

Abstract
“Myxoid glioneuronal tumor, PDGFRA p.K385‐mutant” is a recently described tumor entity of the central nervous system with a predilection for origin in the septum pellucidum and a defining dinucleotide mutation at codon 385 of the PDGFRA oncogene replacing lysine with either leucine or isoleucine (p.K385L/I). Clinical outcomes and optimal treatment for this new tumor entity have yet to be defined. Here, we report a comprehensive clinical, radiologic, and histopathologic assessment of eight cases. In addition to its stereotypic location in the septum pellucidum, we identify that this tumor can also occur in the corpus callosum and periventricular white matter of the lateral ventricle. Tumors centered in the septum pellucidum uniformly were associated with obstructive hydrocephalus, whereas tumors centered in the corpus callosum and periventricular white matter did not demonstrate hydrocephalus. While multiple patients were found to have ventricular dissemination or local recurrence/progression, all patients in this series remain alive at last clinical follow‐up despite only biopsy or subtotal resection without adjuvant therapy in most cases. Our study further supports “myxoid glioneuronal tumor, PDGFRA p.K385‐mutant” as a distinct CNS tumor entity and expands the spectrum of clinicopathologic and radiologic features of this neoplasm.
Keywords: corpus callosum, DNT‐like tumor of the septum pellucidum, dysembryoplastic neuroepithelial tumor (DNT), lateral ventricle, molecular neuro‐oncology, molecular neuropathology, myxoid glioneuronal tumor, PDGFRA, periventricular white matter, platelet‐derived growth factor receptor alpha, septal DNT, septum pellucidum
Introduction
“Myxoid glioneuronal tumor, PDGFRA p.K385‐mutant” is a recently described tumor entity of the central nervous system (CNS) that has a stereotypic location in the septum pellucidum and a characteristic dinucleotide mutation at codon 385 of the PDGFRA oncogene replacing lysine with either leucine or isoleucine (p.K385L/I) in the encoded platelet‐derived growth factor receptor alpha protein 27. These tumors are low‐grade glioneuronal neoplasms with histologic features reminiscent of either dysembryoplastic neuroepithelial tumor (DNT) or rosette‐forming glioneuronal tumor (RGNT), composed of oligodendrocyte‐like cells in a prominent myxoid stroma. However, they uniformly lack the well‐defined mucin‐patterned nodules of cortically based DNT and also lack the BRAF and FGFR1 mutations or rearrangements that genetically characterize DNT, RGNT and other low‐grade neuroepithelial tumor entities 10, 24, 25, 26, 32. This new proposed entity likely includes the majority of cases that were previously described as “dysembryoplastic neuroepithelial tumor‐like neoplasm of the septum pellucidum” and intraventricular DNT 1, 3, 5, 7, 9, 12, 18, 28, 30, 31. A recent study that combined genome‐wide methylation profiling and sequencing analysis on a series of 11 cases of “septal dysembryoplastic neuroepithelial tumor” found that all eight cases harboring PDGFRA p.K385L/I mutation formed a distinct methylation cluster separate from other known CNS tumor entities, while the remaining few cases harboring NF1 or FGFR1 alterations instead clustered with other low‐grade neuroepithelial tumors (eg, pilocytic astrocytoma) 6.
Due to its recent recognition as a distinct tumor entity, the complete spectrum of anatomic site of origin, clinical presentation and patient outcomes has not yet been described. Here, we report our experience with the clinical, radiologic, histopathologic and molecular characteristics of eight cases of myxoid glioneuronal tumor, PDGFRA p.K385‐mutant.
Methods
Patient cohort and tumor samples
Eight patients with low‐grade glioneuronal tumors harboring the defining PDGFRA p.K385I or p.K385L dinucleotide substitution by targeted next‐generation sequencing analysis were included in this study. Patients MGNT #1–4 have previously been reported in part 27. All tumor specimens were fixed in 10% neutral‐buffered formalin and embedded in paraffin. Pathologic review of all tumors was conducted by a group of expert neuropathologists (TT, AWB, AP and DAS).
Imaging review
All brain imaging studies for each patient were reviewed by an expert neuroradiologist (JEV‐M). Tumors were examined for T1 and T2 signal intensity, T2 FLAIR suppression, contrast enhancement, diffusion restriction, perfusion changes, susceptibility artifact, anatomic tumor location, parenchymal tumor involvement, ventricular dissemination, and hydrocephalus.
Immunohistochemistry
Immunohistochemistry was performed on whole formalin‐fixed, paraffin‐embedded tissue sections using the following antibodies: glial fibrillary acidic protein (GFAP, Dako, cat# GA524, polyclonal, 1:3000 dilution); oligodendrocyte transcription factor 2 (OLIG2, Immuno Bio Labs, polyclonal, 1:200 dilution); SOX10 (Cell Marque, cat# AC‐0237, clone EP268, 1:250 dilution); microtubule associated protein 2 (MAP2, Sigma‐Aldrich, cat# M4403, clone HM2, 1:20 000 dilution); synaptophysin (Cell Marque, cat# 336A, polyclonal, 1:100 dilution); neurofilament (Cell Marque, cat# 302M, clone 2F11, undiluted); NeuN (Chemicon, cat# MAB377, clone A60, 1:4000 dilution); CD34 (Leica Biosystems, cat# PA0354, clone QBend/10, pre‐diluted); Ki67 (Dako, cat# GA626, clone MIB1, 1:50 dilution). All immunostaining were performed on a Leica Bond‐III automated stainer. Diaminobenzidine was used as the chromogen, followed by hematoxylin counterstain.
