

Abstract
Immunotherapy, as a novel treatment, has brought new hope to many patients with cancer, including patients with lung cancer. However, the overall cure rate and survival rate of lung cancer are still not satisfactory. The process of evolution has improved the ability of tumors to adapt to immunotherapy, which induces drug resistance. Many studies have focused on immunoresistance and achieved meaningful results. Therefore, it is necessary to have an in-depth understanding of the current research progress in immunoresistance, which will help to achieve good clinical results more efficiently.
Keywords: Lung cancer, immunotherapy, cancer evolution, drug resistance
Introduction
Lung cancer is the leading cause of cancer-related mortality in both men and women, with more than a million attributed deaths each year worldwide.1 Although there have been advances in the treatment of patients with lung cancer, including advances in surgery, chemoradiotherapy, and targeted molecular therapy, the 5-year overall survival rate of non-small cell lung cancer (NSCLC) remains only 18.1% in the United States.2 Since the concept of evolution was applied to tumors in the last century,3 accelerated technological progress has led to extensive study of tumor genomes and further awareness of the complexity of tumor cytogenetics and tumor evolution. The theory of evolution that has developed since Darwin time to understand population genetics is also applicable to understanding the diversity of cancer cells in patients.
A tumor itself has an evolutionary history, and all cells in the tumor, including those with different somatic variations and epigenetic and transcriptional cleavage states, are unique.4 Even in healthy cells, approximately 3 somatic mutations may occur during each cell cycle.5 The regular immune system can recognize and eliminate mutated cells, and most of these changes do not affect cell function; they are only “passengers” in the process of cell evolution. The occurrence of “driver” mutations may create some growth advantages in cells. In the case of unregulated growth, mutated cells are produced and escape the supervision of the immune system, causing damage to the human body.
Tumors are highly heterogeneous: different spatial regions of a single tumor have significant intratumor heterogeneity, and various individual tumors of the same type may experience very different clonal evolution paths.6-8 Specifically, most of the genetic variation is harmful, and the related cells are either eliminated by competition for resources or destroyed by host immune cells. Owing to the pressures of different treatment options, coupled with the fact that tumor cells often cannot effectively maintain genomic integrity,9 the inherent genomic instability of tumor cells makes them prone to genetic changes or damage during the proliferative process, and then, those tumor cells that are most suitable and proliferate fastest will predominate.10 Tumors evolve through genetic changes, respond to changing microenvironments (including related microenvironments such as immunotherapy) and can produce new mutations that confer selective growth advantages at any time.
In recent years, immunotherapy has been established as a breakthrough in tumor therapy. Because of its long-lasting effect and few adverse reactions, it is widely used in the clinic and is expected to open a new model of tumor treatment.11 However, most patients who are initially sensitive eventually develop immune drug resistance. At present, the greatest problem in tumor treatment is drug resistance, which should become an area of focus.12
This review will focus on the evolutionary performance of lung cancer under the pressure of immunotherapy and the possibility of using evolution itself to treat disease. We will introduce the vital role of the immune system in antitumor therapy first, discuss the process of tumor evolution in the context of immunotherapy, and propose different strategies to take advantage of the evolutionary nature of tumors to benefit patients with cancer.
Evolution of Lung Cancer in the Context of Immunotherapy
Immunity and tumors
Tumors are the product of the malignant transformation of healthy cells in the body, which can produce new antigenic markers that are not found in the same kind of healthy cells. Owing to the existence of tumor antigens, tumors are bound to be recognized by the body’s immune system and thus stimulate specific immune responses, including cellular immunity and humoral immunity. In cellular immunity, T lymphocytes, antibody-dependent cell-mediated cytotoxicity, natural killer cells, and macrophages can kill tumor cells. The humoral immunity against a tumor mainly manifests as the destructive effect of antitumor antibodies on tumor cells. Only through the synergistic effects of cellular immunity and humoral immunity can tumor cells be inhibited or eliminated.
Under normal circumstances, the body relies on a complete immune mechanism to effectively monitor and reject cancerous cells, so the vast majority of individuals do not have tumors. If cancerous cells proliferate to a certain extent and manage to evade immune surveillance, the occurrence of a tumor is inevitable. There are stimulating factors and inhibitory factors in the process of antitumor immunity that control the positive and negative regulation of the antitumor immune response, respectively. The immune balance affects the outcome of each tumor, but it is difficult to find a balance in the clinic because of the preexisting immune escape of the tumor.
