

Abstract
Cyanines are indispensable fluorophores that form the chemical basis of many fluorescence-based applications. A feature that distinguishes cyanines from other common fluorophores is an exposed polyene linker that is both crucial to absorption and emission and subject to covalent reactions that dramatically alter these optical properties. Over the past decade, reactions involving the cyanine polyene have been used as foundational elements for a range of biomedical techniques. These include the optical sensing of biological analytes, super-resolution imaging, and near-IR light-initiated uncaging. This review surveys the chemical reactivity of the cyanine polyene and the biomedical methods enabled by these reactions. The overarching goal is to highlight the multifaceted nature of cyanine chemistry and biology, as well as to point out the key role of reactivity-based insights in this promising area.
Brief Description
Reactions involving the covalent modification of the cyanine polyene are enabling emerging approaches in optical sensing, super-resolution imaging, and near-IR uncaging.
Graphical Abstract
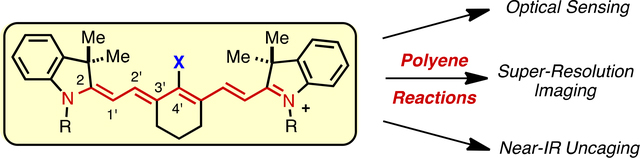
1. Introduction
The rich history of the cyanine class of fluorophores originates with the synthesis of 1 (Fig. 1A) in 1856.1 Over the intervening years new variants have been developed for use as photographic sensitizers,2 rewritable DVD response elements,3 laser dyes,4 analyte-responsive sensors,5 and for biomedical fluorescence techniques.6,7 The indocyanines, which contain a polyene linker connecting 3,3-dialkyl-indolenine heterocycles are generally preferred for biomedical applications. Within this group, the heptamethine cyanines, which contain 7 carbon atoms linking the indolenine heterocycles, are notable because their ~800 nm emission maxima falls in the center of the so-called “near-IR window”. Such wavelengths are advantageous for in vivo applications as autofluorescence is minimized and light penetration through tissue is maximized. The most medically significant of these is indocyanine green (ICG, 2), which was first studied over 50 years ago and is an FDA-approved clinical agent initially developed as a blood-pooling agent.8,9,10 Highlighting the expanding role of near-IR fluorescence in modern diagnostic medicine, new uses for ICG are being examined in over 50 active clinical trials, with especially promising progress achieved in diagnosing lymphoid disorders.11,12 A variety of indocyanines that lack polyene substitution, including Cy3 (4), Cy5 (5), Cy7 (6), and Alexa 647 (7) (Fig. 1B), are commercial products that serve as common biological labels in a range of fluorescence-based experiments.13,14,15,16 In 1992, a new structural class of heptamethine indocyanines of the general structure 3 (Fig. 1A) was introduced where the polyene region is modified with a heteroatom at C42 and a carbocyclic ring linking C32 and C52.17 These compounds exhibit generally improved physical properties, greater stability, and higher fluorescence quantum yields compared to ICG.18,19,20 One commercial derivative, IR800-CW (8, Fig. 1B), is being studied extensively for translation as an imaging agent in clinical settings.21
Fig. 1.

(A) Key background, (B) common commercial fluorophores, and (C) overview of cyanine polyene reactivity and applications (Cy3 and Cy5: GE Life Sciences, Alexa 647: Life Technologies, IR800-CW: LI-COR).
The defining chemical feature, and the key chromophore, of the cyanines is the polyene linker. It is perhaps not surprising that this cationic, sterically accessible olefinic functionality is subject to a range of chemical reactions. Maybe more surprising is the extent to which these reactions have be harnessed for productive use. Cyanine polyene reactions have been applied as central components of biomedical techniques in the fields of optical sensing, super-resolution microscopy, and, most recently, near-IR light-mediated uncaging. Here we survey reactions involving the covalent modification of the indocyanine polyene and the resulting applications. We provide a reactivity-driven perspective focused on mechanistic organic chemistry considerations. We anticipate this approach will facilitate the rational design of future applications. Moreover, organizing cyanine polyene reactivity in this fashion may help identify potentially undesirable cross-reactivity at the design stage. To provide the reader with an initial sense for the scope of cyanine polyene reactivity, Figure 1C includes many of the accessible reaction pathways available from the key heptamethine cyanine scaffold.
This review is organized into three sections: (1) reactions associated with chemical sensing and imaging methods, (2) chemical reactivity involved in photoswitching-dependent super resolution microscopy, and (3) photochemical reactions of cyanines and resulting applications. The scope of this review is limited to reactions that covalently modify the polyene linker region or directly attached atoms. Thus, we exclude a series of elegant optical sensing methods that use remote ligand exchange reactions to modify emissive properties through photophysical processes such as photoinduced electron transfer (PET).22,23,24,25 Aspects of cyanine polyene chemistry have been covered previously in surveys that centered on single-molecule fluorescence,15,26 cyanine synthesis and stability,27,28,29 and optical sensing.22,30,31,32,33,34,35,36,37 This review is the first to systematically organize biologically useful indocyanine polyene reactivity. We hope that this approach will clearly delineate the critical role these reactions play across diverse biological fields and assist in developing future applications of these remarkable molecules.
Section 2. Cyanine Polyene Reactivity for Optical Sensing
2.1. Background
Fluorogenic small molecules that provide a fluorescent signal in response to an enzymatic process or analyte of interest are integral to a variety of laboratory methods.38 With coumarin, fluorescein, or rhodamine fluorophores, the general paradigm involves the covalent modification of a key heteroatom (phenol or aniline) by some analyte or enzyme-specific functional group. This added chemical functionality eliminates, or at least reduces, the fluorescence signal and its removal, mediated by the analyte of interest, restores the characteristic emission. Extending these approaches into the near-IR range would facilitate monitoring analytes in blood, tissues, and whole organisms. As the most common near-IR fluorophore, heptamethine cyanines are an obvious candidate scaffold for developing such methods. However, the general approach discussed above is not applicable to conventional cyanine fluorophores. For example, heteroatoms on the indolenine heterocycle do not dramatically influence optical properties as the indolenine ring is outside of the key polyene chromophore. Consequently, the methods below, which involve modification to the cyanine polyene, are born from the necessity of finding alternatives. It is important to mention that a variety of highly useful strategies that rely on the disruption of FRET pairs or the use of PET strategies lie beyond the scope of this survey.22,39
2.2. ROS Sensing
Altered levels of reactive oxygen species (ROS) are hallmarks of various disease states and inflammatory processes. Consequently the development of fluorogenic strategies that provide real-time readouts of ROS concentrations are of significant interest.34 One such strategy uses hydrocyanines (9), which are easily formed by sodium borohydride reduction of the terminal C2-N bond in the cyanine polyene (Fig. 2A). Oxidation by various cellular ROS restores the fluorescent cyanine, with particularly rapid kinetics in the case of superoxide (O2●−) and hydroxyl radical (HO●).40 The oxidation reaction likely proceeds via sequential electron transfer through cyanine radical cation intermediates, however this has not been proven experimentally. Elevated ROS upon angiotensin-mediated oxidative stress was detected in cells and tissues, and in a mouse model involving the lipopolysaccharide (LPS) endotoxin-induced inflammatory response. It was subsequently observed that mono-deuterated variants of hydrocyanines, formed by addition of NaBD4 to the cyanine, display altered reactivity, with distinct kinetic isotope effects (KIE) between background oxidation (kH/kD = 3.7–4.7) and O2●− oxidation (kH/kD = 2.6–2.8).41 This KIE difference is proposed to improve signal-to-noise ratios and, consequently, provide higher sensitivity for ROS imaging. These molecules are finding extensive use for the detection of ROS in a variety of contexts.42,43,44,45,46,47,48
Fig. 2.

Cyanine polyene reactivity-based optical sensing approaches.
