

Abstract
Purpose
Autoimmune lymphoproliferative syndrome (ALPS) is a non-malignant genetic disorder of lymphocyte homeostasis with defective Fas-mediated apoptosis. Current therapies for ALPS primarily target autoimmune manifestations with non-specific immune suppressants with variable success thus highlighting the need for better therapeutics for this disorder.
Methods
The spectrum of clinical manifestations of ALPS is mirrored by MRL/lpr mice that carry a loss of function mutation in the Fas gene and have proven to be a valuable model in predicting the efficacy of several therapeutics that are front-line modalities for the treatment of ALPS. We evaluated the potential efficacy of tofacitinib, an orally active, pan-JAK inhibitor currently approved for rheumatoid arthritis as a single agent modality against ALPS using MRL/lpr mice.
Results
We demonstrate that a 42-day course of tofacitinib therapy leads to a lasting reversal of lymphadenopathy and autoimmune manifestations in the treated MRL/lpr mice, Specifically, in treated mice the peripheral blood white blood cell counts were reversed to near normal levels with almost a 50 % reduction in the TCRαβ+CD4−CD8−T lymphocyte numbers that coincided with a parallel increase in CD8+ T cells without a demonstrable effect on CD4+ lymphocytes including FoxP3+ regulatory T cells. The elevated plasma IgG and IgA levels were also drastically lowered along with a significant reduction in plasmablasts and plasmacytes in the spleen.
Conclusion
On the basis of these results, it is likely that tofacitinib would prove to be a potent single agent therapeutic modality capable of ameliorating both offending lymphadenopathy as well as autoimmunity in ALPS patients.
Keywords: Autoimmunity, lymphoproliferation, ALPS, tofacitinib, SLE
Introduction
Autoimmune lymphoproliferative syndrome (ALPS) is a non-malignant disorder of defective lymphocyte homeostasis and is characterized by massive accumulation of lymphocytes resulting in splenomegaly, and lymphoadenopathy [1, 2]. The majority of ALPS patients harbor heterozygous germline mutations in the death-receptor encoding TNF receptor super-family, member 6 gene (TNFRSF6 also known as Fas or CD95) that are inherited in an autosomal dominant manner [3,4]. The presence of a large number of TCRαβ+CD4−CD8− double negative (DN) T cells with an obscure ontogeny, both in the peripheral blood and in lymphoid organs is a hallmark feature of ALPS. In addition to near universal autoimmune manifestations in these patients that are most often directed against hematopoietic elements, a heightened incidence of hematologic malignancies is also observed [5, 6].
Most therapeutic approaches for ALPS target autoimmune manifestations with non-specific immune suppressants. However, these agents do not appreciably shrink grossly enlarged lymph nodes or spleens [7]. More recently, sirolimus, has been recognized as a promising treatment modality capable of improving both lymphoproliferation and autoimmunity especially in patients with steroid refractory disease, but requires long-term administration of the drug and close monitoring for toxicities [8]. Thus, there is a clear need for novel, efficacious lympholytic modalities that not only blunt autoimmune complications but also remedy grossly enlarged lymphoid organs, thus mitigating the complications associated with both infiltrative lymphoproliferation and autoimmunity.
Materials and Methods
Mice, Drugs and Devices
MRL/MpJJmsSlc-lpr and C57BL/6JJmsSlc-lpr/lpr mice were obtained from Japan SLC Inc. All animal experiments were approved by the institutional animal care and use committee of Tokyo Metropolitan Institute of Medical Science. Tofacitinib purchased from LC Laboratories, was dissolved in a sterile solution of 50 % DMSO, 10 % PEG 300, and 40 % water. Subcutaneously implanted (dorsally between the scapulae) ALZET mini osmotic pumps from Durect were used for in vivo delivery of tofacitinib at a dose of 30 mg/kg/day for 42 days.
Flow Cytometry
Lymphocytes were isolated by Ficoll density gradient centrifugation either from blood collected from the retro-orbital plexus of living animals or spleens harvested from euthanized mice, prior to surface staining with fluoro-chrome conjugated antibodies. Flow cytometric analysis was performed using a BD FACSCanto II workstation with Flo Jo data analysis software as described previously [9].
