formations is 0% (Fig 2G). Taken together, our data demonstrated that the percentage of cells expressing any combination of two or more TRs is significantly higher in cells isolated from the early‐iPSC colonies (Fig EV3D) and that as the number of co‐expressing TRs is increased, this co‐expression occurs in a progressively diminished percentage of cells. Interestingly, we noticed that cells co‐expressing all nine TRs are generally defined by the co‐expression of Nanog, Gli2, and Ovol1. Of note, Nanog and Ovol1 are expressed in largely different groups of cells, which are intersected by their shared Gli2 expression and that Nanog and Irf6 are expressed in the same population of cells (Fig 2F, see also below). As seen in the figure, Gli2, Taf1c, Tead4, and Rcan1 are co‐expressed in the majority of cells lying within the early‐iPSC colonies and together with the widely expressed Tfap4 generally mark the group of cells in which all nine TRs will be expressed. In contrast to our finding for the cells lying within the early‐iPSC colonies, we found that the rest of the cells (outside colonies) exhibit radically different patterns of co‐expression and strong anti‐correlation gene expression patterns for distinct TRs (Fig 2G). For example, Nanog expression anti‐correlates with the Gli2, Ovol1, Irf6, and Cbfa2t3 expression pattern, whereas the highly overlapping expression of Gli2, Taf1c, Tead4, and Rcan1 as well as Irf6 and Nanog observed in early‐iPSC colonies is generally disturbed in the rest of cells (Fig 2G). Thus, each of the nine TRs is stochastically expressed in different subpopulations of cells outside of the early‐iPSC colonies, but they are coordinatively expressed in cells within the early rising iPSC colonies. These observations further support our conclusion that the coordinated interdependent expression of all nine TRs occurs only in rare subsets of cells lying within the early‐iPSC colonies and it is required for cellular reprogramming. This interdependent expression of the nine TRs in conjunction with their increased co‐expression probability strongly suggests the existence of an OSKM‐activated gene regulatory network (GRN) of transcription factors assembled during reprogramming.
OSKM bind to the regulatory chromatin of the genes encoding the nine TRs
To investigate whether the nine TRs are direct targets of OSKM DNA binding, we carried out chromatin immunoprecipitation (ChIP)‐seq experiments for Oct4, Sox2, Klf4, and c‐Myc using chromatin prepared from MEFs undergoing reprogramming for 18 h, 3 and 5 days, in parallel with control chromatin prepared from naïve MEFs, mESCs, and miPSCs. Figure 3 shows a detailed topographic map of the various dynamic DNA‐binding patterns of individual OSKM factors at the putative regulatory regions surrounding the transcription start sites (TSSs) of the nine TR genes during different time points of cellular reprogramming. Notably, with the exception of Nanog, OSKM DNA‐binding profiles perfectly correlate with the expression patterns of the nine TRs during the entire process of reprogramming, (Fig 3‐vertical red/blue bars on the right of each snapshot and Fig 1C). For example, while OSKM bind with variable kinetics and affinities to the promoters of the Cbfa2t3 and Ovol1 in cells undergoing reprogramming, this binding is abolished in iPSCs and ESCs (Fig 3A and B), a finding consistent with the transient expression of these factors during reprogramming, which is also marked by their low expression in iPSCs/ESCs (unpublished data). Furthermore, we found that although the −5 kb distal Nanog enhancer (Levasseur et al, 2008) is occupied by the OSKM as early as at 18 h of reprogramming, the Nanog gene remains inactive until day 5 (Figs 3C and 1C). However, in ESCs/iPSCs where Nanog is expressed, OSKM associate with the Nanog promoter with high avidity. Thus, OSKM binding at both the Nanog enhancer and promoter correlates with its expression (Fig 3C).
Figure 3. The expression of the nine TRs is regulated by direct binding of the OSKM reprogramming factors to putative regulatory regions.

-
A–IShown are ChIP‐seq big wig files in the IGV browser depicting the binding of Oct4 (red), Sox2 (blue), Klf4 (green), and c‐Myc (black) to putative regulatory regions in the nine TR genes in MEFs undergoing reprogramming (18 h, D3, and D5) and in control MEFs, mESCs, and miPSCs. All peaks have been normalized against input DNA. The relative sizes of the represented genomic loci and the corresponding TSSs are also indicated. The side color bar depicts the expression levels of each TR at the indicated time points.
In the cases of the Cbfa2t3, Ovol1, Gli2, and Irf6 loci (Fig 3A, B, D and E), we discovered that the early Klf4 binding is replaced by subsequent binding events of c‐Myc, a finding consistent with the pleiotropic role of c‐Myc in pluripotent cells (Nie et al, 2012). On the other hand, Rcan1, Taf1c, Tead4, and Tfap4 are stably bound by OSKM throughout the course of reprogramming, whereas their initial basal expression in MEFs correlates with c‐Myc binding (Fig 3F–I). We also discovered that OSKM DNA binding is highly enriched at the endogenous O/S/K/M loci, suggesting the existence of autoregulatory loops formed between O/S/K/M during reprogramming (Fig EV4A). However, with the exception of Oct4 and Sox2 loci, we detected no OSKM binding at the Klf4 and c‐Myc loci in iPSCs and ESCs (Fig EV4A), thus suggesting the dynamic nature of assembly and disassembly of OSKM autoregulatory loops during the process of reprogramming. Taken together, we conclude that the dynamic landscape of OSKM DNA binding across putative regulatory regions of the nine TR genes (Fig 3A–I) correlates with the kinetic transcriptomic analysis (Fig EV1) and with the first and second waves of transcriptional changes in MEFs undergoing reprogramming (Fig EV1F) (Polo et al, 2012).
Figure EV4. Binding of OSKM to the O/S/K/M genes in MEFs and to nine TR genes in human fibroblasts.

- Shown are ChIP‐seq big wig files in IGV browser depicting binding of Oct4 (red), Sox2 (blue), Klf4 (green), and c‐Myc (black) to regulatory regions of the O/S/K/M/ genes in MEFs undergoing reprogramming at the 18 h, day 3, and day 5, as well as in control MEFs, mESCs, and miPSCs. The relative sizes of the represented genomic loci and the corresponding TSSs are also indicated. The side color bar depicts the expression levels of each TR at the indicated time points. All peaks have been normalized against input DNA
- Shown are ChIP‐seq big wig files in IGV browser depicting OSKM binding to regulatory regions of the indicated TRs on day 2 of human fibroblasts undergoing reprogramming. The results shown were derived from meta‐analyses of the published ChIP‐seq data from K.S Zaret Lab (Soufi et al, 2012).
To examine whether the OSKM DNA‐binding events to the nine TR genes are evolutionary conserved between mouse and human cells during reprogramming, we analyzed ChIP‐seq data from earlier studies examining OSKM DNA binding in human fibroblasts undergoing reprogramming (Soufi et al, 2012). Indeed, Fig EV4B demonstrates that OSKM DNA binding to the human homologues of the nine TRs is generally conserved, a result that further underscores the biological significance of our findings in reprogramming.
Next, we compared OSKM DNA binding between iPSCs and ESCs by merging all peaks from the ChIP‐seq experiments and calculated the Pearson's correlation coefficient for all ESC‐iPSC O/S/K/M pairs. The heat map shown in Fig EV5A depicts a high degree of similarity of each of the O/S/K/M‐binding sites between iPSCs and ESCs. The differences observed in Sox2 binding at the promoters of Rcan1, Taf1c, and Tfap4 (Fig 3F, G and I) are most likely due to stochastic binding events and/or to clonal differences between iPSCs and ESCs. This notion is supported by the scatter plot shown in Fig EV5B indicating the extensive similarity of Sox2 genome‐wide binding sites between iPSCs and ESCs.
Figure EV5. Genome‐wide correlation of OSKM binding between mESCs and miPSCs.
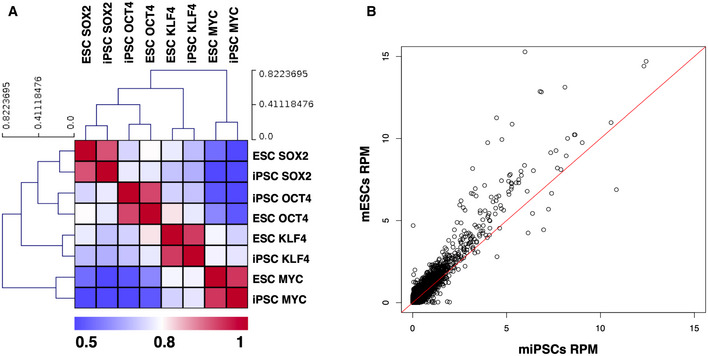
- Shown is a heat map depicting the similarity of O/S/K/M binding between mESCs and iPSCs. All ChIP‐seq peaks were merged and the total number of reads corresponding in each peak was counted, the signal was normalized for sequencing depth (RPM—reads per million), and the Pearson's correlation coefficient was estimated for all ESC‐iPSC and O/S/K/M pairs.
- Scatter plot depicting the extensive similarity of the Sox2 genome‐wide DNA‐binding pattern between iPSCs and ESCs. The red line represents the correlation equal to 1.