The reason for tumor escape lies in the changes in tumor cells themselves and the changes in the tumor microenvironment. The first changes occur in the tumor cells: (1) Loss of tumor-associated antigens and NKG2D ligands reduces immunogenicity.13 (2) The expression of major histocompatibility complex I (MHC-I) molecules and key molecules involved in antigen processing is downregulated or abnormal, resulting in a decrease in the ability of T cells to recognize tumor cells.14 (3) Changes in the apoptosis signaling pathway invalidate the mechanism of apoptosis induced by immune cells or reduce the sensitivity to T cells. The second wave of changes involves the tumor microenvironment: (1) Tumor cells release a variety of immunosuppressive factors, such as vascular endothelial growth factor and transforming growth factor β. (2) Tumor cells overexpress ligand molecules (such as programmed death ligand-1 [PD-L1]) that lead to lymphocyte dysfunction and death or upregulate inhibitory receptors, such as cytotoxic T-lymphocyte-associated protein 4 (CTLA-4). (3) Tumor-infiltrating regulatory T cells and myeloid suppressor cells overexpress immunosuppressive factors and molecules to weaken the function of T cells or induce T cell apoptosis.15 Eliminating the escape of tumor cells and improving the antitumor immune response are the basic strategies of tumor immunotherapy.
The mechanism of tumor drug resistance is complex, and immune escape is inevitable. Basic immunology research provides a theoretical basis for the in-depth understanding of the mechanism of tumor immune escape. This understanding plays an essential supporting role in the further development of new and effective tumor treatment methods. It will be crucial to exploit the mechanisms of tumor resistance, select reasonable treatment modalities, and successfully extend new immunotherapies to more patients with cancer.
Development of tumor immunotherapy
In cancer immunotherapy, traditional treatment methods include bacterial/viral infection to enhance the immune response and tumor vaccination. The earliest tumor immunotherapy treatments can be traced back to 1893, when William Coley 16 accidentally found that suppurative streptococcal infection in patients with osteosarcoma after surgical resection of the tumor could lead to cancer regression, which is an example of infection promoting an immune response to cure cancer. Oncolytic virus therapy has been studied as an immunotherapy strategy for hundreds of years, but it has showed little effect for a long time. At present, there is only 1 oncolytic virus approved by the Food and Drug Administration (FDA), Imlygic (talimogene laherparepvec), which is a genetically modified herpes simplex virus approved for the treatment of lymphoma and melanoma.17 Cancer vaccines usually use dendritic cells (DCs) loaded with tumor-specific antigens to present the antigens and activate host T cells to trigger an antitumor immune response. However, no significant progress has been made in the use of a single antigen (such as melanoma-associated antigen 3) or cancer cell lysates (such as the vaccine made of a tumor cell transfected with granulocyte-macrophage colony-stimulating factor [GVAX]). At present, only 1 therapeutic DC vaccine, sipuleucel-T (Provenge), has been approved for clinical use.18,19 The risk of death has been reduced by 33%, indicating an absolute improvement in survival.20
In recent years, emerging tumor immunotherapy has been an active research area. In 1984, the Steven Rosenberg team 21 used interleukin-2 (IL-2) and lymphokine-activated killer cells to treat 25 patients with tumor and achieved excellent results, which was the first successful example of adoptive cell therapy and brought a glimmer of hope to tumor immunotherapy. However, high-dose IL-2 has considerable side effects, so researchers use in vitro extraction, activation, and reinfusion of autologous/allogeneic cytokine-induced killer cells, tumor-specific T cells, tumor-infiltrating lymphocytes, and other cells for tumor adoptive immunotherapy.22,23 Since 2011, blocking CTLA4 or programmed death 1 (PD-1)/PD-L1 and other new immunotherapies have achieved outstanding clinical efficacy (Figure 1), and checkpoint blockade is considered to be the most promising tumor immunotherapy strategy. In 2011, the FDA approved the first immune checkpoint blocker (ICB; the anti-CTLA-4 monoclonal antibody ipilimumab) for second-line treatment of advanced melanoma, marking a new era of tumor immunotherapy.24 In 2012, Carl H. June used chimeric antigen receptor T (CAR-T) cell therapy to cure a 6-year-old girl with leukemia, Emily, causing a global sensation, and thus far, Emily cancer cells have remained undetectable for 8 years.25 In 2013, Science listed immunotherapy as 1 of the top 10 scientific breakthroughs of the year, which made immunotherapy once again the focus of global research and development.26
Figure 1.
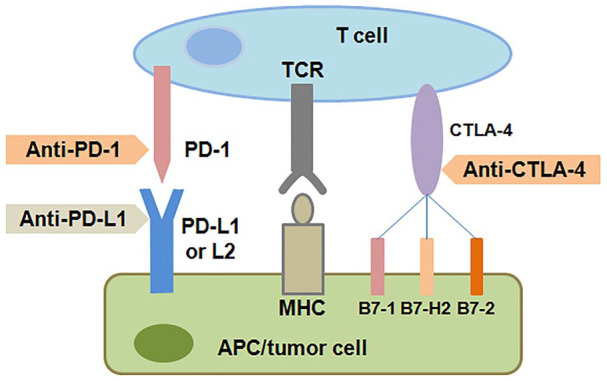
Blockade of CTLA-4 and of PD-1 and PD-L1 to induce antitumor responses. CTLA-4 and PD-1 are both inhibitory immunocheckpoint, and their activation can turn off the antitumor T cell responses. The activation of CTLA-4 can be blocked with anti-CTLA-4 antibodies. The activation of PD-1 can be blocked by anti-PD-1 or anti-PD-L1 antibodies.