Targeted next‐generation sequencing
Targeted next‐generation sequencing was performed using the UCSF500 Cancer Panel as previously described 8, 11, 13, 14, 15, 16, 21, 22, 23, 29. Genomic DNA was extracted from formalin‐fixed, paraffin‐embedded blocks of tumor tissue from the eight tumors using the QIAamp DNA FFPE Tissue Kit (Qiagen). Capture‐based next‐generation DNA sequencing was performed using an assay that targets all coding exons of 479 cancer‐related genes, select introns and upstream regulatory regions of 47 genes to enable detection of structural variants including gene fusions, and DNA segments at regular intervals along each chromosome to enable genome‐wide copy number and zygosity analysis, with a total sequencing footprint of 2.8 Mb (Table S1). Multiplex library preparation was performed using the KAPA Hyper Prep Kit (Roche) according to the manufacturer's specifications using 250 ng of sample DNA. Hybrid capture of pooled libraries was performed using a custom oligonucleotide library (Nimblegen SeqCap EZ Choice). Captured libraries were sequenced as paired‐end 100 bp reads on an Illumina HiSeq 2500 instrument. Sequence reads were mapped to the reference human genome build GRCh37 (hg19) using the Burrows–Wheeler aligner (BWA). Recalibration and deduplication of reads were performed using the Genome Analysis Toolkit (GATK). Coverage and sequencing statistics were determined using Picard CalculateHsMetrics and Picard CollectInsertSizeMetrics. Single nucleotide variant and insertion/deletion mutation calling were performed with Unified Genotyper, Pindel, and Delly. Variant annotation was performed with Annovar. Single nucleotide variants, insertions/deletions, and structural variants were visualized and verified using Integrative Genome Viewer. Genome‐wide copy number analysis based on on‐target and off‐target reads was performed by CNVkit and visualized using Nexus Copy Number (Biodiscovery).
Results
Clinical features
The study included four male and four female patients with a median age at time of initial surgery of 23.6 years (range 6‐65 years) (Table 1). Presenting symptoms were variable, including intermittent headaches and subjective cognitive disturbance (ie, attention deficit and short‐term memory impairment). Four of the eight cases were discovered incidentally upon head imaging performed for an unrelated condition such as orbital cellulitis or following motor vehicle accident. Tumors were centered in the septum pellucidum in four patients, in the genu or rostrum of the corpus callosum in three patients, and in the periventricular white matter of the lateral ventricle in one patient. Of the eight patients, three were found to have disseminated intraventricular disease, two at initial diagnosis and one on serial imaging. Intraoperatively, tumors were soft gelatinous gray masses coating the septum pellucidum or ventricular wall.
Table 1.
Clinical features of the eight patients with myxoid glioneuronal tumor, PDGFRA p.K385‐mutant.
| Patient ID | Age at dx (years) | Sex | Tumor location | Presenting symptoms | Extent of resection | Adjuvant therapy | Recurrence or progression | Additional treatment after recurrence or progression | Clinical status at last follow‐up | Length of follow‐up (years) |
|---|---|---|---|---|---|---|---|---|---|---|
| MGNT #1 | 22 | Male | Septum pellucidum | Cognitive impairment | Gross total | None | Recurrence at 2.0 years after gross total resection | Second gross total resection at 4.8 years | Alive without evidence of disease | 6.2 |
| MGNT #2 | 31 | Male | Septum pellucidum | None, incidental finding | Subtotal | None | None/stable | N/A | Alive with stable disease | 1.1 |
| MGNT #3 | 10 | Male | Septum pellucidum | None, incidental finding | Subtotal | None | Progression at 1.3 years after subtotal resection | Second subtotal resection at 2.8 years | Alive with stable disease | 4.3 |
| MGNT #4 | 8 | Female | Genu of corpus callosum | Headaches | Gross total | None | None | N/A | Alive without evidence of disease | 1.7 |
| MGNT #5 | 65 | Female | Septum pellucidum | Headaches, cognitive impairment | Gross total | None | Recurrence at 1.2 years after gross total resection | Temozolomide (3 cycles and 14 cycles) at 1.2 and 6.0 years, CCNU (5 cycles) at 7.7 years, 50.4 Gy external beam radiation at 9.8 years | Alive with stable disease | 12.1 |
| MGNT #6 | 6 | Female | Genu of corpus callosum | None, incidental finding on follow‐up imaging for prior suboccipital decompression of Chiari malformation | Biopsy only | Sterotactic laser interstitial thermal therapy (LITT) | None | N/A | Alive with stable disease | 0.5 |
| MGNT #7 | 28 | Female | Rostrum of corpus callosum | Headaches | Subtotal | None | None/stable | N/A | Alive with stable disease | 0.4 |
| MGNT #8 | 14 | Male | Periventricular white matter of lateral ventricle | Headaches, ataxia | Biopsy only | 54 Gy fractionated external beam radiation, multiagent systemic chemotherapy targeted at synchronous medulloblastoma | None | N/A | Alive with stable disease | 0.2 |
Imaging features
Pre‐operative magnetic resonance imaging features are listed in Table 2 and representative images are shown in Figures 1, 2, 6, 7, 8. Four tumors were centered in the septum pellucidum (MGNT #1, #2, #3, and #5), three were centered in the genu or rostrum of the corpus callosum (MGNT #4, #6, #7), and one was centered in the periventricular white matter of the posterior left lateral ventricle (MGNT #8). All tumors were T1 hypointense and T2 hyperintense. No contrast enhancement or restricted diffusion was seen in any of the cases. Only one of the eight tumors (MGNT #5) demonstrated artifact on susceptibility‐weighted imaging suggestive of either mineralization or blood products from prior intratumoral hemorrhage (which corresponded histologically with the presence of intratumoral hemosiderin‐laden macrophages). Obstructive hydrocephalus was present in the four patients with tumors centered in the septum pellucidum (MGNT #1, #2, #3 and #5), but was not observed in the four patients with tumors centered in the corpus callosum or periventricular white matter. Widespread ventricular dissemination was seen at time of diagnosis for patient MGNT #2, with nodular studding along the ependymal surface of the lateral and third ventricles. A solitary nodular focus of presumed ventricular dissemination was observed in the inferior horn of the left lateral ventricle for patient MGNT #8.
Table 2.