Potent oxidants can, under certain circumstances, react with the cyanine polyene and abolish all long wavelength absorption and emission. A creative approach to ROS sensing takes advantage of this otherwise deleterious process (Fig. 2B).49 Screening a series of commercially available cyanine fluorophores revealed that the ROS-mediated oxidative reactivity is highly variable and correlates strongly with oxidation potential. For example, IR786S, which bears a thioether at C42, had the lowest measured value (0.303 V) and reacted most readily with a range of ROS, including with O2●− and even with high concentrations of H2O2, the weakest oxidant tested. Conversely, Cy5SO3H with electron-withdrawing sulfonate groups on the indolenine ring system has the highest oxidation potential (0.628 V) and the greatest resistance to ROS reactivity. The covalent linkage of IR786S (ΦF = 0.035 with λex = 771 nm) and Cy5SO3H (ΦF = 0.171 at λex = 645 nm) forms 11, which exhibits dramatically reduced fluorescence (ΦF = 0.009 at λex = 771 nm and ΦF = 0.014 at λex = 645 nm) due to self-quenching. Exposure of 11 to the oxidants peroxynitrite (ONOO−), HO●, hypochlorite (ClO−), O2●−, and singlet oxygen (1O2) provided significant fluorescence enhancements at the Cy5 wavelength. The fluorescence increase arises from the selective oxidation of IR786S to restore the emission of the ROS-resistant fluorophore (Cy5SO3H). Among several applications, O2●− generated by membrane bound NADPH oxidase was detected in HL60 human promyelocytic leukemia cells. ROS production was also imaged in a mouse model of peritonitis. An important concept that emerges from these efforts is the significant variability in ROS-susceptibility between various cyanines. That is, certain cyanines can react with certain biologically relevant ROS, whereas other cyanines, for example those with electron-withdrawing groups on the indolenine rings, are nearly immune to such reactions. A detailed description of these oxidative processes, and particularly the specific features that control differential reactivity, would be of significant utility.
The concept of using a ROS-reactive/ROS-resistant pair of fluorophores was extended using highly stable fluorescent semiconducting polymer nanoparticles (SPN) as the ROS-resistant component.50 SPNs are a new class of arene-based mixed polymeric materials, which provide improved stability as compared to conventional fluorophores or quantum dots. The SPN surface was modified with C42-thiol-substituted heptamethine cyanines, thereby quenching a significant fraction of the SPN fluorescence. Selective oxidation of the heptamethine cyanine by ONOO−, HO●, and ClO− restores much of the lost SPN emission at 678 nm. This approach was used to visualize ROS flux in live murine macrophages and in a mouse model of LPS-induced peritonitis. A hepatocyte-targeted version allowed for real-time monitoring of drug-induced oxidative stress in the liver of live mice.51 A related polymeric system using rhodamine B as the ROS-resistant element has also been reported.52 In addition, the oxidation of heptamethine cyanines has also been used as a sensor for hypochlorous acid.53
In addition to ROS-sensing, the oxidative reactivity of cyanine fluorophores is the basis for an advanced imaging approach developed by GE.54 This method, termed multiplexed fluorescence microscopy, uses formalin-fixed paraffin-embedded samples, which is a standard protocol for archiving clinical samples. Cy3 and Cy5-labeled antibodies are applied, subjected to fluorescence microscopy, and then the cyanines are oxidatively inactivated using a proprietary alkaline oxidant mixture, with apparently minimal damage to the samples. This process can be repeated using various antibodies, allowing up to 61 proteins to be visualized in a single sample.
While fluorophores containing the standard cyanine polyene are by far the most common, other cyanine variants have also been studied. Shabat and coworkers recently introduced cyanines modified with an aryl ring in the center of the polyene.55,56 With conventional heptamethine cyanines, the two alkylated nitrogen atoms linked through a polyene act as a “donor-acceptor” pair, which is the key chromophore that provides the unique optical properties of these molecules. With these aryl ring incorporated variants, protonation or protection of the phenol forms a dicationic polyene, e.g. 13 (an “acceptor/acceptor” pair) with no meaningful red absorption. Unveiling the phenolate form, which adopts a quinone-methide-like tautomer, e.g. 14, introduces the key “donor/acceptor” arrangement of the nitrogen atoms thereby providing useful red absorption and emission. The pKa of the unmasked phenol is 4.5, which means these compounds adopt the fluorescent phenolate form under most biologically relevant conditions. Masking the phenol with a cleavable group creates fluorescent turn-on sensors, which is exemplified here by the selective sensing of H2O2 using compound 13 (Fig. 2C). Thus, non-fluorescent 13 is converted to a fluorescent form, 14, upon boronic acid to phenol oxidation and p-quinone methide elimination sequence. It is important to note that the fluorescent form exhibits optical properties (ΦF = 0.16, ε = 5.2 × 104 M−1 cm−1 at 570 nm) that are quite different than the conventional heptamethine cyanine scaffold, although the large Stokes shift may be ideal for certain applications. Nevertheless, these molecules are very promising and a variety of additional applications have been reported since the initial disclosure in 2011, including sensing of nitroreductase,57 β-galactosidase,58 cathepsin B,59 and thiols.60,61
A conceptually different approach utilizes analyte-specific cleavage of C42-carbamates to provide C42-amino heptamethine cyanines, which are readily distinguishable by virtue of the hyposochromic shift of the amino variants.62 Aryl boronic acid or aryl azide cyanines of the structure 15 enable H2O2 and H2S sensing, respectively, with these analytes producing phenols or anilines that undergo intramolecular 1,6-elimination and CO2 extrusion to provide 16 (Fig. 2D). Simultaneous measurement of the relative fluorescence from the initial carbamate and the product amine enables ratiometric readout. Ratiometric measurement can have certain advantages over simpler turn-on approaches, including greater independence from effects of probe concentration and local environment. Hypsochromic shifts in the absorption and emission of 15a were only observed in the presence of H2O2 among various other ROS, with up to a 22-fold increase in the fluorescence emission. Similar results were obtained with the azide variant 15b, which showed selectivity for H2S over other ROS and thiols (ΔFR/FNIR ~18-fold). The boronic acid derivative 15a was used to detect exogenous H2O2 in fibroblastic NIH 3T3 cells. Treatment of the same cell line with zinc(II) chloride, an H2S scavenger, ablated the intracellular fluorescence signal of azide 15b at the blue-shifted emission wavelength.
The detection of lipid peroxides is of significant interest due to the formation of these species during oxidative stress and in a variety of disease states.63 An unusual approach designed to assess lipid peroxide concentrations was reported using cyanine 17 (Fig. 2E). Compound 17 was formed from a C42-chloro derivative by lithiation with Li(0) and addition of PPh2Cl to form a C42 C-P bond, presumably followed by phosphine lone pair polyene addition.64 Occurrence of a polyene reaction was clearly defined by loss of the characteristic cyanine absorption, although a detailed structural assignment using NMR or X-ray crystallography for this quite unusual product has not been reported. Oxidation to form the phosphine oxide 18 with methyl linoleate hydroperoxide (MeLOOH) restores the cyanine λabs at 747 nm with concurrent increase in emission. The turn-on effect is selective for ROOH over other ROS and metal ions. Exposure to t-Bu and cumene hydroperoxides did not lead to a restoration of cyanine absorption and emission, suggesting that 17 may be particularly selective for unhindered organic hydroperoxides. Oxidative stress induced by stimulation of macrophages with a phorbol ester was analyzed using 17. A related probe involving C42-selenium substitution has also been reported.65,66
The potential for environmental alkyl mercury toxins to induce neurotoxicity have created a need for improved sensing methods.67 One such approach involves a methyl mercury-induced cyclization of the C42 amine, 19, on a pendant thiourea to form a cyclic guanidine (20, Fig. 2F).68 Related mercury-mediated cyclization reactions have been used elsewhere for sensing-applications.69 Mercury salts selectively promote this cyclization, with little competing reactivity from cadmium, silver, copper, and lead salts. The reaction converts the blue-shifted amine derivative (λabs = 670 nm) to a C42-guanidine derivative (λabs = 810 nm), allowing for efficient ratiometric sensing and cellular fluorescence imaging.
2.3. pH Sensing
Many approaches have been explored for the generation of pH-sensitive fluorescent molecules.70 To extend these techniques into the near-IR range, several have used the protonation of C42-amino heptamethine cyanines. This category includes the use of a C42-m-amino-phenol derivative, 21 (Fig. 3A).71 Compound 21 displays a fairly complex pH profile, likely due to the presence of two titratable protons, with a useful 10-fold increase in fluorescence intensity within a pH range of 4.0–6.5. Another method uses C42 piperidine derivatives, such as 23 (Fig. 3B).72 Here, the far-red absorption of the unprotonated form is converted to a near-IR absorption upon protonation. The distinct absorption spectra enabled ratiometric sensing, which increased the sensitivity of these measurements. Compounds 21 and 23 both capitalize on altering the capacity of the central C42-amine lone pair to donate electron density into the cyanine polyene. C42-free amines exhibit an atypical cyanine absorption with a significantly hypsochromically-shifted absorption spectra, whereas the protonated forms more closely resemble other C42 derivatives (e.g. Cl-, O-, C-, or S-). Another recent elegant approach used heptamethine cyanines substituted at C42 with a pyrazole heterocycle.73
Fig. 3.

