

Abstract
Current immunotherapies for cancer seek to modulate the balance among different immune cell populations, thereby promoting antitumor immune responses. However, because these are systemic therapies, they often cause treatment-limiting autoimmune adverse effects. It would be ideal to manipulate the balance between suppressor and effector cells within the tumor without disturbing homeostasis elsewhere in the body. CD4+CD25+Foxp3+ regulatory T cells (Tregs) are well-known immunosuppressor cells that play a key role in tumor immunoevasion and have been the target of systemic immunotherapies. We used CD25-targeted near-infrared photoimmunotherapy (NIR-PIT) to selectively deplete Tregs, thus activating CD8 T and natural killer cells and restoring local antitumor immunity. This not only resulted in regression of the treated tumor but also induced responses in separate untreated tumors of the same cell line derivation. We conclude that CD25-targeted NIR-PIT causes spatially selective depletion of Tregs, thereby providing an alternative approach to cancer immunotherapy.
INTRODUCTION
Cancer immunotherapy, which includes the use of immunomodulatory antibodies, cancer vaccines, and cell-based therapies, has become an important strategy in the control of cancer (1–4). These therapies are minimally invasive, but they still have room for improvement (5, 6). Recently, improved understanding of the interactions between immune cells and cancer cells resulted in the development of immune checkpoint inhibitors, which have shown remarkable therapeutic efficacy (7–9). A variety of strategies have been developed to inhibit immunosuppressive regulatory mechanisms, thus enhancing anticancer immune responses; however, systemic blockade of these same functions in normal organs has produced life-threatening and, therefore, dose-limiting autoimmune side effects (10–13). A method that selectively and locally suppresses regulatory T cells (Tregs) within tumors, but not systemically, could avoid systemic adverse effects.
Near-infrared photoimmunotherapy (NIR-PIT) is a method of treating cancers that uses activation of an antibody-photoabsorber conjugate activated by NIR light to kill cells (14). The antibody binds to the appropriate cell surface antigen, and the photoactivatable silica-phthalocyanine dye (IRDye 700DX) induces lethal damage to the cell membrane after NIR light exposure. The NIR light exposure (690 nm) induces highly selective, necrotic cancer cell death within minutes, without damaging adjoining cells. A phase 1 human study of NIR-PIT using cetuximab-IR700 to target tumor epidermal growth factor receptor is currently under way for the treatment of inoperable head and neck cancers (NCT02422979). NIR-PIT appears to specifically kill target cells while leaving adjacent normal cells unharmed (15–17). Although NIR-PIT has been primarily tested with antibodies directed at cancer antigens, it can also be used to kill cells of any type for which there is an appropriate antibody (18–20). Therefore, NIR-PIT could also be used to deplete a selected subpopulation of immune cells within the tumor tissue.
Within the cancer tissue, T cells and natural killer (NK) cells that recognize cancer cells are often present in large numbers, but their cytotoxic function is suppressed by nearby immunosuppressor cells, such as Tregs (21, 22). Thus, controlling tumor-infiltrating CD4+CD25+Foxp3+ Tregs has been considered an essential step for enhancing anticancer immune reactions (23–25). A variety of approaches have been used to deplete or ablate Tregs, but they have met with limited success (22, 26–28). For instance, depletion of Tregs using systemic anti-CD25 antibodies also depletes CD25-expressing antitumor effector cells, reducing the therapeutic effectiveness of Treg depletion (29). In research settings, elimination of Tregs has been achieved with intratumoral injection of anti-CD4 antibodies (30). Alternatively, genetically engineered animal models have been designed to permit transient conditional ablation of Tregs (31–33). However, this method is impossible to translate clinically. Therefore, a practical technique that enables local, selective depletion of tumor-infiltrating Tregs without depleting other cell types and/or systemic Tregs is highly desirable. Here, we used CD25-targeted NIR-PIT to selectively deplete CD4+CD25+Foxp3+ Tregs within the tumor microenvironment to induce activation of antitumor effector cells in a syngeneic tumor model.
RESULTS
NIR-PIT with Fc-deficient anti-CD25-F(ab’)2-IR700 induces necrotic cell death of CD25-expressing cells in vitro
To avoid Fc-mediated antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) in vivo, F(ab’)2 fragments were generated from an anti-CD25 antibody [anti-CD25-F(ab’)2] and a control immunoglobulin G (IgG) antibody [control-F(ab’)2], Both F(ab’)2 fragments were purified using high-performance liquid chromatography (HPLC) with more than 90% purity (fig. S1A) and conjugated with the IR700 dye [anti-CD25-F(ab’)2–IR700 and control-F(ab’)2–IR700, respectively] (fig. S1, B and C). The anti-CD25-F(ab’)2–IR700 demonstrated specific binding to CD25 expressed on mouse T lymphocyte HT-2 clone A5E (HT-2-A5E) cells (fig. S1D). These results indicated that the bioactivity and specificity of anti-CD25-F(ab’)2 were maintained during digestion, purification, and conjugation. We confirmed the absence of a systemic ADCC or CDC effect by administering anti-CD25-F(ab’)2 (100 μg) to mice and analyzing the splenic CD4+CD25+Foxp3+ Tregs 1 day later. Although administration of the intact anti-CD25-IgG caused a decrease in the frequency of Tregs among CD4 T cells, administration of Fc-deficient anti-CD25-F(ab’)2 did not significantly deplete Tregs (Fig. 1A). In vitro NIR-PIT with anti-CD25-F(ab’)2–IR700 against HT-2-A5E cells induced cellular swelling and bleb formation (Fig. 1B), resulting in necrotic cell death in a light dose-dependent manner (Fig. 1C and fig. S2). We observed no significant cytotoxicity associated with NIR light alone, anti-CD25-F(ab’)2–IR700 incubation alone, or NIR light with control-F(ab’)2–IR700 incubation (Fig. 1C).
Fig. 1. Anti-CD25-F(ab’)2 lacks Treg depletion effect, but NIR-PIT with anti-CD25-F(ab’)2–IR700 kills target cells.
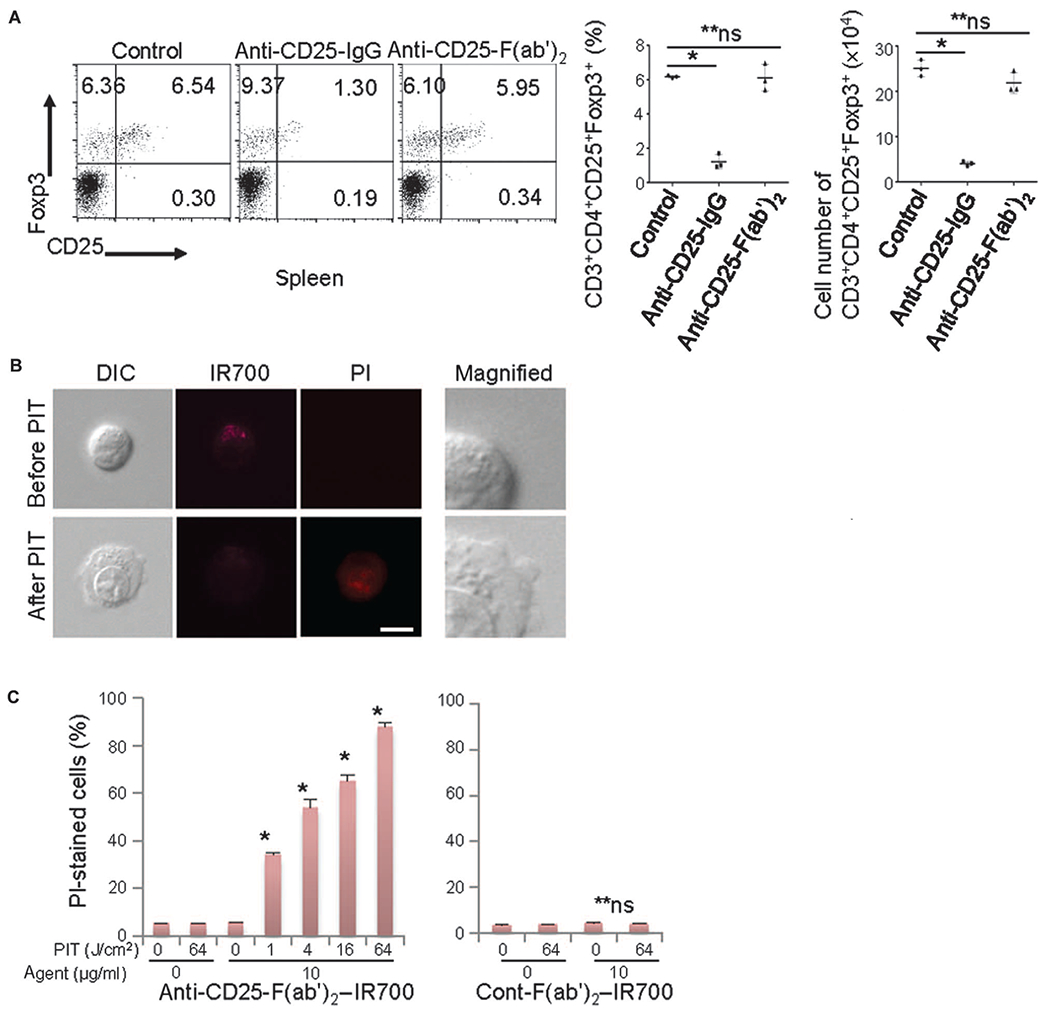
(A) Intravenously injected anti-CD25-IgG (100 μg) systemically depleted CD4+CD25+Foxp3+ Tregs, but anti-CD25-F(ab’)2 (100 μg) did not significantly deplete these cells within the CD4 T cell population 1 day after injection (n = 3) [*P < 0.0001, one-way analysis of variance (ANOVA) with Dunnett’s test; **P = not significant (ns)]. (B) HT-2-A5E cells (mouse T lymphocytes) incubated with anti-CD25-F(ab’)2–IR700 for 6 hours were examined under a microscope before and 0.5 hour after NIR light irradiation (4 J/cm2). NIR light irradiation induced cellular swelling, bleb formation, and necrosis of the cells, as indicated with propidium iodide (PI) staining (scale bar, 10 μm; right magnified view, ×4). DIC, differential interference contrast. (C) Cell necrosis induced by NIR-PIT increased in a NIR light dose–dependent manner, as determined by flow cytometry analysis with PI staining (left graph, n = 3; *P < 0.0001 versus 0 J/cm2, unpaired t test). No significant cell killing was detected when a control-F(ab’)2–IR700 was used (right graph, n = 3; **P = ns versus 0 J/cm2, unpaired t test).