Imaging features at time of initial diagnosis for the eight patients with myxoid glioneuronal tumor, PDGFRA p.K385‐mutant.
| Patient ID | Primary location | T1 signal | T2 signal | FLAIR signal | Contrast enhancement | Diffusion | Perfusion | Mineralization/Hemorrhage | Dissemination | Obstructive hydrocephalus | Parenchymal tumor involvement |
|---|---|---|---|---|---|---|---|---|---|---|---|
| MGNT #1 | Septum pellucidum | Hypointense | Hyperintense | Non‐suppression | None | Facilitated | Not elevated | None | None | Present, lateral ventricles | Absent |
| MGNT #2 | Septum pellucidum | Hypointense | Hyperintense | Non‐suppression | None | Facilitated | Not elevated | None | Intraventricular | Present, lateral ventricles | Present |
| MGNT #3 | Septum pellucidum | Hypointense | Hyperintense | Non‐suppression | None | Facilitated | N/A | None | None | Present, lateral ventricles | Absent |
| MGNT #4 | Genu of corpus callosum | Hypointense | Hyperintense | Non‐suppression | None | Facilitated | Not elevated | None | None | Absent | Absent |
| MGNT #5 | Septum pellucidum | Hypointense | Hyperintense | Non‐suppression | None | Facilitated | Not elevated | Present | None | Present, lateral ventricles | Absent |
| MGNT #6 | Genu of corpus callosum | Hypointense | Hyperintense | Non‐suppression | None | Facilitated | N/A | None | None | Absent | Absent |
| MGNT #7 | Rostrum of corpus callosum | Hypointense | Hyperintense | Non‐suppression | None | Facilitated | N/A | None | None | Absent | Present |
| MGNT #8 | Periventricular white matter of lateral ventricle | Hypointense | Hyperintense | Non‐suppression | None | Facilitated | N/A | None | Intraventricular | Absent | Present |
Figure 1.
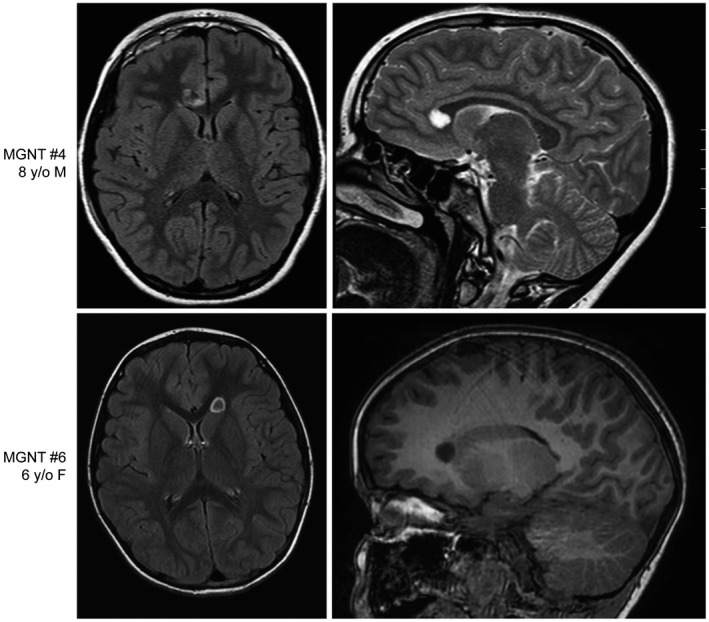
Myxoid glioneuronal tumor, PDGFRA p.K385‐mutant, can also arise in the corpus callosum, in addition to its stereotypic location in the septum pellucidum. Pre‐operative magnetic resonance images for patients MGNT #4 (8‐year‐old female) and MGNT #6 (6‐year‐old female) showing a well‐circumscribed, T2/FLAIR‐hyperintense mass lesion centered in the genu of the corpus callosum.
Figure 2.
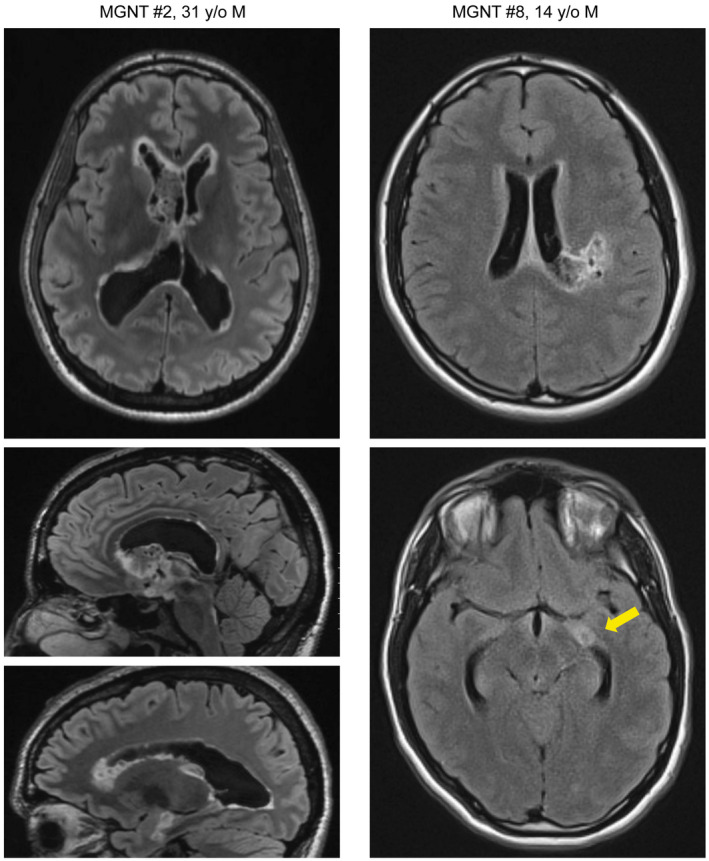
Myxoid glioneuronal tumor, PDGFRA p.K385‐mutant, can be associated with ventricular dissemination. Pre‐operative magnetic resonance images (left) for patient MGNT #2 (31‐year‐old male) showing a primary mass centered in the septum pellucidum with associated ventricular dissemination consisting of nodular studding along the ventricular surfaces. Pre‐operative magnetic resonance images (right) for patient MGNT #8 (14‐year‐old male) showing a primary mass centered in the periventricular white matter of the left lateral ventricle, as well as a solitary nodular focus of presumed ventricular dissemination in the inferior horn of the left lateral ventricle.
Figure 6.