ELISA
Total IgM, IgG, and IgA levels in the sera of mice were measured by a sandwich ELISA as described previously [9].
Histopathologic Studies
Histopathologic studies were performed on kidney tissues after harvesting the kidneys from euthanized mice. Kidney tissues were fixed in 10 % buffered formalin, embedded in paraffin, sectioned at 2 μ, and stained with hemotoxylin and eosine. Stained slides were examined and scored by a veterinary pathologist in a blinded fashion taking into account the extent of glomerular hypercellularity, mesangial expansion, crescent formation, fibrinoid necrosis, glomerular hyalinization, and infiltration of inflammatory cells using a semi quantitative grading system ranging from 0 to 3: 0= none; 1=mild; 2=moderate; 3=severe inflammation.
Statistics
All data are expressed as mean± SD and Student’s t test was used for analysis of data and a P value less than 0.05 was considered significant.
Results and Discussion
The spectrum of clinical manifestations seen in ALPS patients is mirrored by MRL/lpr mice that carry a loss of function mutation in the Fas gene [10]. MRL/lpr mice also display progressive accumulation of TCRαβ+CD4−CD8− DN T lymphocytes thus recapitulating one of the disease-defining hallmark features of ALPS and have proven to be a valuable preclinical model in predicting the efficacy of several therapeutics that are front-line modalities today in the treatment of ALPS [11, 12].
The identification of somatic Fas mutations in a significant subset of ALPS patients, primarily confined to the non-thymic TCRαβ+CD4−CD8− DN T lymphocytes suggests a pathogenic and causal role for these DN T cells in ALPS as opposed to being an epiphenomenon in the disease [13-16]. Therefore, we chose to focus on potentially activated signaling pathways in these DNT to identify targets that can be exploited therapeutically to eliminate these pathogenic T cells. In order to gain insights as to which signaling pathways drive the proliferation of TCRαβ+CD4−CD8− DNT lymphocytes, an initial phospho-protein profiling of these cells was undertaken with a commercially available phospho-antibody microarray platform (Full Moon Biosystems, CA; also see Fig. 1S and Table 1S, supplemental data). The detection of phosphorylated JAK1, JAK2, STAT5 and STAT6 was suggestive of a pivotal role for the JAK-STAT signaling pathway in these pathognomonic TCRαβ+CD4−CD8− DN T lymphocytes.
The JAK family of non-receptor tyrosine kinases plays a pivotal role in lymphoid homeostasis and in the pathophysiology of autoimmunity. These multifactorial processes require the participation of a variety of immune cell elements that are critically dependent on JAK signaling pathways [17, 18]. Therefore, it is conceivable that the interruption of JAK signaling pathways in lymphocytes is likely to ameliorate both gross lymphadenopathy as well as autoimmunity in ALPS patients. Inhibition of multiple Janus kinases with a pan-JAK inhibitor such as tofacitinib, an orally active, small molecule JAK inhibitor with a favorable safety profile that is currently approved for rheumatoid arthritis [19, 20, http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm327152.htm] provides a novel therapeutic approach for a variety of inflammatory and autoimmune disorders by simultaneously inhibiting downstream signaling of a multiplicity of cytokines that act in concert to drive the pathogenesis of such diseases. Thus, we evaluated the potential efficacy of tofacitinib in MRL/lpr mice both as a late-stage therapeutic agent and as an early-stage preventative modality for ALPS. MRL/lpr mice, 25-weeks old with overt clinical manifestations including splenomegaly, hypergammaglobulinemia, and massively expanded TCRαβ+CD4−CD8− T cells, were treated with tofacitinib via subcutaneously implanted mini osmotic pumps that delivered a dose of 30 mg/kg/day of tofacitinib for a period of 42 days and the cell profiles in the peripheral blood were monitored longitudinally for 3 months. We opted to deliver the drug via implanted pumps to minimize frequent handling stress to the animals and the dosage selected was identical that of other published preclinical studies for tofacitinib. Samples evaluated on days 0, 14, 26, 73, and 90 post-treatment showed a gradual decline of white blood cell numbers reaching near normal levels [normal being the white blood cell counts of peripheral blood of age matched C57BL6 mice having a cell count of 138±31×100 per microliter (n=5)] as shown in Fig. 1a. To assess the impact of tofacitinib treatment