CD4+CD25+Foxp3+ Tregs are abundant in tumors
Tregs are abundant in various cancers (22). Flow cytometry analysis of the CD25+Foxp3+ Treg fraction in CD4 T cells indicated an increase in this cell population within LL/2-luc (Lewis lung cancer) and MC38-luc (colon cancer) tumors compared to the normal spleen (Fig. 2A). CD8 T and NK cells were also prevalent within tumors, and they were CD25-negative before activation (fig. S3). The anti-CD25-F(ab’)2–IR700 did not bind to the tumor cells (fig. S4). These data suggest that CD4+CD25+Foxp3+ Tregs are common within tumors and thus are a target of anti-CD25–IR700 NIR-PIT.
Fig. 2. In vivo local CD25-targeted NIR-PIT induces regression of treated LL/2-luc tumors.
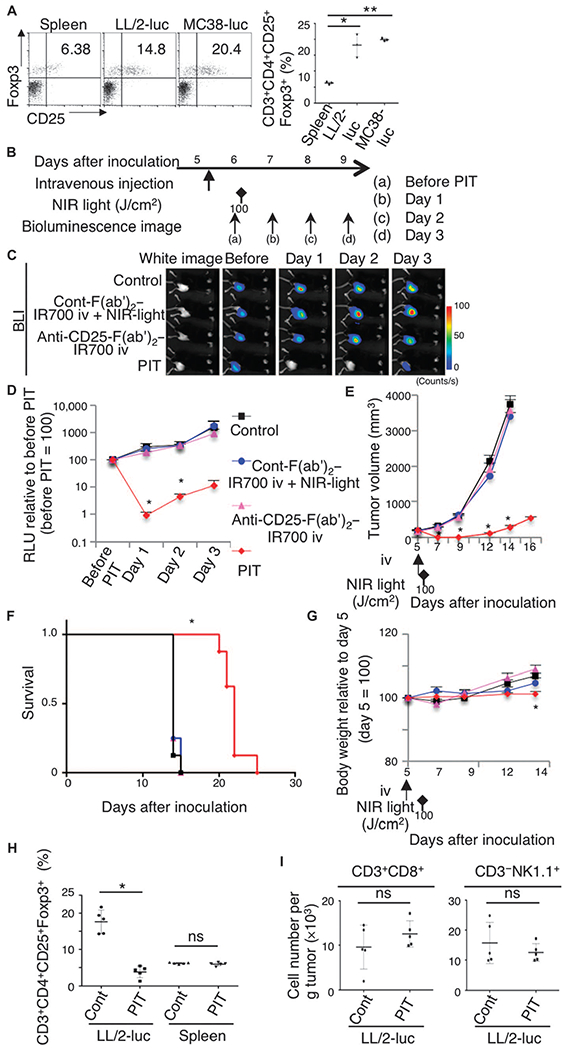
(A) An increased CD4+CD25+Foxp3+ Treg population among CD4 T cells was observed in tumors (LL/2-luc or MC38-luc) compared to that in the spleen (n = 3) (*P < 0.001, **P < 0.001 versus control, one-way ANOVA with Dunnett’s test). (B) Regimen used for local NIR-PIT. (C) In vivo BLI is shown for tumor-bearing mice that were untreated, received control-F(ab’)2–IR700 followed by NIR-PIT, received CD25-F(ab’)2–IR700 alone, or treated with local CD25-targeted NIR-PIT. Before NIR-PIT, tumors sizes were equivalent, exhibiting similar bioluminescence, but only the CD25-targeted NIR-PIT group showed a decrease in BLI. iv, intravenously. (D) Quantitative relative light units (RLU) (before PIT is set to 100) showed a significant RLU decrease in the experimental tumors [n = 6 mice in each group; *P = 0.0217 (day 1) and 0.0243 (day 2), PIT versus control, Tukey’s test with ANOVA]. (E) Local CD25-targeted NIR-PIT reduced tumor volume (n = 8 mice in each group; *P < 0.0001, PIT versus others, Tukey’s test with ANOVA). Treatment schedule indicated below the graph corresponds to that in (B). (F) Local CD25-targeted NIR-PIT prolonged the survival of the mice (n = 8 mice in each group; *P < 0.0001, PIT versus control, log-rank test and Wilcoxon test). (G) The body weight of mice not receiving CD25-targeted NIR-PIT gradually increased because of tumor growth, in contrast to the PIT group showing negligible body weight increase (n = 8 mice in each group) (*P = 0.0128, PIT versus control at day 14). (H) Local CD25-targeted NIR-PIT resulted in a depletion of CD4+CD25+Foxp3+ Tregs in tumors, but not in the spleen (n = 5) (*P < 0.008, Mann-Whitney test; P = ns, Mann-Whitney test). (I) Local CD25-targeted NIR-PIT did not significantly affect the number of CD8 T cells (CD3+CD8+) or NK cells (CD3−NK1.1+) (n = 5; P = ns, Mann-Whitney test).
In vivo NIR-PIT targeting CD4+CD25+Foxp3+ Tregs induces regression of treated tumors
We evaluated the effect of in vivo local NIR-PIT with anti-CD25–IR700 (CD25-targeted NIR-PIT) against intratumoral Tregs in LL/2-luc flank tumors (Fig. 2B). A NIR light dose escalation study indicated that more than 85% depletion of CD25+Foxp3+ Tregs in CD4 T cells occurred with light power (100 J/cm2) within 30 min (fig. S5). With this light dose, a decrease in CD4+Foxp3+ cell number in the tumor was observed (fig. S6), indicating that this observation was not a mere down-regulation of CD25 expression on Tregs, but actual depletion of the cells. Therefore, this light dose was chosen for in vivo treatment. This light dose is considered nonthermal and does not cause harmful local heating upon exposure to the skin or tumor (14–19). The experimental group of mice with LL/2-luc tumors received anti-CD25-F(ab’)2–IR700 injection, followed by NIR light exposure (“PIT” group). This group showed reduction of tumor luciferase activity as indicated by bioluminescence imaging (BLI) at day 1, compared to the three control groups: nontreated mice, mice that received control-F(ab’)2–IR700 with NIR light, and mice that received only anti-CD25-F(ab’)2–IR700 but no light (Fig. 2, C and D). Quantitative analysis of luciferase activity showed significant decreases in relative light units in the PIT group for 2 days after the treatment [*P = 0.0217 (day 1) and 0.0243 (day 2), PIT versus control, Tukey’s test with ANOVA], in contrast to those of the other groups, which gradually increased as the tumors grew (Fig. 2D). Consistent with the BLI data, the growth of tumors was also suppressed in the PIT group (Fig. 2E), significantly prolonging the survival of mice compared to the control groups (*P < 0.0001) (Fig. 2F). The body weight of mice not receiving CD25-targeted NIR-PIT gradually increased because of tumor growth, whereas the PIT group showed negligible body weight increase because of the regression of tumors by the treatment (Fig. 2G; *P = 0.0128, PIT versus control at day 14). Analysis of tumor-infiltrating lymphocytes and splenic lymphocytes at 0.5 hour after the treatment confirmed that the depletion of CD4+CD25+Foxp3+ Tregs was limited to the tumor and that it did not significantly affect CD8 T and NK cells (Fig. 2, H and I). Similar findings were observed when MC38-luc flank tumors were treated with local CD25-targeted NIR-PIT (fig. S7). Furthermore, we also observed increased tumor-infiltrating Tregs in the TRAMP-C2-luc prostate cancer model compared to the normal spleen, and a CD25-targeted NIR-PIT successfully induced antitumor effects in this model as well (fig. S8). Together, these data suggest that NIR-PIT targeting CD4+CD25+Foxp3+ Tregs locally and specifically depletes these cells, followed by tumor reduction and prolonged survival in multiple cancer types.
The depletion of intratumoral Tregs lasted for about 4 days, after which there was a gradual repopulation of Tregs, reaching pretreatment numbers at about 6 days after the therapy (fig. S9A). Gradual tumor regrowth was also observed (Fig. 2E). Repeat treatment with CD25-targeted NIR-PIT was capable of inducing prolonged tumor suppression and survival (fig. S9, B to F).