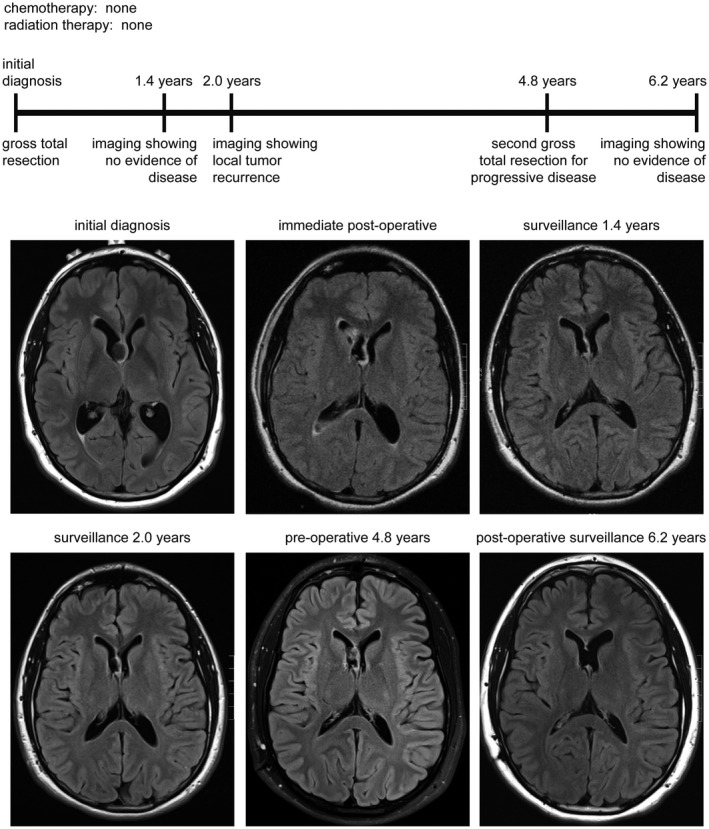
Clinical timeline for patient MGNT #1, a 27‐year‐old man who presented with cognitive disturbance. Following gross total resection of a tumor centered in the septum pellucidum without administration of adjuvant therapy, he subsequently experienced local disease recurrence at 2 years after diagnosis. He is currently alive at 6.2 years following a second resection in the absence of any radiation or chemotherapy.
Figure 7.
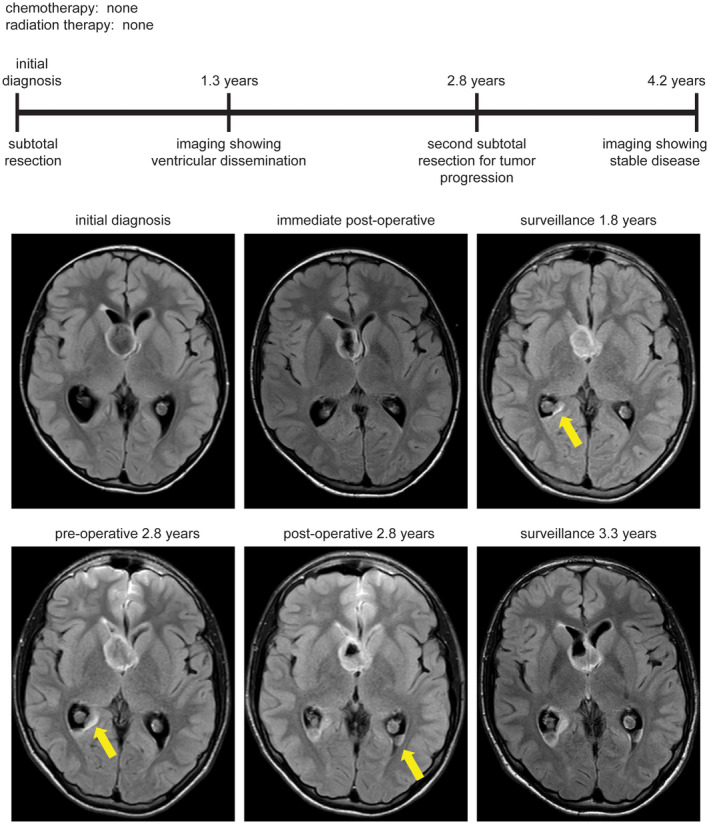
Clinical timeline for patient MGNT #3, a 13‐year‐old boy who presented with orbital cellulitis and was found to have an incidental mass lesion centered in the septum pellucidum. Following subtotal resection without administration of adjuvant therapy, he was found to have local disease progression as well as disseminated disease in the posterior horns of the lateral ventricles at 1.3 years after diagnosis. He is currently alive at 4.2 years following a second subtotal resection in the absence of any radiation or chemotherapy.
Figure 8.
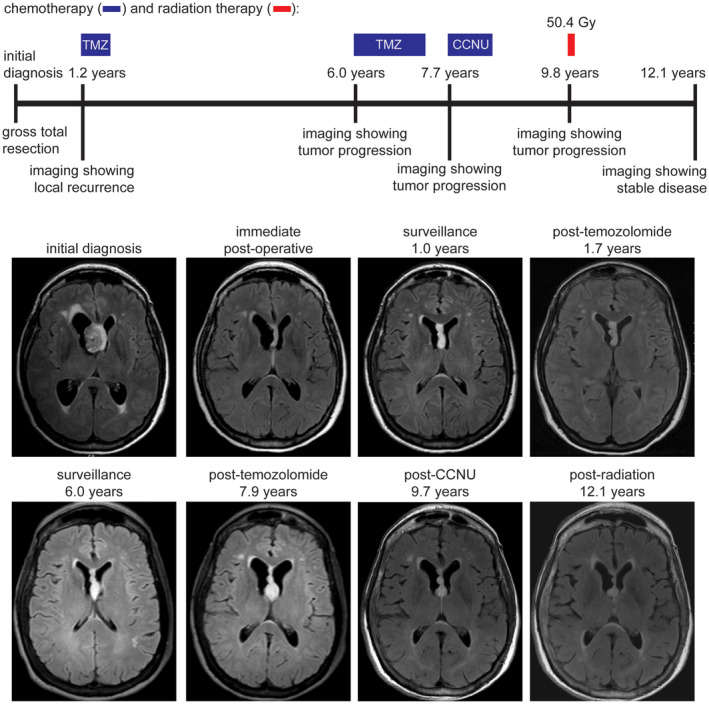
Clinical timeline for patient MGNT #5, a 65‐year‐old woman who presented with headaches and cognitive impairment. Following gross total resection of a tumor centered in the septum pellucidum without administration of adjuvant therapy, she subsequently experienced local disease recurrence at 1.2 years after diagnosis. She is currently alive at 12.1 years following multiple courses of chemotherapy with temozolomide and lomustine (CCNU), as well as fractionated external beam radiation therapy.