Reconstructing a novel gene regulatory network driving cellular reprogramming
So far, we have shown that the nine TRs are co‐expressed in a small percentage of cells within the early rising iPSC colonies, where they synergize to promote cellular reprogramming. To uncover the functional hierarchy of the nine TRs and to provide a comprehensive view of their roles, we performed an integrative analysis of the various types of biological data generated in this study by adapting both data‐driven and knowledge‐based approaches (Fig 4A). Our strategy led to the generation of a gene regulatory network (GRN) representing the various functional interconnections between the nine TRs and their linkage to OSKM in the form of a complex, interconnected molecular circuit providing a novel means for interpreting and predicting their role in cellular reprogramming. To infer transcriptional and genetic interactions, we integrated data derived from gene expression analysis (qPCR and transcriptomics), functional assays such as knockdowns and overexpression, as well as ChIP‐seq experiments for all time points of reprogramming (Fig 4A). Our comprehensive experimental and computational integration processes captured important interactions between critical transcriptional regulators and revealed the dynamic architecture of the gradually assembled nine TR GRN (9TR GRN). These data suggest how the functional interplay between OSKM and the nine TRs drives iPSC generation (Fig 4B), thus providing important insights into the biological logic for cell fate decisions taken to induce cellular reprogramming.
Figure 4. Identification of a transcription gene regulatory network essential for the acquisition of pluripotency.
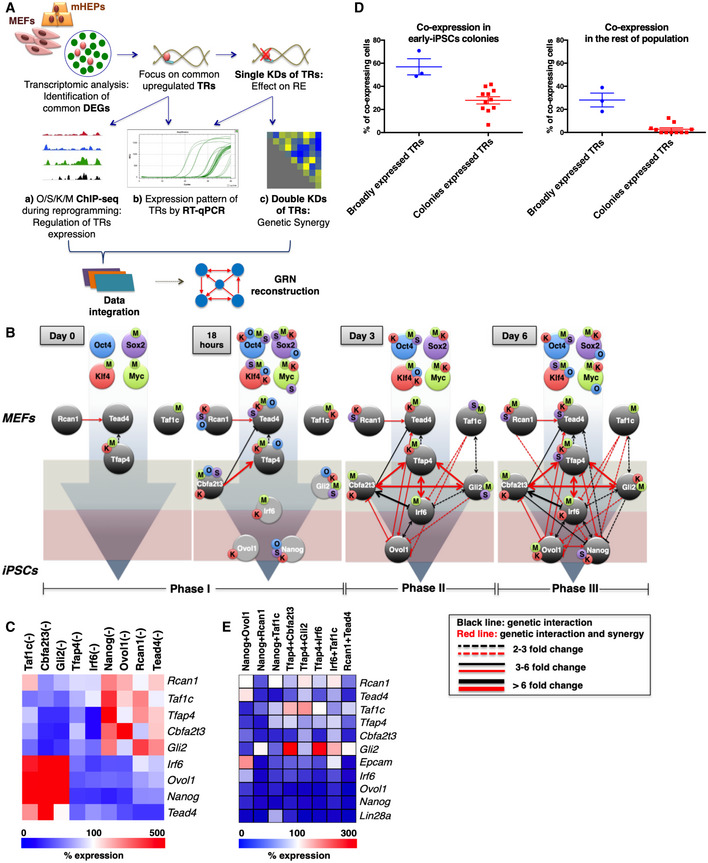
- Schematic representation of the experimental data sets and the integration procedure used to reconstruct the 9TR GRN. DEGs: differentially expressed genes; TRs: transcriptional regulators; KDs: knockdowns; RE: reprogramming efficiency; GRN: gene regulatory network
- The sequential assembly of a novel gene regulatory network required for reprogramming. Each connection (line) represents the regulatory interaction between the indicated nine TRs during the reprogramming course. The red connections indicate functional synergy between the TRs. The light gray circles shown at 18‐h time point represent the TR genes that have not yet been expressed. The O/S/K/M‐labeled beads placed on the gene circles denote the direct O/S/K/M binding at putative gene regulatory regions as deciphered from our ChIP‐seq-binding profiles. O: Oct4, S: Sox2, K: Klf4, and M: c‐Myc.
- Heat map summarizing the effects of each of the nine TR single knockdowns on the expression of the other TRs on day 6; the white color denotes no change in expression levels.
- Scatter plots depicting the extent of co‐expression of either the broadly or early‐iPSC-specifically expressed TRs in individual cells obtained from the early‐iPSC colonies or from the rest of the population (left and right panel, respectively). Each dot depicts the percentage of cells co‐expressing a unique combination of two or three TRs. Data are shown as mean ± SEM of at least two independent experiments.
- Heat map summarizing the most striking effects of double nine TR KDs on the expression of the other TRs and on the expression of the Epcam and Lin28a pluripotency markers; the white color indicates no change in expression levels. The genes examined have been ranked in the heat map according to their order of upregulation during reprogramming; Lin28a is the latest upregulated gene.
Source data are available online for this figure.
Focusing on key connections, we describe below the mechanistic insights of the 9TR GRN, which is reconstructed in three distinct phases and organized into three tiers by its hierarchical assembly. First, we focused on the four TRs that are constitutively expressed in naïve MEFS (phase I). The two constitutively expressed factors Klf4 and c‐Myc (Figs EV1H and EV2B) bind to the Taf1c and Tfap4 (Taf1c is bound by c‐Myc only) regulatory regions and maintain their basal level of expression (Fig 4B, see also Fig 3G and I). The expression of Rcan1 and Tead4 is maintained in naïve MEFs by other yet unidentified factors. The existence of Tfap4, Taf1c, Rcan1, and Tead4 prior to the start of reprogramming forms the foundation of the GRN. In naïve MEFs, we detected two connections only, where Rcan1 and Tfap4 positively regulate Tead4 expression (Fig 4B and C). Notably, the combined knockdown of Rcan1 and Tead4 resulted in a dramatic (85%) reduction in the reprogramming efficiency (Fig 1D), a result consistent with the destruction of the foundation of the 9TR GRN (Fig EV6B). The rapid and extensive gene expression changes induced 18 h upon the beginning of reprogramming lead among others to the activation of Cbfa2t3 by all four OSKM and the establishment of various interactions of Cbfa2t3 with Tfap4, and Tead4, whose expression is also activated and/or maintained by OSKM (Fig 4B). Importantly, at this early time point Oct4, Sox2, Klf4, and c‐Myc bind to the promoters of Gli2, Irf6, Ovol1, and Nanog, without affecting their expression yet (Figs 4B and 3B–E), a result consistent with the property of Oct4, Sox2, and Klf4 to function as pioneer transcription factors, thus setting the stage for the subsequent expression of TRs by controlling local chromatin dynamics and nucleosome remodeling (Soufi et al, 2012, 2015).
Figure EV6. The effect of nine TR knockdown on network integrity.
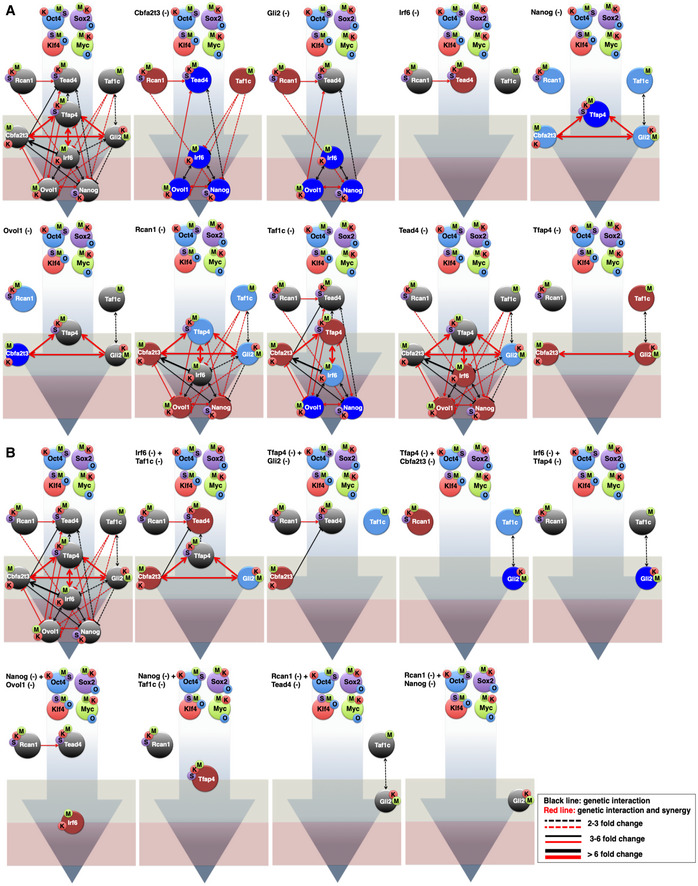
- Shown is a schematic representation of the knockdown (KD) effects of each of the indicated TRs on the expression of the other factors and on network integrity, as determined by RT–qPCR analyses. Dark blue color denotes TRs upregulated more than twofold. Reduction by more than 2 fold in the expression is depicted by the absence of the corresponding TR. The light red and blue colored circles indicate downregulation or upregulation in the range of 1.3 to twofold, respectively, while gray circles indicate no change in gene expression. O: Oct4, S: Sox2, K: Klf4, and M: c‐Myc. The light gray‐green and red boxes depict the upstream and downstream layers of the 9TR GRN. The first panel on the left denotes the native 9TR GRN as shown in Fig 4B (phase III).
- Same as in (A) except that double KDs of the indicated TRs were carried out.
Source data are available online for this figure.
The time‐dependent reconstruction of the GRN continues as at the third day of reprogramming (phase II), when we found that all TRs (Gli2, Irf6, and Ovol1), except Nanog, have been transcriptionally activated and built complex patterns of interdependent and OSKM‐dependent regulatory events (Fig 4B). We observed extensive co‐expression of these TRs with similar expression patterns, but with different relative magnitudes (Fig 1C), thus indicating that the dynamic assembly of the 9TR GRN is marked by the fact that none of the participating TRs appears to be static as they exhibit unique transcriptional behaviors. These findings are consistent with nine TRs’ interdependent regulation to build time‐dependent modules of transcriptionally regulated expression. Phase II is also marked by a densely connected network of interactions in which Tfap4 activates and highly synergizes with most of the other TRs, thus functioning as a key connector for the progressive assembly of the GRN (Figs 1D and 4B and C). A total of 17 interactions were observed of which more are directed to or emanate from Irf6, thus linking the upstream and downstream layers of the network.