on the pathognomonic TCRαβ+CD4−CD8− DN T cells, we compared the numbers of these cells in the peripheral blood before the initiation of treatment to those of 10-week post-initiation therapy by flow cytometry. The mean percentage of TCRαβ+CD4−CD8− T cells in the peripheral blood of animals prior to therapy was 79.4±4.0(SD) (Fig. 1b). However, upon treatment the percentage of these cells dropped to 42.2±5.0 (SD,p<0·001) attesting to the efficacy of tofacitinib in reducing the burden of TCRαβ+CD4−CD8− T cells in the advanced stages of the lymphoproliferative disorder in these MRL/lpr mice. To evaluate the efficacy of tofacitinib in the early stages of the disease, we treated younger 15-week old mice in an identical manner (Fig. 1c). In these mice, the mean percentage of TCRαβ+CD4−CD8− T cells was 69.7±6.5(SD) prior to the initiation of therapy and 10 weeks after tofacitinib treatment this percentage was reduced to 51.1±7.8(SD, p=0·035) suggesting the possibility of initiating effective tofacitinib treatment early on in the disease process. Importantly, tofacitinib treatment had no significant impact on CD4+ T cells at the dosage used (for 25-week old mice a mean value of 15 % versus 18 % or for 15-week old mice 23·7 % versus 23·6 % respectively). In addition, when we specifically examined the impact of tofacitinib therapy on CD4+Foxp3+ regulatory T (Treg) cells in the peripheral blood, the percentage of these cells in the blood did not show any significant differences between the tofacitinib treated group versus sham treated group of mice (see Fig. 2S, supplemental data) thus supporting the emerging view [21] that despite their exquisite IL-2 dependency, Tregs are refractory to pan-JAK inhibitor tofacitinib (in 25-week old mice a mean value of 5.7 % in untreated versus 6.4 % in tofacitinib-treated; p = 0.24) However, as shown in Fig. 1 panels b and c concomitant with the reduction of TCRαβ+CD4−CD8− T cells, there was a robust reappearance of CD8+ T cells (in 25-week old mice a mean value of 6 % in untreated versus 39 % in treated and in 15-week old mice 7 % in untreated versus 25 % in treated).
Fig. 1.

Panel a Tofacitinib treatment results in re-establishment of near normal white blood cell counts in adult MRL/lpr mice with lymphoproliferative disease. Five mice that were 25 weeks of age were implanted with mini osmotic pumps subcutaneously to deliver tofacitinib at a dose of 30 mg/kg/day over a period of 42 days. Blood samples were collected longitudinally with the first sample being collected just prior to initiating treatment. Panel b Tofacitinib treatment results in a dramatic reduction of pathognomonic TCRαβ+CD4−CD8− T cells in 25 week-old mice. Panel c Tofacitinib treatment also reduces pathognomonic TCRαβ+CD4−CD8− T cells in younger 15 week-old mice. Lymphocytes were purified from blood samples collected just prior to initiation of tofacitinib treatment and 10 weeks post initiation of treatment. After surface staining of lymphocytes for CD3e, CD4 and CD8, the modulation of CD4+, CD8+ and CD4−CD8− DN T cell subsets with tofacitinib therapy was examined by flow cytometry after gating on CD3+ lymphocytes using a BD FACSCanto II workstation with Flo Jo data analysis software
The ontogeny of the pathognomonic TCRαβ+CD4−CD8− T cells remains controversial [22-26] and it is unclear as to what cues, signals or cytokines are responsible for their survival or active proliferation in vivo and perhaps IL-17 may play a pivotal role as suggested by a recent report [27]. However, it is these accumulating TCRαβ+CD4−CD8− T cells that lead to gross enlargement of secondary lymphoid organs dictating the clinical symptoms of ALPS. Therefore, we evaluated whether tofacitinib treatment also ameliorated the lymphoid organ enlargement in the treated mice. As shown in Fig. 2a, MRL/lpr mice with advancing age display massive enlargement of secondary lymphoid organs (lymphoid organs from a representative 7-month old mouse out of five such mice is shown). In contrast, mice treated with tofacitinib for a period of 42 days beginning at 10 weeks of age and then examined when they were 7 months old, showed near normal sizes of spleens and lymph nodes, thus illustrating long-lasting therapeutic benefits of tofacitinib therapy in this mouse model of ALPS. Despite this dramatic effect of tofacitinib therapy on the secondary lymphoid organs of MRL/lpr mice, we were unable to see any meaningful impact on the serum IL-17 levels which were low to begin with in MRL/lpr mice studied (data not shown).