APC indicates antigen-presenting cell; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; MHC, major histocompatibility complex; PD-1, programmed death 1; PD-L1, programmed death ligand-1; TCR, T-cell receptor.
In the field of lung cancer, in 2014, the FDA approved the anti-PD-1 antibodies Keytruda (pembrolizumab) and Opdivo (nivolumab), and tumor immunotherapy has since become a hot topic of research and development. On October 24, 2016, the FDA approved pembrolizumab for the treatment of metastatic NSCLC patients with tumor PD-L1 expression. Based on the efficacy and safety data of CheckMate 032,27 nivolumab was approved by the FDA in 2018 as a third-line treatment for small cell lung cancer. 2018 was also the year that data on combined immunotherapy strategies become prominent. Based on the previous Keynote-02128 and IMpower15029 studies, many phase III clinical studies, such as Keynote189, IMpower130,30 and IMpower132 for nonsquamous cell NSCLC and Keynote407 and IMpower13131 for lung squamous cell carcinoma, have suggested that combination of chemotherapy and immunotherapy can improve the objective response rate in NSCLC and significantly reduce the risks of disease progression and death. At present, chemotherapy plus immunotherapy as a first-line treatment option for treating gene-negative advanced NSCLC has been written into the guidelines of the National Comprehensive Cancer Network.
However, there are many kinds of prognostic molecules for predicting the effect of immunotherapy, but more clinical data needed to explore and verify the best oncology marker. In the future, we need to comprehensively evaluate the tumor microenvironment and immune genes to make accurate choices and develop effective treatment strategies.
Evolution and immune escape of lung cancer
The most direct reason why tumors do not respond to immunotherapy is a lack of T cell recognition due to a lack of tumor antigens. Alternatively, cancer cells may have tumor antigens that cannot be presented on the cell surface to T cells due to changes in antigen presentation mechanisms (such as mutations in proteases or transporters associated with antigen processing), β-2-microglobulin (B2M), or MHC. As shown in Figure 2, both tumor cell and immune microenvironment factors contribute to the resistance mechanisms.
Figure 2.

Summary of resistance factors of PD-1/PD-L1. Tumor resistance to PD-1/PD-L1 immunotherapy is mainly related to the influence of the 2 aspects in the figure, each of which can be subdivided into multiple factors to form an interwoven network of mutual influence. Combined multitarget therapy can improve the tumor response rate. The combination of multiple drugs and the development of new drugs are current trends.
ALK indicates anaplastic lymphoma kinase; B2M, β-2-microglobulin; BTLA, B- and T-lymphocyte attenuator; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; EGFR, epidermal growth factor receptor; IDO, indoleamine 2,3-dioxygenase; IFN-γ, interferon-gamma; JAK/STAT, Janus kinase/signal transducer and activator of transcription; LAG-3, lymphocyte activation gene 3; MDSC, myeloid suppressor cells; MHC, major histocompatibility complex; NKG2D, natural killer group 2, member D; PD-1, programmed death 1; PD-L1, programmed death ligand-1; PI3K/Akt, phosphatidylinositol-3 kinase/serine-threonine kinase; PTEN, phosphatase and tensin homolog; RAS/MAPK, RAS-mitogen-activated protein kinase; TGF, transforming growth factor; TIGIT, T cell immunoglobulin and ITIM domain; TIM3, T cell immunoglobulin mucin 3; Treg, tumor-infiltrating regulatory T cell; VEGF, vascular endothelial growth factor.