Having observed the time‐dependent patterns and determined the probability of co‐expression for the nine TRs, we noticed that the broadly expressed Rcan1, Taf1c, Tead4, and Tfap4 are co‐expressed on the average in ~58% of the cells lying within the early‐iPSC colonies and that this percentage of co‐expressing cells drops to ~30% for cells lying outside the early‐iPSC colonies (Fig 4D). These data suggest that the cells co‐expressing the broadly expressed TRs within the early‐iPSC colonies are those in which the rest of the TRs will be stochastically expressed to assemble the GRN. Consistently, Cbfa2t3 and Gli2 positively regulate Tfap4, as their single knockdowns caused an ~85% decrease in the expression of Tfap4 (Fig 4C). Importantly, double knockdowns of Cbfa2t3 and Tfap4 or Gli2 and Tfap4 reduced reprogramming efficiency by more than 70%, thus underscoring the critical role of these connections in reprogramming (Fig 1D). Therefore, the activation of Cbfa2t3 and Gli2 expression along with the elevated levels of Tfap4 forms a three‐component highly synergistic autoregulatory loop (Fig 4B, phase II).
In turn, this loop through the effector Tfap4 activates Irf6 and Ovol1, which reach simultaneously a steady‐state level of expression (Fig 1C) and are fine‐tuned through the negative action of Cbfa2t3 and Gli2. Irf6 and Ovol1 expression levels are presumably stabilized by their ability to regulate each other (Fig 4B and C). This conclusion is in agreement with the previously reported observation that Irf6 activates Ovol1 in order to promote cell cycle exit of keratinocytes from the progenitor cell compartment (Botti et al, 2011). Of note, Ovol1 negatively regulates Cbfa2t3, thus being part of a unique negative feedback loop. This is similar to the previously described repressive feedback loop of Ovol1‐Zeb1 occurring in human cancers, where Ovol1 acts as a balancer between epithelial and mesenchymal states (Roca et al, 2013). A crucial difference between Irf6 and Ovol1 is that although Irf6 activates on its own many of the factors of the 9TR GRN (Figs 4C and EV6A), it does not exhibit a genetic synergy with anyone of them except Tfap4 and Taf1c (Fig EV6B). Remarkably, previous studies had uncovered interesting modes of interaction among members of the AP‐1 (Tfap4 is a member of the AP‐1 family of TFs) and IRF family members during the differentiation of Th17 cells in inflammatory responses (Ciofani et al, 2012). On the other hand, Ovol1 does not significantly activate other TRs on its own, but its combined activity with many TRs of the network is essential for cellular reprogramming (nearly all Ovol1 connections are highly synergistic for cellular reprogramming) (Fig 1D). These data support the notion that Irf6 and Ovol1 may be involved in the activation of distinct sets of genes during reprogramming with the help of other yet unknown factors. These observations are consistent with the general notion that the regulatory network connections are more specific than the nodes (transcription factors) themselves and that the biological specificity of the network is achieved by the context of the regulatory pathways building the network.
The last TR connecting to the GRN is Nanog (Fig 4B, phase III), as a result of multiple positive and negative inputs from all TRs of the network including the direct action of OSKM. Nanog together with Irf6 and Ovol1 forms the second tripartite autoregulatory loop in 9TR GRN, which is connected to the upstream “Cbfa2t3‐Gli2‐Tfap4” loop via Irf6. We estimate that these loops are assembled in ~28% of the cells residing within the dynamically emerged early‐iPSC colonies (Figs 2F and 4D). Within the 9TR GRN, all TRs receive and deliver a multitude of positive and negative inputs, which altogether result in the robustness of their expression and thus stabilizing the network. In summary, our data overall support a model in which the initially constitutively expressed TRs in MEFs, with the help of OSKM‐induced expression of additional TRs, progressively build a complex GRN culminating in the expression of Nanog to pave the route to pluripotency.
9TR GRN validation
We assessed whether our 9TR GRN could be able to accurately predict each TR's expression in the absence or upon overexpression of the other TRs and the effects of all of the above perturbations in reprogramming efficiency. To test the precision and evaluate the prediction performance of the 9TR GRN, we carried out cross‐validation using three approaches. In the first validation round, we tested GRN integrity and function by single and double knockdowns and measured the expression of the rest of the TRs, the reprogramming efficiency of the corresponding knockdown cells (Fig 1D), and the expression of the two key markers Epcam and Lin28A, which are suggestive for the route to pluripotency. As shown in Figs 4E and EV6 , nearly all combinations of pairwise knockdown of TRs eliminated the expression of most of the other TRs, including the reprogramming‐induced expression of Epcam and Lin28A, whereas their single knockdowns had, as expected, a weaker effect (Figs 4C and EV6Α). The 9TR GRN model is also successful in predicting the critical function of the two central nodes of the 9TR GRN. Indeed, the single or double knockdown of Tfap4 and Irf6 dramatically affected the integrity of the entire 9TR GRN (Figs 4C and E, and EV6). The validity, accuracy, and function of the 9TR GRN are strongly supported by the fact that the combinatorial knockdown of Nanog and Ovol1 (the most downstream nodes of the 9TR GRN) did not significantly affect the expression of Epcam (Fig 4E), but it dramatically reduced reprogramming efficiency (Fig 1D). This is explained by the fact that Epcam becomes activated earlier in reprogramming, that is, prior to Nanog and Ovol1 expression, and thus lies upstream of the end points of the 9TR GRN (Figs 1C and EV1H). In sharp contrast, the pairwise knockdowns of Nanog with either Rcan1 or Taf1c, both of which are expressed prior to Epcam, led to a dramatic decrease in the expression of this epithelial marker (Fig 4E). Again, this result underscores and further validates the hierarchical construction of the 9TR GRN. Therefore, downregulation of the early expressed TRs leads to a broad reduction in expression of all subsequently activated GRN components, thus causing destruction of the network (Fig EV6), as well as reduced expression of pluripotency markers, followed by a significant decrease in reprogramming efficiency (see also Fig 1D).
In the second validation round of the network, we performed TR overexpression experiments to examine whether the predicted targets of the TRs are functionally dependent or downstream of their regulator. Consistent with our described network architecture, Nanog overexpression did not significantly affect the expression of any of the other 9TR GRN components (Fig 5A), but it caused a substantial increase in reprogramming efficiency (Fig 5B). These results strongly support our network hierarchical architecture showing that Nanog is the end point of the 9TR GRN, thus strongly suggesting that at least one of GRN's role is to ensure the proper Nanog expression in order to trigger reprogramming. Another important finding derived from our reconstructed GRN is that Tfap4 and Irf6 occupy strategic positions within the network by connecting the two autoregulatory loops, Tfap4‐Cbfa2t3‐Gli2‐Irf6 and Irf6‐Ovol1‐Nanog, both of which form the core of the 9TR GRN. Therefore, to test our predictions we overexpressed Tfap4 and Irf6 and measured whether their combined high‐level expression would increase reprogramming efficiency. Indeed, Fig 5B shows that the overexpression of Tfap4 and Irf6 increased reprogramming efficiency through the super‐induction of the other TRs including Nanog (Fig 5A). Consistent with this finding is the high‐level Epcam expression (unpublished data), which is a major marker of the mesenchymal‐to‐epithelial transition (MET). These overexpression data are also supported by our observation that the double knockdown of Irf6 and Tfap4 caused severe destruction of the 9TR GRN and a dramatic reduction in reprogramming efficiency (Figs 1D and EV6B), thus highlighting the significance of their synergy for the induction of pluripotency. Of note, Tfap4 overexpression also appears to support the induction of pluripotency through the direct upregulation of Nanog, since its overexpression caused its strong upregulation (Fig 5A).
Figure 5. Validation of the 9TR GRN.
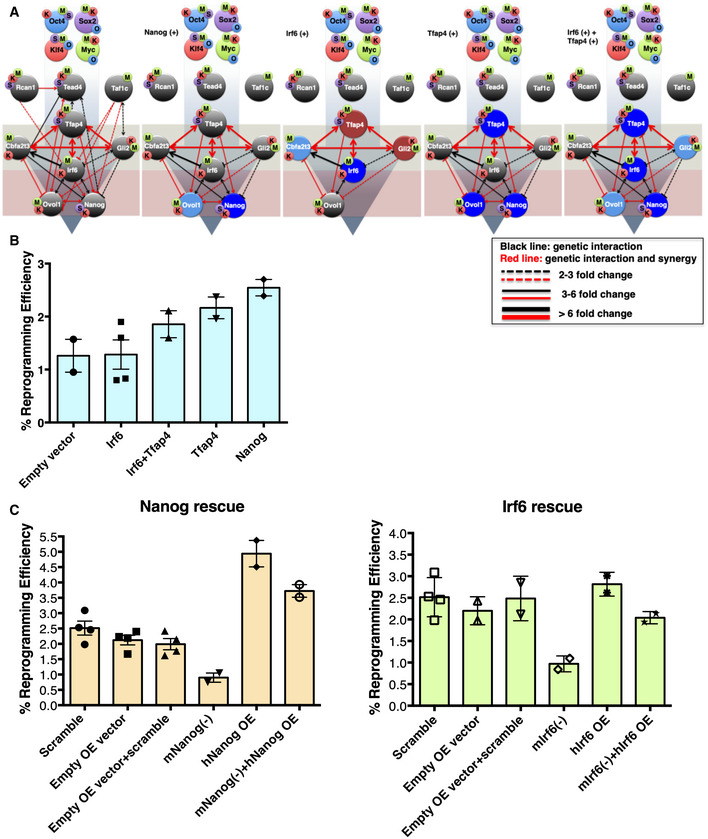
- The effect of overexpression of the indicated TRs (Nanog, Irf6, Tfap4, and Irf6 + Tfap4) on the expression of the other TRs of the network was determined by RT–qPCR analyses. Dark blue color denotes TRs upregulated more than twofold. Reduction by more than twofold in the expression is depicted by the absence of the corresponding TR. The light red and blue colored circles indicate downregulation or upregulation in the range of 1.3 to twofold, respectively, while gray circles indicate no change in gene expression. O: Oct4, S: Sox2, K: Klf4, and M: c‐Myc. The light gray‐green and red boxes depict the upstream and downstream layers of the 9TR GRN. The first panel on the left denotes the native 9TR GRN as shown in Fig 4B (phase III).