Fig. 2.

Panel aTofacitinib treatment dramatically reduces secondary lymphoid organ enlargement in MRL/lpr mice. Ten-week old mice were either sham treated with formulation vehicle or tofacitinib for a period of 42 days. Mice were sacrificed when they reached 7 months of age and their secondary lymphoid organs harvested for evaluation. Organs are representative of five such animals. Panel b Assessment of the impact of tofacitinib treatment on serum immunoglobulin subtypes in MRL/lpr mice. Serum samples were collected just prior to initiating treatment when mice were 10 weeks old and the second serum samples were collected just prior to euthanasia for organ harvest at 7 months of age (n=5). Serum immunoglobulin subtypes were determined by sub-type specific ELISA. For comparison, blood samples from age matched Balb/c were also included in the assay. Y-axis denotes OD readings at 450 nm. Panel c tofacitinib treatment results in reduction of CD3−B220brightCD138+ plasmablasts and CD3−B220dimCD138+ differentiated plasma cells in the spleen. Twenty five-week old MRL/lpr mice were treated as described in the legend to Fig. 1 and control mice were identically sham-treated with the vehicle formulation. Lymphocytes were purified from harvested spleens 10 weeks post treatment. Ficoll gradient isolated lymphocytes were further subjected to a round of negative selection using anti-CD43 microbeads to eliminate contaminating T lymphocytes. Flow cytometric analysis was performed using a BD FACSCanto II workstation with Flo Jo data analysis software. Data shown is representative of five similarly treated and five untreated mice
Hypergammaglobulinemia is a consistent feature in ALPS patients and is recapitulated in MRL/lpr mice. Therefore we assessed the impact of tofacitinib therapy on serum immunoglobulin levels in treated mice. Mice treated with tofacitinib displayed reduced levels of IgG, IgA, and IgM (Fig. 2b). In order to ascertain whether the reduced immunoglobulin levels were due to a direct reduction in the plasma cell numbers, we assessed the impact of tofacitinib treatment on splenic plasma cells by flow cytometry. The B220brightCD138+ plasmablasts (0·79 % in sham-treated versus 0·05 % in tofacitinib-treated) as well as B220dimCD138+ plasma cells (0·71 % in sham-treated versus 0·49 % in tofacitinib-treated) were reduced in the tofacitinib-treated MRL/lpr mice (Fig. 2c). Plasmablasts are critically dependent on IL-21 for proliferation and survival whereas mature plasma cells are less IL-21 dependent [28] and JAK/STAT pathways are pivotal in mediating IL-21 signaling. Therefore, the sensitivity of plasmablasts and plasma cells to tofacitinib that we observed in our study is not surprising although the effects of tofacitinib on B cell subsets in the treated patients still remain to be explored in depth as the clinical use of this drug continues to expand.
Moreover, consistent with the above observations of reduced TCRαβ+CD4−CD8− T lymphocyte numbers and the reversal of lymphoid organ enlargement, we also noted a parallel decline which was significant (p=0.01) in soluble Fas ligand (sFasL) levels, a bio-marker of proliferating TCRαβ+CD4−CD8− T lymphocytes, in the sera of tofacitinib-treated mice as shown in Fig. 3.
Fig. 3.