Histologic features
Microscopic examination of the eight tumors uniformly demonstrated a low‐grade proliferation of oligodendrocyte‐like cells with monotonous round to oval nuclei, small nucleoli and scant to moderate eosinophilic cytoplasm (Figure 3 and Table 3). All cases demonstrated a prominent myxoid stroma, with some cases additionally containing microcysts filled with basophilic mucin. A fine capillary network reminiscent of DNT or oligodendroglioma was uniformly present, sometimes with angiocentric clustering or linear arrangement of the oligodendrocyte‐like tumor cells along vessels. A neuronal element composed of individual neurons floating in the mucinous stroma was seen at least focally in all tumors. Additionally, a subset of cases demonstrated neurocytic rosettes (MGNT #2 and #3), with tumor cells surrounding cores of eosinophilic fibrillar material that stained with synaptophysin. In contrast to cortically based DNT, multinodular architecture with well‐defined mucin‐patterned nodules was not appreciated in any of the tumors. While most of the tumors lacked Rosenthal fibers or eosinophilic granular bodies, one tumor (MGNT #5) demonstrated intermixed areas of piloid morphology with tumor cells showing elongate bipolar cytoplasmic processes and contained both Rosenthal fibers and eosinophilic granular bodies. Calcifications were not observed in any of the cases. Mitotic figures were uniformly inconspicuous (either absent or less than 1 mitosis per 10 high power fields in all cases). Neither necrosis nor glomeruloid microvascular proliferation was seen in any of the tumors.
Figure 3.
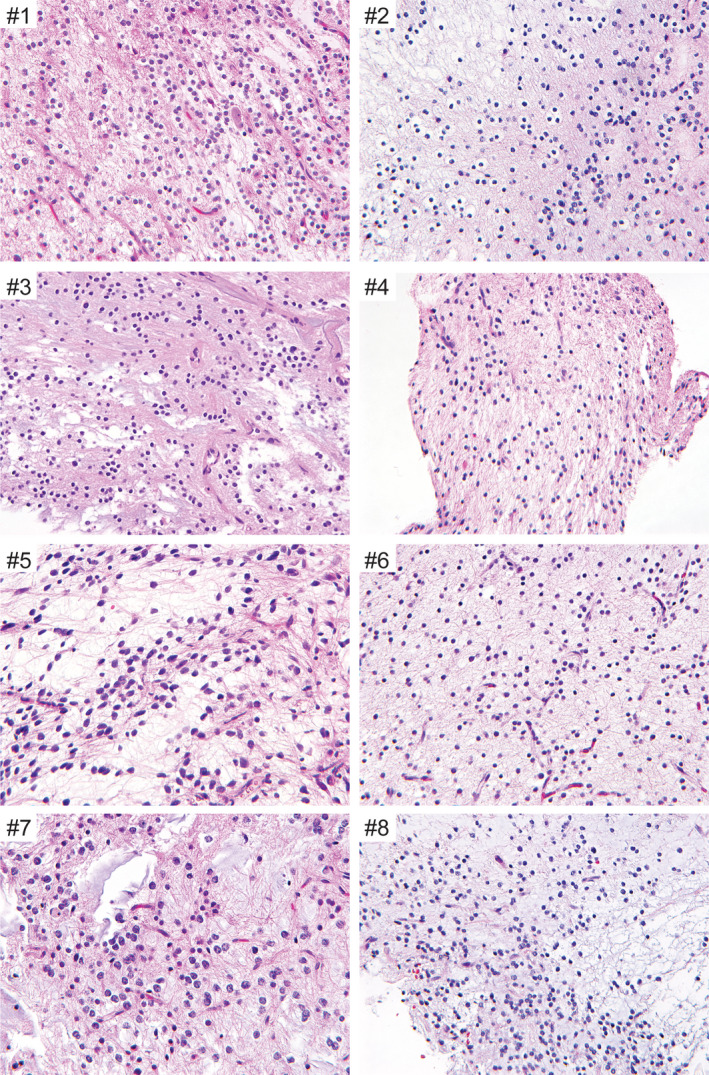
Histologic features of the eight myxoid glioneuronal tumors, PDGFRA p.K385‐mutant. Shown are representative hematoxylin and eosin stained sections from cases MGNT #1‐8. All cases demonstrate a low cellularity proliferation of oligodendrocyte‐like cells with monotonous round to oval nuclei, small nucleoli, and scant to moderate eosinophilic cytoplasm within a mucin‐rich stroma. Scattered floating neurons and neurocytic rosettes are occasionally seen. Mitotic activity is uniformly inconspicuous, while neither necrosis nor glomeruloid microvascular proliferation are present in any of the tumors.
Table 3.
Histologic features of the eight myxoid glioneuronal tumors, PDGFRA p.K385‐mutant.
| Tumor ID | Morphology of glial component | Myxoid background | Multinodular architecture | Floating neurons | Neurocytic rosettes | Eosinophilic granular bodies | Rosenthal fibers | Calcifications | Mitotic activity | Necrosis | Microvascular proliferation |
|---|---|---|---|---|---|---|---|---|---|---|---|
| MGNT #1 | Oligodendroglial | Present | Absent | Present | Absent | Absent | Absent | Absent | Absent | Absent | Absent |
| MGNT #2 | Oligodendroglial | Present | Absent | Present | Present | Absent | Absent | Absent | Absent | Absent | Absent |
| MGNT #3 | Oligodendroglial | Present | Absent | Present | Present | Absent | Absent | Absent | Absent | Absent | Absent |
| MGNT #4 | Oligodendroglial | Present | Absent | Present | Absent | Absent | Absent | Absent | Absent | Absent | Absent |
| MGNT #5 | Piloid to oligodendroglial | Present | Absent | Present | Absent | Present | Present | Absent | Absent | Absent | Absent |
| MGNT #6 | Oligodendroglial | Present | Absent | Present | Absent | Absent | Absent | Absent | Absent | Absent | Absent |
| MGNT #7 | Oligodendroglial | Present | Absent | Present | Absent | Absent | Absent | Absent | Absent | Absent | Absent |
| MGNT #8 | Oligodendroglial | Present | Absent | Present | Absent | Absent | Absent | Absent | Absent | Absent | Absent |
Immunohistochemical features
Diffuse OLIG2 and SOX10 nuclear positivity was observed in the oligodendrocyte‐like tumor cells (Figure 4 and Table 4). Immunostaining for GFAP was strongly positive in all evaluated cases. Two cases were evaluated for MAP2 expression that revealed positivity in the majority of the oligodendrocyte‐like tumor cells. Neurofilament staining highlighted background axons and rare floating neurons, but was negative in the oligodendrocyte‐like tumor cells. There was weak granular synaptophysin staining throughout the background neuropil, with limited to absent cytoplasmic staining of the oligodendrocyte‐like tumor cells. Additionally, the synaptophysin staining highlighted the occasional floating neurons and the neuropil cores of neurocytic rosettes (MGNT #2 and #3). NeuN staining was only positive in rare floating neurons. CD34 immunoreactivity was limited to vascular endothelial cells only, with no CD34‐positive ramified cells identified. The Ki67 labeling index was uniformly low, ranging from 1% to 4%.
