

Abstract
Desmoplastic small round cell tumors (DSRCTs) are highly aggressive sarcomas that most commonly occur intra‐abdominally, and are defined by EWSR1‐WT1 gene fusion. Intracranial DSRCTs are exceptionally rare with only seven previously reported fusion‐positive cases. Herein, we evaluate the clinical, morphologic, immunohistochemical and molecular features of five additional examples. All patients were male (age range 6–25 years; median 11 years), with four tumors located supratentorially and one within the posterior fossa. The histologic features were highly variable including small cell, embryonal, clear cell, rhabdoid, anaplastic and glioma‐like appearances. A prominent desmoplastic stroma was seen in only two cases. The mitotic index ranged from <1 to 12/10 HPF (median 5). While all tumors showed strong desmin positivity, epithelial markers such as EMA, CAM 5.2 and other keratins were strongly positive in only one, focally positive in two and negative in two cases. EWSR1‐WT1 gene fusion was present in all cases, with accompanying mutations in the TERT promoter or STAG2 gene in individual cases. Given the significant histologic diversity, in the absence of genetic evaluation these cases could easily be misinterpreted as other entities. Desmin immunostaining is a useful initial screening method for consideration of a DSRCT diagnosis, prompting confirmatory molecular testing. Demonstrating the presence of an EWSR1‐WT1 fusion provides a definitive diagnosis of DSRCT. Genome‐wide methylation profiles of intracranial DSRCTs matched those of extracranial DSRCTs. Thus, despite the occasionally unusual histologic features and immunoprofile, intracranial DSRCTs likely represent a similar, if not the same, entity as their soft tissue counterpart based on the shared fusion and methylation profiles.
Keywords: desmin positivity, desmoplastic small round cell tumor, desmoplastic stroma, EWSR1‐WT1 fusion, intracranial, polyphenotypic
Introduction
Desmoplastic small round cell tumors (DSRCTs) are malignant mesenchymal neoplasms of uncertain histogenesis that most often occur intra‐abdominally, with a male to female ratio of approximately 4:1. They are defined by EWSR1‐WT1 gene fusion and display a polyphenotypic immunoprofile with co‐expression of epithelial (EMA, cytokeratins), mesenchymal (desmin) and neuronal markers (NSE, synaptophysin) 6, 21, 22, 23, 24, 25, 35, 36, 41, 56. Prior synonyms have emphasized the tumor's unique multilineage immunoprofile and predilection to occur within the abdomen, including desmoplastic small cell tumor with divergent differentiation, polyphenotypic small round cell tumor and intra‐abdominal desmoplastic round cell tumor 6, 25.
The characteristic histologic features of DSRCTs include clusters of small uniform oval cells with hyperchromatic nuclei and scant cytoplasm embedded within a prominent desmoplastic stroma. However, non‐classic patterns exist in approximately one third of cases 42. Alternative appearances include epithelioid, spindled or signet ring‐like cells, cellular pleomorphism with marked nuclear atypia, tight paraganglioma‐like “Zellballen” nests, Homer Wright‐like rosettes, gland formation, papillary growth or solid sheet‐like growth 2, 3, 6, 14, 42. Prominent desmoplasia may be absent in rare examples 2, 42.
DSRCTs are less frequently reported in other locations such as the pleura 31, lung 49, parotid gland 26, kidney 12, pancreas 43, paratesticular region 3, 44 or bone and soft tissues of the extremities 1, 3, 55. Seven intracranial DSRCT cases with confirmation of EWSR1‐WT1 fusion have been reported previously 3, 9, 40, 50, 52. Two other cases reported based on characteristic histology and immunostaining alone 54, or in combination with EWSR1 rearrangement by FISH 3 may also represent the same entity. Clinical prognosis for patients with abdominal DSRCT has been poor, with a median overall survival of approximately 26 months and 5 years overall survival of 18% 18, 29, 48. Multimodality therapy options utilizing modern surgical and chemotherapy delivery techniques continue to be explored 18, 27.
Materials and Methods
Five cases of intracranial DSRCT, each from a different institution, were evaluated for clinical, morphologic, immunohistochemical and molecular features. Study inclusion criteria required demonstration of EWSR1‐WT1 fusion either by next‐generation sequencing of genomic DNA, or RT‐PCR detection of the mRNA fusion transcript. Cases 1–4 had sufficient tumor tissue for genetic evaluation on the UCSF500 Cancer Panel, which assesses approximately 500 cancer‐associated genes for mutations, copy number alterations and structural variants including gene fusions (17, 32, 37 and Supporting Information Table 1). Paired tumor‐normal sequencing was performed for patient #1 using a buccal swab sample, whereas analysis of tumor tissue only was conducted for patients #2–4. Genomic DNA was extracted from formalin‐fixed, paraffin‐embedded tumor tissue using the QIAamp DNA FFPE Tissue Kit (Qiagen #56404). Methylation profiling of all five intracranial DSRCT cases was assessed at the University Medical Center Utrecht in the Netherlands using the Illumina EPIC (850k) array and was analyzed by the DKFZ sarcoma classifier for generation of calibrated scores. A detailed list of the entities included in the sarcoma classifier reference cohort is available online (molecularneuropathology.org). For visualization of intracranial DSRCT DNA‐methylation data as compared to abdominal/soft tissue DSRCTs and other relevant tumor entities, t‐distributed stochastic neighbor embedding (t‐SNE) analysis was performed. For case #5, there was only sufficient material for genetic evaluation by RT‐PCR and methylation profiling. Copy number changes were evaluated based on results from the UCSF500 Cancer Panel.
Results
The clinical features of the five cases of intracranial DSRCTs are summarized in Table 1. All patients were male (age range 6–25 years; median 11 years). Presenting symptoms were variable and included seizures, headaches, numbness and weakness. Four tumors were located supratentorially, while one was located within the posterior fossa. Magnetic resonance imaging features ranged from variably enhancing heterogeneous masses with a small cystic component, to multilocular predominantly cystic masses with enhancing solid nodules (Table 1 and Figure 1). All tumors were predominantly parenchymal, with extension to the leptomeninges without overlying dural changes noted in four cases (case #1, #3, #4 and #5). Susceptibility consistent with microhemorrhages was present in case #1, #3 and #4. Case #3 demonstrated reduced diffusion peripherally. For patients #1 and #4, whole‐body PET‐CT or PET‐MRI results were available, which showed no evidence of extracranial malignancy. Patients #4 and #5 underwent gross total resection followed by chemotherapy and radiation, with no evidence of residual disease at 13 months and 8 years after diagnosis, respectively. Patient #4 received six cycles of vincristine and cyclophosphamide with 55.8 Gy of radiation therapy delivered in 31 fractions (Table 1). Patient #5 received chemotherapy according to the Memorial Sloan Kettering Cancer Center P6 protocol 34, and 55.8 Gy of radiation therapy with a daily fraction of 1.8 Gy. One patient showed no evidence of disease 16 months after gross total resection and 6 weeks of radiation therapy only. One patient died within 1 month of surgery, of unspecified causes.
Table 1.