At present, many kinds of tumor drug resistance mechanisms have been found, including (1) signal transduction and/or loss of PTEN expression through the mitogen-activated protein kinase pathway, thus enhancing the PI3 K signaling pathway; (2) activation of components of the Wnt/b-catenin signaling pathway; (3) loss of the interferon-gamma (IFN-γ) signaling pathway; and (4) loss of the T cell response caused by loss of tumor antigen expression. These mechanisms are related to primary and adaptive tumor resistance to immunotherapy. One of the hallmarks of tumor immunotherapy is a lasting antitumor response. With extension of the treatment cycle, initially, sensitive patients will gradually develop drug resistance, known as acquired drug resistance. Primary and adaptive resistance and acquired drug resistance have similar underlying mechanisms. For example, IFN-γ binds to IFN receptors on the surface of tumor cells and activates the downstream Janus kinase (JAK)/signal transducer and activator of transcription signaling pathway. At the same time, JAK1/2 mutation can also lead to a decrease in IFN-γ-mediated adaptive PD-L1 expression, which is not mediated through the IFN-γ receptor pathway, resulting in a lack of tumor cell sensitivity to anti-PD-1 antibody therapy.32-34 After the pressure exerted by PD-1/PD-L1 treatment, a tumor can reduce its expression of human leukocyte antigen-I, which leads to the failure of treatment.35-37 Chimeric antigen receptor T cells are not limited by MHC and can be modified to enhance T cell function, which has a significant effect on the treatment of acute leukemia and non-Hodgkin lymphoma. However, in solid tumors, studies have shown that CAR-T cells can reduce the invasiveness and diffusivity of pancreatic cancer.38 However, in lung cancer and other solid tumors, there are still many obstacles to the application of CAR-T cell therapy in solid tumors.39 What needs to be addressed is that both CAR-T cell therapy and anti-PD-1/PD-L1 therapy require tumors to express specific neoantigens. Owing to the treatment pressure and competition among tumor subsets, the expression of tumor antigens tends to decrease during the course of treatment.40-42
The emergence of acquired drug resistance is related to the loss or mutation of tumor-specific neoantigens, which occurs not only through the elimination of tumor subclones but also through the loss of chromosomes. In tumor samples analyzed during acquired drug resistance, most of the mutations eliminated are typically highly expressed genes in lung cancer, and the neoantigens encoded are predicted to either provide high-affinity MHC binding or affect T-cell receptor contact residues.42 The process of antigen presentation is the initial stage of an immune response. Antigen loss affects the immune recognition of tumor cells, and disorder in antigen presentation leads to the immune system being unable to kill. For example, the B2M protein is involved in the transfer of a tumor antigen from MHC-I to CD8+ T cells.43 β-2-microglobulin gene mutation is recognized as 1 of the mechanisms of tumor immune escape.34,36 Similarly, a decrease in or loss of MHC-I expression promotes immune escape by reducing the ability to present antigens.35,36,44 The significant roles of IFN-γ signaling pathway–related molecules in the process of antigen presentation also involve mutation of these molecules in the decrease in sensitivity to anti-PD-1 antibody therapy and the occurrence of acquired drug resistance.43,45
In addition, immunomodulatory factors play essential roles in acquired drug resistance. T cell immunoglobulin mucin 3 (TIM3) and PD-1 are both negative regulators of the immune system. T cell immunoglobulin mucin 3 can bind to its ligand galactose lectin 9 (galectin 9) and inhibit the activation and proliferation of antitumor immunity–related T cells.46 In a study, after failure of first-line chemotherapy, an anti-PD-1 antibody was used to treat lung adenocarcinoma with disease progression. The expression levels of TIM3 and galectin 9 were significantly upregulated, indicating that the selective upregulation of TIM3 expression and the activation of the galectin 9/TIM3 pathway were involved in the occurrence of acquired resistance to the anti-PD-1 antibody, which was a prominent manifestation of tumor evolution.47
The study of tumor evolution plays a vital role in revealing the mechanism of immune drug resistance. Acquired drug resistance is the manifestation of tumor evolution under treatment pressure, and tumor enlargement and distant metastasis are the final stages of evolution. At present, there is no standardized solution to the problem of ICB drug resistance. In-depth study of the mechanism of drug resistance to ICBs, monitoring the process of tumor evolution, and then reducing and avoiding the occurrence of drug resistance are bound to promote the rapid development of tumor immunotherapy and achieve more long-term survival benefits in patients with tumor.
Can the evolution of lung cancer be controlled?
The core of Darwin theory of evolution is natural selection of living things. There is competition among tumor cell populations. Traditional antitumor drugs cannot destroy all tumor cells; they kill only sensitive tumor cells but allow drug-resistant cancer cells to grow unconstrained. For patients who exhibit sensitivity at the initial stage of antitumor therapy, drug resistance mutations may exist before drug treatment. The immune microenvironment during drug therapy breaks the competitive balance and promotes the growth of the drug-resistant subgroup, which becomes a new dominant subgroup. The emergence of dominant subgroups often indicates a weak curative effect.
In the face of evolving tumors, do we have the ability to achieve the goal of treating tumors by controlling the evolutionary process? Understanding how to make good use of the competitive balance among tumor cell subsets will contribute to long-term benefits in patients. For example, can low-dose continuous antineoplastic drugs maintain the balance of competition among tumor cells? Can competition among tumor cell subsets be regulated by alternating the use of chemotherapy, targeted therapy, and immunotherapy? Can we achieve the goal of radical tumor cure through combination of multiple drugs? At present, low-dose maintenance regimens have been well evaluated in clinical practice,48 but there is a lack of evidence for control of tumor evolution in patients by rhythmic methods. The alternating use of targeted therapy and chemotherapy exerts an excellent antitumor effect on the mechanism of multidrug resistance.49 Multidrug combinations have achieved outstanding results in the clinic,29 but they still cannot avoid the emergence of drug resistance and the dilemma that there is no drug available after drug resistance develops. Tumors are in the process of continuous evolution, and new mutations produce new tumor cell subsets, which is illustrated as the constant branching of a tree.50 If a treatment can directly attack a trunk mutation, it may be the most effective approach. However, at present, many mutations do not have targeted drugs.