In vivo local CD25-targeted NIR-PIT induces rapid activation of tumor-infiltrating CD8 T and NK cells and activation of antigen-presenting cells
To elucidate how the specific depletion of tumor-infiltrating Tregs with local CD25-targeted NIR-PIT stimulated tumor regression, we examined whether intratumoral CD8 T and NK cells became activated after NIR-PIT. As early as 1.5 hours after the treatment, CD8 T and NK cells began to produce interferon-γ (IFN-γ) and interleukin-2 (IL-2) and expose CD107a, indicating activation and killing of tumor cells (Fig. 3A). At 1 day after the treatment, up-regulation of CD69 and CD25 and production of IL-2 were observed on these effector cells (Fig. 3B). However, the local CD25-targeted NIR-PIT did not deplete effector cells, which were CD25-negative before NIR-PIT and only began to express CD25 after the NIR treatment (Figs. 2I and 3B). Thus, Treg depletion by NIR-PIT resulted in rapid activation of tumor-infiltrating CD8 T and NK cells accompanied by tumor regression (Fig. 2E). Comparison of CD25 expression before and after local CD25-targeted NIR-PIT in a repeated NIR-PIT regimen showed up-regulation of CD25 after each PIT treatment (fig. S10, A to C). CD25 was already down-regulated to the pretreatment levels by the time the second PIT was performed after the 4-day interval from the first PIT.
Fig. 3. In vivo local CD25-targeted NIR-PIT induces rapid activation and cytotoxicity of intratumoral CD8 T and NK cells.
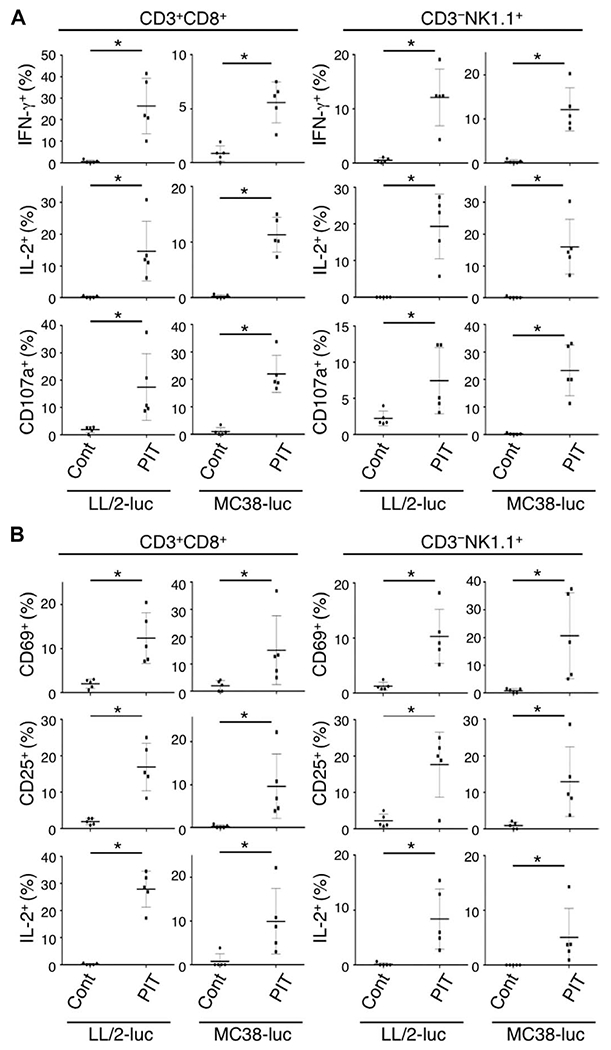
(A) Cytotoxic action of CD8 T and NK cells infiltrating LL/2-luc or MC38-luc tumors was examined by flow cytometry with or without local CD25-targeted NIR-PIT. CD8 T and NK cells collected 1.5 hours after PIT were producing IFN-γ and IL-2 and had CD107a exposed on the cell surface, whereas the cells from nontreated tumors did not (n = 5; *P < 0.008, Mann-Whitney test). (B) One day after local CD25-targeted NIR-PIT, up-regulation of activation markers, CD69 and CD25, and continued production of IL-2 in both CD8 T and NK cells were observed (n = 5; *P < 0.008, Mann-Whitney test).
We also examined whether antigen-presenting cells (APCs) within the tumor were activated upon PIT of Tregs. Dendritic cells found in the tumor expressed relatively high amounts of the major histocompatibility complex I molecule; however, local CD25-targeted NIR-PIT induced its further up-regulation (fig. S11A). This up-regulation was accompanied by up-regulation of costimulatory molecules, such as CD86 and CD40 (fig. S11A). This activation may have been caused by their exposure to the components of dead Tregs and also to the components of tumor cells killed by CD8 T and NK cells. Other APCs such as B cells, monocytes, and macrophages also showed up-regulation of CD69, indicating their activation after local CD25-targeted NIR-PIT (fig. S11, B to D). These findings suggest that after the Treg-targeting PIT, tumor cells were killed by the effector cells, which likely had further induced APC-mediated vaccine effects via various antigens derived from killed tumor cells. An increase of granulocytes inside the treated tumor was also detected after the therapy (fig. S12).
Local CD25-targeted NIR-PIT induces a systemic and intratumoral cytokine storm
We investigated changes of both serum and intratumoral cytokine and chemokine concentrations induced by local CD25-targeted NIR-PIT. First, we confirmed that the effect of NIR light irradiation on the tumor itself was negligible (fig. S13, A and B). At 1.5 hours after the therapy, a broad range of cytokines and chemokines increased in the serum and tumors (fig. S14, A and B). The cytokines and chemokines that were elevated after NIR-PIT broadly overlapped with those reported in clinical “cytokine storm” (34, 35). One day after the therapy, the concentrations of cytokines and chemokines abruptly decreased except for granulocyte colony-stimulating factor (G-CSF) (fig. S14, C and D). The changes in concentration of IFN-γ, IL-6, and G-CSF in serum and tumor are presented separately (fig. S14, E to J). These data indicate that a local CD25-targeted NIR-PIT caused a rapid but transient release of cytokines and chemokines into the serum, which abated by day 1 after treatment. Lymphocytes in the lungs of mice undergoing local CD25-targeted NIR-PIT showed no sign of IFN-γ production at day 1 after treatment (fig. S15), and IFN-γ concentration was below the detectable level in both the lungs and intestines at 1 day after the treatment.
The therapeutic effects of CD25-targeted NIR-PIT extend to distant nontreated tumors in a tumor-specific manner
We hypothesized that the rapid antitumor immune activation and regression of the PIT-treated tumor might enable activated cytotoxic effector cells to attack other tumor locations distant from the NIR-PIT–treated lesion. Local CD25-targeted NIR-PIT was performed only on the right-sided tumors in mice bearing bilateral LL/2-luc flank tumors, whereas the left-sided tumors were shielded from light (Fig. 4, A and B). This shield was confirmed with a fluorescence image before NIR-PIT, demonstrating that IR700 fluorescence was present in both tumors, but the fluorescence in the PIT-treated side decreased immediately after the irradiation because of bleaching (fig. S16). On the untreated left side, IR700 fluorescence was maintained after contralateral NIR-PIT. However, tumor bioluminescence significantly decreased both in the right-sided (PIT-treated side) tumor and in the left-sided tumor that had received no NIR light [Fig. 4, C and D; *P < 0.001, **P < 0.01, ***P = 0.0197 (PIT:R) and 0.0142 (PIT:L), PIT versus cont-F(ab’)2–IR700 iv:R, Tukey’s test with ANOVA]. This untreated tumor followed a similar slowing growth curve compared to the PIT-treated tumor (Fig. 4, C to E). Survival of the mice was significantly prolonged in the local CD25-targeted NIR-PIT group compared with control groups (*P < 0.0001) (Fig. 4F). Body weight measurement showed that local CD25-targeted NIR-PIT–treated mice gained weight 1 to 3 days after the treatment (Fig. 4G), which was likely due to edema in both flanks (Fig. 4H), but this resolved at about day 4 (Fig. 4G). This edema was more severe than that observed in mice bearing one tumor and subjected to local PIT. The tumor-infiltrating CD4+CD25+Foxp3+ Tregs decreased only in the NIR-PIT–treated tumor (Fig. 4I). In an animal model mimicking multiple metastases, the local CD25-targeted NIR-PIT directed at one tumor induced antitumor effects in distant nontreated tumors of the same cell type. Edema was also noted at these sites (Fig. 4J and fig. S17). Moreover, the growth of tumor inoculated on the contralateral side of PIT-treated tumor 1 day after the NIR-PIT was inhibited compared to that in animals treated with control-F(ab’)2–IR700 and NIR light irradiation (fig. S18, A and B). When the mouse was inoculated with a tumor of a different cell type (MC38-luc) in the left flank and NIR-PIT was directed at the right-sided LL/2-luc tumor, minimal changes in the MC38-luc tumor were observed (Fig. 4K). These data suggest that local CD25-targeted NIR-PIT has cell type-specific antitumor effects on distant tumors.
Fig. 4. The therapeutic effects of local CD25-targeted NIR-PIT extend to distant nonirradiated tumors.