Clinical and radiologic features of the five intracranial DSRCTs within our series. Available clinical and radiologic information, including the status at last follow‐up and length of follow‐up interval, is depicted. The Memorial Sloan Kettering Cancer Center (MSKCC) P6 protocol has seven courses of chemotherapy. Courses 1, 2, 3 and 6 included cyclophosphamide 4200 mg/m2, doxorubicin 75 mg/m2 and vincristine. Courses 4, 5 and 7 consisted of ifosfamide 9 g/m2 and etoposide 500 mg/m2 for previously untreated patients 34. The symbol “–” is used to indicate when selected information was not available. Abbreviations: XRT = radiation therapy; Gy = gray; MSKCC = Memorial Sloan Kettering Cancer Center.
| Case # | Age (years) | Gender | Tumor location | Size (cm) | Presenting symptoms | Imaging findings at presentation | Extent of resection | Adjuvant chemotherapy | Adjuvant radiation therapy | Clinical status | Length of follow‐up |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 13 | M | Right temporal | 4.3 | Seizures | Solid and cystic mass, heterogeneous nodular enhancement | Gross total | None | 6 weeks of radiation therapy (XRT) | No evidence of disease | 16 months |
| 2 | 6 | M | Left occipital | 6.3 | – | Heterogeneous mass (contrast not administered) | – | – | – | – | – |
| 3 | 25 | M | Left cerebellum | 4.7 | Left hand numbness, headaches | Heterogeneous mass, minimal nodular enhancement | Gross total | None | None | Deceased | 1 month |
| 4 | 11 | M | Left parietal | 8.5 | Progressive right‐sided weakness | Multilocular cystic mass with enhancing nodules | Gross total | 6 cycles of vincristine and cyclophosphamide | XRT, 55.8 Gy in 31 fractions | no evidence of disease | 13 months |
| 5 | 8 | M | Right frontal | – | Headaches | T1 hyperintense cystic mass with focal enhancement | Gross total | MSKCC P6 protocol | XRT, 55.8 Gy with a daily fraction of 1.8 Gy | no evidence of disease | 8 years |
Figure 1.
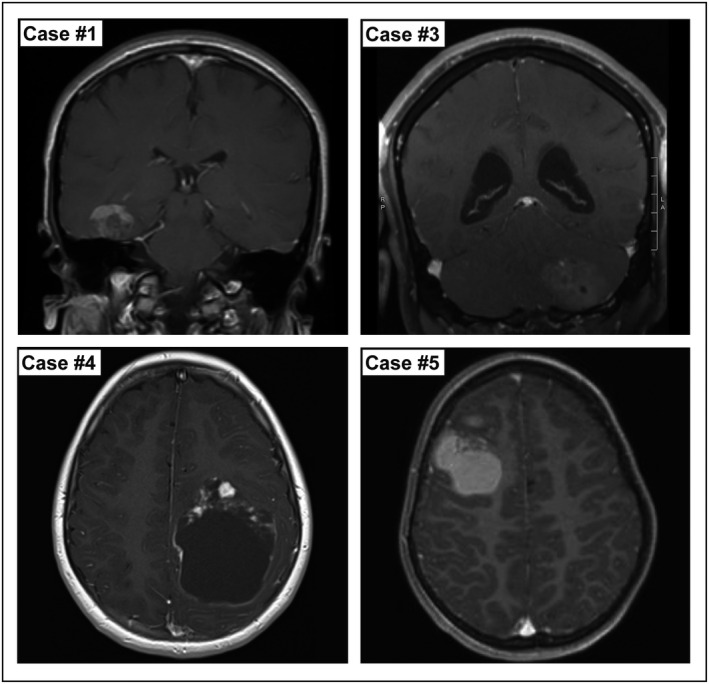
Representative pre‐operative imaging from four cases of intracranial DSRCTs, showing T1 weighted MRIs with contrast. Images demonstrate the variety of radiologic findings including supratentorial and infratentorial locations, the variable degree of a cystic component and relative degree of enhancement. Case #1: A right temporal heterogeneously enhancing mass. Case #3: A left cerebellar heterogeneous minimally enhancing mass. Case #4: A left parietal cystic mass with enhancing nodules. Case #5: A right frontal intrinsically T1 hyperintense mass with only focal nodular enhancement.
In all five cases the diagnosis of intracranial DSRCT was made after genetic evaluation demonstrated an EWSR1‐WT1 fusion. Initial diagnostic impressions included anaplastic medulloblastoma, astroblastoma‐like neoplasm, low‐grade tumor with glioneuronal features, malignant tumor NOS and small round blue cell tumor (Table 2 and Figure 2). Despite the entity's name, a prominent desmoplastic stroma was present in only two cases (cases #1 and #4), with the remaining three cases representing the less common “non‐desmoplastic variant” or solid‐pattern 2, 42. The growth pattern within the brain was either solid, or mixed solid and infiltrative with entrapped axons at the periphery. Cytologic features included cells with hyperchromatic angulated nuclei and indistinctive cell boundaries, clear cells with small bland oval nuclei, small epithelioid cells with amphophilic cytoplasm and oval nuclei, loose areas of low cellularity with spindled tapering bipolar cells, cells with large hyperchromatic pleomorphic nuclei and cells with small hyperchromatic oval to irregular nuclei within a fibrillar background.
Table 2.
Histologic and immunohistochemical features of the five intracranial DSRCTs within our series.
| Case # | Initial diagnostic impression | Desmoplastic stroma | Mitotic index | Ki‐67 LI | Necrosis | Myogenic markers | Epithelial markers | Neuronal markers |
|---|---|---|---|---|---|---|---|---|
| 1 | Glial neoplasm with an astroblastoma‐like pattern | Markedly desmoplastic stroma | Less than 1/10 HPFs | 2% | Not present | Desmin: patchy strong positivity | EMA: focal, CAM 5.2: negative Cytokeratin, MCK: negative | Synaptophysin: focal, NeuN: patchy, Neurofilament: negative, CD56: positive |
| 2 | Malignant tumor, not otherwise specified (NOS) | Only focal desmoplasia | 10/10 HPFs | – | Foci of necrosis | Desmin: diffusely positive | EMA: negative, CAM 5.2: negative, AE1/AE3: negative | Synaptophysin: negative |
| 3 | Favor anaplastic medulloblastoma, WHO grade IV | Not present | 12/10 HPFs | 80% | Foci of necrosis | Desmin: diffusely positive, SMA: negative | EMA: extensively positive, CAM 5.2: patchy, CK7: focal, CK20: focal | Synaptophysin: patchy, NeuN: patchy, Neurofilament: patchy |
| 4 | Low‐grade glioneuronal tumor | Markedly desmoplastic stroma | Less than 1/10 HPFs | 8% | Not present | Desmin: diffusely positive, Myogenin: negative | EMA: focal, CAM 5.2: negative, Pan‐cytokeratin: focal | Synaptophysin: focal, NeuN: patchy, Neurofilament: negative, CD56: positive |
| 5 | Small round blue cell tumor | Only focal desmoplasia | – | – | – | Desmin: strongly positive, Myogenin: negative, SMA: focally positive | EMA: negative, CAM 5.2: negative, Pan‐cytokeratin: negative | Synaptophysin: focal, CD56: positive |
Figure 2.