- Shown is a bar graph depicting the effect of overexpression of the indicated TRs in reprogramming efficiency. Data are shown as mean ± SEM of at least two independent experiments. The y‐axis labeled as % reprogramming efficiency refers to the AP+ colonies scored as iPSCs.
- Shown is a bar graph depicting the effects of rescue experiments in reprogramming efficiency. MEFs undergoing reprogramming were knocked down using shRNAs for the endogenous Nanog or Irf6 expression and were co‐infected with a vector expressing an shRNA‐resistant human homologue of Nanog or Irf6 (hNanog OE, hIrf6 OE). Data are shown as mean ± SEM of two independent experiments.
Source data are available online for this figure.
In the third round of 9TR GRN validation, we carried out rescue experiments in which the corresponding endogenous Irf6 and Nanog genes were knocked down, whereas simultaneously in the same cells we overexpressed an shRNA‐resistant human homologue of the corresponding knockdown gene. Figure 5C shows that knockdown of the endogenous mouse Irf6 or Nanog genes reduced the reprogramming efficiency of MEFs, whereas the overexpression of human Irf6 or Nanog genes in MEFs had the opposite effect. Interestingly, the overexpression of the human Irf6 or Nanog genes in knockdown MEFs for the endogenous Irf6 or Nanog genes, respectively, restored their reprogramming potential. These experiments when taken together with the data presented above not only validate our 9TR GRN predictions, but they also strongly suggest that the network is functionally important in promoting cellular reprogramming.
Discussion
Our study highlights the complex transcriptional regulatory circuits driving cellular reprogramming by providing a more comprehensive picture of the mechanisms initiating the process. We identified and characterized transcriptional regulators that are directly activated by OSKM. These regulators work as “middle” factors building a gene regulatory network, the 9TR GRN, through a stepwise process across at least two different cell types in mouse and human cells. Τhe OSKM‐induced cascade of dynamic transcriptional events culminates in the stochastic co‐expression of the nine TRs in a small unpredictable fraction of cells. The 9TR GRN, once assembled, triggers the next phase of reprogramming, which seems to be controlled by more deterministic processes, orchestrated by pluripotent factors such as Esrrb, Sall4, Lin28A, and Nanog (Buganim et al, 2012; Zhang et al, 2008a; Wu et al, 2006; Yu et al, 2007; Chambers et al, 2007; Silva et al, 2009). We have demonstrated that Nanog, a milestone for pluripotency, is the end node of the 9TR GRN. Thus, the route to pluripotency consists of layers of determinism and non‐determinism (stochasticity). Completion of the 9TR GRN assembly occurs just before the time of the appearance of early‐iPSC colonies, and we consider that this step is required for the subsequent cascade of deterministic transcriptional events converting the early‐iPSC colonies to mature iPSC (Fig 6). We also provide an explanation for the paradoxical observation that although transduced OSKM factors bind directly to Nanog remote regulatory elements immediately upon their overexpression in MEFs, they cannot activate Nanog transcription at this time. Instead, Nanog expression is turned on 5–6 days later mostly in the context of the 9TR GRN and only when OSKM bind to its promoter, presumably because of alterations in local chromatin structure (Schwarz et al, 2018).
Figure 6. A model depicting the reconstruction of the 9TR GRN during cellular reprogramming.

Day 0: The TRs Rcan1, Taf1c, Tead4, and Tfap4 are expressed in naïve MEFs. Approximately 50% of naïve cells express all 4 factors. 18 h: OSKM induce the expression of Cbfa2t3 in ~40% of the cells. Only a fraction of the population expresses all 5 TRs. Day 3: The expression of Gli2, Irf6, and Ovol1 has been induced by direct and indirect OSKM effects. Various interconnections between the TRs are established, thus stabilizing their expression. All eight TRs are expressed in a limited number of cells found exclusively in early‐iPSC colonies. Day 6: Nanog expression is induced in ~40% of the cells within the early‐iPSC colonies. All nine TRs are expressed only in ~6% of the cells found exclusively within these early formations. The nine TRs reconstruct the 9TR GRN, which is required to promote cellular reprogramming. iPSCs: The 9TR GRN is dissolved in iPSCs because Ovol1 and Cbfa2t3 expression is dramatically reduced to undetectable levels.
Besides reconstructing the GRN to induce the formation of iPSCs, the nine TRs could also generate a cell fate continuum during reprogramming, that is, cells with different developmental trajectories. We discovered populations of cells expressing different combinations of the nine TRs suggesting that these cells may possess unique biological programs related to distinct states and tissues. This is supported by previous observations showing that the nine TRs are involved in various biological functions unrelated to the achievement or maintenance of pluripotency such as hematopoiesis, epidermal and mesoderm development, and MET transition of cancer cells (see Table EV1).
Our network analysis strongly suggests that the nine TRs participate in regulatory process leading to pluripotency through the establishment of highly specialized connections and regulatory pathways. We propose that although the nodes of the 9TR GRN were not identified as transcription factors specific for cellular reprogramming, we determined cell‐specific connections (edges) that appear to be unique in promoting the corresponding cells to be reprogrammed. In other words, the network described here relies on the strong synergy between few TRs that share a relatively broad expression pattern in naïve MEFs (Rcan1, Taf1c, Tead4, and Tfap4) with TRs (Cbfa2t3, Gli2, Irf6, Nanog, and Ovol1) whose co‐expression is stochastically activated in a small number of cells within the early‐iPSC colonies. Another interesting result derived from our analysis is that the expression of individual TRs poorly correlates with cellular reprogramming. Thus, it is the regulatory network connections and not the specificity of TR expression per se, which instructs cellular reprogramming.
We also found that c‐Myc can be replaced in the reprogramming cocktail by selected members of the 9TR GRN. We showed that Cbfa2t3, Ovol1, and Gli2, in the absence of c‐Myc together with Oct4, Sox2, and Klf4, fully re‐establish the kinetics of cellular reprogramming, whereas the rest of the TRs had no effect. These findings suggest that these factors might share common target genes with c‐Myc and/or participate with c‐Myc in common networks. Nevertheless, the 9TR GRN is not a simple flow diagram representing epistatic relationships between OSKM and the implicated transcription factors. Rather, it represents the unique integration of components, which through the formation of a dense network of positive and negative interactions produce a distinct biological output. This fine balance determining cellular reprogramming depends on multiple layers of genetic and epigenetic regulation. We propose that the probabilistic assembly of the 9TR GRN is the result of rare highly dynamic molecular events that progressively define and remodel a small poised subpopulation of cells that becomes appropriately “equipped” to be reprogrammed (Fig 6). This subpopulation of poised cells appears at low frequency as it is the outcome of stochastic processes, and it is subsequently marked by the deterministic function of the 9TR GRN. These “rare” 9TR GRN‐bearing cells could be related to the day 3 and day 6 “efficient” cells described before that have high potential to become reprogrammed (Schwarz et al, 2018).
As OSKM expression is characterized by autoregulatory loops, which, as we showed, are directly connected to the 9TR GRN, we propose that these two independent circuits can integrate into a highly robust and synergistic composite network driving cellular reprogramming. Conceptually, the progressive co‐expression of the 9TR GRN members during reprogramming in an increasingly restricted number of cells (Fig 6) resembles the basic aspects of the combinatorial mode of gene expression according to which the simultaneous co‐expression of a defined set of widely expressed and tissue‐ or signal‐enriched transcription factors instructs the specific activation of only those genes bearing accessible binding sites for these transcription factors. The complex regulatory mechanisms described herein ensure that the appropriate switch to specific gene expression programs occurs only in a subset of the starting cell population to acquire pluripotency, thus explaining the stochastic nature of cellular reprogramming.
Materials and Methods
Generation of lentiviral particles for cellular reprogramming
Cellular reprogramming experiments were carried out using a lentiviral conditional OSKM co‐expression system consisting of FUW‐M2rtTA (FUW‐M2rtTA was a gift from Rudolf Jaenisch (Addgene plasmid #20342; http://n2t.net/addgene:20342; RRID:Addgene_20342)) and TetO‐FUW‐OSKM (TetO‐FUW‐OSKM was a gift from Rudolf Jaenisch (Addgene plasmid # 20321; http://n2t.net/addgene:20321; RRID:Addgene_20321)) constructs. Reconstitution of lentiviruses was carried out in human embryonic kidney cells 293T (HEK293T) by standard calcium phosphate DNA transfection protocols using pMD2.G (pMD2.G was a gift from Didier Trono (Addgene plasmid # 12259; http://n2t.net/addgene:12259; RRID:Addgene_12259)) and psPAX2 (psPAX2 was a gift from Didier Trono (Addgene plasmid # 12260; http://n2t.net/addgene:12260; RRID:Addgene_12260)) packaging plasmids. Three days upon medium change in transfected HEK293T, lentivirus‐containing supernatants were collected for further use.