pH-Sensing approaches.
The cellular internalization of many tumor-targeting agents is accompanied by significant reduction of the local pH as the targeting agent/receptor complex is trafficked through endosomal compartments to the lysosome. An approach to selectively report on ligand/receptor internalization was recently disclosed by Achilefu and coworkers. These studies employ a heptamethine cyanine labeled the with the αvβ3 integrin-avid peptide cRGD, 25, that undergoes fluorescence enhancement in acidic environments (Fig. 3C).74 The pH sensitivity of 25 is derived from the norcyanine scaffold, which bears unsubstituted, as opposed to alkylated, indolenine nitrogen atoms. The molecule exhibits a characteristic heptamethine cyanine absorption and emission only upon protonation of the indolenine nitrogen (pKa ~ 4.7). At neutral pH, the absorption of the uncharged cyanine is dramatically shifted to the visible region (λabs ≈ 520 nm). The cRGD peptide-αvβ3 integrin receptor interaction led to selective trafficking to late endosomal/lysosomal compartments in MDA-MB-435 and 4T1/luc cells in vitro. Productive imaging was further demonstrated in a 4T1/luc orthotopic tumor model. A related approach has also been investigated using pentamethine cyanines.75
2.4. Thiol Sensing
Thiols, particularly cysteine (Cys), homocysteine (Hcy), and glutathione (GSH), play important roles in redox homeostasis, and diminished thiol regulation is implicated in several disorders and disease states.76 A variety of approaches to thiol sensing have been developed using the unique chemistry of the C42 position of heptamethine cyanines. One such approach for selective sensing of intracellular Cys uses 27, which is functionalized with an acrylate ester at C42 (Fig. 4A).77 Cysteine addition forms a Michael-addition adduct that rapidly cyclizes/eliminates to afford the C42-ketone 28. The absorption spectrum of the product 28 is hypsochromically-shifted relative to 27 and, consequently, can be measured almost independently with the resulting ratio representing reaction progress. The rate of ketone formation with Cys is approximately tenfold greater than that of Hcy, likely owing to increased kinetics for forming the cyclic intermediate (seven-membered ring formation faster than eight). Intracellular Cys levels were examined in vitro using MCF-7 cells cultured in standard and glucose-deficient environments, which elevates both GSH and its precursor, Cys, due to oxidative stress. 78 A conceptually related strategy for H2S sensing using 29 also was recently reported (Fig. 4B).79 Nucleophilic addition of hydrogen sulfide to the aldehyde triggers a second nucleophilic addition to the ester to provide the corresponding C42-ketone 30. The probe displays selectivity toward H2S over other thiol-containing molecules, such as Cys and GSH, and exhibits a linear correlation between fluorescence enhancement at 625 nm and H2S concentration with a detection limit of 10 nM. Neither of these C42 cyanine esters have been applied in animal models, and it is possible that premature ester-to-ketone conversion, perhaps through hydrolysis or esterase activity, precludes live animal applications. Notably, the pKa of the ketone product, ~4.5, should be considered when contemplating the reactivity of these compounds.80 Along these lines, a simple C42-acetylated derivative was used as an esterase-activated probe.81 A nearly identical molecule has also been reported to be a hydrazine sensor.82
Fig. 4.

Thiol sensing methods.
Another approach to thiol sensing sought to address the key role of mitochondrial thiol regulation, an important area given the central role of mitochondria in ROS generation and cellular redox signaling.83 In particular, the mitochondrial glutathione (mGSH) pool is a critical antioxidant reservoir that protects mitochondrial proteins from oxidative stress generated during normal aerobic metabolism.84 Therefore, a near-IR fluorescent probe capable of localizing to mitochondria and distinguishing mGSH from other thiols (e.g. Cys and Hcy) would be of significant utility. A heptamethine cyanine, 31, was recently reported as a probe for selective sensing of mGSH (Fig. 4C).85 In this design, the cationic heptamethine system of 31 in concert with the relatively hydrophobic nBu esters leads to selective mitochondrial targeting. Meanwhile, the C42-diaryldiazoether serves as a fluorescence quencher via PET that is interrupted by GSH-selective reactivity to form the GSH-incorporated product 32. A measureable “turn-on” effect was observed upon exposure to GSH at concentrations as low as 26 nM. NMR analysis using β-mercaptoethanol support a mechanism in which C42-thiolate nucleophilic substitution leads to loss of the diaryldiazoether functionality, thereby interrupting PET quenching. Evidence is provided that, unlike thiol addition in organic solvents (which proceeds through a SRN1 mechanism),17 the thiolate nucleophilic substitution reaction in protic conditions proceeds through an ionic reaction pathway. Specifically, a 10-fold rate enhancement was observed in protic solvent (methanol) over aprotic solvent (DMSO), while addition of a radical scavenger (O2) and an electron scavenger (nitrobenzene) had no effect. The fluorescence signal from 31 at 810 nm increased approximately 450-fold in the presence of GSH, while exposure to two other biologically relevant thiols, Cys and Hcy, shifted the emission maximum to 747 nm and only resulted in a 10- and 15-fold fluorescence enhancement, respectively. This dramatic selectivity results from the C42-substitution adducts formed from Cys and Hcy undergoing subsequent intramolecular cyclization to provide a C42-N-linked product, 33 (Fig. 4C). A recent synthesis-oriented study observed a similar intramolecular cyclization.86 Notably, mitochondrial localization was derived specifically from the n-Bu ester functional group, as the corresponding carboxylic acids and N-(2-morpholinoethyl)amides induced cytosolic and lysosomal accumulation, respectively. The sensing reaction was shown to be dependent on mGSH levels, as “turn-on” fluorescence intensity decreased in the presence of GSH scavengers and increased in the presence of GSH enhancers. A related approach has been reported for hydrogen sulfide sensing using the conversion of a C42-chloro cyanine, which displays significant fluorescence, to the nonfluorescent C42-free thiol form.87
A fundamental understanding regarding the timing of cellular internalization and drug release could provide key insights useful for drug development. A fluorogenic strategy using 34 provided a fluorescent readout for intracellular accumulation and drug release (Fig. 4D) through a method based on the elevated cellular thiol concentrations.88 The design of this approach relies on another instance of distinguishing a C42-amino fluorophore from one with a less π-donating C42 substituent. In this case, the amine exhibits a significant detectable emission when excited at 615 nm, whereas the carbamate variant has only modest emission when excited at the same wavelength. Compound 34 contains a C42-carbamate linked through a disulfide to a carbamate modified version of gemcitabine, a nucleoside analogue currently used as a broad spectrum chemotherapeutic.89 The cyanine scaffold was also appended with a folate-targeting ligand to enable selective uptake in cells over-expressing the folate receptor. Intracellular disulfide cleavage and intramolecular cyclization cleaves the C42-carbamate to provide the C42-amine, 36, creating the fluorescence “turn-on” effect (Fig. 4D). Concurrently, a similar process unmasks the 52-hydroxyl of gemcitabine, 35, affording the active drug molecule. The disulfide linkage is cleaved by thiols, with an approximately 40-fold fluorescence enhancement observed for 34 in the presence of GSH and negligible increase in the presence of nonthiol-containing amino acids and metal ions. Reduction of intracellular thiols using N-ethylmaleimide, a thiol-reactive electrophile, attenuates the turn-on effect. This approach was used to evaluate the receptor-mediated cellular internalization of 34 in folate receptor (FR)-positive KB cells and not FR-negative cells in both in vitro and in vivo experiments.
2.5. Other Methods
While the majority of cyanine polyene chemistry useful for sensing applications involves heptamethine cyanines, there are isolated examples of reactions in other systems. In particular, it has been observed that hybrid indocyanine/coumarin dyes react with biologically relevant nucleophiles at the electrophilic indolenine position. One such approach involves compound 37, which was applied towards the detection of mitochondrial H2S (Fig. 5A).90 Nucleophilic addition of HS− at the C2-indolenine carbon to form 38, which was confirmed by NMR, abolishes merocyanine emission while leaving the coumarin emission unaffected. The resulting ratiometric measurement was quite selective for H2S (pKa ~7) over Cys, GSH, Hcy, and bovine serum albumin (pKas ≥ 8.5). 37 preferentially localizes to the mitochondria in MCF-7 cells and was used for the ratiometric visualization of exogenous H2S. A closely related strategy has been explored for cyanide sensing.91,92
Fig. 5.