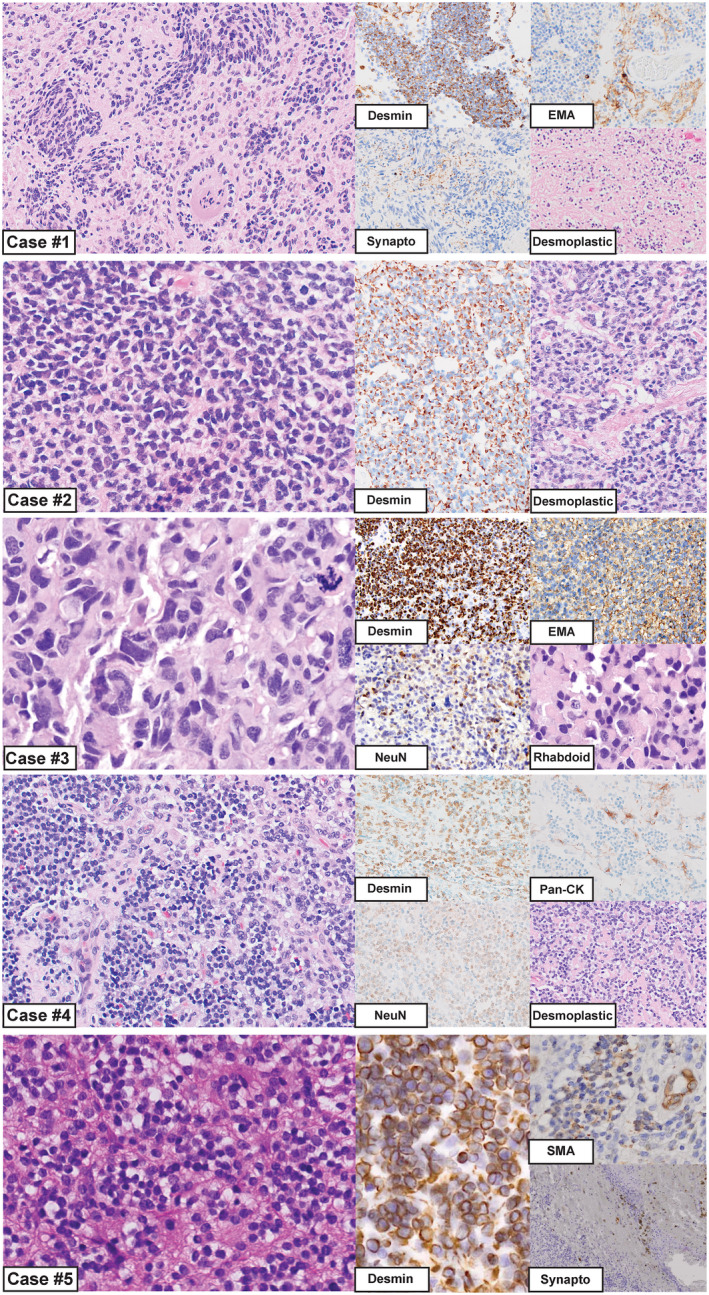
Morphologic appearance of intracranial DSRCTs for cases 1–5. Case #1: Regions of this tumor resembled an astroblastoma‐like glial neoplasm, while in other areas there was a markedly desmoplastic stroma with interspersed small round cells. Immunostaining for desmin, EMA and synaptophysin are shown. Case #2: This tumor had a uniform solid appearance with sheets of hyperchromatic nuclei, and only focal areas of mild desmoplasia. Desmin was strong and diffusely positive. Case #3: The histology of this case resembled an anaplastic medulloblastoma, including cell‐wrapping, large cells and nuclear molding. Rhabdoid features were appreciated focally; however, a desmoplastic stroma was not present in this case. Immunostaining for desmin, EMA and NeuN are shown, depicting the globular desmin staining that is often described for DSRCTs. This was the only case in our series with extensive epithelial marker expression. Case #4: This tumor appeared low grade with prominent areas of desmoplasia. Immunostaining for desmin, pan‐cytokeratin and NeuN are shown. Case #5: These tumor cells contained small round blue nuclei, and there were focal areas of desmoplasia. Immunostaining for desmin, EMA and synaptophysin are shown.
In case #1 (Table 2 and Figure 2), the majority of the tumor appeared glial; there were hyalinized vessels and structures resembling astroblastic pseudorosettes with broad perivascular processes. GFAP staining for this case showed patchy positivity. Other regions contained small round cells embedded in a desmoplastic stroma, indicating a more classical DSRCT appearance at least focally.
Case #3 was located in the posterior fossa, and was diagnostically challenging as the histologic and immunohistochemical features had a remarkable resemblance to an anaplastic medulloblastoma. The tumor cells had large pleomorphic nuclei with abundant mitoses, and features often seen in anaplastic medulloblastomas such as cell‐wrapping, apoptotic lakes and nuclear molding were present (Table 2 and Figure 2). Immunohistochemical studies for medulloblastoma subtyping were performed, which showed strong and diffuse YAP1 staining, moderate GAB1 positivity, cytoplasmic beta‐catenin staining and p53 positivity in 60% of tumor nuclei. A diagnosis of “anaplastic medulloblastoma, SHH‐activated and likely TP53‐mutant, WHO grade IV” was favored before the case underwent genetic characterization. Rhabdoid features were present focally, a finding that is not uncommon in DSRCTs 22, 42.
Case #2 contained solid sheets of tumor cells with hyperchromatic nuclei, only focal desmoplasia, a high mitotic index and a malignant appearance. YAP1 immunohistochemistry was also performed in this case, and showed strong diffuse positivity. In contrast, case #4 appeared low grade (ie, cytologically bland with low proliferative activity) with prominent areas of desmoplasia. Case #5 displayed the typical “small round blue cell” appearance; however, this case contained only focal areas of desmoplasia.
The mitotic index ranged from <1 to 12/10 HPF (median 5), and the Ki‐67 labeling index ranged from 2% to 80%. The myogenic differentiation marker desmin was strongly positive in all five cases, with the globular staining pattern often described for DSRCTs 25, 41 present in case #3. While most cases had strong and diffuse desmin staining, case #1 had strong patchy desmin staining. Staining for the epithelial markers EMA and CAM 5.2 showed strong positivity in only one case (case #3), focal positivity in two cases and was negative in two cases (Table 2). Staining for neuronal markers showed patchy or focal synaptophysin in 4/5 cases, patchy NeuN in 3/3 cases, and strong CD56 staining in 3/3 cases. GFAP immunostaining performed in four cases (cases #1, 3, 4 and 5) showed patchy or focal positivity.
EWSR1‐WT1 fusion was detected by targeted next‐generation DNA sequencing (4/5) or RT‐PCR (1/5) (Table 3). For all cases evaluated by the UCSF500 Cancer panel (cases #1, 2, 3 4), the fusion junction occurred between intron 8–9 of the EWSR1 gene (NM_013986) on chromosome 22q12 and intron 7–8 of the WT1 gene (NM_024426) on chromosome 11p33. This fusion is predicted to result in an in‐frame fusion protein where the N‐terminal portion is composed of exons 1–8 (codons 1–270) of EWSR1 and the C‐terminal portion is composed of exons 8–10 (codons 418–517) of WT1. This fusion is identical to the most common EWSR1‐WT1 fusion found in extracranial DSRCTs 5, 20, 24, 35, 39, and only antibodies against the C‐terminus of WT1 would be expected to detect WT1 protein expression 39, 56. RT‐PCR evaluation of case #5 detected an EWSR1‐WT1 mRNA fusion transcript.
Table 3.