The process of tumor evolution is complicated. Early focal resection is the most direct way to eliminate the original mutation, which can significantly prolong the time to disease recurrence, and surgically reducing the tumor load has been proven to reduce the incidence of tumor metastasis.51 Postoperative specimens provide a complete tumor mutation map, which provides a basis for the tumor evolutionary tree and screening for cancer neoantigens. Targeting trunk mutations or multiple antigens through personalized vaccines or immunotherapy will significantly reduce the risk of drug resistance. Circulating tumor DNA (ctDNA) analysis can be used to monitor changes in tumor heterogeneity, and the response to treatment in real time and drugs can be adjusted accordingly to achieve the best effect. The emergence of clustered regularly interspaced short palindromic repeats (CRISPR) technology in 2012 completely changed life science.52 Clustered regularly interspaced short palindromic repeats gene editing has great potential in gene modification, and experimental methods based on CRISPR technology have made a significant contribution to the study of the evolutionary process.53 This technology is also an auspicious, inexpensive, and simple tool for the development of effective cancer treatments. In 2016, CRISPR-Cas9 technology was used for the first time to knock out the gene encoding PD-1 in T cells, which were then reinfused into patients for the treatment of lung cancer.54 Recently, the first phase I clinical trial of CRISPR for cancer immunotherapy was reported to be successful.55 With the in-depth study of tumor evolution, CRISPR technology is expected to become a powerful tool to regulate the evolutionary process.
The application of CRISPR technology provides technical support for the development of effective cancer therapies. Real-time assessment of ctDNA tracks tumor evolution and allows clinicians to choose the right treatment at the right time. It should be emphasized that tumors have temporal and spatial heterogeneity,56 each tumor is unique, and current technical problems limit our ability to predict tumor evolution. The study of tumor evolution through multiregion sequencing and liquid biopsy provides new clues for us to understand the carcinogenic process and the mechanism of tumor escape during treatment. However, ethical issues are the bottom line that cannot be crossed. In the later stage, more in-depth research still needs to be performed to achieve technological progress and cost reduction.
The important role and clinical significance of lung cancer evolution
Study on tumor evolution at the gene level undoubtedly provides a solid theoretical basis for accurate tumor therapy. From the point of view of tumor evolution theory, if we can realize dynamic monitoring of the evolutionary process and develop treatments that evolve over time, we can improve treatment and prognosis. For example, the overexpression of CD38 after anti-PD-1/PD-L1 treatment increases the level of extracellular adenosine, which may be related to acquired resistance and can be overcome by blocking CD38.57 Dynamic samples are needed to realize proactive monitoring of gene expression levels. Tissue sampling is traumatic, it is difficult to perform biopsies repeatedly, and a sample is single and cannot be used to comprehensively monitor tumor evolution. Multipoint sampling can fully reflect the process of tumor evolution, and blood sampling provides an alternative to achieve early intervention.
In 2014, the United Kingdom established the Tracking non-small cell lung cancer evolution through therapy (Rx) (TRACERx) Lung Cancer Research Alliance, which aims to track the evolution of lung cancer in time and space through multiregional and longitudinal sampling of lesions.58 Recently, a coalition study confirmed that pulmonary venous circulating tumor cells are an independent predictor of lung cancer recurrence.59 The importance of circulating samples is emphasized again. Circulating tumor DNA, as a DNA fragment secreted by a small number of tumor cells in the blood, can relatively accurately predict the development of tumors because its half-life is only approximately 2 hours. For example, ctDNA levels in patients who are clinically responsive to treatment were shown to decrease after the start of treatment, whereas ctDNA levels in nonresponding patients did not change or increase significantly. In patients with an initial response who developed acquired drug resistance to treatment, ctDNA levels initially decreased and then increased.60 If clonal evolution eventually leads to therapeutic resistance, then detection through tissue and liquid biopsies may play a key role in guiding the most effective treatment routes.
Circulating tumor DNA can be used to monitor the heterogeneity of tumor cells in real time, and the measurement of clonal evolution enables clinicians to choose and adjust appropriate strategies to control tumor evolution. For example, chromosomal instability and mutations are markers of cancer and are also considered to promote cancer, enhancing the clonal evolution of cancer cells by accumulating gene copy number changes, rearrangements, and mutations. However, these markers also provide targeted vulnerabilities that are relatively specific to cancer cells and can be exploited clinically through the use of DNA damage response (DDR) inhibitors in combination with anti-PD-1 or anti-PD-L1 antibodies.9 The combination of immune checkpoint inhibitors and DDR inhibitors is currently being evaluated in clinical trials.61 Identifying targeted trunk changes can reduce the probability of clone branches that lack targeted changes escaping. Prophylactic combination therapy, such as alternate use of drug cycles 49 and treatments targeting different branches of a tumor, may allow the elimination of drug-resistant cells before further drug resistance mechanisms emerge. For example, the first-line use of third-generation epidermal growth factor receptor (EGFR)-tyrosine kinase inhibitors for T790M mutations has increased survival benefits to patients.62
The immune response can be used to guide evolution by selecting and infusing tumor-specific lymphocytes recognizing trunk changes to resist evolution or promote the immune response; this process works through triggering evolution by increasing the number of neoantigens. For example, EGFR-CAR-T cell therapy is a promising strategy to improve the effectiveness of NSCLC adoptive immunotherapy.63 Advanced intrahepatic cholangiocarcinoma patients with hepatitis B virus infection show a better response to anti-PD-1 treatment than noninfected patients.64
Dynamic monitoring of tumor evolution can guide clinicians to formulate appropriate treatment methods and adjust the dose and timing of each drug to minimize adverse events and maximize benefits, which will also be key to optimizing patient prognosis.