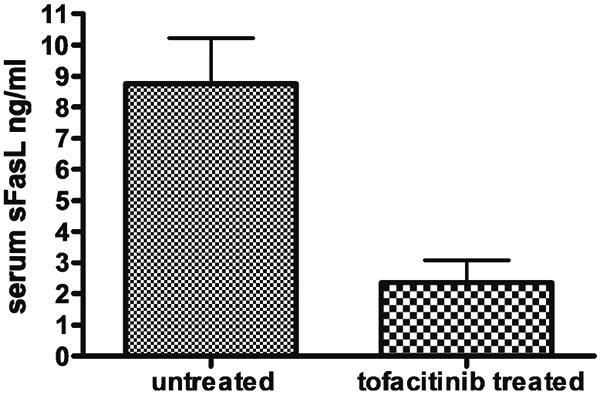
Tofacitinib treatment reduces serum soluble Fas ligand (sFasL) levels in MRL/lpr mice. Ten-week old mice (n=5, for each group) were either sham treated with formulation vehicle or tofacitinib for a period of 42 days. The serum sFasL levels were measured when they reached 7 months of age using a commercial ELISA kit from R&D systems (Minneapolis, MN) according to the manufacturer’s instructions
Although immune-mediated kidney damage is a relatively rare occurrence in ALPS, immune-complex mediated glomerulonephritis is a consistent pathologic feature in MRL/lpr mice making these mice also a valid model for systemic lupus erythematosus (SLE). Ten-week old mice that were either sham treated with formulation vehicle or tofacitinib for a period of 42 days were sacrificed when they reached 7 months and their kidneys harvested for histopathologic evaluation (three mice from each group). Shown in Fig. 4, are representative renal tissue specimens from a mouse treated with tofacitinib, and a mouse that was sham-treated. Kidney tissue derived from the tofacitib-treated animals displayed minimal vascular or kidney parenchymal damage (all three renal tissue specimens from the tofacitinib-treated group displayed a pathologic score of 1) whereas in sham-treated mice there were widespread mononuclear cell infiltrative tubular/interstitial lesions (three specimens from the sham-treated group displayed a pathologic score of 3) thus further demonstrating the efficacy of tofacitinib in reversing the immune complex mediated tissue damage in the kidneys. Concordant with this observation, we also detected a significant reduction (p=0.02) in the circulating anti-DNA antibodies in the sera of tofacitinib-treated animals as shown in Fig. 5.
Fig. 4.

Tofacitinib treatment reverses immune-mediated kidney damage in MRL/lpr mice. Representative H&E stained kidney sections of mice described in Fig. 2a. Magnification, 100×. The extent of glomerular hypercellularity, mesangial expansion, crescent formation, fibrinoid necrosis, glomerular hyalinization, and infiltration of inflammatory cells was scored using a semi quantitative grading system ranging from 0 to 3: 0=none; 1=mild; 2=moderate; 3=severe inflammation
Fig. 5.