(A) Coumarin/cyanine hybrid for H2S sensing and (B) in situ cyanine formation with engineered CRABPII protein.
Fluorescent complexes formed from the combination of encoded proteins and exogenously supplied small molecules can provide unique optical properties and a level of temporal and spatial control not possible with conventional fluorescent proteins alone.93 For example, SNAP- or Halo-tag approaches allow suitably modified fluorophores to be affixed to nearly any protein through the genetic incorporation of the tagging complex.94 An exciting contribution to this rapidly expanding field involves the nucleophilic addition of a lysine residue on an engineered protein to a non-fluorescent cyanine precursor, 39, to form a fluorescent cyanine/protein complex in situ (Fig. 5B). 95 As one might expect, fluorescence requires the protonated form of the Schiff base, which predominates under most biologically relevant conditions due to its pKa of 9.6. The protein reengineering process used cellular retinoic acid binding protein II (CRABPII), a retinoic acid carrier protein, as the starting point. The design of this approach was stimulated by the structural homology between the cyanines and the retinoids, and capitalized on recent studies from the same group dealing with rational modification of CRABPII/retinal complexes.96,97
Section 3. Cyanine Polyene-Based Switching for Super Resolution Microscopy
Super resolution methods that exceed the diffraction limit are revolutionizing fluorescence microscopy.98,99,100,101 Among several general approaches, one set of methods relies on imaging single molecules undergoing controlled switching between dark and emissive states. By collecting photons from a few single-molecule emitters, which represent only a fraction of all the labels in the sample, sub-diffraction limit resolution can be achieved by calculating the center of the single-molecule emission pattern (generally assumed to be a simple Gaussian distribution) prior to its return to a dark state. Repeated activation, localization, and deactivation forms a high-resolution pattern (Fig. 6A). The two most common methods of this type are referred to as photoactivated localization microscopy (PALM), which generally involves photoactivateable fluorescent proteins, and stochastic optical reconstruction microscopy (STORM), which usually employs cyanine fluorophores (particularly Alexa 647). While key aspects of these methods span numerous scientific disciplines, with many details residing beyond the scope of this review, a critical component is the chemistry controlling the pivotal on/off events.102,103 The chemistry of cyanine photoswitching, which featured prominently in the early methodological developments and is still a pillar of these approaches, has been elucidated to a detailed molecular level and is reviewed below.
Fig. 6.

(A) General scheme for switching-dependent super resolution microscopy and (B) cyanine switching reactivity.
The propensity of cyanines, particularly pentamethine variants, to reversibly enter relatively long-lived dark states at the single molecule level was observed in early single molecule spectroscopy studies.104,105 These blinking events can be controlled by altering between 488 nm (on) and 633 nm (off) light, and require efficient oxygen removal and high concentrations (mM) of a primary thiol. The utility of this observation for super resolution imaging (as STORM) was subsequently developed by Zhuang and coworkers.106,107 Subsequently, the blinking phenomenon was shown to involve light-mediated primary thiolate addition to C22 of the pentamethine cyanine polyene (Fig. 6Ba).108 Thus, illumination of a pentamethine cyanine derivative (41) at 633 nm in the presence of β-mercaptoethanol (βME) decreased the near-IR absorption at 650 nm due to the formation of a thiol-cyanine adduct (42). UV irradiation restores the intact cyanine and its 650 nm absorption. Preferential thiol addition to C22 on a model pentamethine cyanine was demonstrated by MS/MS analysis. Mechanistically, the reaction is proposed to involve initial formation of a cyanine-thiolate encounter complex, excitation, and final thiolate addition, with the key thiolate addition step likely involving an initial electron transfer followed by thiyl radical addition. The rate of switching to the dark state initially increases linearly (first-order kinetics) and saturates above 1 M βME concentration (zero-order kinetics), and the reaction rate is reduced in the presence of a radical quencher (isoascorbate). Consistent with the overall reaction model, blinking is also effective using other primary thiols (β-mercaptoethylamine and Cys derivatives), and, in recent work, the high intracellular GSH concentration has enabled live-cell imaging without the addition of thiol-containing buffers.109 This process is compatible with pentamethine cyanines and heptamethine cyanines, but not trimethine cyanines. A recent detailed analysis provided a comprehensive examination of the applicability of various commercial fluorophores to this method.110 Of note, while thiol adducts of heptamethine cyanines have been observed using mass spectral analysis,108 the site of reactivity (i.e. which carbon is modified in the polyene chain) has not been determined. Critically, with other classes of fluorophores, and possibly with certain cyanines, dark states arise through redox events to form non-fluorescent radical intermediates.
It has also been demonstrated that relatively stable dark states can be generated through phosphine addition to the cyanine polyene (Fig. 6Bb).111 Exposure of Cy5 to millimolar concentrations of tris(2-carboxyethyl)phosphine (TCEP), a widely used reducing agent, leads to rapid formation of a phosphine adduct, 43. In contrast to the thiol addition discussed above, this reaction occurs spontaneously in the absence of light, with UV irradiation in this case restoring the intact cyanine polyene. Kinetic analysis supports a reversible bimolecular reaction model, with Keq = 0.91 mM-1. Convincing mass spectral and 1H NMR evidence for TCEP addition to C22 of the pentamethine polyene has been obtained. As above, other pentamethine and heptamethine variants were also reversibly quenched by TCEP, however trimethine cyanines were again unaffected. This approach enabled two-color STORM imaging using a combination of Alexa 647 and Alexa 750.
Another method to generate stable dark states for super resolution imaging uses the reduced form of cyanines, the hydrocyanines, which were discussed in Section 2.2 for ROS sensing.112 Treatment of a variety of cyanine dyes with sodium borohydride results in hydride addition to the indolenine carbon, causing a large blue shift that leaves the resulting hydrocyanine (44) in an effective “dark” state (Fig. 6Bc). Subsequent illumination with UV or violet light recovers a variable percentage of the reduced dye molecules to the fluorescent state, enabling STORM imaging. The rate of the oxidation process can be enhanced through the use of riboflavin, which acts as a photosensitizer in the presence of UV light to generate cyanine-activating ROS. Low recovery yields represent a limitation of this approach, but this can be somewhat compensated for by increasing the degree of labeling. The biologically compatible oxidation of hydrocyanines to cyanines illustrates how useful reaction-based insights can be. These two different applications, ROS-sensing (discussed in Section 2.2) and STORM activation, are totally distinct at a cursory glance but, nevertheless, involve the same enabling chemical transformation.
An active area of research is the development of small molecules designed to photoswitch spontaneously. One chemical approach towards this goal is based on hybrid coumarin/cyanines such as 45 (Fig 6Bd).113 The pendant phenol, which preferentially forms the ring-closed oxazine form 46, opens (on-state, 45) upon UV illumination on a subnanosecond timescale and spontaneously reverts to the ring-closed isomer (off-state) on a submicrosecond timescale. The ring-open state can be trapped by protonation of the resulting phenolate. The concept of using a pendant nucleophile for photoswitching was also recently reported on the rhodamine scaffold.114
4. Photochemistry of the Cyanine Polyene and Resulting Applications
4.1. Cyanine Photooxidation - Background
As with most fluorophores, cyanines are susceptible to light-dependent decomposition or photobleaching, which can be deleterious for a variety of fluorescence experiments. Starting with a seminal study by Byers at the Kodak laboratories in 1976, a several studies examined the chemical basis of this photodegradation process.115 As observed across several structurally desperate cyanine scaffolds, photolysis results in oxidative C-C cleavage occurring at certain positions along the polyene chain.116,117,118,119,120,121,122,123 Representative of this cleavage reaction is the conversion of ICG to products 47 to 50 (Fig. 7A). The most common mechanism to describe this process involves photosensitization to form 1O2, dioxetane formation, and final dioxetane cleavage to form carbonyl products (Fig. 7B). Key pieces of evidence to support this pathway include: (1) a dependence on oxygen concentration,117 (2) decreased, though not completely ablated, kinetics in the presence of 1O2 quenchers (e.g. NaN3),120 (3) accelerated kinetics in deuterated solvents which extend the lifetime of singlet oxygen,124 (4) the use of singlet oxygen trapping agents,118,124 and (5) the direct observation of dioxetane intermediates by mass spectrometry (+32 mass units).120,123 It is important to note that cyanine photobleaching may involve alternative pathways to that presented in Figure 7.118,122,125 The most likely alternative mechanism involves electron transfer, rather than energy transfer, from the triplet state. Electron transfer with oxygen would produce both O2●−, which may ultimately generate HO● or other ROS, and the potentially reactive cyanine-radical cation. Critically, the products resulting from this alternative pathway, and the substrates or circumstances that favor it, remain to be defined. A detailed understanding of the interplay between these two possible mechanisms, as well as a quantitative accounting of the products formed through photolysis, is still required.