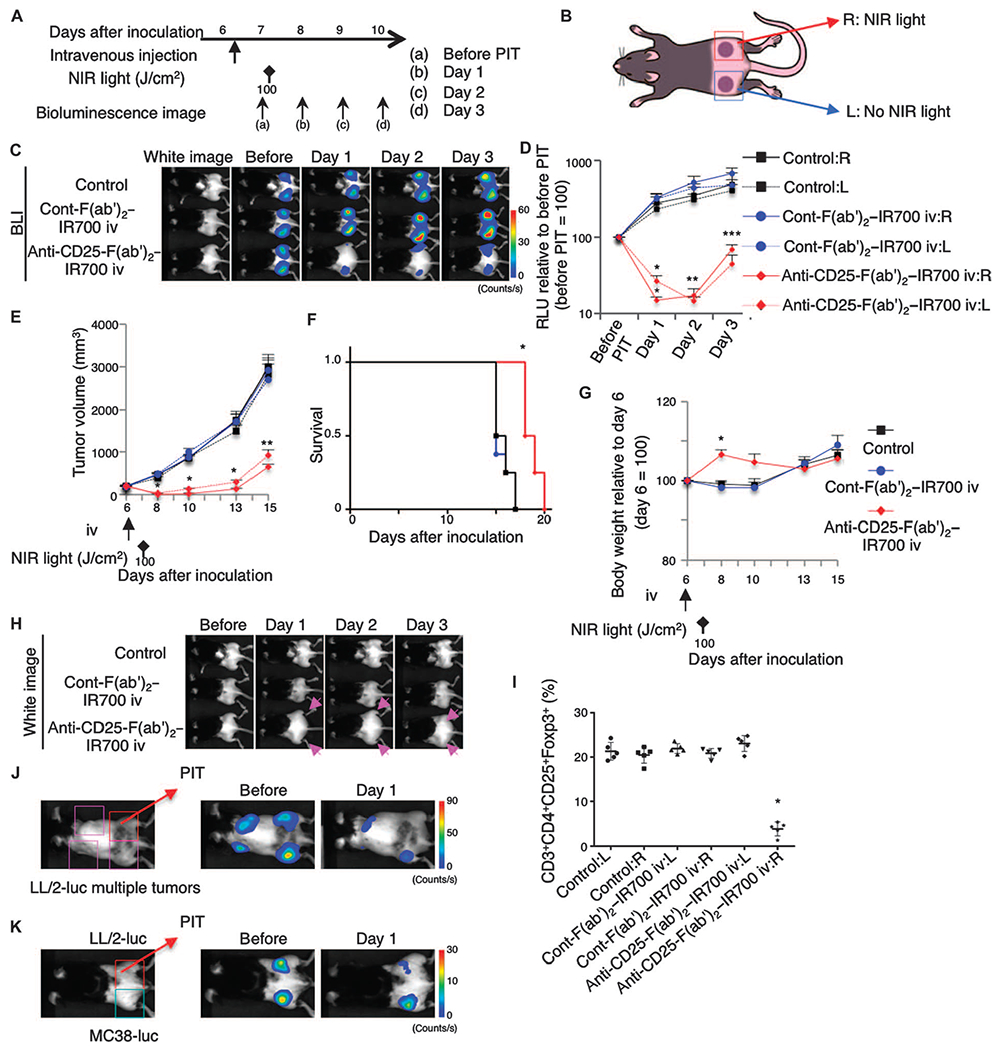
(A) Regimen of NIR-PIT. (B) Mice with bilateral flank tumors were either not injected (control) or injected with control-F(ab’)2–IR700 or anti-CD25-F(ab’)2–IR700, followed by NIR light irradiation of only the right tumor. (C) In vivo BLI showed changes in bioluminescence signals in the tumors in response to local CD25-targeted NIR-PIT only. Before NIR-PIT, tumors were about the same size and exhibited similar bioluminescence. (D) Quantitative RLU showed a significant decrease in signal in NIR-PIT–treated right-side tumors and even in nonirradiated left-side tumors [n = 6 mice in each group; *P < 0.001, **P <0.01, ***P = 0.0197 (PIT:R) and 0.0142 (PIT:L), PIT versus cont-F(ab’)2–IR700 iv:R, Tukey’s test with ANOVA]. (E) Local CD25-targeted NIR-PIT reduced the size of NIR-PIT–treated right tumors and nonirradiated left tumors (n = 8 mice in each group; *P < 0.0001, **P < 0.0005, PIT versus others, Tukey’s test with ANOVA). Time of treatments is indicated below the graph. (F) Local CD25-targeted NIR-PIT prolonged the survival of the mice (n = 8 mice in each group; *P < 0.0001, PIT versus control, log-rank test and Wilcoxon test). (G) Body weight changes of tumor-bearing mice were followed. After NIR-PIT, both right and left dorsa became edematous and mice gained weight (n = 8 mice in each group; *P < 0.001, PIT versus others, Tukey’s test with ANOVA), which started to disappear by day 10. (H) Local CD25-targeted NIR-PIT of the right dorsal tumor caused edema bilaterally (arrow). (I) NIR-PIT depleted CD4+CD25+Foxp3+ Tregs within the irradiated tumor on the right dorsum, but not Tregs in the left nonirradiated tumors. Mice not injected (control) or injected with control-F(ab’)2–IR700 showed no significant difference in Treg populations between the right and left tumors (n = 5 in each group; *P < 0.0001, Tukey’s test with ANOVA). (J) Local CD25-targeted NIR-PIT of the right dorsal LL/2-luc tumor caused regression of multiple additional LL/2-luc tumors by 1 day after the treatment. (K) Local CD25-targeted NIR-PIT of the right dorsal LL/2-luc tumor had negligible antitumor effects on the left dorsal MC38-luc tumor 1 day after PIT.
CD8 T and NK cells expressing activation markers are present in the nonirradiated tumor after CD25-targeted NIR-PIT
We next examined whether the nonirradiated tumor contained activated CD8 T and NK cells after CD25-targeted NIR-PIT was performed on the contralateral tumor. One day after local CD25-targeted NIR-PIT, CD8 T and NK cells producing IFN-γ and IL-2 and expressing up-regulated activation markers CD69 and CD25 were identified within the nonirradiated tumor (Fig. 5, A to D). Analysis of the cytokine and chemokine concentrations in the nonirradiated tumors also indicated increases in G-CSF, IFN-γ, IL-2, MCP-1 (monocyte chemoattractant protein–1), MIP-2 (macrophage inflammatory protein–2), and MIP-1α (fig. S19, A to C). Collectively, the immune responses triggered by CD25-targeted NIR-PIT on the right side induced similar changes in nontreated tumors located on the opposite side.
Fig. 5. Activated CD8 T and NK cells are present in the contralateral tumor after ipsilateral CD25-targeted NIR-PIT.
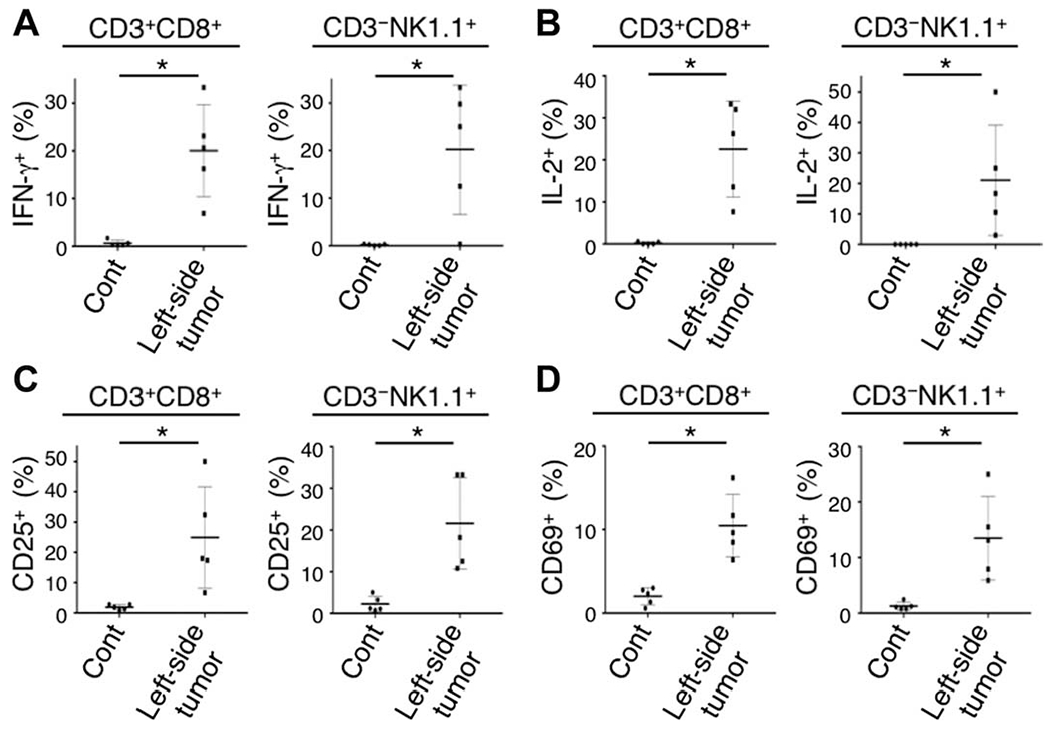
(A to D) CD8 T and NK cells collected from nonirradiated left dorsal tumors in mice receiving local CD25-targeted NIR-PIT of the right dorsal tumors were analyzed for their expression of activation markers 1 day after the treatment. CD8 T and NK cells producing IFN-γ (A) and IL-2 (B) and with up-regulated CD25 (C) and CD69 (D) expression were present in the nonirradiated left tumor after CD25-targeted NIR-PIT of the right-side tumor [n = 5; *P = 0.0021 (A: CD8 T cells), P = 0.0023 (B: CD8 T cells), P = 0.0012 (D: CD8 T cells), P < 0.008 (others), Mann-Whitney test].
Antitumor effects of CD25-targeted NIR-PIT depend partially on CD8 T and NK cells and IFN-γ production
To further elucidate the role of effector cells in the therapeutic efficacy of local CD25-targeted NIR-PIT, we depleted NK or CD8 T cells using repeated systemic administration of an anti-NK1.1 or anti-CD8 antibody or we neutralized IFN-γ using repeated injections of an anti–IFN-γ antibody (Fig. 6A). Both NK and CD8 T cell depletion and IFN-γ neutralization attenuated the efficacy of CD25-targeted NIR-PIT, as demonstrated by BLI with quantitative luciferase activity as well as by tumor growth and mouse survival data (Fig. 6, B to E). Body weight measurement showed no difference between the groups (Fig. 6F). The inhibitory effect of the IFN-γ–neutralizing antibody on CD25-targeted NIR-PIT was reproduced when IFN-γ–deficient (IFN-γ–KO) mice were used (fig. S20, A to F). These data suggested that antitumor efficacy of local CD25-targeted NIR-PIT was mediated, at least in part, by NK cells, CD8 T cells, and IFN-γ production and likely by a combination of all of these factors.