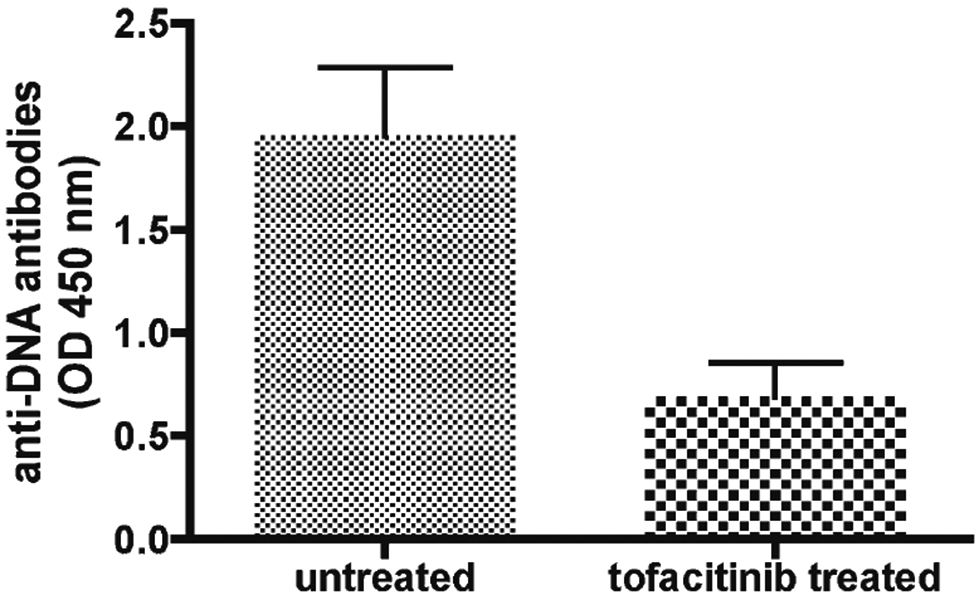
Tofacitinib treatment reduces serum anti-DNA antibody levels in MRL/lpr mice. Ten-week old mice (n=5, for each group) were either sham treated with formulation vehicle or tofacitinib for a period of 42 days. The serum anti-DNA antibody levels were measured when they reached 7 months of age using a commercial ELISA kit from Alpha Diagnostic International (San Antonio, TX) according to the manufacturer’s instructions
Collectively, using four separate cohorts of mice totaling 20 animals altogether, we have assessed the impact of tofacitinib treatment in the prodromal, early clinical as well as late clinical phases of the disease in MRL/lpr mice and found tofacitinib therapy to be efficacious in improving the lymphoadenopathy and autoimmune manifestations irrespective of the stage of the disease at which treatment was initiated. However, one could argue, a potential limitation of the present study relates to the use of MRL/lpr mice, a mouse strain that is lupus-prone genetically and our findings may possibly have been influenced by this lupus-prone genetic background in which excess type 1 interferons play a central pathogenetic role. However, our preliminary studies in non-lupus prone C57BL/6JJmsSlc-lpr/lpr mice indicate that the effects of tofacitinib we observed in MRL/lpr mice were independent of their lupus-prone genetic background as we have seen a 53 % reduction of DN CD3+CD4−CD8− T cells in C57BL/6JJmsSlc-lpr/lpr mice treated with tofacitinib for a period of 4 weeks.
Unlike most other therapeutic modalities that are currently being utilized in the treatment of ALPS patients, tofacitinib diminishes both DN TCRαβ+CD4−CD8− T cells and hypergammaglobulinemia thereby favorably impacting both T and B lymphocyte-mediated pathologic processes. It is likely that the clinical amelioration of the disease we have observed in MRL/lpr mice with tofacitinib is a confluence of its effects on both DN TCRαβ+CD4−CD8− T as well as B cells. In this regard, it is noteworthy that Stranges et al. had reported [29] conditional knockout of Fas in B cells or other antigen presenting cells such as dendritic cells also results in autoimmune lymphoproliferative disease except that the accumulation DN TCRαβ+CD4−CD8− T cells is not seen in such mice. On the basis of these results, it is likely that tofacitinib would prove to be a potent single agent therapeutic modality capable of ameliorating both offending lymphadenopathy as well as autoimmunity in ALPS patients than the current front line therapeutic agents. Additionally, we believe that this drug may also prove to be potently effective in treating systemic lupus erythematosus (SLE) patients because of its ability to simultaneously impact both activated T cells and immunoglobulin-secreting CD3−B220dimCD138+ plasma cells as well as immature plasmablasts (CD3−B220brightCD138+) together with a reversal of immune-complex mediated kidney damage in the tofacitinib-treated MRL/lpr mice.
Supplementary Material
Acknowledgments
This research was supported in part by the intramural programs of the National Cancer Institute and a Grant-in-Aid for 2009 Multidisciplinary Research Project from MEXT in Japan from the Ministry of Education, Science, Sports, and Culture of Japan (T. Hiroi). L.P. Perera gratefully acknowledges the receipt of an invitational fellowship from the Japan Society for the Promotion of Science.