Fig. 7.

(A) ICG photobleaching products and (B) general singlet-oxygen photooxidation mechanism.
4.2. Approaches to Circumvent Cyanine Photooxidation
Several strategies have been developed to reduce the photobleaching rate of cyanines.126 One general approach is to introduce electron-withdrawing groups that remove electron density from the polyene, presumably to reduce the kinetics of 1O2 reactions. Perfluorination of the aromatic rings of thiadicarbocyanine 51 to provide 52 appreciably reduced aggregation and photobleaching (by ~3.5-fold) compared to the all protio counterpoint, although with somewhat reduced molar absorptivity (Fig. 8A).121 Alternatively, the cyano-substituted merocyanine 54 displayed a reduced rate (~39-fold) of photobleaching versus the analogue, 53, that lacks the electron-withdrawing functionality (Fig. 8B).122 Exogenously generated 1O2 (formed by methylene blue photosensitization) reacted with the original merocyanine 53, but not with 54. The fact that 54 photobleaches at all, despite being apparently immune to exogenously-generated 1O2, is evidence for 1O2 independent pathways in the photobleaching of, at least some, cyanines.
Fig. 8.

Strategies for improving cyanine photostability.
Stable emission profiles are particularly critical for single molecule fluorescence experiments and a variety of efforts have addressed cyanine photobleaching in this context. Oxygen removal, through enzymatic methods, reduces the rate of irreversible bleaching but leads to rapid (ms) intensity fluctuations (blinking) due to long-lived triplet states.26 Triplet quenchers (such the hydroquinone, Trolox, and cyclooctatetraene) have been added to mounting media to mitigate blinking. These triplet quenchers both reduce dark states and decrease the rate of photobleaching.127 Building on the use of these molecules as antifade additives, Blanchard and coworkers recently introduced the concept of “self-healing” cyanines, which involves the covalent linkage of a triplet state-quenching group (TSQ) tethered to a pentamethine cyanine (56, Fig. 8C).125,128,129 It was demonstrated, in both bulk and single molecule fluorescence experiments, that the appended TSQ (nitrobenzyl, cyclooctatetraene, or Trolox) improves photostability. In the absence of oxygen removal in bulk solution there was approximately a 7-fold improvement with nitrobenzyl and cyclooctatetraene modification and approximately a 2-fold improvement with Trolox modification, with greater stability improvements observed with enzymatic oxygen depletion. The ability of proximate triplet quenchers to improve cyanine photostability has also been investigated using DNA templating.130,131
4.3. Cyanine Photooxidation for Near-IR Uncaging
Much as near-IR fluorescence is required for many live-animal imaging applications, near-IR photocaging methods are required to advance the photocaged/uncaged logic into organismal contexts. There has been significant progress using two-photon approaches, which are ideally suited for rapid uncaging in the small focal volumes achieved with pulsed femtosecond laser sources, and in thermal-release methods usually involving metallic nanoparticles. 132,133,134,135 Existing single-photon photochemistry initiated by light of longer wavelengths is quite sparse, especially when compared to the variety of available lower wavelength approaches, with only a handful of reports just appearing in recent years. 136,137,138,139,140,141
The photochemical reactivity of heptamethine cyanines discussed above, coupled with their well-established biocompatibility, suggested to us that these fluorophores might have a second use as a platform for near-IR uncaging. A first foray into this area led to an approach using C42-amine-linked heptamethine cyanines of the general type 57 (Fig. 9).123 We projected that the key bond breaking event would occur through the selective hydrolysis of the C42-N bond in photooxidation products 58 and 59 but not in the starting compound 57. The motivating notion was that altered π-conjugation would increase the electrophilic reactivity of the key C42-N bond (i.e. through the increased iminium character). As shown in Figure 9A, the overall reaction sequence comprises photosensitization to form 1O2, formation of dioxetanes, dioxetane cleavage to from the hydrolytically labile intermediates 58 and 59, and final hydrolysis/cyclization to provide the uncaged phenol. The release reaction was characterized by NMR, absorption, fluorescence, and mass spectrometry methods. Remarkably, each of the key intermediates are observable by mass spectral analysis, and time-course studies validated the general mechanistic model. These compounds display excellent dark stability, the photochemical reactions can be initiated by a convenient, low-energy 690 nm LED light source, and the chemistry proceeds readily under a variety of conditions. In proof-of-concept applications, this method was used to inhibit cell viability through the light-dependent release of the estrogen receptor antagonist, 4-hydroxycyclofen, using cyanine 57C (Fig. 9B,C). We also demonstrated, through uncaging of the same compound, near-IR light mediated control of gene expression in a ligand-dependent CreERT/LoxP-reporter cell line.142 One advantageous component of this method is that the fluorescence of these compounds can be used to evaluate pre-uncaging localization with low light fluence, prior to applying high fluence for small molecule delivery. Thus, this photooxidation of heptamethine cyanines, originally associated only with fluorophore degradation, has a promising second career as the centerpiece of a new approach for near-IR uncaging.
Fig. 9.

(A) Near-IR cyanine uncaging mechanism, (B) caged compounds, and (C) applications.
Conclusion
Molecules containing the cyanine scaffold are fundamental components of many experimental biological procedures and clinical diagnostic protocols. As detailed above, reactions involving the exposed cyanine polyene play pivotal roles in a number of emerging biomedical methods. While cyanines themselves are not new, reactions of the cyanine polyene are almost all quite recent. A promising component of many of these emerging techniques is the use of heptamethine cyanines. The near-IR optical properties of these molecules render these approaches excellent candidates for translation into complex animal and, ultimately, clinical settings. The recent clinical success of fluorescence-guided surgery clearly underscores the potential of near-IR optical methods.143
The biological utility of these molecules rests, by necessity, on a chemical foundation. Quite notably, several of the methods discussed above were discovered by identifying a productive use for molecular events that could have easily been seen as only detrimental to standard imaging applications (e.g. oxidative polyene cleavage for ROS sensing, single molecule blinking for super resolution microscopy, and photooxidation for near-IR uncaging). The conversion of these undesirable, typically loss of signal events, into useful procedures was facilitated by a mechanistic understanding of the underlying chemistry. Future efforts will need to define approaches that modulate and distinguish between the various, likely sometimes competing, reaction pathways. Consequently, interdisciplinary efforts that blend physical organic chemistry, complex molecule synthesis, and advanced chemical biology and biomedical techniques are essential.
Acknowledgments
We thank Rolf Swenson (NIH, NHLBI) and members of the Chemical Biology Laboratory for helpful suggestions. This work was supported by the Intramural Research Program of the National Institutes of Health, Center for Cancer Research, and the National Cancer Institute, National Institutes of Health.
Biographies
Alexander Gorka earned his B.S. in Chemistry in 2008 from Monmouth University, NJ. He went on to obtain a Ph.D. in Chemistry under Prof. Paul Roepe at Georgetown University, where he studied drug pharmacology and resistance in Plasmodium falciparum malaria. He is currently a postdoctoral fellow with Dr. Martin Schnermann in the Chemical Biology Laboratory at the National Cancer Institute, working on developing and applying near-IR light-based uncaging strategies for drug delivery and control of gene expression.
Roger Nani graduated from Boston College in 2006 with a B.S. degree in Biochemistry. After working at Amgen in Cambridge, MA he joined Professor Sarah E. Reisman’s group at the California Institute of Technology, earning a Ph.D. in Organic Chemistry in 2013. He is currently engaged in postdoctoral research with Dr. Martin J. Schnermann in the Chemical Biology Laboratory at the National Cancer Institute, working on the discovery of near-IR light uncaging methods using heptamethine cyanine photochemistry.