Fig. 6. Antitumor effect of local CD25-targeted NIR-PIT partially depends on each of NK cells, CD8 T cells, and IFN-γ production.
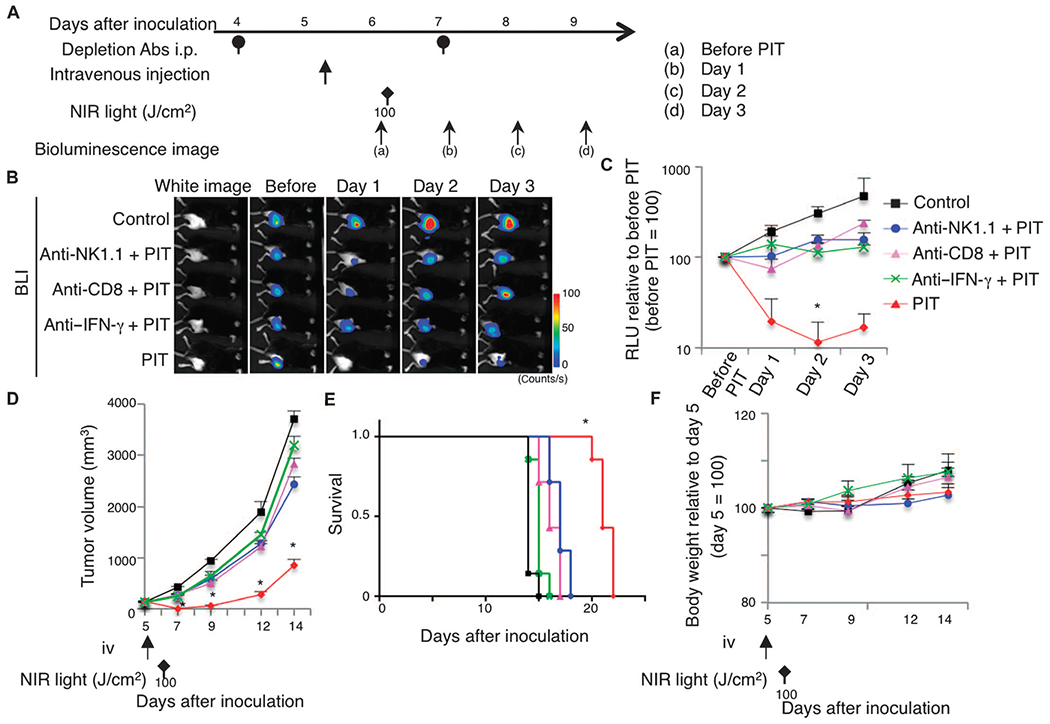
(A)Regimen of NIR-PIT against LL/2-luc tumors subjected to depletion of NK or CD8 T cells or neutralization of IFN-γ (Depletion Abs i.p. at 4 and 7 days after tumor inoculation). (B and C) In vivo BLI (B) and quantitative RLU (C) showed a significant decrease of bioluminescence signals in NIR-PIT–treated tumors; however, depletion of NK (anti-NK1.1) or CD8 T (anti-CD8) cells or neutralization of IFN-γ (anti–IFN-γ) reduced the effect (n = 5 mice in each group; *P = 0.0158, PIT versus anti-NK1.1 + PIT; *P < 0.0001, PIT versus control, Tukey’s test with ANOVA). (D and E) Similarly, the effect of local CD25-targeted NIR-PIT in suppressing tumor growth was inhibited by adding the depletion or neutralization antibodies (n = 7 mice in each group; *P < 0.0001, PIT versus others, Tukey’s test with ANOVA; treatments are indicated below the graph) (D), resulting in shorter survival of these groups of mice compared to the PIT group (n = 7 mice in each group; *P < 0.0001 versus control, log-rank test and Wilcoxon test) (E). (F) Body weights showed no significant difference among groups (n = 7 mice in each group; Tukey’s test with ANOVA).
DISCUSSION
In cancer treatment, systemic administration of drugs inhibiting immunosuppressive cells and immune checkpoint inhibitors provides therapeutic benefit for some patients but often also introduce severe side effects, such as autoimmune-like disease and acute interstitial pneumonia–acute respiratory distress syndrome (36, 37). An alternative approach is to inhibit or kill immunosuppressive cells only within the tumor but not systemically. However, methods that can selectively remove such cells from the tumor microenvironment without damaging effector cells are limited. Here, we used CD25-targeted NIR-PIT to selectively deplete CD4+CD25+Foxp3+ Tregs infiltrating the microenvironment of the tumors. This minimally invasive method used an antibody-photoabsorber conjugate that targeted CD25. The conjugate was administered systemically, but it was only activated at sites where the antibody-photoabsorber conjugate was bound to the cells and exposed to NIR light; both conditions had to be met for effective killing of CD4+CD25+Foxp3+ Tregs. Meanwhile, because this method only depleted Tregs locally, Tregs were maintained normally in other organs. The killing of Tregs in the tumor resulted in the activation of CD8 T and NK cells already present in the tumor but suppressed by the Tregs. At the time of NIR-PIT, the existing intratumoral CD8 T and NK cells did not express CD25. Thus, CD25-targeted NIR-PIT selectively killed Tregs but left tumor-infiltrating nonactivated CD8 T and NK cells unharmed. After local CD25-targeted NIR-PIT, intratumoral CD8 T and NK cells quickly became activated and exhibited cytotoxicity against the tumor. Only at this point, well after NIR-PIT had been applied, did these cells up-regulate CD25. Moreover, in cases of repeated NIR-PIT application, CD25 on intratumoral effector cells was already down-regulated before the PIT was repeated after a 4-day interval. Thus, local CD25-targeted NIR-PIT enables activation of effector cells without systemically eliminating suppressor cells. These findings stand in contrast to previously reported methods of whole-body Treg depletion with systemic administration of anti-CD25 antibodies or IL-2-toxin conjugates, which deplete both Tregs and activated effector cells because of the long half-life of such antibodies (29, 38). Consequently, methods using anti-CD25 antibodies to systemically deplete Tregs only prevented engraftment of tumors but did not interfere with the growth of established tumors and additionally caused autoimmune side effects (22). Consistent with previous studies (27, 39), the antitumor effect of our local CD25-targeted NIR-PIT was partially abrogated when CD8 T or NK cells were depleted or IFN-γ was neutralized.
An attempt to deplete the subpopulation of mature Tregs, rather than eliminating all Treg populations, to maintain self-tolerance has been made in patients with adult T cell leukemia (28). Systemic administration of an antibody targeted against a C-C chemokine receptor, CCR4, which is highly expressed on terminally differentiated Tregs prevalent in the tumors as well as on the leukemic cells, was used as a treatment. Although TH2 (T helper 2) cells and some central memory CD8 T cells also express CCR4, this strategy induced a reduction of leukemic cell number, whereas CCR4-negative naïve and precursor Tregs were spared. By locally targeting CD25-positive cells including mature and naïve Tregs within the tumor microenvironment, CD25-targeted NIR-PIT can produce more potent and prolonged elimination of immunosuppressor function in the tumor microenvironment than the CCR4-targeting strategy, without inducing systemic autoimmune side effects. Looking toward the future, local CD25-targeted NIR-PIT could also be used in combination with other immunomodulatory therapies such as immune checkpoint inhibitors or cancer type-specific therapies, or it could be modified to involve two different anti-CD25 clones (40) or CD103-targeted NIR-PIT for effector Treg depletion (41, 42), to augment the therapeutic efficacy of the method.
Unlike conventional NIR-PIT, which requires a tumor-specific antibody-photoabsorber conjugate for each tumor, CD25-targeted NIR-PIT is effective against a broad range of tumors, because Tregs expressing high amounts of CD25 are often abundant in tumors regardless of their type (22). Thus, rather than developing a whole host of target conjugates, anti-CD25–IR700 conjugates might be useful in many different types of tumors (22,26). For instance, in this study, local CD25-targeted NIR-PIT using the same conjugates was effective in three different cancer models (lung, colon, and prostate).
CD25-targeted NIR-PIT resulted in rapid tumor killing by activated CD8 T and NK cells and likely induced activation of multiple immune cell types in the tumor. We observed a cytokine storm (34,35) not only within tumors but also in the serum within a few hours after the treatment, suggesting the possibility of additional activation of immune responses outside of the treated tumor. Cytokines and chemokines that were highly increased included those with proinflammatory and antiinflammatory characteristics, some of which may have been of macrophage origin. Release of cytokines, such as IFN-γ and IL-2, resulting in full activation of effector cells, partly explains the antitumor effects in distant tumors that were not directly treated with NIR-PIT. One potential concern is the presence of a cytokine storm after treatment, which could cause severe, if temporary, side effects. Fortunately, the elevated cytokines and chemokines, including IL-6, began to decrease within 1 day of therapy, and thus, the cytokine storm was self-limited. We believe that the clinical side effects are likely to be modest and short-lived on the basis of the temporary elevation of cytokines/chemokines observed. Anticytokine treatments such as tocilizumab for IL-6 could be used if symptoms were to develop after NIR-PIT treatment in patients (43).
A phase 1 study of NIR-PIT in inoperable head and neck cancers using cetuximab-IR700 (NCT02422979) is currently under way. Antihuman CD25 antibodies such as basiliximab (chimeric mouse-human IgG1) and daclizumab (humanized IgG1) are available and are approved by the U.S. Food and Drug Administration. The existence of approved anti-CD25 antibodies considerably reduces the barriers to clinical translation of this method.