Abbreviations
- ALPS
Autoimmune lymphoproliferative syndrome
- DN
Double negative
- SLE
Systemic lupus erythematosus
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10875-015-0203-z) contains supplementary material, which is available to authorized users.
Conflict of Interest The authors declare no competing financial interests.
References
- 1.Canale VC, Smith CH. Chronic lymphadenopathy simulating malignant lymphoma. J Pediatr. 1967;70:891–9. [DOI] [PubMed] [Google Scholar]
- 2.Sneller MC, Straus SE, Jaffe ES, Jaffe JS, Fleisher TA, Stetler-Stevenson M, et al. A novel lymphoproliferative/autoimmune syndrome resembling murine lpr/gld disease. J Clin Invest. 1992;90: 334–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rieux-Laucat F, Le Deist F, Hivroz C, Roberts IA, Debatin KM, Fischer A, et al. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science. 1995;268: 1347–9. [DOI] [PubMed] [Google Scholar]
- 4.Fisher GH, Rosenburg FJ, Straus SE, Dale JK, Middleton LA, Lin AY, et al. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell. 1995;81:935–46. [DOI] [PubMed] [Google Scholar]
- 5.Straus SE, Jaffe ES, Puck JM, Dale JK, Elkon KB, Rösen-Wolff A, et al. The development of lymphomas in families with autoimmune lymphoproliferative syndrome with germline Fas mutations and defective lymphocyte apoptosis. Blood. 2001;98:194–200. [DOI] [PubMed] [Google Scholar]
- 6.Madkaikar M, Mhatre S, Gupta M, Ghosh K. Advances in autoimmune lymphoproliferative syndromes. Eur J Haematol. 2011;87:1–9. [DOI] [PubMed] [Google Scholar]
- 7.Rao VK, Oliveira JB. How I treat autoimmune lymphoproliferative syndrome. Blood. 2011;118:5741–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Teachey DT, Greiner R, Seif A, Attiyeh E, Bleesing J, Choi J, et al. Treatment with sirolimus results in complete responses in patients with autoimmune lymphoproliferative syndrome. Br J Haematol. 2009;145:101–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yokoyama S, Takada K, Hirasawa M, Perera LP, Hiroi T. Transgenic mice that overexpress human IL-15 in enterocytes recapitulate both B and T cell-mediated pathologic manifestations of celiac disease. J Clin Immunol. 2011;31:1038–44. [DOI] [PubMed] [Google Scholar]
- 10.Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356:314–7. [DOI] [PubMed] [Google Scholar]
- 11.Teachey DT, Obzut DA, Axsom K, Choi JK, Goldsmith KC, Hall J, et al. Rapamycin improves lymphoproliferative disease in murine autoimmune lymphoproliferative syndrome (ALPS). Blood. 2006;108:1965–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dowdell KC, Pesnicak L, Hoffmann V, Steadman K, Remaley AT, Cohen JI, et al. Valproic acid (VPA), a histone deacetylase (HDAC) inhibitor, diminishes lymphoproliferation in the Fas-deficient MRL/lpr(−/−) murine model of autoimmune lymphoproliferative syndrome (ALPS). Exp Hematol. 2009;37:487–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holzelova E, Vonarbourg C, Stolzenberg MC, Arkwright PD, Selz F, Prieur AM, et al. Autoimmune lymphoproliferative syndrome with somatic Fas mutations. N Engl J Med. 2004;351:1409–18. [DOI] [PubMed] [Google Scholar]
- 14.Magerus-Chatinet A, Neven B, Stolzenberg MC, Daussy C, Arkwright PD, Lanzarotti N, et al. Onset of autoimmune lymphoproliferative syndrome (ALPS) in humans as a consequence of genetic defect accumulation. J Clin Invest. 2011;121:106–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hauck F, Magerus-Chatinet A, Vicca S, Rensing-Ehl A, Roesen-Wolff A, Roesler J, et al. Somatic loss of heterozygosity, but not haploinsufficiency alone, leads to full-blown autoimmune lymphoproliferative syndrome in 1 of 12 family members with FAS start codon mutation. Clin Immunol. 2013;147:61–8. [DOI] [PubMed] [Google Scholar]