Martin Schnermann is an investigator in the Chemical Biology Laboratory in the intramural program of the National Cancer Institute (NCI). He attended Colby College and graduated in 2002 with degrees in Chemistry (research with Prof. Dasan Thamattoor) and Physics. After a year at Pfizer, he was a graduate student Prof. Dale Boger at the Scripps Research Institute and obtained his Ph.D. in 2008. He then completed an NIH-postdoctoral fellowship with Prof. Larry Overman at the University of California, Irvine. In 2012, Dr. Schnermann joined the NCI where his research focuses on the development of new chemical approaches for drug delivery and imaging, as well as the synthesis of natural products of relevance to cancer imaging and therapy.
Picture

Roger Nani (Right), Alex Gorka (Center), Martin Schnermann (Left)
References
- 1.Williams CHG, Trans. Roy. Soc. Edinburgh 1856, 21, 377. [Google Scholar]
- 2.Lenhard JR, Hein BR and Muenter AA, J. Phys. Chem, 1993, 97, 8269. [Google Scholar]
- 3.Mustroph H, Stollenwerk M and Bressau V, Angew. Chem. Int. Ed, 2006, 45, 2016. [DOI] [PubMed] [Google Scholar]
- 4.Fabian J, Nakazumi H and Matsuoka M, Chem. Rev, 1992, 92, 1197. [Google Scholar]
- 5.Yuan L, Lin WY, Zheng KB, He LW and Huang WM, Chem. Soc. Rev, 2013, 42, 622. [DOI] [PubMed] [Google Scholar]
- 6.Luo SL, Zhang EL, Su YP, Cheng TM and Shi CM, Biomaterials, 2011, 32, 7127. [DOI] [PubMed] [Google Scholar]
- 7.Pansare VJ, Hejazi S, Faenza WJ and Prud’homme RK, Chem. Mater, 2012, 24, 812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Busch DR and Chance B, Diffuse Optical Tomography and Spectroscopy in Molecular Imaging Principles and Practice, Weissleder R, Ross BD, Rehemtulla A and Gambhir SS, Eds.; Peoples: Connecticut, 2010; p. 214–215. [Google Scholar]
- 9.Frangioni JV, Curr. Opin. Chem. Biol 2003, 7, 626. [DOI] [PubMed] [Google Scholar]
- 10.Alford R, Simpson HM, Duberman J, Hill GC, Ogawa M, Regino C, Kobayashi H and Choyke PL, Mol. Imaging, 2009, 8, 341. [PubMed] [Google Scholar]
- 11.From https://clinicaltrials.gov, search term ICG.
- 12.Marshall MV, Rasmussen JC, Tan IC, Aldrich MB, Adams KE, Wang X, Fife CE, Maus EA, Smith LA and Sevick-Muraca EM, Open. Surg. Oncol. J, 2010, 2, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ernst LA, Gupta RK, Mujumdar RB and Waggoner AS, Cytometry, 1989, 10, 3. [DOI] [PubMed] [Google Scholar]
- 14.Mujumdar RB, Ernst LA, Mujumdar SR, Lewis CJ and Waggoner AS, Bioconjug. Chem, 1993, 4, 105. [DOI] [PubMed] [Google Scholar]
- 15.Stennett EMS, Ciuba MA and Levitus M, Chem. Soc. Rev, 2014, 43, 1057. [DOI] [PubMed] [Google Scholar]
- 16.Ye Y, Li WP, Anderson CJ, Kao J, Nikiforovich GV and Achilefu S, J. Am. Chem. Soc, 2003, 125, 7766. [DOI] [PubMed] [Google Scholar]
- 17.Strekowski L, Lipowska M and Patonay G, J. Org. Chem, 1992, 57, 4578. [Google Scholar]
- 18.Patonay G, Kim JS, Kodagahally R and Strekowski L, Appl. Spectrosc, 2005, 59, 682. [DOI] [PubMed] [Google Scholar]
- 19.Strekowski L, Lipowska M, Gorecki T, Mason JC and Patonay G, J. Heterocyclic. Chem, 1996, 33, 1685. [Google Scholar]
- 20.Tarazi L, George A, Patonay G and Strekowski L, Talanta, 1998, 46, 1413. [DOI] [PubMed] [Google Scholar]
- 21.Marshall MV, Draney D, Sevick-Muraca EM and Olive DM, Mol. Imaging Biol, 2010, 12, 583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuan L, Lin WY, Zheng KB, He LW and Huang WM, Chem. Soc. Rev, 2013, 42, 622. [DOI] [PubMed] [Google Scholar]
- 23.Que EL, Domaille DW and Chang CJ, Chem. Rev, 2008, 108, 1517. [DOI] [PubMed] [Google Scholar]
- 24.Sasaki E, Kojima H, Nishimatsu H, Urano Y, Kikuchi K, Hirata Y and Nagano T, J. Am. Chem. Soc, 2005, 127, 3684. [DOI] [PubMed] [Google Scholar]
- 25.Li Y, Sun Y, Li J, Su Q, Yuan W, Dai Y, Han C, Wang Q, Feng W and Li F, J. Am. Chem. Soc, 2015, DOI: 10.1021/jacs.5b04097. [DOI] [PubMed] [Google Scholar]
- 26.Levitus M and Ranjit S, Q. Rev. Biophys, 2011, 44, 123. [DOI] [PubMed] [Google Scholar]
- 27.Henary M and Mojzych M, Stability and Reactivity of Polymethine Dyes in Heterocyclic Polymethine Dyes: Synthesis, Properties and Applications; Strekowski L Ed.; Topics in Heterocyclic Chemistry 14; Springer-Verlag: Berlin; 2008, pp. 221. [Google Scholar]
- 28.Panigrahi M, Dash S, Patel S and Mishra BK, Tetrahedron, 2012, 68, 781. [Google Scholar]
- 29.Mishra A, Behera RK, Behera PK, Mishra BK and Behera GB, Chem. Rev, 2000, 100, 1973. [DOI] [PubMed] [Google Scholar]
- 30.Yin J, Hu Y and Yoon J, Chem. Soc. Rev, 2015, DOI 10.1039/C4CS00275J. [DOI] [Google Scholar]
- 31.Guo ZQ, Park S, Yoon J and Shin I, Chem. Soc. Rev, 2014, 43, 16. [DOI] [PubMed] [Google Scholar]
- 32.You Y and Nam W, Chem. Sci, 2014, 5, 4123. [Google Scholar]
- 33.Lippert AR, J. Inorg. Biochem, 2014, 133, 136. [DOI] [PubMed] [Google Scholar]
- 34.Chen X, Tian X, Shin I and Yoon J, Chem. Soc. Rev, 2011, 40, 4783. [DOI] [PubMed] [Google Scholar]
- 35.Lin VS, Chen W, Xian M, Chang CJ, Chem. Soc. Rev, 2015, DOI 10.1039/C4CS00298A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singha S, Kim D, Seo H, Cho SW and Ahn KH, Chem. Soc. Rev, 2015, DOI: 10.1039/C4CS00328D [DOI] [PubMed] [Google Scholar]
- 37.Singha S, Kim D, Moon H, Wang T, Kim KH, Shin YH, Jung J, Seo E, Lee S-J and Ahn KH, Anal. Chem, 2015, 87, 1188. [DOI] [PubMed] [Google Scholar]
- 38.Grimm JB, Heckman LM and Lavis LD, Prog. Mol. Biol. Transl. Sci, 2013, 113, 1. [DOI] [PubMed] [Google Scholar]
- 39.Fan J, Hu M, Zhan P and Peng X, Chem. Soc. Rev, 2013, 42, 29. [DOI] [PubMed] [Google Scholar]
- 40.Kundu K, Knight SF, Willett N, Lee S, Taylor WR and Murthy N, Angew. Chem. Int. Ed, 2009, 48, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kundu K, Knight SF, Lee S, Taylor WR and Murthy N, Angew. Chem. Int. Ed, 2010, 49, 6134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim JY, Choi WI, Kim YH and Tae G, J. Control. Release, 2011, 156, 398. [DOI] [PubMed] [Google Scholar]
- 43.Yuan L, Lin W and Song J, Chem. Commun, 2010, 46, 7930. [DOI] [PubMed] [Google Scholar]