Isolation of mouse embryonic fibroblasts (MEFs)
E13.5 C57BL/6 mouse embryos were surgically removed from pregnant female mice and placed in PBS (1X). The uterine tissue was cut, and each yolk sac was removed to separate the embryos. Each embryo was transferred to a new dish with fresh PBS (1X), where the head, heart, and liver were removed. The rest of the embryonic tissue was chopped up and minced with a razor blade followed by trypsinization in 0.05% trypsin–EDTA for 5 min at 37°C. Next, upon addition of MEF medium (high‐glucose DMEM, 15% FBS, GlutaMAX, P/S, and NEAA) the tissue suspension passed through an 18G and then a 21G syringe in order to become dissociated into single cells, followed by 1‐day incubation, medium replacement, and cell freezing using standard conditions.
Isolation of mouse hepatocytes (mHeps)
Primary murine hepatocytes (mHeps) were isolated from 12‐ to 15‐day‐old mice. Isolated livers were minced and placed in liver digestion medium [10 mM HEPES, 0.7 mM Na2HPO4, 2 mM KCl, 136 mM NaCl, 5 mM CaCl2 (pH 7.65)] containing 0.05% collagenase and 0.1 μg/μl DNase I (≥ 400 KU/mg, DN25, Sigma). The extracted tissues were mechanically disrupted and then incubated at 37°C for 10 min followed by centrifugation, cell harvesting, and resuspension in hypotonic solution for red blood cell lysis [16 mM Tris, 100 mM NH4Cl (pH 7.6)] coupled with incubation at room temperature for 5 min. The samples were centrifuged again, and the hypotonic treatment was repeated once more. All cell suspensions were then washed once with DMEM containing 10% FBS, centrifuged again, resuspended in hepatocyte culture medium [DMEM, 10% FBS, 1X antibiotic/antimycotic (15240062, Thermo Fisher Scientific), 1X insulin/selenium/transferrin (41400045, Thermo Fisher Scientific), and 10−7 M dexamethasone (D4902, Sigma)], and seeded on collagen‐coated plates (5 μg collagen/cm2, C3867, Sigma). Two to three hours later, the cultures were mildly washed to remove cell debris, and the culture medium was replaced every 2 days thereafter. The endodermal nature of the isolated mHeps was verified by transcriptomic analysis confirming their molecular identity and by immunofluorescence experiments using antibodies against hepatic markers (unpublished data).
Transduction of MEFs and mHeps for kinetic experiments of cellular reprogramming
~500,000 of C57BL/6 early‐passage MEFs (passages 1–3) or mHeps were co‐transduced overnight with lenti‐supernatants of FUW‐M2rtTA and TetO‐FUW‐OSKM followed by medium change [MEF medium (high‐glucose DMEM, 15% FBS, GlutaMAX, P/S, NEAA)] and recovery of the culture for an additional day. The transduced cells were expanded and then passaged in 0.1% gelatin pre‐coated 6‐cm dishes. The induction of OSKM overexpression was initiated 24 h later by the addition of doxycycline (DOX) at 2 μg/ml final concentration. Half of the dishes were treated with doxycycline (+DOX), while the other half was left untreated (‐DOX). Cells were harvested at selected time points during reprogramming course followed by total RNA extraction and microarray hybridization assays (mm430A 2.0 Affymetrix Chip). Transduction of human fibroblasts (hFBs) for kinetic experiments of cellular reprogramming was performed as previously described (Pliatska et al, 2018).
RNAi functional assays in MEFs undergoing reprogramming
For the KD studies, we screened several pLKO.1 backbone constructs (pLKO.1‐TRC cloning vector was a gift from David Root (Addgene plasmid # 10878; http://n2t.net/addgene:10878; RRID:Addgene_10878)) of cloned sequences (Table EV2) encoding for gene‐specific shRNAs. The pLKO.1 constructs were obtained either from the TRC libraries (The RNAi Consortium library) or constructed “in house” with sequences generated using the TRC algorithms. C57BL/6 MEFs were co‐transduced with the two reprogramming lentiviral supernatants and the selected shRNA(s). After 24 h, DOX was added (2 μg/ml final concentration), and on day 6, the transduced cultures were trypsinized to single‐cell suspensions and ~75,000 cells from each culture were seeded onto pre‐coated dishes with mitomycin C‐treated MEFs used as feeder layers. The cultures were maintained in miPSC medium [high‐glucose DMEM, 20% KnockOut Serum Replacement (10828028, Thermo Fisher Scientific), GlutaMAX, P/S, NEAA] and 10 ng/ml mLIF (mBA‐FL, sc‐4378, Santa Cruz Biotechnology). On days 18 to 21, all cultures were stained for alkaline phosphatase (AP) activity using NBT/BCIP substrate solution (11681451001, Roche Life Sciences) in NTMT buffer [100 mM Tris–HCl, 100 mM NaCl, 50 mM MgCl2 0.1% Tween 20, pH 9.5] and counted. RE (%) was calculated by dividing the total number of AP‐stained formations with the number of trypsinized cells seeded on day 6 on the feeder layers (~75,000 cells) and multiplying by 100. All single and double knockdown experiments were carried out at least in two biological replicates. We did not succeed in obtaining reproducible knockdowns for Phox2a and Cphx, and therefore, the role of these TRs in reprogramming remains unknown.
Overexpression functional studies, rescue experiments, and c‐Myc substitution experiment
TetO‐FUW constructs harboring the full‐length coding sequences (CDS) of the 9 murine TRs (engineered in house) (Table EV6) were co‐transduced using the same stoichiometry of FUW‐M2rtTA, TetO‐FUW‐OSKM, and each one of the TetO‐FUW‐TR overexpression (OE) constructs. 24 hours later, the induction of OSKM and TetO‐FUW transgenes was initiated by DOX addition (2 μg/ml final concentration). On day 6, approximately 10,000 cells from each trypsinized cell suspension were seeded in new 6‐well culture dishes pre‐coated with feeder layers. All cultures were maintained in miPSC medium with mLIF (10 ng/ml final concentration) until the final alkaline phosphatase (AP) staining and RE (%) calculation as described above.
For the rescue experiment, MEFs bearing an OKSM cassette under the control of a tet‐responsive element (TetO) inserted in the 3'UTR of the Col1a1 locus (originating from the transgenic murine strain: B6;129S4‐Col1a1 tm1(tetO‐Pou5f1,‐Klf4,‐Sox2,‐Myc)Hoch/J, Jackson ID #011001) were co‐transduced with lentiviral supernatants of FUW‐M2rtTA, mouse Irf6, or Nanog shRNA constructs (Table EV2), as well as the corresponding OE constructs of human Irf6 (engineered in house based on the LeGO‐iT2 backbone) (Table EV6) or human Nanog (FUW‐tetO‐loxP‐hNANOG was a gift from Rudolf Jaenisch (Addgene plasmid #60849; http://n2t.net/addgene:60849; RRID:Addgene_60849)) and were processed as above. All experiments were carried out in two biological replicates.
For the c‐Myc substitution experiment, MEFs containing an OSK‐mCherry cassette under the control of the tet‐responsive element (tetO) in the 3'UTR of the Col1a1 locus and the rtTA activator gene in the ROSA26 locus were transduced with lentiviral supernatants of the TetO‐FUW constructs supporting the individual OE of the nine TRs (Table EV6). The c‐Myc overexpression (TetO‐FUW‐c‐Myc was a gift from Rudolf Jaenisch (Addgene plasmid # 20324; http://n2t.net/addgene:20324; RRID:Addgene_20324)) was used as reference control. On day 6, cultures were examined for the emergence of early‐iPSC colonies under the inverted microscope (brightfield, phase contrast; DM IRE2, Leica) equipped with an ORCA‐Flash4.0 LT (Hamamatsu) camera (HCImage Live software). All pictures were taken at a 10× magnification. OE experiments were carried out in two biological replicates.
cDNA synthesis and Real‐Time qPCR
cDNA synthesis was carried out using 1 μg of total RNA following the instructions of ImProm‐II Reverse transcriptase (M314A, Promega). Real‐time quantitative PCRs (RT–qPCRs) were set up in duplicates using 5 ng of cDNA per reaction amplified by SYBR FAST qPCR Master Mix (KM4114, Kapa Biosystems) under optimized cycling conditions. The efficiency of each pair of primers was assessed prior to any application using dilutions series of the template. Primers’ sequences are listed in Table EV3. All Ct values were filtered through thresholds and normalized to the Ct values of endogenous Gapdh (ΔCt method).
RNA in situ hybridization
MEFs grown on coverslips were fixed in 4% formaldehyde/5% acetic acid buffer at room temperature for 15 min, and endogenous alkaline phosphatase was inactivated at 65°C for 30 min. Prior to hybridizations, all coverslips were rehydrated through ethanol washes and the cells were permeabilized in PBT followed by pre‐hybridization at 65°C for 1 h. Hybridization of DIG‐labeled RNA probes was carried out overnight in a humidified chamber. Sequence‐specific RNA probes were synthesized following standard in vitro transcription protocols: DIG‐11‐UTP (digoxigenin‐11‐uridine‐5’‐triphosphate) labeling mix (11209256910, Roche) with T7 or SP6 RNA polymerases, PCRII‐TOPO vector (TOPO TA Cloning Kit, Dual Promoter, 450640, Thermo Fisher Scientific). Primers’ sequences are listed in Table EV4. Upon overnight hybridization, extensive washes were performed, followed by a 2‐h blocking step with 2% sheep serum (S3772, Sigma‐Aldrich) and 2 μg/μl BSA (bovine serum albumin) (B900S, NEB) in MAB. The cells were incubated overnight at 4°C with anti‐DIG‐AP conjugate antibody (Fab fragments) (1:2,000 dilution) (11093274910, Roche Life Sciences), and next day, the detection was carried out using NBT/BCIP substrate for alkaline phosphatase (11681451001, Roche Life Sciences). All images were captured in the upright microscope (brightfield; DM LS2, Leica) equipped with a DFC500 (Leica) camera (LAS V4.6 software) at a 20× magnification and were analyzed in ImageJ software (NIH).