- 16.Rensing-Ehl A, Völkl S, Speckmann C, Lorenz MR, Ritter J, Janda A, et al. Abnormally differentiated CD4+ or CD8+ T cells with phenotypic and genetic features of double negative T cells in human Fas deficiency. Blood. 2014;124:851–60. [DOI] [PubMed] [Google Scholar]
- 17.Pesu M, Laurence A, Kishore N, Zwillich SH, Chan G, O’Shea JJ. Therapeutic targeting of Janus kinases. Immunol Rev. 2008;223: 132–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ghoreschi K, Jesson MI, Li X, Lee JL, Ghosh S, Alsup JW, et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550). J Immunol. 2011;186:4234–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fleischmann R, Kremer J, Cush J, Schulze-Koops H, Connell CA, Bradley JD, et al. Placebo-controlled trial of tofacitinib monotherapy in rheumatoid arthritis. N Engl J Med. 2012;367:495–507. [DOI] [PubMed] [Google Scholar]
- 20.Garbe K Pfizer’s JAK inhibitor sails through phase 3 in rheumatoid arthritis. Nat Biotechnol. 2011;29:467–8. [DOI] [PubMed] [Google Scholar]
- 21.Sewgobind VD, Quaedackers ME, van der Laan LJ, Kraaijeveld R, Korevaar SS, Chan G, et al. The Jak inhibitor CP-690,550 preserves the function of CD4CD25FoxP3 regulatory T cells and inhibits effector T cells. Am J Transplant. 2010;10:1785–95. [DOI] [PubMed] [Google Scholar]
- 22.Bristeau-Leprince A, Mateo V, Lim A, Magerus-Chatinet A, Solary E, Fischer A, et al. Human TCR alpha/beta+CD4−CD8− double-negative T cells in patients with autoimmune lymphoproliferative syndrome express restricted Vbeta TCR diversity and are clonally related to CD8+ T cells. J Immunol. 2008;181:440–8. [DOI] [PubMed] [Google Scholar]
- 23.Bleesing JJ, Brown MR, Dale JK, Straus SE, Lenardo MJ, Puck JM, et al. TCR-alpha/beta(+) CD4(−)CD8(−) T cells in humans with the autoimmune lymphoproliferative syndrome express a novel CD45 isoform that is analogous to murine B220 and represents a marker of altered O-glycan biosynthesis. Clin Immunol. 2001;100: 314–24. [DOI] [PubMed] [Google Scholar]
- 24.Marlies A, Udo G, Juergen B, Bernd S, Herrmann M, Haas JP. The expanded double negative T cell populations of a patient with ALPS are not clonally related to CD4+ or to CD8+ T cells. Autoimmunity. 2007;40:299–301. [DOI] [PubMed] [Google Scholar]
- 25.Oliveira JB, Bleesing JJ, Dianzani U, Fleisher TA, Jaffe ES, Lenardo MJ, et al. Revised diagnostic criteria and classification for the autoimmune lymphoproliferative syndrome (ALPS): report from the 2009 NIH International Workshop. Blood. 2010;116:e35–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Price S, Shaw PA, Seitz A, Joshi G, Davis J, Niemela JE, et al. Natural history of autoimmune lymphoproliferative syndrome associated with FAS gene mutations. Blood. 2014;123:1989–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boggio E, Clemente N, Mondino A, Cappellano G, Orilieri E, Gigliotti CL, et al. IL-17 protects T cells from apoptosis and contributes to development of ALPS-like phenotypes. Blood. 2014;123:1178–86. [DOI] [PubMed] [Google Scholar]
- 28.Chavele KM, Merry E, Ehrenstein MR. Cutting edge: circulating plasmablasts induce the differentiation of human T follicular helper cells via IL-6 production. J Immunol. 2015;194:2482–24855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stranges PB, Watson J, Cooper CJ, Choisy-Rossi CM, Stonebraker AC, Beighton RA, et al. Elimination of antigen-presenting cells and autoreactive T cells by Fas contributes to prevention of autoimmunity. Immunity. 2007;26:629–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.