- 44.Swanson II PA, Kumar A, Samarin S, Vijay-Lumar M, Kundu K, Murthy N, Hansen J, Nusrat A and Neish AS, Proc. Natl. Acad. Sci. U S A, 2011, 108, 8803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Selvam S, Kundu K, Templeman KL, Murthy N and J Garcia A, Biomaterials, 2011, 32, 7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Magalotti S, Gustafson TP, Cao Q, Abendschein DR, Pierce RA, Berezin MY and Akers WJ, Mol. Imaging. Biol, 2013, 15, 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goodson P, Kumar A, Jain L, Kundu K, Murthy N, Koval M and Helms MN, Am. J. Physiol. Lung Cell. Mol. Physiol, 2012, 302, L410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Suri S, Lehman SM, Selvam S, Reddie K, Maity S, Murthy N and Garcia AJ, J. Biomed. Mater. Res. A, 2015, 103A, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oushiki D, Kojima H, Terai T, Arita M, Hanaoka K, Urano Y and Nagano T, J. Am. Chem. Soc, 2010, 132, 2795. [DOI] [PubMed] [Google Scholar]
- 50.Pu K, Shuhendler AJ and Rao J, Angew. Chem. Int. Ed, 2013, 52, 10325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shuhendler AJ, Pu K, Cui L, Uetrecht JP and Rao J, Nat. Biotechnol, 2014, 32, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen G, Song F, Wang J, Yang Z, Sun S, Fan J, Qiang X, Wang X, Dou B and Peng X, Chem. Commun, 2012, 48, 2949. [DOI] [PubMed] [Google Scholar]
- 53.Lou Z, Li P, Song P and Han K, Analyst, 2013, 138, 6291. [DOI] [PubMed] [Google Scholar]
- 54.Gerdes MJ, et al. , Proc. Natl. Acad. Sci. U S A, 2013, 110, 11982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Karton-Lifshin N, Segal E, Omer L, Portnoy M, Satchi-Fainaro R and Shabat D, J. Am. Chem. Soc, 2011, 133, 10960. [DOI] [PubMed] [Google Scholar]
- 56.Karton-Lifshin N, Albertazzi L, Bendikov M, Baran PS and Shabat D, J. Am. Chem. Soc, 2012, 134, 20412. [DOI] [PubMed] [Google Scholar]
- 57.Shi YM, Zhang SC and Zhang XR, Analyst, 2013, 138, 1952. [DOI] [PubMed] [Google Scholar]
- 58.Karton-Lifshin N, Vogel U, Sella E, Seeberger PH, Shabat D and Lepenies B, Org. Biomol. Chem, 2013, 11, 2903. [DOI] [PubMed] [Google Scholar]
- 59.Kisin-Finfer E, Ferber S, Blau R, Satchi-Fainaro R and Shabat D, Bioorg. Med. Chem. Lett, 2014, 24, 2453. [DOI] [PubMed] [Google Scholar]
- 60.Maity D and Govindaraju T, Org. Biomol. Chem, 2013, 11, 2098. [DOI] [PubMed] [Google Scholar]
- 61.Redy-Keisar O, Kisin-Finfer E, Ferber S, Satchi-Fainaro R and Shabat D, Nat. Protocol, 2014, 9, 27. [DOI] [PubMed] [Google Scholar]
- 62.Zhu D, Li G, Xue L and Jiang H, Org. Biomol. Chem, 2013, 11, 4577. [DOI] [PubMed] [Google Scholar]
- 63.Davies SS and Guo L, Chem. Phys. Lipids, 2014, 181, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li P, Tang B, Xing Y, Li P, Yang G and Zhang L, Analyst, 2008, 133, 1409. [DOI] [PubMed] [Google Scholar]
- 65.Wang B, Li P, Yu F, Chen J, Qu Z and Han K, Chem. Commun, 2013, 49, 5790. [DOI] [PubMed] [Google Scholar]
- 66.Tian J, Chen H, Zhuo L, Xie Y, Li N and Tang B, Chem. Eur. J, 2011, 17, 6626. [DOI] [PubMed] [Google Scholar]
- 67.Farina M, Rocha JBT and Aschner M, Life Sci, 2011, 89, 555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guo Z, Zhu WH, Zhu MM, Wu XM and Tian H, Chem. Eur. J, 2010, 16, 14424. [DOI] [PubMed] [Google Scholar]
- 69.Kim HN, Ren WX, Kim JS and Yoon J, Chem. Soc. Rev, 2012, 41, 3210. [DOI] [PubMed] [Google Scholar]
- 70.Han JY and Burgess K, Chem. Rev, 2010, 110, 2709. [DOI] [PubMed] [Google Scholar]
- 71.Tang B, Liu X, Xu K, Huang H, Yang G and An L, Chem. Commun, 2007, 3726. [DOI] [PubMed] [Google Scholar]
- 72.Myochin T, Kiyose K, Hanaoka K, Kojima H, Terai T and Nagano T, J. Am. Chem. Soc, 2011, 133, 3401. [DOI] [PubMed] [Google Scholar]
- 73.Lee H, Berezin MY, Tang R, Zhegalova N and Achilefu S, Photochem. Photobiol, 2013, 89, 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee H, Akers W, Bhushan K, Bloch S, Sudlow G, Tang R and Achilefu S, Bioconjug. Chem, 2011, 22, 777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hilderbrand SA, Kelly KA, Niedre M and Weissleder R, Bioconjug Chem, 2008, 19, 1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Reddie KG and Carroll KS, Curr. Opin. Chem. Biol, 2008, 12, 746. [DOI] [PubMed] [Google Scholar]
- 77.Guo Z, Nam S, Park S and Yoon J, Chem. Sci, 2012, 3, 2760. [Google Scholar]
- 78.Lee YJ, Chen JC, Amoscato AA, Bennouna J, Spitz DR, Suntharalingam M and Rhee JG, J. Cell. Sci, 2001, 114, 677. [DOI] [PubMed] [Google Scholar]
- 79.Wang X, Sun J, Zhang W, Ma X, Lv J, and Tang B, Chem. Sci, 2013, 4, 2551. [Google Scholar]
- 80.Strekowski L, Mason JC, Lee H, Say M and Patonay G, J. Heterocyclic Chem, 2004, 41, 227. [Google Scholar]
- 81.Kim YM, Choi YD, Weissleder R and Tung CH, Bioorg. Med. Chem. Lett, 2007, 17, 5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hu C, Sun W, Cao JF, Gao P, Wang JY, Fan JL, Song FL, Sun SG and Peng XJ, Org. Lett, 2013, 15, 4022. [DOI] [PubMed] [Google Scholar]
- 83.Bak DW and Weerapana E, Mol. Biosyst, 2015, 11, 678. [DOI] [PubMed] [Google Scholar]
- 84.Montserrat M, Morales A, Colell A, Garcia-Ruiz C, Kaplowitz N and Fernandez-Checa JC, Biochim. Biophys. Acta, 2013, 1830, 3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lim SY, Hong KH, Kim DI, Kwon H and Kim HJ, J. Am. Chem. Soc, 2014, 136, 7018. [DOI] [PubMed] [Google Scholar]
- 86.Nani RR, Shaum JB, Gorka AP and Schnermann MJ, Org. Lett, 2015, 17, 302–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wu X, Shi J, Yang L, Han J and Han S, Bioorg. Med. Chem. Lett, 2014, 24, 314. [DOI] [PubMed] [Google Scholar]
- 88.Yang Z, Lee JH, Jeon HM, Han JH, Park N, He Y, Lee H, Hong KS, Kang C and Kim JS, J. Am. Chem. Soc, 2013, 135, 11657. [DOI] [PubMed] [Google Scholar]
- 89.Moysan E, Bastiat G and Benoit J-P, Mol. Pharm, 2013, 10, 430. [DOI] [PubMed] [Google Scholar]
- 90.Chen Y, Zhu C, Yang Z, Chen J, He Y, Jiao Y, He W, Qiu L, Cen J and Guo Z, Angew. Chem. Int. Ed, 2013, 52, 1688. [DOI] [PubMed] [Google Scholar]
- 91.Yang Z, Liu Z, Chen Y, Wang X, He W and Lu Y, Org. Biomol. Chem, 2012, 10, 5073. [DOI] [PubMed] [Google Scholar]
- 92.Shiraishi Y, Nakamura M, Yamamoto K and Hirai T, Chem. Commun, 2014, 50, 11583. [DOI] [PubMed] [Google Scholar]