Molecular features of intracranial DSRCTs, including methylation profiling calibrated scores as analyzed by the DKFZ sarcoma classifier (molecularneuropathology.org), fusions, pathogenic mutations and copy number alterations. For cases 1–4, the EWSR1‐WT1 gene fusion was detected by the UCSF500 Cancer Panel; for case #5, the EWSR1‐WT1 fusion transcript was detected by RT‐PCR. The symbol “–” is used to indicate when selected information was not available.
| Case # | Methylation profile DKFZ Sarcoma Classifier | Fusions | Pathogenic mutations | Copy number alterations |
|---|---|---|---|---|
| 1 | Desmoplastic small round cell tumor (calibrated score of 0.99) | EWSR1‐WT1 gene fusion, NM_013986, NM_024426, intron 8–9 of EWSR1 to intron 7–8 of WT1, 334 reads over fusion junction | None | Loss of 8p |
| 2 | Desmoplastic small round cell tumor (calibrated score of 0.99) | EWSR1‐WT1 gene fusion, NM_013986, NM_024426, intron 8–9 of EWSR1 to intron 7–8 of WT1, 229 reads over fusion junction | None | None |
| 3 | Desmoplastic small round cell tumor (calibrated score of 0.99) | EWSR1‐WT1 gene fusion, NM_013986, NM_024426, intron 8–9 of EWSR1 to intron 7–8 of WT1, 191 reads over fusion junction | TERT promoter hotspot mutation c.‐124C > T NM_198253 796 reads, 28% MAF | Gains of proximal 1p, 1q, 2, 5, 7, proximal 11p, 11q, 15, 18, 19, 20, 21, and distal 22q Losses of distal 1p, 16, and 17 |
| 4 | Desmoplastic small round cell tumor (calibrated score of 0.99) | EWSR1‐WT1 gene fusion, NM_013986, NM_024426, intron 8–9 of EWSR1 to intron 7–8 of WT1, 389 reads over fusion junction | STAG2 splice site mutation c.3278‐1G > A NM_001042749 480 reads, 19% MAF | Gain of 1q and loss of 20p |
| 5 | Desmoplastic small round cell tumor (calibrated score of 0.99) | EWSR1‐WT1 fusion transcript | – | – |
Accompanying pathogenic mutations included a TERT promoter hotspot mutation in case #3, and a subclonal STAG2 splice site mutation in case #4 predicted to disrupt gene function (Table 3). Though case #3 and case #4 both showed staining for p53 in greater than 60% of tumor nuclei, this did not correlate with the presence of an identifiable TP53 mutation in either case. Case #3 showed multiple chromosomal copy number alterations, while the remaining cases evaluated by the UCSF500 Cancer Panel demonstrated two or fewer chromosomal copy number changes.
Using the DKFZ sarcoma classifier and reference cohort, the methylation profiles of intracranial DSRCTs matched the profiles of extracranial DSRCTs with a calibrated score of 0.99 for all cases. The intracranial and extracranial DSRCT methylation profiles also clustered together by t‐SNE analysis (Figure 3).
Figure 3.
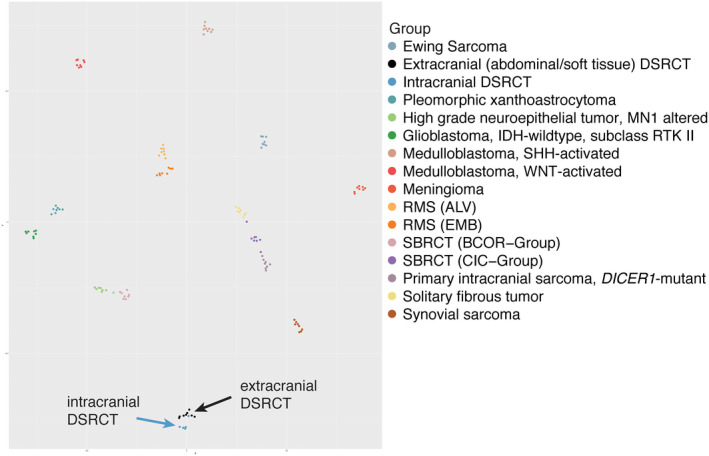
DNA‐methylation profiling data visualized by t‐SNE, for comparison of DSRCTs located intracranially (dark blue circles) to those occurring extracranially within the abdomen and soft tissues (black circles). Other relevant CNS and soft tissue tumor entities are also shown. Abbreviations: DSRCT: Desmoplastic small round cell tumor, RMS (ALV): alveolar rhabdomyosarcoma, RMS (EMB): embryonal rhabdomyosarcoma, SBRCT (BCOR − Group): Small blue round cell tumor with BCOR alteration, SBRCT (CIC − Group): Small blue round cell tumor with CIC alteration.
Discussion
Though DSRCT morphology is characteristically that of a “small round blue cell tumor” with a desmoplastic stroma, non‐classic morphologies are known to exist in a subset of cases 2, 3, 6, 14, 42. Therefore, it is important to consider DSRCT in the differential diagnosis not only for “small round blue cell tumors” of the CNS 15, 40, but also for CNS cases with other morphologies, especially those with a polyphenotypic immunoprofile. We encountered one case in particular that remarkably resembled an anaplastic medulloblastoma, which lacked the desmoplastic stroma usually seen in DSRCT cases (Figure 2, Case #3). This case also illustrated how immunostaining results typically utilized for medulloblastoma subtyping can be misleading if a definitive medulloblastoma diagnosis is not established. This case was positive for YAP1 and GAB1, with p53 positivity in 60% of tumor nuclei, mimicking an anaplastic medulloblastoma, SHH‐activated and likely TP53 mutant.
NSE is the neuronal marker typically used during diagnostic evaluation of extracranial DSRCTs, with synaptophysin being less sensitive. Markers such as NSE and CD56 are less often utilized within the CNS due to their lack of specificity. Nevertheless, patchy NeuN and synaptophysin positivity was often found in our intracranial DSRCTs. Polyphenotypic expression is an inherent quality of DSRCTs encountered within the abdomen and soft tissues, with expression of EMA in 94% of 79 cases 21, 41, and expression of CAM 5.2 or AE1/AE2 in 88% of 149 cases 21, 36, 41. Cytokeratin staining in extracranial DSRCT cases usually show diffuse cytoplasmic staining, occasional dot‐like staining, and is rarely only focally positive 36, 41. Of the five intracranial DSRCTs within our series, only one was strongly positive for an epithelial lineage marker. If we consider the previously published intracranial DSCRTs with confirmed fusion, only 6 of 12 (50%) have shown strong epithelial antigen positivity. While not all abdominal DSRCTs express epithelial antigens 53, the absence of epithelial antigen positivity is an infrequent finding.
Due to some of the atypical morphologic patterns encountered within this series of intracranial DSRCTs, the suggestion of decreased immunopositivity for epithelial lineage marker expression as compared to intra‐abdominal cases, and the unique location of our intracranial EWSR1‐WT1 fusion tumors, we considered the possibility that these cases may represent a distinct entity. Despite sharing the same EWSR1‐WT1 fusion as abdominal DSRCT cases, we postulated that the fundamental biology of intracranial cases could be unique, and thus account for the observed phenotypic differences.
As the methylation profiles of extracranial DSRCTs have been established 33, we compared the methylation profiles of intracranial DSRCT cases to those of extracranial cases, to serve as an additional assessment of similarities in tumor biology. The methylation profiles of intracranial and extracranial DSRCTs were almost identical, with calibrated scores of 0.99. In contrast to our initial hypothesis that these could be distinctive entities with identical genetics and differences in other aspects of their pathology, the shared epigenetic methylation profiles supports that these two groups represent a similar, if not the same, entity at both sites.