Single‐cell sorting and RT–qPCR assay
MEFs were reprogrammed, and on day 6, the first early‐iPCS colonies were dissected side‐by‐side with cells residing at the rest of MEFs (cells outside of the colonies) using 0.2‐ to 10‐μl tips. Each cell aggregate was independently trypsinized, and single cells were resuspended in sorting buffer [5% FBS, 1 mM EDTA in PBS], filtered through a 70‐μm cell strainer, and stained with DAPI for the exclusion of dead cells. FACS was performed in BD Bioscience FACS Aria II Device (Becton, Dickinson and Company). Individual cells were sorted and collected into separate wells on a qPCR 96‐well plate, containing Master Mix (2X Reaction Mix; Superscript III RT/ Platinum Taq Mix; pooled TaqMan Probes) of the Superscript III Platinum One‐Step qRT–PCR Kit (11732‐020, Thermo Fisher Scientific). Briefly, cell lysis and sequence‐specific reverse transcription were carried out at 50°C for 15 min, followed by the inactivation of reverse transcriptase at 95°C for 2 min. Subsequent gene‐specific amplification was performed, in the same well, by pooled TaqMan probes (Table EV5) for 40 cycles of denaturation and amplification. The combination of TaqMan probes labeled with different fluorophores (Cy5, FAM, Texas Red, HEX) facilitates the simultaneous detection of multiple genes per cell. All single‐cell RT–qPCRs and data acquisition were performed in a CFX96 real‐time PCR device (Bio‐Rad). The Ct values of tested genes and of endogenous Gapdh obtained from qPCRs were filtered through standard thresholds.
Chromatin Immunoprecipitation (ChIP)
MEFs undergoing reprogramming and control cells (naïve MEFs, mESCs, miPSCs) were cultured under optimum conditions and were fixed at a high‐confluence stage at room temperature for 10 min using 1% formaldehyde in fixing buffer, followed by quenching with 0.125 M glycine at room temperature for 5 min. Upon extensive washes, cells were resuspended in lysis buffer [50 mM Hepes (pH 7.9), 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP‐40, 0.25% Triton X‐100] and then in sonication buffer [0.1% SDS, 1 mM EDTA, 10 mM Tris (pH 8.1)]. Chromatin shearing was carried out in the Covaris S2 sonicator using the Covaris TC12 × 12 mm tubes (Tube AFA Fiber and Cap, Covaris) for 12 min (200 cycles per burst) allowing the shearing of chromatin within a range of 250–500 base pairs DNA fragments. Triton X‐100 and NaCl were then added in the sheared chromatin to final concentrations of 1% and 150 mM, respectively. The chromatin was then centrifuged, and the harvested supernatants were filtered throughout a 0.2‐μm syringe. ChIPs were carried out by incubating 75 μg of chromatin (corresponding to approximately 5 × 107 cells) with 2–10 μg of antibody per ChIP reaction [anti‐mOct4 (19857, Abcam), anti‐mSox2 (2748S, Cell Signaling), anti‐mKlf4 (H‐180, SC‐20691, Santa Cruz), anti‐mc‐Myc (N‐262, SC‐764, Santa Cruz), and rabbit IgG (crude serum)] overnight at 4°C. Next, protein G‐Dynabeads (10004D, Thermo Fisher Scientific) pre‐equilibrated in IP buffer [0.1% SDS, 1 mM EDTA, 10 mM Tris (pH 8.1), 1% Triton X‐100, 150 mM NaCl] were incubated with the chromatin‐antibody solution in an orbital mixer at 4°C for 2 h. The recovered resin was subsequently washed with low and high salt buffers and LiCl buffer and the captured chromatin fragments were subjected to Proteinase K (03115828001, Roche Life Sciences) digestion at 50°C for 15 min, followed by overnight incubation with RNase A at 65°C. All DNA present in each sample was purified with AMPure XP beads and eluted in TE buffer.
Preparation of DNA libraries for ChIP‐sequencing
Next‐generation sequencing (NGS) libraries were prepared using 1–15 ng of ChIP DNA and TruSeq adapters as described previously (Ford et al, 2014). Briefly, DNA was blunt‐ended by End Repair Enzyme Mix (T4 DNA polymerase, Klenow fragment, T4 DNA polynucleotide kinase) followed by “A” tailing of 3’ ends and ligation with the annealed TruSeq Adapters (Illumina). Conversion of the Y‐shaped adapters to dsDNA occurred prior to the library size selection through 2.5% Metaphor/SeaKem LE (3:1 ratio) agarose gel electrophoresis. Each library was purified using MinElute columns (Qiagen) and then subjected to pre‐amplification. The final quantification of DNA libraries was carried out according to the Quantification Standards of Illumina on an Agilent Technologies 2100 Bioanalyzer (Agilent Technologies).
Western blots
Whole‐cell extracts from non‐transduced MEFs, iPSCs, and different time points of MEFs undergoing reprogramming were resuspended in sample buffer (200 mM Tris pH 6.8, 8% SDS, 0.4% bromophenol blue, 40% glycerol, 400 mM DTT), and ~25 × 104 cells were loaded on a 10% SDS–PAGE gel. For Irf6, Rcan1, Taf1c, Tead4, Cbfa2t3, and Tfap4, ~50 × 104 of non‐transduced MEFs and day 6 MEFs were loaded per lane. The post‐run nitrocellulose membrane was blocked in 5% milk/TBST for 2 h at room temperature and subsequently incubated overnight at 4°C while agitating with the primary antibodies against Oct‐3/4 (C‐10, sc‐5279, Santa Cruz), Sox2 (2748S, Cell Signaling Technology), Klf4 (H‐180, sc‐20691, Santa Cruz), c‐Myc (N‐262, sc‐764 Santa Cruz), AP‐4 (A‐B, sc377042, Santa Cruz), and DCSR1 (Rcan1) (G‐2, sc377507, Santa Cruz). GAPDH (AM4300, Ambion), b‐tubulin (D3U1W, Cell Signaling Technology), or mTOR (7C10, Cell Signaling Technology) were used as loading controls. For Irf6, Tead4, Taf1c, and Cbfa2t3, custom‐made antibodies were used (see in separate section “Antibody Production” for immunization). Next day, the membrane was incubated with secondary IgG antibodies conjugated with HRP (goat anti‐rabbit HRP antibody, 1706515, Bio‐Rad; goat anti‐mouse HRP antibody sc‐2055, Santa Cruz) for 1 h at room temperature followed by extensive washes with 2% milk/TBST. Developing was carried out using ECL (Thermo Scientific), and all experiments were normalized using housekeeping genes as loading control samples.
Antibody production
Part of CDS of mouse Cbfa2t3, Irf6, Taf1c, and Tead4 were amplified from the corresponding OE constructs (Table EV6). The amplified DNA fragments were digested with a defined combination of restriction enzymes (EcoRI‐XhoI) and were subcloned into the pGEX‐5X‐1 GST expression vector. The primers used for the amplification of the targets and the restriction enzymes used for the digestions are presented in Table EV7. BL21 CodonPlus (pRIP) Escherichia coli cells were transformed with the above‐mentioned constructs, and the antigens were produced upon a 3‐h IPTG (50 mM) induction in bacterial cell cultures. Cells expressing the GST‐fused antigens were lysed, and the soluble fraction was loaded on 1 ml glutathione–agarose beads (Pierce–Thermo Scientific) pre‐packed column. Elution was performed with 10 mM reduced glutathione, and the eluate was dialyzed in 10 mM Tris–HCl pH 8 and 100 mM NaCl buffer. The purified antigens were sent for immunization at Davids Biotechnologie, Germany. The antibodies were purified from the rabbit serums through ammonium sulfate precipitation (40% w/v ammonium sulfate), DEAE chromatography, and affinity chromatography against the antigens, which were covalently bound at CNBr beads. Elution of the antibodies was carried out with glycine 20 mM solution, pH 2.5. The eluate was dialyzed in 10 mM Tris–HCl pH 8 and 100 mM NaCl buffer. The quality and specificity of antibodies were evaluated using HEK293T cell lines transduced with lentiviral particles overexpressing the Cbfa2t3, Irf6, Taf1c, and Tead4 genes (unpublished data).