There are some limitations to this study. Tregs are not the only mechanism for cancer evasion of host antitumor immunity. In cancers where Tregs play smaller roles in escaping from the immune system, CD25-targeted NIR-PIT could be less effective than in cancers where Tregs play major roles in immunoevasion. Moreover, one treatment by CD25-targeted NIR-PIT induced only transient Treg depletion at the tumor, and the influx of Tregs from circulation could reconvert the immune balance to an immunosuppressive state, allowing the tumors to regrow. Therefore, CD25-targeted NIR-PIT may need to be repeated upon tumor regrowth, which usually occurs months or years later in patients, in contrast to the experimental mouse tumor models. Further, prolonged cancer control would provide sufficient time for anticancer memory T cells to be generated and protect patients from recurrence of the cancer.
In conclusion, we demonstrate that local CD25-targeted NIR-PIT can selectively deplete tumor-infiltrating Tregs without eliminating local effector cells or Tregs in other organs. This results in rapid activation of CD8 T and NK cells and subsequent cell-mediated cancer killing. Local CD25-targeted NIR-PIT also produces tumor type–specific, systemic antitumor effects, which alter the growth of distant tumors of the same type (Fig. 7). Thus, local CD25-targeted NIR-PIT may be a viable method for reducing immunosuppression caused by Tregs and thus augmenting effector cell–mediated tumor killing.
Fig. 7. Scheme indicates the proposed mechanism of local CD25-targeted NIR-PIT–induced immunotherapy.
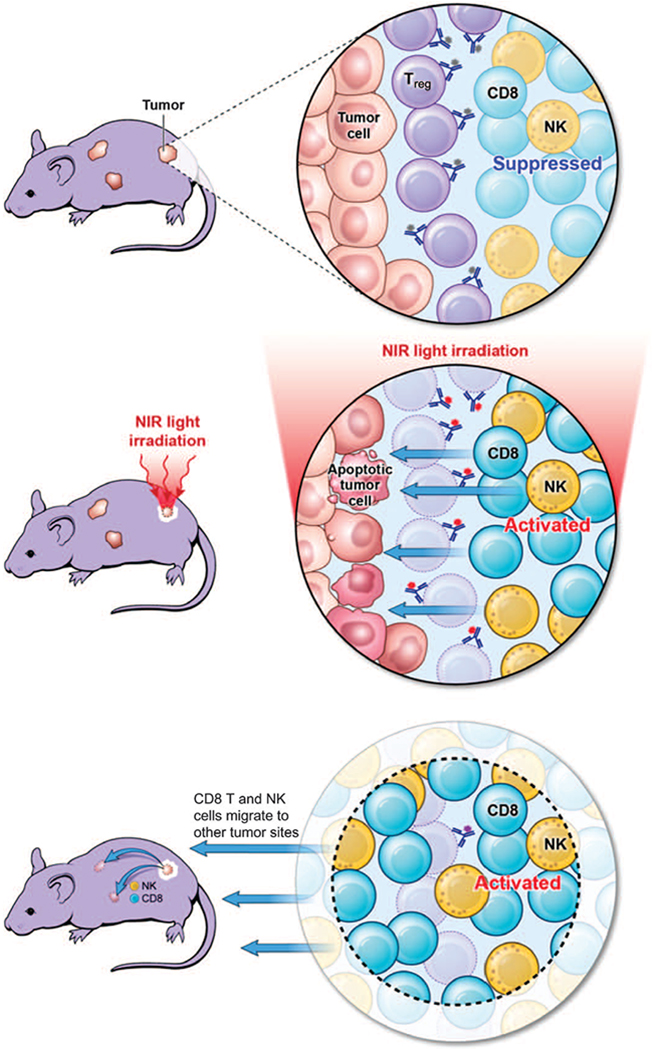
Tregs suppress CD8 T cell and NK cell activation, providing a permissive environment for tumor growth (upper panel). After Tregs are selectively depleted by NIR-PIT, CD8 T and NK cells are activated against the tumor (middle panel). Activated CD8 T and NK cells can also leave the treated tumor to attack distant tumors, accompanied by released cytokines and chemokines (lower panel).
MATERIALS AND METHODS
Study design
Our primary research objective was to establish a method of local immunomodulation for cancer treatment. We focused on Treg depletion–mediated cancer therapy with anti-CD25 antibody. The overall study design was a series of controlled laboratory experiments as indicated in the sections below. Animals were assigned to each group such that luciferase activity was as similar as possible for each group. At least five mice were used in each group. All in vivo procedures were conducted in compliance with the Guide for the Care and Use of Laboratory Animal Resources (1996) and U.S. National Research Council, and they were approved by the National Institutes of Health (NIH) Animal Care and Use Committee.
Reagents
Water-soluble, silica-phthalocyanine derivative IRDye 700DX NHS ester was purchased from LI-COR Biosciences. Anti-mouse CD25 antibody (PC-61.5.3) and rat IgG1 (HRPN) were obtained from Bio X Cell. All other chemicals were of reagent grade.
Cell culture
Luciferase-expressing MC38 (colon cancer), LL/2 (Lewis lung carcinoma), and TRAMP-C2 (prostate cancer) mouse cell lines [American Type Culture Collection (ATCC)] were established by transducing them with RediFect Red-FLuc lentiviral particles (PerkinElmer). Their high luciferase expression was confirmed through 10 passages. IL-2–dependent CD25-expressing mouse T lymphocyte HT-2 clone A5E cells (HT-2-A5E) were purchased from ATCC. Cells were cultured in RPMI 1640 medium (Thermo Fisher Scientific Inc.) supplemented with 10% fetal bovine serum and penicillin (100 IU/ml)–streptomycin (100 μg/ml) (Thermo Fisher Scientific Inc.). For HT-2-A5E culture, 0.05 mM 2-mercaptoethanol and 0.1 nM human IL-2 (Roche) were also added.
Preparation of anti-CD25-F(ab’)2 and control-F(ab’)2
F(ab’)2 fragments of the anti-mouse CD25 antibody [PC-61.5.3, anti-CD25-F(ab’)2] and rat IgG1 as a control [control-F(ab’)2] were generated by digesting the whole antibody using immobilized ficin (Thermo Fisher Scientific Inc.) in 10 mM citrate buffer with 4 mM cysteine and 5 mM EDTA (pH 6.0) at 37°C for 26 hours. After the digestion, the F(ab’)2 was purified by HPLC (fig. S1A) using a G2000SWxL column and phosphate-buffered saline (PBS) as the eluent (flow rate, 0.5 ml/min).
Conjugation of IR700 to the anti-CD25-F(ab’)2 or control-F(ab’)2
Anti-CD25-F(ab’)2 or control-F(ab’)2 (9.1 nmol) was incubated with IR700 NHS ester (45.5 nmol, LI-COR Biosciences) in 0.3 ml of 0.1 M Na2HPO4 (pH 8.6) at room temperature for 1 hour. The mixture was purified with a Sephadex G-25 column (PD-10; GE Healthcare). The protein concentration was determined using the Coomassie Plus Protein Assay Kit (Thermo Fisher Scientific Inc.) by measuring the absorption at 595 nm using 8453 Value System (Agilent Technologies). The concentration of IR700 was determined by measuring the absorption at 689 nm, and the number of fluorophore molecules conjugated to each F(ab’)2 molecule was calculated. The conjugation was performed such that an average of three IR700 molecules was bound to a single F(ab’)2. We performed SDS–polyacrylamide gel electrophoresis to confirm the integrity of IR700-conjugated F(ab’)2, and the bioactivity was confirmed by examining its binding to HT-2-A5E cells. The cells (1 × 105) were incubated with anti-CD25-F(ab’)2–IR700 (10 μg/ml) in medium for 1 to 6 hours at 37°C. To validate the specificity of the binding, a competition assay was performed by adding excess untreated anti-CD25 antibody (50 μg). Cells were analyzed by flow cytometry (FACSCalibur, BD Biosciences) using the CellQuest software (BD Biosciences).
Fluorescence microscopy
Anti-CD25-F(ab’)2–IR700 was incubated with 10,000 HT-2-A5E cells at 10 μg/ml for 6 hours and washed with PBS, and PI (Life Technologies) was added at 2 μg/ml for 30 min. The cells were then exposed to NIR light (4 J/cm2), and serial images were obtained using a fluorescence microscope (IX61; Olympus America) with a filter set for IR700 fluorescence (590- to 650-nm excitation filter; 665- to 740-nm band-pass emission filter). The images were analyzed using the ImageJ software (http://rsb.info.nih.gov/ij/).
In vitro NIR-PIT
One hundred thousand HT-2-A5E cells were seeded into 24-well plates and incubated with anti-CD25-F(ab’)2–IR700 at 10 μg/ml for 6 hours at 37°C. After washing the cells with PBS, we added culture medium without phenol red. Then, the cells were irradiated with a NIR light-emitting diode, which emits light at 670- to 710-nm wavelength (L690-66-60; Marubeni America Co.). The actual power density (mW/cm2) was measured with an optical power meter (PM100, Thorlabs). To determine the cytotoxic effects of NIR-PIT, PI staining was performed as described above 1 hour after irradiation, and cells were analyzed by flow cytometry.