As is the case for many neoplasms, the cell of origin for intracranial DSRCTs is not well established. However, one could speculate that a mesencyhmal progenitor cell associated with the meninges, the vasculature or possibly located within the brain parenchyma due to abnormal developmental differentiation or migration, could acquire genetic and epigenetic alterations resulting in sarcoma tumorigenesis. For intra‐abdominal DSRCTs a mesothelial or submesothelial origin has been considered 22, 25, which is supported to some degree by expression of desmin and WT1 in both DSRCT and mesothelial cells, and by the predilection for DSRCT formation within mesothelial‐lined cavities. However, DSRCTs also occur in extra‐abdominal locations which are not associated with a mesothelial lining, do not show ultrastructural evidence of mesothelial differentiation and are generally regarded to be of uncertain histogenesis 3, 25, 41, 56.
It may be that the actual number of intracranial DSRCT cases is greater than currently recognized. Due to the variety of morphologic features and radiologic appearances, with both supratentorial and infratentorial locations possible, many cases could go unrecognized. In the absence of genetic evaluation, these cases could easily be misinterpreted as other entities. Since most DSRCTs are strongly desmin positive, desmin immunostaining could be used as an initial screening method to consider the diagnosis.
Desmin positivity, in the context of a polyphenotypic immunoprofile and particularly if there is a globular staining pattern 25, 41, is highly suggestive of a DSRCT diagnosis. However, a polyphenotypic immuneprofile with desmin positivity is not specific to DSRCT, and can also be encountered in angiomatous fibrous histiocytoma, intracranial myxoid mesenchymal tumors and sarcomas with EWSR1‐PATZ1 gene fusion 7, 11, 19, 30. We encountered two such cases while establishing our DSRCT study cohort, which were desmin‐positive but demonstrated an EWSR1‐ATF1 gene fusion on molecular evaluation, one of these cases also had a prominent desmoplastic stroma. Desmin positivity could be very useful as an initial screening test, which would then initiate additional confirmatory molecular testing to evaluate for the presence of an EWSR1‐WT1 gene fusion, a matching methylation profile or surrogate indication of the fusion by WT1 immunostaining.
Utilizing a WT1 antibody that specifically recognizes the C‐terminus of WT1 detects nuclear positivity in many cases of DSRCT with confirmed EWSR1‐WT1 gene fusion 39, 56, with sensitivity ranging from 70% to 100% depending on the study 8, 28, 56. Specificity for DSRCT was also high when compared to peripherally located neoplasms including rhabdomyosarcoma, Ewing sarcoma, neuroblastoma and rhabdoid tumors of the kidney, with the exception being that nephroblastomas also show nuclear WT1 positivity 8, 28. However, caution must be utilized in relying on immunohistochemistry alone, as it is important to recognize that WT1 antibodies directed against the N‐terminus of WT1 will not recognized the nuclear fusion protein generated by transcription/translation of an EWSR1‐WT1 gene fusion containing the C‐terminal exons of WT1 39, 56. A WT1 antibody appropriately targeted against the C‐terminus is a less expensive and rapid analysis option for immunohistochemical indication of EWSR1‐WT1 fusion in medical centers without molecular testing techniques.
Demonstrating the presence of an EWSR1‐WT1 fusion provides a definitive diagnosis of DSRCT. An important caveat is that establishing the presence of an EWSR1 gene rearrangement by break‐apart FISH probes alone is insufficient, as other EWSR1 fusion tumors can closely resemble a DSRCT by both histologic features and immunostaining. This is especially relevant for angiomatous fibrous histiocytoma, and for intracranial myxoid mesenchymal tumors with EWSR1‐CREB family gene fusions 4, 7, 19, 30. As previously mentioned, these tumors can have a desmoplastic stroma and show polyphenotypic differentiation with desmin positivity, and would also be positive for EWSR1 gene rearrangement by break‐apart FISH. Intra‐abdominal sarcomas with EWSR1‐PATZ1 gene fusion have also shown similar histologic features and immunostaining results, and similarly would show an EWSR1 rearrangement by break‐apart FISH 11.
In addition to the EWSR1‐WT1 gene fusion within our five cases, accompanying pathogenic mutations of the TERT promoter or STAG2 gene were demonstrated in individual cases. STAG2 encodes a core subunit of the cohesin complex that regulates chromatid cohesion, segregation and architecture 38, 47. Inactivating mutations in STAG2 are the most frequent accompanying somatic mutation in Ewing sarcoma, occurring in approximately 17% of cases, and are associated with poor outcome 10, 51. To the best of our knowledge, among the relatively few cases of DSRCT that have either undergone extensive genetic sequencing 13, 16, 46 or sequencing focused specifically on accompanying STAG2 or TP53 mutations 45, only one additional case with a STAG2 mutation has been reported 45. This patient's EWSR1‐WT1 gene fusion and accompanying inactivating STAG2 splice site mutation were detected within tumor tissue and within plasma derived cell free DNA 45. The prognostic implication of accompanying STAG2 mutations within DSRCT is of uncertain significance. Within our review of DSRCT sequencing studies available in the literature, whole exome sequencing analysis was performed on seven tumors 13, 16, a targeted panel of cancer related genes were assessed in one tumor 46, and 6 tumors were evaluated specifically for accompanying STAG2 or TP53 mutations using a targeted cancer panel 45. Mutation of the TERT promoter was either not identified or not assessed in these studies, and would not be detected by sequencing limited to the exome.
Though prognosis for patients with abdominal DSRCTs has been well studied 18, 27, 29, 48, prediction of clinical outcomes for intracranial DSRCTs is currently limited by the small number of reported cases. Of the 12 genetically confirmed cases within the literature including this series 3, 9, 40, 50, 52, four patients died at 1 month, 20 months, 2 years and 2.6 years after diagnosis 3, 40, 50. One patient was alive with progressive disease at 27 months 40. Four patients were alive with no evidence of disease at 13 months, 16 months, 18 months 9, 50 and 8 years. One patient was alive with no clinical signs of relapse at approximately 3 years 50, 52. For two patients, clinical follow‐up was not available. Among these 12 patients, one developed spinal dissemination, one patient who had leptomeningeal deposits at presentation developed progressive disease 40, one patient developed recurrent disease with lymph node metastases 50, and one had vertebral metastatic disease 3. A summary of the clinical, histologic and molecular findings for the seven previously reported cases of EWSR1‐WT1 fusion‐positive intracranial DSRCTs is provided within Table 4 3, 9, 40, 50, 52.
Table 4.
A brief summary of the available clinical, radiologic, histologic, immunohistochemical and molecular features for the seven previously reported cases of EWSR1‐WT1 fusion‐positive intracranial DSRCTs is provided within Table 4 3, 9, 40, 50, 52, which is adapted from Thondam et al. Two other cases reported in the literature based on characteristic histology and immunostaining alone 54, or in combination with EWSR1 rearrangement by FISH 3 may also represent the same entity. The Memorial Sloan Kettering Cancer Center (MSKCC) P6 protocol has seven courses of chemotherapy. Courses 1, 2, 3 and 6 included cyclophosphamide 4200 mg/m2, doxorubicin 75 mg/m2 and vincristine. Courses 4, 5 and 7 consisted of ifosfamide 9 g/m2 and etoposide 500 mg/m2 for previously untreated patients 34. The symbol “–” is used to indicate when selected information was not available. Abbreviation: NSE = neuron‐specific enolase.