Figure 4.
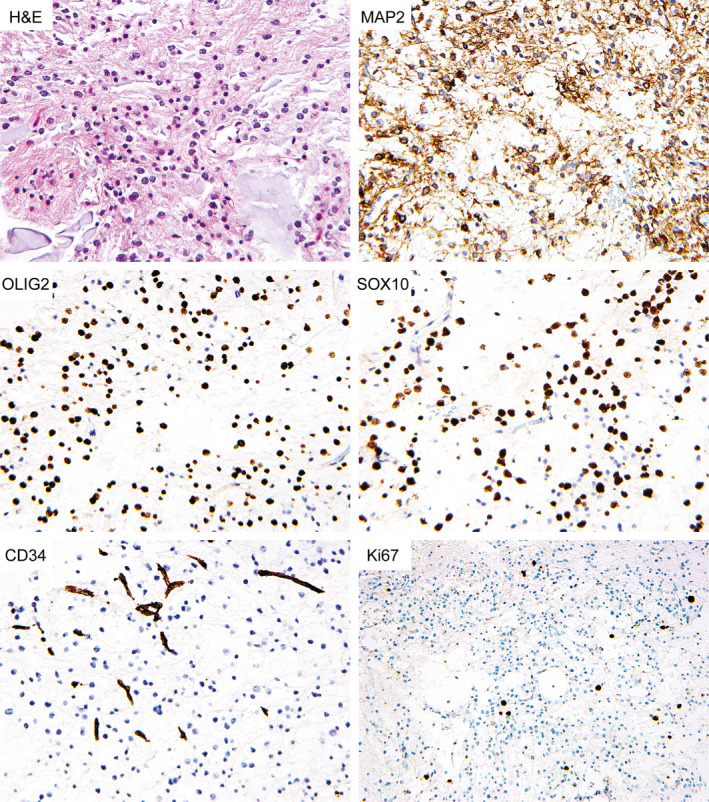
Immunohistochemical features of myxoid glioneuronal tumor, PDGFRA p.K385‐mutant. Shown are representative immunohistochemical stains from case MGNT #7 demonstrating cytoplasmic positivity for MAP2 in most tumor cells, along with diffuse nuclear positivity for OLIG2 and SOX10. CD34 staining is limited to endothelial cells. The Ki67 labeling is low.
Table 4.
Immunohistochemical features of the eight myxoid glioneuronal tumors, PDGFRA p.K385‐mutant.
| Tumor ID | GFAP | OLIG2 | SOX10 | MAP2 | Synaptophysin | Neurofilament | NeuN | CD34‐positive ramified cells | Ki67 |
|---|---|---|---|---|---|---|---|---|---|
| MGNT #1 | Strongly positive | N/A | N/A | Positive | Weak positivity; also highlights background neuropil | Negative; highlights background axonal processes | N/A | Absent | 1% |
| MGNT #2 | Strongly positive | Strongly positive | N/A | N/A | Weak positivity; also highlights background neuropil | Negative; highlights background axonal processes | Negative; rare positive neuronal cells | N/A | 3% |
| MGNT #3 | Strongly positive | Strongly positive | N/A | N/A | Weak positivity; also highlights background neuropil | Negative; highlights background axonal processes | Negative; rare positive neuronal cells | Absent | 3% |
| MGNT #4 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
| MGNT #5 | Strongly positive | N/A | N/A | N/A | Weak positivity; also highlights background neuropil | Negative; highlights background axonal processes | N/A | Absent | 1% |
| MGNT #6 | Strongly positive | N/A | N/A | N/A | Weak positivity; also highlights background neuropil | Negative; highlights background axonal processes | N/A | Absent | 4% |
| MGNT #7 | Strongly positive | Strongly positive | Strongly positive | Positive | Weak positivity; also highlights background neuropil | N/A | N/A | Absent | 2% |
| MGNT #8 | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A |
Targeted next‐generation sequencing results
Targeted next‐generation sequencing of approximately 500 cancer‐associated genes and genome‐wide copy number analysis was performed on the eight tumors. All eight cases harbored a dinucleotide substitution at codon 385 of the PDGFRA oncogene, resulting in either a lysine to leucine change (p.K385L, n = 6) or a lysine to isoleucine change (p.K385I, n = 2) (Figure 5 and Table S2). The 20%–44% allele frequencies of these PDGFRA p.K385L/I mutations are consistent with being clonal heterozygous somatic variants in each case. This p.K385L/I mutation localizes within exon 8 that encodes one of the immunoglobulin‐like C2 domains in the extracellular ligand‐binding domain portion of the receptor tyrosine kinase (Figure 5B). The PDGFRA mutation was the solitary somatic pathogenic alteration identified in all cases. One tumor (MGNT #5) harbored trisomy 12q as the solitary copy number alteration observed. The remaining seven cases demonstrated a balanced diploid genome without chromosomal gains, losses or focal amplifications or deletions (Table S3).
Figure 5.