Animals and tumor models
All in vivo procedures were performed in accordance with a protocol approved by the NIH Animal Care and Use Committee. All mice were purchased from The Jackson Laboratory. Eleven- to 15-week-old C57BL/6 mice or IFN-γ–deficient (IFN-γ–KO) mice were inoculated with 6 million MC38-luc or LL/2-luc cells or 8 million TRAMP-C2-luc cells into the right or left dorsum. Mice with tumors measuring about 150 mm3 (6 to 7 mm in diameter) were used for the experiments. Mice were shaved at the tumor sites for irradiation and image analysis. C57BL/6 albino mice were used for repeated local NIR-PIT because C57BL/6 mice started to have skin pigmentation within 4 days after shaving and were not suitable for the experiments to detect luciferase activities. Mice were monitored daily, and tumor volumes were measured three times a week until the tumor (any tumor when mice had multiple tumors) diameter reached 2 cm, whereupon the mice were euthanized with carbon dioxide.
In vivo BLI
For BLI, d-luciferin (15 mg/ml, 200 μl) was injected intraperitoneally, and the mice were analyzed on a BLI system (PhotonIMAGER; Biospace Lab) for luciferase activity. Regions of interest were set on the entire tumors to quantify the luciferase activities.
In vivo IR700 fluorescence imaging
IR700 fluorescence images before and after therapy were acquired with a fluorescence imager (Pearl Imager, LI-COR Biosciences).
In vivo local CD25-targeted NIR-PIT
Local CD25-targeted NIR-PIT at the tumor was performed 6 days after tumor inoculation for mice bearing one tumor or at day 7 for mice bearing multiple tumors. The mice were injected with 100 μg of anti-CD25-F(ab’)2–IR700 or control-F(ab’)2–IR700 and irradiated with NIR light at 100 J/cm2 unless otherwise specified to the right tumor on the following day.
Analysis of tumor-infiltrating and splenic and lung lymphocytes
To determine the systemic effect of anti-CD25-F(ab’)2 administration on CD4+CD25+Foxp3+ Tregs, 100 μg of anti-CD25-F(ab’)2 or anti-CD25-IgG was injected intravenously into mice, and splenocytes were analyzed for CD4+CD25+Foxp3+ Tregs 1 day later. To determine the effects of anti-CD25-F(ab’)2–IR700 NIR-PIT on various lymphocytes, the tumors and spleen were harvested at the indicated time after NIR-PIT. To examine the potential side effects of local CD25-targeted NIR-PIT, lungs were harvested 1 day after the therapy. Single-cell suspensions were prepared by passing the fragmented tissues through 70-μm filters, followed by Ficoll centrifugation. The cells were stained with antibodies against CD3e (145-2C11), CD8a (53-6.7), CD4 (RM4-5), CD25 (3C7), NK1.1 (PK136), CD19 (1D3), CD11c (N418), CD11b (M1/70), Ly-6C (HK1.4), Ly-6G (1A8-Ly6g), CD86 (GL1), CD40 (3/23), and H-2Kb (AF6-88.5). Foxp3 and intracellular cytokine staining were performed using Foxp3/Transcription Factor Fixation/ Permeabilization Concentrate and Diluent (Affymetrix) and antibodies against Foxp3 (FJK-16s), IFN-γ (XMG1.2), and IL-2 (JES6-5H4), according to the manufacturer’s instructions. All antibodies were purchased from Affymetrix. The stained cells were applied to a flow cytometer, and data were analyzed with the FlowJo software (FlowJo LLC).
Serum, tissue, and intratumoral cytokine analysis
Tumor inoculation and treatment were performed as described above. Serum was serially collected from the mice before and 1.5 hours and 1 day after the local CD25-targeted NIR-PIT at the LL/2-luc tumor. Tumors were harvested, and single-cell suspensions were prepared by passing the cut tissues through 70-μm filters in PBS with cOmplete Protease Inhibitor Cocktail (Roche). Next, they were centrifuged at 5000 rpm for 15 min, and the supernatant was collected and filtered at 0.2 μm. The lungs or intestines were harvested, and the samples were prepared in the same way. Protein concentrations of all samples were normalized using BCA Assay (Thermo Fisher Scientific Inc.), and concentrations of various cytokines and chemokines were analyzed with Mouse Cytokine Array/Chemokine Array by Eve Technologies.
Immune depletion of NK and CD8 T cells and neutralization of IFN-γ in vivo
Anti-NK1.1 (PK136)– or anti-CD8a (2.43)–depleting antibodies or anti-IFN-γ (XMG1.2)–neutralizing antibody was injected intraperitoneally every 2 days starting from 2 days before the PIT at doses of 25, 50, and 100 μg, respectively, until the mice were euthanized (see the regimen in Fig. 6A). All antibodies were purchased from Bio X Cell.
Statistics
Data are expressed as means ± SEM from a minimum of four experiments, unless otherwise indicated. Statistical analyses were performed with a statistics program (GraphPad Prism; GraphPad Software). For two-group comparisons, Mann-Whitney test or unpaired t test was used. For multiple-group comparisons, a one-way ANOVA with Tukey’s test or Dunnett’s test was used. The cumulative probability of survival, defined as the tumor diameter failing to reach 2 cm, was estimated in each group with the Kaplan-Meier survival curve analysis, and the results were compared with the log-rank test and Wilcoxon test. P < 0.05 was considered to indicate a statistically significant difference.
Supplementary Material
Fig. S1. Anti-CD25-F(ab’)2 and control-F(ab’)2 are generated, characterized, and conjugated with IR700 dye.
Fig. S2. NIR-PIT with anti-CD25-F(ab’)2–IR700 induces necrosis of CD25-expressing cells in a NIR light dose–dependent manner.
Fig. S3. Tumor-infiltrating CD8 T and NK cells do not express CD25.
Fig. S4. Anti-CD25-F(ab’)2–IR700 does not bind to the tumor cells.
Fig. S5. Depletion of tumor-infiltrating CD4+CD25+Foxp3+ Tregs with local CD25-targeted NIR-PIT is NIR light dose–dependent.
Fig. S6. Tumor-infiltrating CD4+Foxp3+ cells are depleted by local CD25-targeted NIR-PIT.
Fig. S7. Local CD25-targeted NIR-PIT of an MC38-luc tumor induces regression of the tumor.
Fig. S8. Local CD25-targeted NIR-PIT induces regression of TRAMP-C2-luc flank tumors.
Fig. S9. Repeated local CD25-targeted NIR-PIT enables prolonged suppression of tumor growth.
Fig. S10. Repeated local CD25-targeted NIR-PIT induces up-regulation of CD25 on CD8 T and NK cells at each treatment.
Fig. S11. Local CD25-targeted NIR-PIT induces activation of tumor-infiltrating dendritic cells and other APCs.
Fig. S12. Local CD25-targeted NIR-PIT increases the number of granulocytes in treated tumors.
Fig. S13. NIR light irradiation of tumors induces negligible production of cytokines and chemokines.
Fig. S14. Local CD25-targeted NIR-PIT induces systemic and intratumoral cytokine storm.
Fig. S15. IFN-γ production by CD8 T and NK cells in the lungs is not detected 1 day after CD25-targeted NIR-PIT.
Fig. S16. CD25-targeted NIR-PIT reduces IR700 fluorescence of the treated tumor, but not that of the contralateral nonirradiated tumor.
Fig. S17. Local CD25-targeted NIR-PIT of the right dorsal tumor induces reduction of multiple tumors at distant sites.
Fig. S18. Local CD25-targeted NIR-PIT inhibits the growth of tumor challenged on the contralateral side.
Fig. S19. The concentrations of cytokines and chemokines in the contralateral nonirradiated tumor are increased after CD25-targeted NIR-PIT.
Fig. S20. Antitumor effect of local CD25-targeted NIR-PIT is at least partly IFN-γ–dependent.
Acknowledgments:
We thank W. Telford for providing assistance in flow cytometry experiments.