| Case # | Publication | Age (years) | Gender | Tumor location | Presenting symptoms | Imaging findings | Extent of resection | Adjuvant chemotherapy | Adjuvant radiation therapy | Clinical status | Length of follow‐up | Histology | Immuno‐histochemistry | Molecular findings |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Tison et al 52 | 24 | M | Left posterior fossa | Headache, emesis, vertigo, impaired hearing | 4 cm mass adherent to the tentorium and petrous portion of the temporal bone, with displacement of left cerebellar hemisphere; no extracranial malignancy on CT/MRI total body scans | Subtotal | 3 cycles consisting of PCNU, cisplatin, and VP‐16 intracranial methotrexate every 40 days | yes | Alive with no clinical signs of relapse at time of publication | ~3 years | Compact nests of small uniform round and oval cells with hyperchromatic nuclei and scarce cytoplasm, separated by a desmoplastic stroma, infrequent mitoses, with areas of necrosis | Desmin: positive, EMA: positive, Keratin: positive, NSE: positive | PCR performed using EWSR1 and WT1 primers, EWSR1‐WT1 gene fusion detected by Southern blot |
| 2 | Bouchireb et al 9 | 6 | F | Right temporal | Headaches, complex partial seizure | Well‐dermarcated heterogeneously enhancing mass; PET‐CT was negative for extracranial malignancy | Gross total | MSKCC P6 protocol | Focal conformal irradiation to the tumor bed with a 2 cm margin at 54 Gy | No evidence of disease | 18 months | Small round cell tumor with hyperchromatic nuclei and eosinophilic cytoplasm embeded in a fibromyxoid stroma, mitoses were infrequent | Desmin: positive, EMA: negative, AE1/AE3: negative, Synaptophysin: positive | EWSR1‐WT1 gene fusion detected by RT‐PCR |
| 3 | Neder et al 40 | 37 | M | Left cerebellopontine angle | Left‐sided hearing loss and tinnitus | Heterogenouly enhancing mass, initial imaging suggested an acoustic neuroma; no extracranial malignancy on CT/MRI total body scans | Subtotal | After subsequent debulking of intradural spinal nodules, patient received carboplatinum and temozolomide | Sterotactic irradiation to CPA tumor bed after subsequent debulking of intradural spinal nodules received brain and spinal irradiation, with radiosurgery to CPA | Recurrent disease with spinal dissemination at 6 months, died at 2 years | 2 years | Sheets of small to medium sized cells with hyperchromatic nuclei and inconspicuous nucleoli, with a desmoplastic stroma, foci of necrosis mitotic index 5/10 HPFs | Desmin: positive, EMA: positive, CAM 5.2: positive, Synaptophysin: negative, Neurofilament: negative Ki‐67, LI: 17.6% | EWSR1‐WT1 gene fusion detected by RT‐PCR |
| 4 | Neder et al 40 | 39 | M | Posterior fossa | Gait imbalance, bilateral lower limb weakness, with subsequent fall and hypotonic paraparesis | Left cerebellar hemisphere, and CPA lesions with spinal leptomeningeal deposits at presentation, multiple patchy enhancing lesions | Decompression of spinal cord | 3 cycles of cisplatin, etoposide, and Holoxan | Yes | Alive with progressive disease | 27 months | Viable perivascular tumor cells seperated by necrosis, oval to irregular nuclei with coarse chromatin and scant cytoplasm, mitotic index 5/10 HPFs | Desmin: positive, EMA: positive, CAM 5.2: positive, Synaptophysin: negative, Neurofilament: negative Ki‐67, LI: 11.5% | EWSR1‐WT1 gene fusion detected by RT‐PCR |
| 5 | Thondam et al 50 | 27 | M | Suprasellar | Panhypopituitarism, with subsequent development of bitemporal hemianopia one year later after missing follow‐up appointments | Heterogenously enhancing suprasellar mass extending into third ventricle and the pituitary fossa; whole‐body imaging was negative for extracranial malignancy | Near total resection | Palliative chemotherapy was considered, but the patient's clinical condition deteriorated | Fractionated conformal radiotherapy was initiated at recurrence but discontinued due to clinical deterioration | Tumor recurred at 4 months, followed by cervical and mediastinal lymph node metastases, patient died at 20 months | 20 months | Nests and cords of fairly uniform cells with hyperchromatic nuclei and indistinct cytoplasm distributed in a desmpolastic stroma, with calcifications and necrosis | Desmin: positive, CAM 5.2: positive, EMA: patchy, CK7: focal, CK20: focal, NSE: focal, Neurofilament: patchy | EWSR1‐WT1 gene fusion detected by RT‐PCR |
| 6 | Al‐Ibraheemi et al 3 | 6 | M | Intracranial, infratemporal fossa | – | – | – | – | – | – | – | “Ewing sarcoma‐like,” small cell, desmoplasia present | Desmin: positive, Cytokeratin: focal | EWSR1‐WT1 gene fusion detected by RT‐PCR |
| 7 | Al‐Ibraheemi et al 3 | 37 | M | Cerebellopontine angle | – | – | – | Yes | Yes | Vertebral metastasis, died with disease | 32 months | “Ewing sarcoma‐like,” desmoplasia present | Desmin: positive, Cytokeratin: positive | EWSR1‐WT1 gene fusion detected by RT‐PCR |
In summary, the histologic features of intracranial DSRCTs can be highly variable, and therefore a high index of suspicion is required for cases that lack the classical appearance. Intracranial DSRCTs with EWSR1‐WT1 fusion may not have the same degree of epithelial lineage marker expression as seen in intra‐abdominal cases. Despite differences in the morphologic appearance and immunostaining profile, methylation analysis supports that intracranial DSRCTs represent a similar, if not the same, entity as DSRCTs seen elsewhere in the body.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Table S1. List of the 479 genes targeted for sequencing on the UCSF500 Cancer Panel. All coding exons were captured for sequencing from each of these genes, with those highlighted genes also having select intronic or upstream regulatory regions that were captured for sequencing to enable detection of structural variants.
Acknowledgments
This study was supported in part by the NIH Director's Early Independence Award (DP5 OD021403) to D.A.S. We thank the staff of the UCSF Clinical Cancer Genomics Laboratory for assistance with genetic profiling, and the staff of the Molecular Pathology Laboratory of the University Medical Center Utrecht for their help with methylation profiling.