Microarray analysis
Affymetrix Human Genome 133 A 2.0 version was used for gene expression profiling of the reprogramming course. RNA was isolated from mESCs, miPSCs, control (naïve) MEFs and MEFs undergoing reprogramming at days 0.5, 1, 1.5, 2, 2.5, 3, 6, 12, and 18 of reprogramming. Additional RNA was isolated from mHeps at days 0, 3, and 6 of reprogramming. Background adjustment was done with MAS5.0 algorithm followed by median normalization per chip. Probes with raw signal intensity below 50 were considered as “ABSENT”. MAS5.0 flagging system was used in addition to characterize probes and thus genes ABSENT as “non‐expressed” and PRESENT / MARGINAL as “expressed”. Multiple probes corresponding to the same gene were filtered out, and only those with the highest signal intensity across all time points were kept for further analysis and evaluation. In order to characterize a gene as differentially expressed during the reprogramming course, it should fulfill the following criteria:
For upregulated genes:
Fold change time T dox+>= 2 (Signal intensity at time point T dox+/ Signal intensity at time point 0 {MEFs} >=2) &
Flag at time point T dox+ = Present &
Fold Change time T dox‐ <2 (Signal intensity at time point T dox‐/ Signal intensity at time point 0 {MEFs} <2)
For downregulated genes:
Fold change time T dox+<= 0.5 (Signal intensity at time point T dox+/ Signal intensity at time point 0 {MEFs} <=0.5) &
Flag at time point 0 dox+ = Present &
Fold Change time T dox‐ > 0.5 (Signal intensity at time point T dox‐/ Signal intensity at time point 0 {MEFs} > 0.5)
Gene Ontology analysis and classification of genes into transcription regulators were performed with the use of Ingenuity Pathway Analysis software and DAVID Knowledgebase (Huang et al, 2009).
ChIP‐sequencing data analysis
ChIP‐seq processing of ChIP‐DNA libraries was performed at the Greek Genome Center (GGC) of BRFAA and in the Genomics Core Facility at EMBL. Both NextSeq 500 and HiSeq 2000 Illumina sequencers were used to produce 75‐bp and 50‐bp single‐end reads, respectively. FastQC software was used for accessing the quality of the fastq files generated from the sequencers. Alignment of the data to the mm9 genome version was performed with bowtie algorithm (1.0.0 version) (Langmead et al, 2009) with the use of ‐v 2 ‐m 1 parameters. Duplicate reads were removed using samtools, and bed files with unique mapped reads generated from samtools and bedtools (Quinlan & Hall, 2010) were used for peak calling. At least 15 millions uniquely mapped reads for each O/S/K/M were generated. MACS14 algorithm (Zhang et al, 2008b) 1.4.2 version was used to identify OSKM‐binding sites by comparing the ChIP bed file with the corresponding input bed file at each time point. MACS14 with P‐value 1e‐5 was used for MYC‐ and KLF4‐binding sites in MEF cells. 1e‐4 was used for O/S/K/M‐binding sites in ESCs and iPSCs and 1e‐3 for O/S/K/M‐binding sites identification in MEF cells undergoing reprogramming at day 3 and day 5. The comparison between the O/S/K/M‐binding sites was performed with the intersectBed command. Visualization of the read density for O/S/K/M was performed in the IGV browser using bigwig files.
Single‐cell analysis
We calculated the expected and observed probability (P) of the 9 TR co‐expression based on single, double, and triple single‐cell qPCR experiments, as shown below:
A) Expected probability of 9 TR co‐expression based on single single‐cell qPCR
B) Observed probability of 9 TR co‐expression based on triple and double single‐cell qPCR
We measured the probability of 9 TR co‐expression taking into account the correlation of their expression per combinations (double and triple single‐cell experiments). To calculate the observed probability of 9TR co‐expression, we used all combinations of triple and double single‐cell co‐expression frequencies including all TRs once.
Statistical analyses
Pearson's correlation coefficient was used for correlation analysis with STATA software. Clustering analysis using Pearson's correlation analysis and average linkage was performed with the use of tMEV software (Saeed et al, 2003).
Data are shown as mean ± SEM. We performed F‐test to check whether the standard deviation between the groups tested was significant. If F‐test P‐value was < 0.05, we performed unpaired Welch's t‐test. For F‐test P‐values > 0.05, we performed unpaired Student's t‐test. P‐values < 0.05 were considered significant.
Author contributions
DT conceived the experiments and wrote the manuscript, MPa conceived and conducted experiments and wrote the manuscript, SAT conceived and conducted experiments and wrote the manuscript, APP conceived experiments and analyzed the data, DP conducted experiments, DV conducted experiments, EK conducted experiments, PK conducted experiments, MPl conducted experiments, IT conceived experiments, and MA conceived experiments and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Table EV7
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We thank Tom Maniatis, Stavros Lomvardas, Spyros Georgatos, Theodore Fotsis, Carol Murphy, George Mosialos, George Panayotou, Apostolos Klinakis, and members of the Thanos Lab for critical reading of the manuscript and useful advice during the work. We also thank Effie Apostolou for the OSK expressing mouse. This work was supported by grants to DT from the Greek General Secretariat for Research and Technology (GSRT) (Cooperative Grants Synergasia I #969, Excellence Award Aristeia I #1567), European Committee FP7 projects (Integer, Nanoma, Predicta, and Biofos), from the European Economic Area (EL0084), and from the KMW offsets program. MA was supported by the Bodossaki Foundation. SAT was supported by a research grant from the EU Initial Training Network (Project number: 214902) and a postdoctoral research grant fellowship from the Greek General Secretariat for Research and Technology (GSRT) (Project Number: LS2‐3765). DV was supported by a scholarship from the Bodossaki Foundation and EK from the State Scholarships Foundation (Operational Programme MIS‐5000432) and from the BIOIMAGING.GR (MIS‐5002755).
The EMBO Journal (2021) 40: e102236
Contributor Information
Marios Agelopoulos, Email: magelo@bioacademy.gr.
Dimitris Thanos, Email: thanos@bioacademy.gr.
Data availability
Microarray data: Gene Expression Omnibus GSE114581 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE114581)
ChIP‐seq data: Gene Expression Omnibus GSE114581: (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE114581)
References
- Apostolou E, Stadtfeld M (2018) Cellular trajectories and molecular mechanisms of iPSC reprogramming. Curr Opin Genet Dev 52: 77–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botti E, Spallone G, Moretti F, Marinari B, Pinetti V, Galanti S, De Meo PD, De Nicola F, Ganci F, Castrignanò T et al (2011) Developmental factor IRF6 exhibits tumor suppressor activity in squamous cell carcinomas. Proc Natl Acad Sci USA 108: 13710–13715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buganim Y, Faddah DA, Cheng AW, Itskovich E, Markoulaki S, Ganz K, Klemm SL, van Oudenaarden A, Jaenisch R (2012) Single‐cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell 150: 1209–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buganim Y, Faddah DA, Jaenisch R (2013) Mechanisms and models of somatic cell reprogramming. Nat Rev Genet 14: 427–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Xu Z, Xie J, Ham A‐JL, Koury MJ, Hiebert SW, Brandt SJ (2009) Eto2/MTG16 and MTGR1 are heteromeric corepressors of the TAL1/SCL transcription factor in murine erythroid progenitors. Biochem Biophys Res Commun 390: 295–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers I, Colby D, Robertson M, Nichols J, Lee S, Tweedie S, Smith A (2003) Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell 113: 643–655 [DOI] [PubMed] [Google Scholar]
- Chambers I, Silva J, Colby D, Nichols J, Nijmeijer B, Robertson M, Vrana J, Jones K, Grotewold L, Smith A (2007) Nanog safeguards pluripotency and mediates germline development. Nature 450: 1230–1234 [DOI] [PubMed] [Google Scholar]
- Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J et al (2008) Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133: 1106–1117 [DOI] [PubMed] [Google Scholar]
- Chen J, Chen X, Li M, Liu X, Gao Y, Kou X, Zhao Y, Zheng W, Zhang X, Huo Y et al (2016) Hierarchical Oct4 Binding in Concert with Primed Epigenetic Rearrangements during Somatic Cell Reprogramming. Cell Rep 14: 1540–1554 [DOI] [PubMed] [Google Scholar]
- Choi SS, Diehl AM (2009) Epithelial‐to‐mesenchymal transitions in the liver. Hepatology 50: 2007–2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chronis C, Fiziev P, Papp B, Butz S, Bonora G, Sabri S, Ernst J, Plath K (2017) Cooperative Binding of Transcription Factors Orchestrates Reprogramming. Cell 168: 442–459.e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, Agarwal A, Huang W, Parkhurst CN, Muratet M et al (2012) A validated regulatory network for Th17 cell specification. Cell 151: 289–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford E, Nikopoulou C, Kokkalis A, Thanos D (2014) A method for generating highly multiplexed ChIP‐seq libraries. BMC Res Notes 7: 312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna J, Saha K, Pando B, van Zon J, Lengner CJ, Creyghton MP, van Oudenaarden A, Jaenisch R (2009) Direct cell reprogramming is a stochastic process amenable to acceleration. Nature 462: 595–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heix J, Zomerdijk JC, Ravanpay A, Tjian R, Grummt I (1997) Cloning of murine RNA polymerase I‐specific TAF factors: conserved interactions between the subunits of the species‐specific transcription initiation factor TIF‐IB/SL1. Proc Natl Acad Sci USA 94: 1733–1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57 [DOI] [PubMed] [Google Scholar]
- Hussein SMI, Puri MC, Tonge PD, Benevento M, Corso AJ, Clancy JL, Mosbergen R, Li M, Lee D‐S, Cloonan N et al (2014) Genome‐wide characterization of the routes to pluripotency. Nature 516: 198–206 [DOI] [PubMed] [Google Scholar]