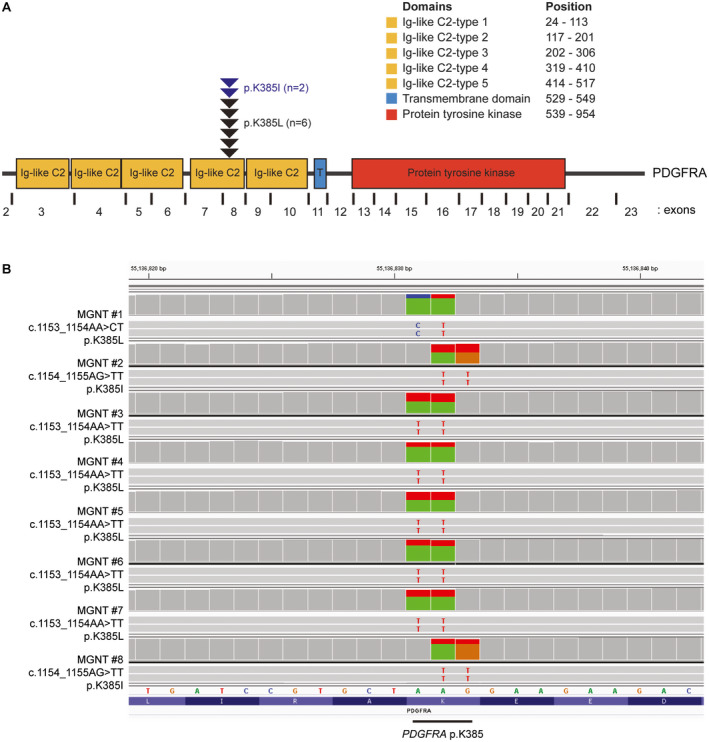
PDGFRA mutations identified in the eight myxoid glioneuronal tumors, PDGFRA p.K385‐mutant. A. Diagram of the human PDGFRA protein with the location of the recurrent p.K385L/I mutations within the extracellular ligand‐binding domain. UniProt ID P16234. B. Snapshots from the Integrative Genome Viewer showing sequencing reads containing a dinucleotide substitution at codon 385 of the PDGFRA gene in all 8 cases causing a lysine to leucine or isoleucine substitution (p.K385L/I). RefSeq transcript NM_006206.
Notably, patient MGNT #8 is a 14‐year‐old boy who initially presented with headaches, nausea and ataxia. Imaging revealed both a solid enhancing mass centered in the left cerebellar hemisphere and a separate periventricular lesion surrounding the lateral ventricle in the left parietal lobe. He underwent resection of the left cerebellar mass revealing large cell/anaplastic medulloblastoma. Two weeks later, he underwent biopsy of the periventricular lesion to rule out the possibility of metastatic/disseminated medulloblastoma. Instead, pathologic analysis revealed a low‐grade myxoid glioneuronal tumor. Genetic analysis revealed a germline truncating nonsense mutation in the BRCA2 tumor suppressor gene at heterozygous allele frequency. No loss of heterozygosity or a second somatic mutation inactivating the remaining wild‐type allele was identified in either the medulloblastoma or myxoid glioneuronal tumor. The medulloblastoma was identified to harbor focal amplification of the MYCN oncogene and homozygous/biallelic deletion of the TP53 tumor suppressor gene, overall consistent with an SHH‐activated and TP53‐mutant molecular subtype. The myxoid glioneuronal tumor from the left periventricular region lacked the MYCN amplification and TP53 deletion observed in the medulloblastoma, but instead harbored PDGFRA p.K385I mutation which was not present in the medulloblastoma. While the medulloblastoma demonstrated marked fragmentation of the genome suggestive of an underlying deficiency of homologous recombination potentially attributable to BRCA deficiency, the myxoid glioneuronal tumor demonstrated a balanced diploid genome. Thus, the contribution of the germline BRCA2 inactivating mutation to the development of the myxoid glioneuronal tumor, PDGFRA p.K385‐mutant, in this child is uncertain.
Clinical outcomes
The complete clinical data including extent of resection, treatment regimen and outcome from the eight patients are presented in Table 1. Clinical follow‐up for this cohort ranged from 0.2 to 12.1 years after initial surgery (median 1.4 years). One patient underwent biopsy followed by laser interstitial thermal therapy (MGNT #6), one underwent biopsy followed by 54 Gy of external beam radiation (MGNT #8), three underwent subtotal resection followed by observation (MGNT #2, #3 and #7), and three underwent gross total resection followed by observation (MGNT #1, #4 and #5). Two of the patients had imaging at time of initial diagnosis showing ventricular dissemination (MGNT #2 and #8). Three patients experienced local disease recurrence or progression (MGNT #1, #3 and #5) at 2.0, 1.3 and 1.2 years after initial surgical intervention, all of which had undergone either subtotal (MGNT #3) or gross total (MGNT #1 and #5) resection followed by observation without adjuvant therapy. One of these patients (MGNT #3) was additionally found to have ventricular dissemination on surveillance imaging at 1.3 years after initial surgical intervention. The remaining five patients (MGNT #2, #4, #6, #7 and #8) did not show evidence of disease recurrence or progression during the follow‐up interval of 1.1, 1.7, 0.5, 0.4 and 0.2 years, respectively.
Among the three patients who experienced disease progression/recurrence, one (MGNT #1) underwent a second gross total resection at 4.8 years after initial surgical intervention and remains alive with no evidence of disease progression at last clinical follow‐up (6.2 years) (Figure 6). Another patient (MGNT #3) underwent a second subtotal resection followed by observation and shows stable disease (both local and disseminated throughout the lateral ventricles) at last clinical follow‐up (4.2 years) (Figure 7). The third patient (MGNT #5) was treated with temozolomide for local recurrence in the septum pellucidum at 1.2 years after initial surgical intervention, which was discontinued after three cycles due to thrombocytopenia (Figure 8). Her residual disease was stable on follow‐up imaging over the next 5 years until local disease progression was again observed. She was treated with temozolomide for another 14 cycles. However, imaging 3 months later was concerning for continued tumor growth, and she was then treated with five cycles of lomustine (CCNU), which was discontinued due to hepatic toxicity. The tumor remained stable for the next 15 months when imaging again demonstrated thickening of the septum worrisome for tumor progression. She was then treated with 50.4 Gy of fractionated external beam radiation therapy at 9.8 years after initial surgical intervention. She currently has stable disease without any evidence of local disease progression or intraventricular dissemination, now 12.1 years after her initial diagnosis and 2.3 years after radiation therapy.