Presented in abstract format at the United States and Canadian Academy of Pathology meeting in National Harbor, Maryland, March 16–21, 2019.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Adsay V, Cheng J, Athanasian E, Gerald W, Rosai J (1999) Primary desmoplastic small cell tumor of soft tissues and bone of the hand. Am J Surg Pathol 23:1408–1413. [DOI] [PubMed] [Google Scholar]
- 2. Ali A, Mohamed M, Chisholm J, Thway K (2017) Solid‐pattern desmoplastic small round cell tumor. Int J Surg Pathol 25:158–161. [DOI] [PubMed] [Google Scholar]
- 3. Al‐Ibraheemi A, Broehm C, Tanas MR, Horvai AE, Rubin BP, Cheah AL et al (2019) Desmoplastic small round cell tumors with atypical presentations: a report of 34 cases. Int J Surg Pathol 27:236–243. [DOI] [PubMed] [Google Scholar]
- 4. Antonescu CR, Dal Cin P, Nafa K, Teot LA, Surti U, Fletcher CD, Ladanyi M (2007) EWSR1‐CREB1 is the predominant gene fusion in angiomatoid fibrous histiocytoma. Genes Chromosomes Cancer 46:1051–1060. [DOI] [PubMed] [Google Scholar]
- 5. Antonescu CR, Gerald WL, Magid MS, Ladanyi M (1998) Molecular variants of the EWS‐WT1 gene fusion in desmoplastic small round cell tumor. Diagn Mol Pathol 7:24–28. [DOI] [PubMed] [Google Scholar]
- 6. Antonescu CR, Ladanyi M (2012) Desmoplastic small round cell tumor. In: WHO Classification of Tumours of Soft Tissue and Bone. Fletcher DM, Bridge JA, Hogendoorn CW, Mertens F (eds), pp. 225–227. International Agency for Research on Cancer: Lyon. [Google Scholar]
- 7. Bale TA, Oviedo A, Kozakewich H, Giannini C, Davineni PK, Ligon K, Alexandrescu S (2018) Intracranial myxoid mesenchymal tumors with EWSR1‐CREB family gene fusions: myxoid variant of angiomatoid fibrous histiocytoma or novel entity? Brain Pathol 28:183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Barnoud R, Sabourin JC, Pasquier D, Ranchère D, Bailly C, Terrier‐Lacombe MJ, Pasquier B (2000) Immunohistochemical expression of WT1 by desmoplastic small round cell tumor: a comparative study with other small round cell tumors. Am J Surg Pathol 24:830–836. [DOI] [PubMed] [Google Scholar]
- 9. Bouchireb K, Auger N, Bhangoo R, Di Rocco F, Brousse N, Delattre O et al (2008) Intracerebral small round cell tumor: an unusual case with EWS‐WT1 translocation. Pediatr Blood Cancer 51:545–548. [DOI] [PubMed] [Google Scholar]
- 10. Brohl AS, Solomon DA, Chang W, Wang J, Song Y, Sindiri S et al (2014) The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet 10:e1004475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chougule A, Taylor MS, Nardi V, Chebib I, Cote GM, Choy E et al (2019) Spindle and round cell sarcoma with EWSR1‐PATZ1 gene fusion: a sarcoma with polyphenotypic differentiation. Am J Surg Pathol 43:220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Collardeau‐Frachon S, Ranchère‐Vince D, Delattre O, Hoarau S, Thiesse P, Dubois R et al (2007) Primary desmoplastic small round cell tumor of the kidney: a case report in a 14‐year‐old girl with molecular confirmation. Pediatr Dev Pathol 10:320–324. [DOI] [PubMed] [Google Scholar]
- 13. Devecchi A, De Cecco L, Dugo M, Penso D, Dagrada G, Brich S et al (2018) The genomics of desmoplastic small round cell tumor reveals the deregulation of genes related to DNA damage response, epithelial‐mesenchymal transition, and immune response. Cancer Commun 38:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dorsey BV, Benjamin LE, Rauscher F III, Klencke B, Venook AP, Warren RS, Weidner N (1996) Intra‐abdominal desmoplastic small round‐cell tumor: expansion of the pathologic profile. Mod Pathol 9:703–709. [PubMed] [Google Scholar]
- 15. Dunham C (2015) Uncommon pediatric tumors of the posterior fossa: pathologic and molecular features. Childs Nerv Syst 31:1729–1737. [DOI] [PubMed] [Google Scholar]
- 16. Ferreira EN, Barros BD, de Souza JE, Almeida RV, Torrezan GT, Garcia S et al (2016) A genomic case study of desmoplastic small round cell tumor: comprehensive analysis reveals insights into potential therapeutic targets and development of a monitoring tool for a rare and aggressive disease. Hum Genomics 10:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ferris SP, Velazquez Vega J, Aboian M, Lee JC, Van Ziffle J, Onodera C et al (2020) High‐grade neuroepithelial tumor with BCOR exon 15 internal tandem duplication‐a comprehensive clinical, radiographic, pathologic, and genomic analysis. Brain Pathol 30:46–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gani F, Goel U, Canner JK, Meyer CF, Johnston FM (2019) A national analysis of patterns of care and outcomes for adults diagnosed with desmoplastic small round cell tumors in the United States. J Surg Oncol 119:880–886. [DOI] [PubMed] [Google Scholar]
- 19. Gareton A, Pierron G, Mokhtari K, Tran S, Tauziède‐Espariat A, Pallud J et al (2018) ESWR1‐CREM fusion in an intracranial myxoid angiomatoid fibrous histiocytoma‐like tumor: a case report and literature review. J Neuropathol Exp Neurol 77:537–541. [DOI] [PubMed] [Google Scholar]
- 20. Gerald WL, Haber DA (2005) The EWS‐WT1 gene fusion in desmoplastic small round cell tumor. Semin Cancer Biol 15:197–205. [DOI] [PubMed] [Google Scholar]
- 21. Gerald WL, Ladanyi M, de Alava E, Cuatrecasas M, Kushner BH, LaQuaglia MP, Rosai J (1998) Clinical, pathologic, and molecular spectrum of tumors associated with t(11;22)(p13;q12): desmoplastic small round‐cell tumor and its variants. J Clin Oncol 16:3028–3036. [DOI] [PubMed] [Google Scholar]
- 22. Gerald WL, Miller HK, Battifora H, Miettinen M, Silva EG, Rosai J (1991) Intra‐abdominal desmoplastic small round‐cell tumor. Report of 19 cases of a distinctive type of high‐grade polyphenotypic malignancy affecting young individuals. Am J Surg Pathol 15:499–513. [PubMed] [Google Scholar]
- 23. Gerald WL, Rosai J (1989) Case 2. Desmoplastic small cell tumor with divergent differentiation. Pediatr Pathol 9:177–183. [DOI] [PubMed] [Google Scholar]
- 24. Gerald WL, Rosai J, Ladanyi M (1995) Characterization of the genomic breakpoint and chimeric transcripts in the EWS‐WT1 gene fusion of desmoplastic small round cell tumor. Proc Natl Acad Sci U S A 92:1028–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goldblum JR, Folpe AL, Weiss SW (eds) (2014) Desmoplastic small round cell tumor. In: Enzinger & Weiss's Soft Tissue Tumors, Chapter 33, pp. 1079–1084. Elsevier Saunders: Philadelphia. [Google Scholar]
- 26. Hatanaka KC, Takakuwa E, Hatanaka Y, Suzuki A, IIzuka S, Tsushima N et al (2019) Desmoplastic small round cell tumor of the parotid gland‐report of a rare case and a review of the literature. Diagn Pathol 14:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hayes‐Jordan AA, Coakley BA, Green HL, Xiao L, Fournier KF, Herzog CE et al (2018) Desmoplastic small round cell tumor treated with cytoreductive surgery and hyperthermic intraperitoneal chemotherapy: results of a phase 2 trial. Ann Surg Oncol 25:872–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hill DA, Pfeifer JD, Marley EF, Dehner LP, Humphrey PA, Zhu X, Swanson PE (2000) WT1 staining reliably differentiates desmoplastic small round cell tumor from Ewing sarcoma/primitive neuroectodermal tumor. An immunohistochemical and molecular diagnostic study. Am J Clin Pathol 114:345–353. [DOI] [PubMed] [Google Scholar]