Summary and Future Directions
Malignant tumors are composed of cell populations, which are affected by genetic drift, selection, and resource competition in a dynamic microenvironment. The continuous changes made during new DNA production in the processes of tumorigenesis and development determine the trends in individual tumor development, and acquired drug resistance is inevitable. Gene diversity improves the adaptability of tumors and is the basis of tumor tolerance of drug therapy. Drug resistance and metastasis are the worst consequences of tumor evolution and the most common causes of cancer-related death.
As an evolving genetic disease, cancer can be difficult to control with chemotherapy and targeted molecular drugs after the formation of complex subclonal lesions. The mutant protein produced after the gradual accumulation of genomic changes, as neoantigens, will become the target of immunotherapy in the future. The combination of multiple drugs and the development of new drugs are current trends. If the side effects of drugs can be controlled, then combining different types of treatment with immunotherapy is a promising treatment strategy. With the progress made in high-throughput whole-exome sequencing, the evolution of tumors has been preliminarily verified by studying the genome of tumor cells. Circulating tumor DNA can be used to evaluate the process of tumor evolution by dynamic monitoring and become a powerful tool for disease monitoring in the future, but it cannot be used to construct a complete gene sequence. It is difficult to obtain tissue samples, and single sampling limits the comprehensive study of tumor evolution. Multisite, multiregion, and multistage sampling is theoretically feasible but difficult to implement. The study of tumor evolution through multiregion sequencing and liquid biopsy provides us with new clues to understand the process of tumor evolution and the mechanism of tumor drug resistance. Dynamic intervention in the evolutionary process can lead to continuous therapeutic responses. Our immune system is constantly changing, and cancer cells are mutating all the time. We need to constantly pay attention to the evolution of cancer, and more research should be invested in this area.
Footnotes
Funding:The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the Science and Technology Program of Henan Province (162102310327), the Medical Science and Technology Program of Henan Province (201702249), the Natural Science Foundation of Henan Province (182300410297, 182300410376, 162300410300), and the 51282 project Leading Talent of Henan Provincial Health Science and Technology Innovation Talents ([2016]32). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Declaration of conflicting interests:The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: All the authors participated in the discussion and development of consensus management approaches, contributed to correcting the draft manuscript and providing additional recommendations, and read and approved the final manuscript.
ORCID iD: Sheng Yu  https://orcid.org/0000-0003-4274-9805
https://orcid.org/0000-0003-4274-9805
References
- 1. Global Burden of Disease Cancer Collaboration, Fitzmaurice C, Akinyemiju TF, Al Lami FH, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 29 cancer groups, 1990 to 2016: a systematic analysis for the global burden of disease study. JAMA Oncol. 2018;4:1553-1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Siegel RL, Miller KD, Jemal A. Cancer statistics. CA. 2017;67:7-30. [DOI] [PubMed] [Google Scholar]
- 3. Nowell PC. The clonal evolution of tumor cell populations. Science (New York, NY). 1976;194:23-28. [DOI] [PubMed] [Google Scholar]
- 4. Fittall MW, Van Loo P. Translating insights into tumor evolution to clinical practice: promises and challenges. Genome Med. 2019;11:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baslan T, Hicks J. Unravelling biology and shifting paradigms in cancer with single-cell sequencing. Nat Rev Cancer. 2017;17:557-569. [DOI] [PubMed] [Google Scholar]
- 6. Hardiman KM, Ulintz PJ, Kuick RD, et al. Intra-tumor genetic heterogeneity in rectal cancer. Lab Invest. 2016;96:4-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Løvf M, Zhao S, Axcrona U, et al. Multifocal primary prostate cancer exhibits high degree of genomic heterogeneity. Eur Urol. 2019;75:498-505. [DOI] [PubMed] [Google Scholar]
- 8. Suda K, Kim J, Murakami I, et al. Innate genetic evolution of lung cancers and spatial heterogeneity: analysis of treatment-naïve lesions. J Thorac Oncol. 2018;13:1496-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pilié PG, Tang C, Mills GB, Yap TA. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol. 2019;16:81-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ben-David U, Beroukhim R, Golub TR. Genomic evolution of cancer models: perils and opportunities. Nat Rev Cancer. 2019;19:97-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Topalian SL, Taube JM, Pardoll DM. Neoadjuvant checkpoint blockade for cancer immunotherapy. Science (New York, NY). 2020;367:eaax0182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rebecca C. Cancer drug resistance needs urgent attention, says research chief. BMJ. 2019;365:l1934. [DOI] [PubMed] [Google Scholar]