- Jackstadt R, Röh S, Neumann J, Jung P, Hoffmann R, Horst D, Berens C, Bornkamm GW, Kirchner T, Menssen A et al (2013) AP4 is a mediator of epithelial‐mesenchymal transition and metastasis in colorectal cancer. J Exp Med 210: 1331–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Chu J, Shen X, Wang J, Orkin SH (2008) An extended transcriptional network for pluripotency of embryonic stem cells. Cell 132: 1049–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koche RP, Smith ZD, Adli M, Gu H, Ku M, Gnirke A, Bernstein BE, Meissner A (2011) Reprogramming factor expression initiates widespread targeted chromatin remodeling. Cell Stem Cell 8: 96–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert SA, Jolma A, Campitelli LF, Das PK, Yin Y, Albu M, Chen X, Taipale J, Hughes TR, Weirauch MT (2018) The Human Transcription Factors. Cell 175: 598–599 [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory‐efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levasseur DN, Wang J, Dorschner MO, Stamatoyannopoulos JA, Orkin SH (2008) Oct4 dependence of chromatin structure within the extended Nanog locus in ES cells. Genes Dev 22: 575–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Drnevich J, Akraiko T, Band M, Li D, Wang F, Matoba R, Tanaka TS (2013) Gene expression profiling reveals the heterogeneous transcriptional activity of Oct3/4 and its possible interaction with Gli2 in mouse embryonic stem cells. Genomics 102: 456–467 [DOI] [PubMed] [Google Scholar]
- Li M, Belmonte JCI (2017) Ground rules of the pluripotency gene regulatory network. Nat Rev Genet 18: 180–191 [DOI] [PubMed] [Google Scholar]
- Loh Y‐H, Wu Q, Chew J‐L, Vega VB, Zhang W, Chen X, Bourque G, George J, Leong B, Liu J et al (2006) The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet 38: 431–440 [DOI] [PubMed] [Google Scholar]
- Merika M, Thanos D (2001) Enhanceosomes. Curr. Opin. Genet. Dev. 11: 205–208 [DOI] [PubMed] [Google Scholar]
- Moore AC, Amann JM, Williams CS, Tahinci E, Farmer TE, Martinez JA, Yang G, Luce KS, Lee E, Hiebert SW (2008) Myeloid translocation gene family members associate with T‐cell factors (TCFs) and influence TCF‐dependent transcription. Mol Cell Biol 28: 977–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa M, Koyanagi M, Tanabe K, Takahashi K, Ichisaka T, Aoi T, Okita K, Mochiduki Y, Takizawa N, Yamanaka S (2008) Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol 26: 101–106 [DOI] [PubMed] [Google Scholar]
- Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R et al (2012) c‐Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 151: 68–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama A, Sharov AA, Piao Y, Amano M, Amano T, Hoang HG, Binder BY, Tapnio R, Bassey U, Malinou JN et al (2013) Systematic repression of transcription factors reveals limited patterns of gene expression changes in ES cells. Sci Rep 3: 1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H (2014) The pluripotency transcription factor network at work in reprogramming. Curr Opin Genet Dev 28: 25–31 [DOI] [PubMed] [Google Scholar]
- Niwa H (2018) The principles that govern transcription factor network functions in stem cells. Development 14: 5 [DOI] [PubMed] [Google Scholar]
- O'Malley J, Skylaki S, Iwabuchi KA, Chantzoura E, Ruetz T, Johnsson A, Tomlinson SR, Linnarsson S, Kaji K (2013) High‐resolution analysis with novel cell‐surface markers identifies routes to iPS cells. Nature 499: 88–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pliatska M, Kapasa M, Kokkalis A, Polyzos A, Thanos D (2018) The Histone Variant MacroH2A Blocks Cellular Reprogramming by Inhibiting Mesenchymal‐to‐Epithelial Transition. Mol Cell Biol 3: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Po A, Ferretti E, Miele E, De Smaele E, Paganelli A, Canettieri G, Coni S, Di Marcotullio L, Biffoni M, Massimi L et al (2010) Hedgehog controls neural stem cells through p53‐independent regulation of Nanog. EMBO J 29: 2646–2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo JM, Anderssen E, Walsh RM, Schwarz BA, Nefzger CM, Lim SM, Borkent M, Apostolou E, Alaei S, Cloutier J et al (2012) A molecular roadmap of reprogramming somatic cells into iPS cells. Cell 151: 1617–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, Tan M, Hui C‐C, Qiu M (2003) Gli2 is required for normal Shh signaling and oligodendrocyte development in the spinal cord. Mol Cell Neurosci 23: 440–450 [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahimov F, Marazita ML, Visel A, Cooper ME, Hitchler MJ, Rubini M, Domann FE, Govil M, Christensen K, Bille C et al (2008) Disruption of an AP‐2alpha binding site in an IRF6 enhancer is associated with cleft lip. Nat Genet 40: 1341–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson RJ, Dixon J, Malhotra S, Hardman MJ, Knowles L, Boot‐Handford RP, Shore P, Whitmarsh A, Dixon MJ (2006) Irf6 is a key determinant of the keratinocyte proliferation‐differentiation switch. Nat Genet 38: 1329–1334 [DOI] [PubMed] [Google Scholar]
- Roca H, Hernandez J, Weidner S, McEachin RC, Fuller D, Sud S, Schumann T, Wilkinson JE, Zaslavsky A, Li H et al (2013) Transcription factors OVOL1 and OVOL2 induce the mesenchymal to epithelial transition in human cancer. PLoS ONE 8: e76773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothermel B, Vega RB, Yang J, Wu H, Bassel‐Duby R, Williams RS (2000) A protein encoded within the Down syndrome critical region is enriched in striated muscles and inhibits calcineurin signaling. J Biol Chem 275: 8719–8725 [DOI] [PubMed] [Google Scholar]
- Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M et al (2003) TM4: a free, open‐source system for microarray data management and analysis. Biotechniques 34: 374–378 [DOI] [PubMed] [Google Scholar]
- Schiebinger G, Shu J, Tabaka M, Cleary B, Subramanian V, Solomon A, Gould J, Liu S, Lin S, Berube P et al (2019) Optimal‐Transport Analysis of Single‐Cell Gene Expression Identifies Developmental Trajectories in Reprogramming. Cell 176: 1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz BA, Cetinbas M, Clement K, Walsh RM, Cheloufi S, Gu H, Langkabel J, Kamiya A, Schorle H, Meissner A et al (2018) Prospective Isolation of Poised iPSC Intermediates Reveals Principles of Cellular Reprogramming. Cell Stem Cell 23: 289–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SH, Kim D, Hwang J, Kim MK, Kim JC, Sung YK (2014) OVO homolog‐like 1, a target gene of the Wnt/β‐catenin pathway, controls hair follicle neogenesis. J. Invest. Dermatol. 134: 838–840 [DOI] [PubMed] [Google Scholar]
- Silva J, Nichols J, Theunissen TW, Guo G, van Oosten AL, Barrandon O, Wray J, Yamanaka S, Chambers I, Smith A (2009) Nanog is the gateway to the pluripotent ground state. Cell 138: 722–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soufi A, Donahue G, Zaret KS (2012) Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell 151: 994–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soufi A, Garcia MF, Jaroszewicz A, Osman N, Pellegrini M, Zaret KS (2015) Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 161: 555–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadhouders R, Vidal E, Serra F, Di Stefano B, Le Dily F, Quilez J, Gomez A, Collombet S, Berenguer C, Cuartero Y et al (2018) Transcription factors orchestrate dynamic interplay between genome topology and gene regulation during cell reprogramming. Nat Genet 50: 238–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M, Maherali N, Breault DT, Hochedlinger K (2008) Defining molecular cornerstones during fibroblast to iPS cell reprogramming in mouse. Cell Stem Cell 2: 230–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126: 663–676 [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131: 861–872 [DOI] [PubMed] [Google Scholar]
- Tu S, Narendra V, Yamaji M, Vidal SE, Rojas LA, Wang X, Kim SY, Garcia BA, Tuschl T, Stadtfeld M et al (2016) Co‐repressor CBFA2T2 regulates pluripotency and germline development. Nature 534: 387–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernig M, Meissner A, Cassady JP, Jaenisch R (2008) c‐Myc is dispensable for direct reprogramming of mouse fibroblasts. Cell Stem Cell 2: 10–12 [DOI] [PubMed] [Google Scholar]
- Wu Q, Chen X, Zhang J, Loh Y‐H, Low T‐Y, Zhang W, Zhang W, Sze S‐K, Lim B, Ng H‐H (2006) Sall4 interacts with Nanog and co‐occupies Nanog genomic sites in embryonic stem cells. J Biol Chem 281: 24090–24094 [DOI] [PubMed] [Google Scholar]
- Wu Z, Li Y, MacNeil AJ, Junkins RD, Berman JN, Lin T‐J (2013) Calcineurin‐Rcan1 interaction contributes to stem cell factor‐mediated mast cell activation. J. Immunol. 191: 5885–5894 [DOI] [PubMed] [Google Scholar]
- Yagi R, Kohn MJ, Karavanova I, Kaneko KJ, Vullhorst D, DePamphilis ML, Buonanno A (2007) Transcription factor TEAD4 specifies the trophectoderm lineage at the beginning of mammalian development. Development 134: 3827–3836 [DOI] [PubMed] [Google Scholar]
- Yamanaka S (2009) Elite and stochastic models for induced pluripotent stem cell generation. Nature 460: 49–52 [DOI] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga‐Otto K, Antosiewicz‐Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R et al (2007) Induced pluripotent stem cell lines derived from human somatic cells. Science 318: 1917–1920 [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhang J, Wang T, Esteban MA, Pei D (2008a) Esrrb activates Oct4 transcription and sustains self‐renewal and pluripotency in embryonic stem cells. J Biol Chem 283: 35825–35833 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W et al (2008b) Model‐based analysis of ChIP‐Seq (MACS). Genome Biol 9: R137 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Table EV6
Table EV7
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5
Data Availability Statement
Microarray data: Gene Expression Omnibus GSE114581 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE114581)
ChIP‐seq data: Gene Expression Omnibus GSE114581: (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE114581)