- 29. Honoré C, Amroun K, Vilcot L, Mir O, Domont J, Terrier P et al (2015) Abdominal desmoplastic small round cell tumor: multimodal treatment combining chemotherapy, surgery, and radiotherapy is the best option. Ann Surg Oncol 22:1073–1079. [DOI] [PubMed] [Google Scholar]
- 30. Kao YC, Sung YS, Zhang L, Chen CL, Vaiyapuri S, Rosenblum MK, Antonescu CR (2017) EWSR1 fusions With CREB family transcription factors define a novel myxoid mesenchymal tumor with predilection for intracranial location. Am J Surg Pathol 41:482–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Karavitakis EM, Moschovi M, Stefanaki K, Karamolegou K, Dimitriadis E, Pandis N et al (2007) Desmoplastic small round cell tumor of the pleura. Pediatr Blood Cancer 49:335–338. [DOI] [PubMed] [Google Scholar]
- 32. Kline CN, Joseph NM, Grenert JP, van Ziffle J, Talevich E, Onodera C et al (2017) Targeted next‐generation sequencing of pediatric neuro‐oncology patients improves diagnosis, identifies pathogenic germline mutations, and directs targeted therapy. Neuro‐Oncol 19:699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Koelsche C, Hartmann W, Schrimpf D, Stichel D, Jabar S, Ranft A et al (2018) Array‐based DNA‐methylation profiling in sarcomas with small blue round cell histology provides valuable diagnostic information. Mod Pathol 31:1246–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kushner BH, LaQuaglia MP, Wollner N, Meyers PA, Lindsley KL, Ghavimi F et al (1996) Desmoplastic small round‐cell tumor: prolonged progression‐free survival with aggressive multimodality therapy. J Clin Oncol 14:1526–1531. [DOI] [PubMed] [Google Scholar]
- 35. Ladanyi M, Gerald W (1994) Fusion of the EWS and WT1 genes in the desmoplastic small round cell tumor. Cancer Res 54:2837–2840. [PubMed] [Google Scholar]
- 36. Lae ME, Roche PC, Jin L, Lloyd RV, Nascimento AG (2002) Desmoplastic small round cell tumor: a clinicopathologic, immunohistochemical, and molecular study of 32 tumors. Am J Surg Pathol 26:823–835. [DOI] [PubMed] [Google Scholar]
- 37. Lee JC, Villanueva‐Meyer JE, Ferris SP, Sloan EA, Hofmann JW, Hattab EM et al (2019) Primary intracranial sarcomas with DICER1 mutation often contain prominent eosinophilic cytoplasmic globules and can occur in the setting of neurofibromatosis type 1. Acta Neuropathol 137:521–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mondal G, Stevers M, Goode B, Ashworth A, Solomon DA (2019) A requirement for STAG2 in replication fork progression creates a targetable synthetic lethality in cohesin‐mutant cancers. Nat Commun 10:1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Murphy AJ, Bishop K, Pereira C, Chilton‐MacNeill S, Ho M, Zielenska M, Thorner PS. A new molecular variant of desmoplastic small round cell tumor: significance of WT1 immunostaining in this entity. Hum Pathol 39:1763–1770. [DOI] [PubMed] [Google Scholar]
- 40. Neder L, Scheithauer BW, Turel KE, Arnesen MA, Ketterling RP, Jin L et al (2009) Desmoplastic small round cell tumor of the central nervous system: report of two cases and review of the literature. Virchows Arch 454:431–439. [DOI] [PubMed] [Google Scholar]
- 41. Ordóñez NG (1998) Desmoplastic small round cell tumor: II: an ultrastructural and immunohistochemical study with emphasis on new immunohistochemical markers. Am J Surg Pathol 22:1314–1327. [DOI] [PubMed] [Google Scholar]
- 42. Ordóñez NG (1998) Desmoplastic small round cell tumor: I: a histopathologic study of 39 cases with emphasis on unusual histological patterns. Am J Surg Pathol 22:1303–1313. [DOI] [PubMed] [Google Scholar]
- 43. Ryan A, Razak A, Graham J, Benson A, Rowe D, Haugk B, Verrill M (2007) Desmoplastic small round‐cell tumor of the pancreas. J Clin Oncol 25:1440–1442. [DOI] [PubMed] [Google Scholar]
- 44. Sedig L, Geiger J, Mody R, Jasty‐Rao R (2017) Paratesticular desmoplastic small round cell tumors: a case report and review of the literature. Pediatr Blood Cancer 64:e26631. [DOI] [PubMed] [Google Scholar]
- 45. Shukla NN, Patel JA, Magnan H, Zehir A, You D, Tang J et al (2017) Plasma DNA‐based molecular diagnosis, prognostication, and monitoring of patients with EWSR1 fusion‐positive sarcomas. JCO Precis Oncol 1:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Silva JG, Corrales‐Medina FF, Maher OM, Tannir N, Huh WW, Rytting ME, Subbiah V (2015) Clinical next generation sequencing of pediatric‐type malignancies in adult patients identifies novel somatic aberrations. Oncoscience 2:187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Solomon DA, Kim T, Diaz‐Martinez LA, Fair J, Elkahloun AG, Harris BT et al (2011) Mutational inactivation of STAG2 causes aneuploidy in human cancer. Science 333:1039–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stiles ZE, Dickson PV, Glazer ES, Murphy AJ, Davidoff AM, Behrman SW et al (2018) Desmoplastic small round cell tumor: a nationwide study of a rare sarcoma. J Surg Oncol 117:1759–1767. [DOI] [PubMed] [Google Scholar]
- 49. Syed S, Haque AK, Hawkins HK, Sorensen PH, Cowan DF (2002) Desmoplastic small round cell tumor of the lung. Arch Pathol Lab Med 126:1226–1228. [DOI] [PubMed] [Google Scholar]
- 50. Thondam SK, du Plessis D, Cuthbertson DJ, Das KS, Javadpour M, MacFarlane IA et al (2015) Intracranial desmoplastic small round cell tumor presenting as a suprasellar mass. J Neurosurg 122:773–777. [DOI] [PubMed] [Google Scholar]
- 51. Tirode F, Surdez D, Ma X, Parker M, Le Deley MC, Bahrami A et al (2014) Genomic landscape of Ewing sarcoma defines an aggressive subtype with co‐association of STAG2 and TP53 mutations. Cancer Discov 4:1342–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tison V, Cerasoli S, Morigi F, Ladanyi M, Gerald WL, Rosai J (1996) Intracranial desmoplastic small‐cell tumor. Report of a case. Am J Surg Pathol 20:112–117. [DOI] [PubMed] [Google Scholar]
- 53. Trupiano JK, Machen SK, Barr FG, Goldblum JR (1999) Cytokeratin‐negative desmoplastic small round cell tumor: a report of two cases emphasizing the utility of reverse transcriptase‐polymerase chain reaction. Mod Pathol 12:849–853. [PubMed] [Google Scholar]
- 54. Yachnis AT, Rorke LB, Biegel JA, Perilongo G, Zimmerman RA, Sutton LN (1992) Desmoplastic primitive neuroectodermal tumor with divergent differentiation. Broadening the spectrum of desmoplastic infantile neuroepithelial tumors. Am J Surg Pathol 16:998–1006. [DOI] [PubMed] [Google Scholar]
- 55. Yoshida A, Edgar MA, Garcia J, Meyers PA, Morris CD, Panicek DM (2008) Primary desmoplastic small round cell tumor of the femur. Skeletal Radiol 37:857–862. [DOI] [PubMed] [Google Scholar]
- 56. Zhang PJ, Goldblum JR, Pawel BR, Fisher C, Pasha TL, Barr FG (2003) Immunophenotype of desmoplastic small round cell tumors as detected in cases with EWS‐WT1 gene fusion product. Mod Pathol 16:229–235. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of the 479 genes targeted for sequencing on the UCSF500 Cancer Panel. All coding exons were captured for sequencing from each of these genes, with those highlighted genes also having select intronic or upstream regulatory regions that were captured for sequencing to enable detection of structural variants.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.