- 93.Jing CR and Cornish VW, Acc. Chem. Res, 2011, 44, 784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Crivat G and Taraska JW, Trends Biotechnol, 2012, 30, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yapici I, Lee KS, Berbasova T, Nosrati M, Jia X, Vasileiou C, Wang W, Santos EM, Geiger JH and Borhan B, J. Am. Chem. Soc, 2015, 137, 1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang W, Nossoni Z, Berbasova T, Watson CT, Yapici I, Lee KS, Vasileiou C, Geiger JH and Borhan B, Science, 2012, 338, 1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Berbasova T, Nosrati M, Vasileiou C, Wang W, Lee KS, Yapici I, Geiger JH and Borhan B, J. Am. Chem. Soc, 2013, 135, 16111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Huang B, Bates M and Zhuang XW, Annu. Rev. Biochem, 2009, 78, 993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schermelleh L, Heintzmann R and Leonhardt H, J. Cell Biol, 2010, 190, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sengupta P, Van Engelenburg S and Lippincott-Schwartz J, Dev. Cell, 2012, 23, 1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sengupta P, van Engelenburg SB and Lippincott-Schwartz J, Chem. Rev, 2014, 114, 3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chozinski TJ, Gagnon LA and Vaughan JC, FEBS Lett, 2014, 588, 3603. [DOI] [PubMed] [Google Scholar]
- 103.van de Linde S and Sauer M, Chem. Soc. Rev, 2014, 43, 1076. [DOI] [PubMed] [Google Scholar]
- 104.Heilemann M, Margeat E, Kasper R, Sauer M and Tinnefeld P, J. Am. Chem. Soc, 2005, 127, 3801. [DOI] [PubMed] [Google Scholar]
- 105.Bates M, Blosser TR and Zhuang XW, Phys. Rev. Lett, 2005, 94, 108101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rust MJ, Bates M and Zhuang XW, Nat. Methods, 2006, 3, 793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bates M, Huang B, Dempsey GT and Zhuang XW, Science, 2007, 317, 1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dempsey GT, Bates M, Kowtoniuk WE, Liu DR, Tsien RY and Zhuang XW, J. Am. Chem. Soc, 2009, 131, 18192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shim SH, Xia CL, Zhong GS, Babcock HP, Vaughan JC, Huang B, Wang X, Xu C, Bi GQ and Zhuang XW, Proc. Natl. Acad. Sci. U S A, 2012, 109, 13978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dempsey GT, Vaughan JC, Chen KH, Bates M and Zhuang X, Nat. Methods, 2011, 8, 1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Vaughan JC, Dempsey GT, Sun E and Zhuang XW, J. Am. Chem. Soc, 2013, 135, 1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Vaughan JC, Jia S and Zhuang XW, Nat. Methods, 2012, 9, 1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Deniz E, Tomasulo M, Cusido J, Yildiz I, Petriella M, Bossi ML, Sortino S and Raymo FM, J. Phys. Chem. C, 2012, 116, 6058. [Google Scholar]
- 114.Uno S, Kamiya M, Yoshihara T, Sugawara K, Okabe K, Tarhan MC, Fujita H, Funatsu T, Okada Y, Tobita S and Urano Y, Nat. Chem, 2014, 6, 681. [DOI] [PubMed] [Google Scholar]
- 115.Byers GW, Gross S and Henrichs PM, Photochem. Photobiol, 1976, 23, 37. [DOI] [PubMed] [Google Scholar]
- 116.Samanta A, Vendrell M, Das R and Chang YT, Chem. Commun, 2010, 46, 7406. [DOI] [PubMed] [Google Scholar]
- 117.Lepaja S, Strub H and Lougnot DJ, Z Naturforsch A, 1983, 38, 56. [Google Scholar]
- 118.Chen P, Li J, Qian ZG, Zheng DS, Okasaki T and Hayami M, Dyes Pigments, 1998, 37, 213. [Google Scholar]
- 119.Kanofsky JR and Sima PD, Photochem. Photobiol, 2000, 71, 361. [DOI] [PubMed] [Google Scholar]
- 120.Engel E, Schraml R, Maisch T, Kobuch K, Koenig B, Szeimies RM, Hillenkamp J, Baumler W and Vasold R, Invest. Ophth. Vis. Sci, 2008, 49, 1777. [DOI] [PubMed] [Google Scholar]
- 121.Renikuntla BR, Rose HC, Eldo J, Waggoner AS and Armitage BA, Org. Lett, 2004, 6, 909. [DOI] [PubMed] [Google Scholar]
- 122.Toutchkine A, Nguyen DV and Hahn KM, Org. Lett, 2007, 9, 2775. [DOI] [PubMed] [Google Scholar]
- 123.Gorka AP, Nani RR, Zhu JJ, Mackem S and Schnermann MJ, J. Am. Chem. Soc, 2014, 136, 14153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Toutchkine A, Kraynov V and Hahn KJ, J. Am. Chem. Soc, 2003, 125, 4132. [DOI] [PubMed] [Google Scholar]
- 125.Zheng QS, Jockusch S, Zhou Z and Blanchard SC, Photochem Photobiol, 2014, 90, 448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wu X and Zhu W, Chem. Soc. Rev. 2015, DOI 10.1039/C4CS00152D. [DOI] [Google Scholar]
- 127.Widengren J, Chmyrov A, Eggeling C, Lofdahl PA and Seidel CA, J. Phys. Chem. A, 2007, 111, 429. [DOI] [PubMed] [Google Scholar]
- 128.Altman RB, Zheng QS, Zhou Z, Terry DS, Warren JD and Blanchard SC, Nat. Methods, 2012, 9, 626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zheng Q, Juette MF, Jockusch S, Wasserman MR, Zhou Z, Altman RB and Blanchard SC, Chem. Soc. Rev, 2014, 43, 1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.van der Velde JHM, Ploetz E, Hiermaier M, Oelerich J, de Vries JW, Roelfes G and Cordes T, Chemphyschem 2013, 14, 4084. [DOI] [PubMed] [Google Scholar]
- 131.van der Velde JHM, Oelerich J, Huang JY, Smit JH, Hiermaier M, Ploetz E, Herrmann A, Roelfes G and Cordes T, J. Phys. Chem. Lett, 2014, 5, 3792. [DOI] [PubMed] [Google Scholar]
- 132.Warther D, Gug S, Specht A, Bolze F, Nicoud JF, Mourot A and Goeldner M, Bioorg. Med. Chem, 2010, 18, 7753. [DOI] [PubMed] [Google Scholar]
- 133.Dore TD and Wilson HC, Chromophores for the Delivery of Bioactive Molecules with Two-Photon Excitation In Photosensitive Molecules for Controlling Biological Function; Chambers JJ, and Kramer RH, R. H., Eds.; Springer Science: New York, 2011; Vol. 55, pp 57–92. [Google Scholar]
- 134.Yang YM, Shao Q, Deng RR, Wang C, Teng X, Cheng K, Cheng Z, Huang L, Liu Z, Liu XG and Xing BG, Angew. Chem. Int. Ed, 2012, 51, 3125. [DOI] [PubMed] [Google Scholar]
- 135.Viger ML, Sheng W, Dore K, Alhasan AH, Carling CJ, Lux J, de Gracia Lux C, Grossman M, Malinow R and Almutairi A, ACS Nano, 2014, 8, 4815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Brieke C, Rohrbach F, Gottschalk A, Mayer G and Heckel A, Angew. Chem. Int. Ed, 2012, 51, 8446. [DOI] [PubMed] [Google Scholar]
- 137.Ruebner A, Yang ZW, Leung D and Breslow R, Proc. Natl. Acad. Sci. U S A, 1999, 96, 14692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jiang MY and Dolphin D, J. Am. Chem. Soc, 2008, 130, 4236. [DOI] [PubMed] [Google Scholar]
- 139.Hossion AML, Bio M, Nkepang G, Awuah SG and You Y, ACS Med. Chem. Lett, 2013, 4, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Rotaru A and Mokhir A, Angew. Chem. Int. Ed, 2007, 46, 6180. [DOI] [PubMed] [Google Scholar]
- 141.Shell TA, Shell JR, Rodgers ZL and Lawrence DS, Angew. Chem. Int. Ed, 2014, 53, 875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Muzumdar MD, Tasic B, Miyamichi K, Li L and Luo L, Genesis, 2007, 45, 593. [DOI] [PubMed] [Google Scholar]
- 143.Nguyen QT and Tsien RY, Nat. Rev. Cancer, 2013, 13, 653. [DOI] [PMC free article] [PubMed] [Google Scholar]