Funding: This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Competing interests: K.S., N.S., P.L.C., and H.K. have filed a patent application for the local PIT strategy to deplete immunosuppressor cells as a cancer treatment. All other authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.Chen DS, Mellman I, Oncology meets immunology: The cancer-immunity cycle. Immunity 39, 1–10 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Childs RW, Carlsten M, Therapeutic approaches to enhance natural killer cell cytotoxicity against cancer: The force awakens. Nat. Rev. Drug Discov 14, 487–498 (2015). [DOI] [PubMed] [Google Scholar]
- 3.June CH, Riddell SR, Schumacher TN, Adoptive cellular therapy: A race to the finish line. Sci. Transl. Med 7, 280ps7 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Melero I, Berman DM, Aznar MA, Korman AJ, Gracia JLP, Haanen J, Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat. Rev. Cancer 15,457–472 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Lesokhin AM, Callahan MK, Postow MA, Wolchok JD, On being less tolerant: Enhanced cancer immunosurveillance enabled by targeting checkpoints and agonists of T cell activation. Sci. Transl. Med 7, 280sr1 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Sharma P, Allison JP, The future of immune checkpoint therapy. Science 348, 56–61 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Mahoney KM, Rennert PD, Freeman GJ, Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov 14, 561–584 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Miller JFAP, Sadelain M, The journey from discoveries in fundamental immunology to cancer immunotherapy. Cancer Cell 27, 439–449 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Topalian SL, Drake CG, Pardoll DM, Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 27, 450–461 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zitvogel L, Galluzzi L, Smyth MJ, Kroemer G, Mechanism of action of conventional and targeted anticancer therapies: Reinstating immunosurveillance. Immunity 39, 74–88 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Smyth MJ, Ngiow SF, Ribas A, Teng MWL, Combination cancer immunotherapies tailored to the tumour microenvironment. Nat. Rev. Clin. Oncol 13, 143–158 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Motz GT, Coukos G, Deciphering and reversing tumor immune suppression. Immunity 39, 61–73 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goldberg MS, Immunoengineering: How nanotechnology can enhance cancer immune-therapy. Cell 161, 201–204 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Mitsunaga M, Ogawa M, Kosaka N, Rosenblum LT, Choyke PL, Kobayashi H, Cancer cell-selective in vivo near infrared photoimmunotherapy targeting specific membrane molecules. Nat. Med 17, 1685–1691 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sato K, Nakajima T, Choyke PL, Kobayashi H, Selective cell elimination in vitro and in vivo from tissues and tumors using antibodies conjugated with a near infrared phthalocyanine. RSC Adv. 5, 25105–25114 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sato K, Nagaya T, Nakamura Y, Harada T, Choyke PL, Kobayashi H, Near infrared photoimmunotherapy prevents lung cancer metastases in a murine model. Oncotarget 6, 19747–19758 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sato K, Nagaya T, Mitsunaga M, Choyke PL, Kobayashi H, Near infrared photo-immunotherapy for lung metastases. Cancer Lett. 365, 112–121 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sato K, Watanabe R, Hanaoka H, Harada T, Nakajima T, Kim I, Paik CH, Choyke PL, Kobayashi H, Photoimmunotherapy: Comparative effectiveness of two monoclonal antibodies targeting the epidermal growth factor receptor. Mol. Oncol 8, 620–632 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sato K, Hanaoka H, Watanabe R, Nakajima T, Choyke PL, Kobayashi H, Near infrared photoimmunotherapy in the treatment of disseminated peritoneal ovarian cancer. Mol. Cancer Ther 14, 141–150 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shirasu N, Yamada H, Shibaguchi H, Kuroki M, Kuroki M, Potent and specific antitumor effect of CEA-targeted photoimmunotherapy. Int. J. Cancer 135, 2697–2710 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M, Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol 155, 1151–1164 (1995). [PubMed] [Google Scholar]
- 22.Colombo MP, Piconese S, Regulatory-T-cell inhibition versus depletion: The right choice in cancer immunotherapy. Nat. Rev. Cancer 7, 880–887 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Sakaguchi S, Miyara M, Costantino CM, Hafler DA, FOXP3+ regulatory T cells in the human immune system. Nat. Rev. Immunol 10, 490–500 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Facciabene A, Motz GT, Coukos G, T-regulatory cells: Key players in tumor immune escape and angiogenesis. Cancer Res. 72, 2162–2171 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishikawa H, Sakaguchi S, Regulatory T cells in cancer immunotherapy. Curr. Opin. Immunol 27, 1–7 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Zou W, Regulatory T cells, tumour immunity and immunotherapy. Nat. Rev. Immunol 6, 295–307 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Ko K, Yamazaki S, Nakamura K, Nishioka T, Hirota K, Yamaguchi T, Shimizu J, Nomura T, Chiba T, Sakaguchi S, Treatment of advanced tumors with agonistic anti-GITR mAb and its effects on tumor-infiltrating Foxp3+CD25+CD4+ regulatory T cells. J. Exp. Med 202, 885–891 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sugiyama D, Nishikawa H, Maeda Y, Nishioka M, Tanemura A, Katayama I, Ezoe S, Kanakura Y, Sato E, Fukumori Y, Karbach J, Jager E, Sakaguchi S, Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc. Natl. Acad. Sci. U.S.A 110, 17945–17950 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E, Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor a monoclonal antibody. Cancer Res. 59, 3128–3133 (1999). [PubMed] [Google Scholar]
- 30.Yu P, Lee Y, Liu W, Krausz T, Chong A, Schreiber H, Fu Y-X, Intratumor depletion of CD4+ cells unmasks tumor immunogenicity leading to the rejection of late-stage tumors. J. Exp. Med 201, 779–791 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li X, Kostareli E, Suffner J, Garbi N, Hämmerling GJ, Efficient Treg depletion induces T-cell infiltration and rejection of large tumors. Eur. J. Immunol 40, 3325–3335 (2010). [DOI] [PubMed] [Google Scholar]
- 32.Teng MWL, Ngiow SF, von Scheidt B, McLaughlin N, Sparwasser T, Smyth MJ, Conditional regulatory T-cell depletion releases adaptive immunity preventing carcinogenesis and suppressing established tumor growth. Cancer Res. 70, 7800–7809 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Bos PD, Plitas G, Rudra D, Lee SY, Rudensky AY, Transient regulatory T cell ablation deters oncogene-driven breast cancer and enhances radiotherapy. J. Exp. Med 210,2435–2466 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Freeman CL, Morschhauser F, Sehn L, Dixon M, Houghton R, Lamy T, Fingerle-Rowson G, Wassner-Fritsch E, Gribben JG, Hallek M, Salles G, Cartron G, Cytokine release in patients with CLL treated with obinutuzumab and possible relationship with infusion-related reactions. Blood 126, 2646–2649 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ganesh S, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N, Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med 355, 1018–1028 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Michot JM, Bigenwald C, Champiat S, Collins M, Carbonnel F, Postel-Vinay S, Berdelou A, Varga A, Bahleda R, Hollebecque A, Massard C, Fuerea A, Ribrag V, Gazzah A, Armand JP, Amellal N, Angevin E, Noel N, Boutros C, Mateus C, Robert C, Soria JC, Marabelle A, Lambotte O, Immune-related adverse events with immune checkpoint blockade: A com-prehensive review. Eur. J. Cancer 54, 139–148 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Nishino M, Sholl LM, Hodi FS, Hatabu H, Ramaiya NH, Anti–PD-1–related pneumonitis during cancer immunotherapy. N. Engl. J. Med 373, 288–290 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Attia P, Maker AV, Haworth LR, Rogers-Freezer L, Rosenberg SA, Inability of a fusion protein of IL-2 and diphtheria toxin (Denileukin Diftitox, DAB389 IL-2, ONTAK) to eliminate regulatory T lymphocytes in patients with melanoma. J. Immunother 28, 582–592 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Teng MWL,Swann JB, von Scheidt B,Sharkey J, Zerafa N, McLaughlin N,Yamaguchi T, Sakaguchi S, Darcy PK, Smyth MJ, Multiple antitumor mechanisms downstream of prophylactic regulatory T-cell depletion. Cancer Res. 70, 2665–2674 (2010). [DOI] [PubMed] [Google Scholar]
- 40.Couper KN, Blount DG, de Souza JB, Suffia I, Belkaid Y, Riley EM, Incomplete depletion and rapid regeneration of Foxp3+ regulatory T cells following anti-CD25 treatment in malaria-infected mice. J. Immunol 178, 4136–4146 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anz D, Mueller W, Golic M, Kunz WG, Rapp M, Koelzer VH, Ellermeier J, Ellwart JW, Schnurr M, Bourquin C, Endres S, CD103 is a hallmark of tumor-infiltrating regulatory T cells. Int. J. Cancer 129, 2417–2426 (2011). [DOI] [PubMed] [Google Scholar]
- 42.Lin Y-C, Chang L-Y, Huang C-T, Peng H-M, Dutta A, Chen T-C, Yeh C-T, Lin C-Y, Effector/memory but not naive regulatory T cells are responsible for the loss of concomitant tumor immunity. J. Immunol 182, 6095–6104 (2009). [DOI] [PubMed] [Google Scholar]
- 43.Tanaka T, Narazaki M, Kishimoto T, IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol 6, a016295 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Anti-CD25-F(ab’)2 and control-F(ab’)2 are generated, characterized, and conjugated with IR700 dye.
Fig. S2. NIR-PIT with anti-CD25-F(ab’)2–IR700 induces necrosis of CD25-expressing cells in a NIR light dose–dependent manner.
Fig. S3. Tumor-infiltrating CD8 T and NK cells do not express CD25.
Fig. S4. Anti-CD25-F(ab’)2–IR700 does not bind to the tumor cells.
Fig. S5. Depletion of tumor-infiltrating CD4+CD25+Foxp3+ Tregs with local CD25-targeted NIR-PIT is NIR light dose–dependent.
Fig. S6. Tumor-infiltrating CD4+Foxp3+ cells are depleted by local CD25-targeted NIR-PIT.
Fig. S7. Local CD25-targeted NIR-PIT of an MC38-luc tumor induces regression of the tumor.
Fig. S8. Local CD25-targeted NIR-PIT induces regression of TRAMP-C2-luc flank tumors.
Fig. S9. Repeated local CD25-targeted NIR-PIT enables prolonged suppression of tumor growth.
Fig. S10. Repeated local CD25-targeted NIR-PIT induces up-regulation of CD25 on CD8 T and NK cells at each treatment.
Fig. S11. Local CD25-targeted NIR-PIT induces activation of tumor-infiltrating dendritic cells and other APCs.
Fig. S12. Local CD25-targeted NIR-PIT increases the number of granulocytes in treated tumors.
Fig. S13. NIR light irradiation of tumors induces negligible production of cytokines and chemokines.
Fig. S14. Local CD25-targeted NIR-PIT induces systemic and intratumoral cytokine storm.
Fig. S15. IFN-γ production by CD8 T and NK cells in the lungs is not detected 1 day after CD25-targeted NIR-PIT.
Fig. S16. CD25-targeted NIR-PIT reduces IR700 fluorescence of the treated tumor, but not that of the contralateral nonirradiated tumor.
Fig. S17. Local CD25-targeted NIR-PIT of the right dorsal tumor induces reduction of multiple tumors at distant sites.
Fig. S18. Local CD25-targeted NIR-PIT inhibits the growth of tumor challenged on the contralateral side.
Fig. S19. The concentrations of cytokines and chemokines in the contralateral nonirradiated tumor are increased after CD25-targeted NIR-PIT.
Fig. S20. Antitumor effect of local CD25-targeted NIR-PIT is at least partly IFN-γ–dependent.