- 13. Paczulla AM, Rothfelder K, Raffel S, et al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature. 2019;572:254-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Burr ML, Sparbier CE, Chan KL, et al. An evolutionarily conserved function of polycomb silences the MHC class I antigen presentation pathway and enables immune evasion in cancer. Cancer Cell. 2019;36:385-401.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168:707-723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Coley WB. The diagnosis and treatment of bone sarcoma. Glasgow Med J. 1936;126:128-164. [PMC free article] [PubMed] [Google Scholar]
- 17. Andtbacka RH, Kaufman HL, Collichio F, et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33:2780-2788. [DOI] [PubMed] [Google Scholar]
- 18. Kantoff PW, Higano CS, Shore ND, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. New Eng J Med. 2010;363:411-422. [DOI] [PubMed] [Google Scholar]
- 19. Drake CG. Prostate cancer as a model for tumour immunotherapy. Nat Rev Immunol. 2010;10:580-593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Higano CS, Schellhammer PF, Small EJ, et al. Integrated data from 2 randomized, double-blind, placebo-controlled, phase 3 trials of active cellular immunotherapy with sipuleucel-T in advanced prostate cancer. Cancer. 2009;115:3670-3679. [DOI] [PubMed] [Google Scholar]
- 21. Mulé JJ, Shu S, Schwarz SL, Rosenberg SA. Adoptive immunotherapy of established pulmonary metastases with LAK cells and recombinant interleukin-2. Science (New York, NY). 1984;225:1487-1489. [DOI] [PubMed] [Google Scholar]
- 22. Fujita K, Ikarashi H, Takakuwa K, et al. Prolonged disease-free period in patients with advanced epithelial ovarian cancer after adoptive transfer of tumor-infiltrating lymphocytes. Clin Cancer Res. 1995;1:501-507. [PubMed] [Google Scholar]
- 23. Kimura H, Yamaguchi Y. A phase III randomized study of interleukin-2 lymphokine-activated killer cell immunotherapy combined with chemotherapy or radiotherapy after curative or noncurative resection of primary lung carcinoma. Cancer. 1997;80:42-49. [PubMed] [Google Scholar]
- 24. Sharma P, Wagner K, Wolchok JD, Allison JP. Novel cancer immunotherapy agents with survival benefit: recent successes and next steps. Nat Rev Cancer. 2011;11:805-812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Grupp SA, Kalos M, Barrett D, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. New Eng J Med. 2013;368:1509-1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Breakthrough of the year 2013. How we did in 2013 and. Science (New York, NY). 2013;342:1442. [DOI] [PubMed] [Google Scholar]
- 27. Antonia SJ, López-Martin JA, Bendell J, et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, phase 1/2 trial. Lancet Oncol. 2016;17:883-895. [DOI] [PubMed] [Google Scholar]
- 28. Langer CJ, Gadgeel SM, Borghaei H, et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol. 2016;17:1497-1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Socinski MA, Jotte RM, Cappuzzo F, et al. Atezolizumab for first-line treatment of metastatic nonsquamous NSCLC. N Eng J Med. 2018;378:2288-2301. [DOI] [PubMed] [Google Scholar]
- 30. West H, McCleod M, Hussein M, et al. Atezolizumab in combination with carboplatin plus nab-paclitaxel chemotherapy compared with chemotherapy alone as first-line treatment for metastatic non-squamous non-small-cell lung cancer (IMpower130): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019;20:924-937. [DOI] [PubMed] [Google Scholar]
- 31. Zhang Y, Zhou H, Zhang L. Which is the optimal immunotherapy for advanced squamous non-small-cell lung cancer in combination with chemotherapy: anti-PD-1 or anti-PD-L1? J Immun Cancer. 2018;6:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang X, Schoenhals JE, Li A, et al. Suppression of type I IFN signaling in tumors mediates resistance to anti-PD-1 treatment that can be overcome by radiotherapy. Cancer Research. 2017;77:839-850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shin DS, Zaretsky JM, Escuin-Ordinas H, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017;7:188-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zaretsky JM, Garcia-Diaz A, Shin DS, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Eng J Med. 2016;375:819-829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McGranahan N, Rosenthal R, Hiley CT, et al. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell. 2017;171:1259.e11-1271.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gettinger S, Choi J, Hastings K, et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov. 2017;7:1420-1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rosenthal R, Cadieux EL, Salgado R, et al. Neoantigen-directed immune escape in lung cancer evolution. Nature. 2019;567:479-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Raj D, Yang MH, Rodgers D, et al. Switchable CAR-T cells mediate remission in metastatic pancreatic ductal adenocarcinoma. Gut. 2019;68:1052-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kiesgen S, Chicaybam L, Chintala NK, Adusumilli PS. Chimeric antigen receptor (CAR) T-cell therapy for thoracic malignancies. J Thorac Oncol. 2018;13:16-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Amirouchene-Angelozzi N, Swanton C, Bardelli A. Tumor evolution as a therapeutic target [published online ahead of print July 20, 2017]. Cancer Discov. doi: 10.1158/2159-8290.CD-17-0343. [DOI] [PubMed] [Google Scholar]
- 41. Mehta A, Kim YJ, Robert L, et al. Immunotherapy resistance by inflammation-induced dedifferentiation. Cancer Discov. 2018;8:935-943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Anagnostou V, Smith KN, Forde PM, et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discov. 2017;7:264-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sade-Feldman M, Jiao YJ, Chen JH, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun. 2017;8:1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. HLA loss facilitates immune escape. Cancer Discov. 2018;8:8. [DOI] [PubMed] [Google Scholar]
- 45. Jacquelot N, Yamazaki T, Roberti MP, et al. Sustained Type I interferon signaling as a mechanism of resistance to PD-1 blockade. Cell Res. 2019;29:846-861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gonzalez-Gugel E, Saxena M, Bhardwaj N. Modulation of innate immunity in the tumor microenvironment. Cancer Immunol Immunother. 2016;65:1261-1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Koyama S, Akbay EA, Li YY, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Munzone E, Colleoni M. Clinical overview of metronomic chemotherapy in breast cancer. Nat Rev Clin Oncol. 2015;12:631-644. [DOI] [PubMed] [Google Scholar]
- 49. Romano E, Pradervand S, Paillusson A, et al. Identification of multiple mechanisms of resistance to vemurafenib in a patient with BRAFV600E-mutated cutaneous melanoma successfully rechallenged after progression. Clin Cancer Res. 2013;19:5749-5757. [DOI] [PubMed] [Google Scholar]
- 50. Willyard C. Cancer therapy: an evolved approach. Nature. 2016;532:166-168. [DOI] [PubMed] [Google Scholar]
- 51. Turajlic S, Xu H, Litchfield K, et al. Tracking cancer evolution reveals constrained routes to metastases: TRACERx renal. Cell. 2018;173:581-94e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jinek M, Chylinski K, Fonfara I, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science (New York, NY). 2012;337:816-821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nakamura T, Gehrke AR, Lemberg J, Szymaszek J, Shubin NH. Digits and fin rays share common developmental histories. Nature. 2016;537:225-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cyranoski D. Chinese scientists to pioneer first human CRISPR trial. Nature. 2016;535:476-477. [DOI] [PubMed] [Google Scholar]
- 55. Stadtmauer EA, Fraietta JA, Davis MM, et al. CRISPR-engineered T cells in patients with refractory cancer. Science (New York, NY). 2020;367:eaba7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Angelova M, Mlecnik B, Vasaturo A, et al. Evolution of metastases in space and time under immune selection. Cell. 2018;175:751-65e16. [DOI] [PubMed] [Google Scholar]
- 57. Mittal D, Vijayan D, Smyth MJ. Overcoming acquired PD-1/PD-L1 resistance with CD38 blockade. Cancer Discov. 2018;8:1066-1068. [DOI] [PubMed] [Google Scholar]
- 58. Jamal-Hanjani M, Hackshaw M, Ngai A, et al. Tracking genomic cancer evolution for precision medicine: the lung TRACERx study. PLoS Biol. 2014;12:e1001906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chemi F, Rothwell DG, McGranahan N, et al. Pulmonary venous circulating tumor cell dissemination before tumor resection and disease relapse. Nat Med. 2019;25:1534-1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Anagnostou V, Forde PM, White JR, et al. Dynamics of tumor and immune responses during immune checkpoint blockade in non-small cell lung cancer. Cancer Res. 2019;79:1214-1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Thomas A, Vilimas R, Trindade C, et al. Durvalumab in combination with olaparib in patients with relapsed SCLC: results from a phase II study. J Thoracic Oncol. 2019;14:1447-1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Aguiar PN, Haaland B, Park W, et al. Cost-effectiveness of osimertinib in the first-line treatment of patients with EGFR-mutated advanced non-small cell lung cancer. JAMA Oncol. 2018;4:1080-1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li H, Huang Y, Jiang DQ, et al. Antitumor activity of EGFR-specific CAR T cells against non-small-cell lung cancer cells in vitro and in mice. Cell Death Disease. 2018;9:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lu JC, Zeng HY, Sun QM, et al. Distinct PD-L1/PD1 profiles and clinical implications in intrahepatic cholangiocarcinoma patients with different risk factors. Theranostics. 2019;9:4678-4687. [DOI] [PMC free article] [PubMed] [Google Scholar]