Discussion
PDGFRA activation via amplification, mutation, or intragenic deletion/rearrangement is known to be a frequent genetic driver among a select group of human tumor types including gastrointestinal stromal tumors, both pediatric and adult glioblastomas, and myxoid glioneuronal tumor. In gastrointestinal stromal tumors lacking KIT mutations, there are frequent PDGFRA activating missense mutations that localize at a couple different mutational hotspots within the intracellular kinase domain (p.D842V and p.V561D). In both pediatric and adult glioblastomas, there is frequent amplification and overexpression of PDGFRA, which is commonly accompanied by intragenic deletions or various missense mutations on the amplified alleles. The most common intragenic deletion eliminates exons 8‐9 that encode a portion of the extracellular ligand‐binding domain 19. A number of different recurrent missense mutations in PDGFRA have been identified in pediatric and adult glioblastomas (including p.C235Y, p.E229K and p.Y288C), which also perturb the coding sequence of the extracellular ligand‐binding domain 20. However, mutations at codon p.K385 of PDGFRA appear to be highly specific to “myxoid glioneuronal tumor, PDGFRA p.K385‐mutant,” as only five tumors among the greater than 83 000 tumors with sequencing data for the PDGFRA gene in the version 89 release of the Catalog of Somatic Mutations In Cancer (COSMIC) database harbor somatic variants at this codon. These five tumors in the current version of the COSMIC database with PDGFRA p.K385I (n = 1) or p.K385M (n = 4) mutations are all reportedly pediatric gliomas, with precise anatomic location and histologic diagnosis not clearly specified. While PDGFRA amplification and mutation/rearrangement are common in high‐grade diffuse gliomas in both children and adults, these PDGFRA alterations invariably co‐occur with other pathogenic alterations such as p.K27M mutation of H3F3A or HIST1H3B genes in diffuse midline gliomas 17, IDH1 p.R132H mutation in IDH‐mutant glioblastomas 4, or TERT promoter mutation and CDKN2A deletion in IDH‐wild‐type glioblastomas 2. PDGFRA mutation has not been identified as the solitary genetic driver in any CNS tumor entity to date other than “myxoid glioneuronal tumor, PDGFRA p.K385‐mutant.”
This p.K385L/I mutation in PDGFRA localizes within exon 8 that encodes one of the immunoglobulin‐like C2 domains in the extracellular ligand‐binding domain portion of the receptor tyrosine kinase. These mutations result in substitution from a basic amino acid (lysine) to a hydrophobic amino acid (leucine or isoleucine). While the precise functional consequence of this recurrent p.K385L/I mutation in PDGFRA has yet to be elucidated, it is predicted to be an oncogenic, gain‐of‐function mutation that causes constitutive activation of the intracellular kinase domain in the absence of ligand. It remains to be determined if these myxoid glioneuronal tumors with PDGFRA p.K385 mutation might perhaps be sensitive to small molecule tyrosine kinase inhibitors such as imatinib or dasatinib.
Together with the two prior reports 6, 27, this study provides further support for “myxoid glioneuronal tumor, PDGFRA p.K385‐mutant” representing a distinct CNS tumor entity. However, this study also indicates that the septum pellucidum is not the exclusive anatomic site of involvement and should not be considered as a defining characteristic. This neoplasm likely has a distinct cell of origin located within the septum pellucidum, corpus callosum or periventricular white matter. It can present in a wide age range, including both young children and older adults. It demonstrates histologic features that are somewhat reminiscent of either DNT or RGNT, but lacks the well‐defined mucin‐patterned nodules that characterize DNT in the cerebral cortex. Furthermore, this tumor entity demonstrates a distinct genetic signature and a distinct methylation profile compared to all other CNS tumor entities that have been described to date 6, 27. Based on the small number of patients with long‐term follow‐up studied to date, this tumor entity appears to follow a benign or indolent disease course that is comparable to other tumor entities assigned a grade I designation by the WHO Classification of Tumors of the Central Nervous System. Unfortunately, many of the cases previously described as “dysembryoplastic neuroepithelial tumor‐like neoplasm of the septum pellucidum” and intraventricular DNT have not been molecularly characterized 1, 3, 5, 7, 9, 12, 18, 28, 30, 31, though it is likely that the majority represent “myxoid glioneuronal tumor, PDGFRA p.K385‐mutant.” They have similarly been associated with a relatively indolent behavior, albeit with occasional ventricular or leptomeningeal dissemination, similar to our series. Further study of this emerging tumor entity is warranted to best determine appropriate grading and treatment guidelines.
Conflict of Interest
None of the authors have any conflicts of interest to disclose.
Supporting information
Table S1. List of the 479 genes targeted for sequencing on the UCSF500 Cancer Panel.
Table S2. PDGFRA mutations identified in the eight myxoid glioneuronal tumors, PDGFRA p.K385‐mutant.
Table S3. Chromosomal copy number alterations identified in the eight myxoid glioneuronal tumors, PDGFRA p.K385‐mutant.
Acknowledgments
D.A.S is supported by NIH Director's Early Independence Award (DP5 OD021403). We thank the staff of the UCSF Clinical Cancer Genomics Laboratory for assistance with genetic profiling.
Data Availability Statement
Scanned image files of H&E‐stained sections from the eight tumors from which representative images are presented are available for downloading and viewing at the following link: https://figshare.com/projects/Myxoid_glioneuronal_tumor_PDGFRA_p_K385-mutant/35651. Sequencing data files are available from the authors upon request.
References
- 1. Baisden BL, Brat DJ, Melhem ER, Rosenblum MK, King AP, Burger PC (2001) Dysembryoplastic neuroepithelial tumor‐like neoplasm of the septum pellucidum: a lesion often misdiagnosed as glioma: report of 10 cases. Am J Surg Pathol 25:494–499. [DOI] [PubMed] [Google Scholar]
- 2. Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR et al (2013) The somatic genomic landscape of glioblastoma. Cell 155:462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Campos AR, Clusmann H, von Lehe M, Niehusmann P, Becker AJ, Schramm J et al (2009) Simple and complex dysembryoplastic neuroepithelial tumors (DNT) variants: clinical profile, MRI, and histopathology. Neuroradiology 51:433–443. [DOI] [PubMed] [Google Scholar]
- 4. Cancer Genome Atlas Research Network , Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR et al (2015) Comprehensive, integrative genomic analysis of diffuse lower‐grade gliomas. N Engl J Med 372:2481–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cervera‐Pierot P, Varlet P, Chodkiewicz JP, Daumas‐Duport C (1997) Dysembryoplastic neuroepithelial tumors located in the caudate nucleus area: report of four cases. Neurosurgery 40:1065–1070. [DOI] [PubMed] [Google Scholar]
- 6. Chiang JCH, Harreld JH, Tanaka R, Li X, Wen J, Zhang C et al (2019) Septal dysembryoplastic neuroepithelial tumor: a comprehensive clinical, imaging, histopathologic, and molecular analysis. Neuro Oncol 21:800–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Daghistani R, Miller E, Kulkarni AV, Widjaja E (2013) Atypical characteristics and behavior of dysembryoplastic neuroepithelial tumors. Neuroradiology 55:217–224. [DOI] [PubMed] [Google Scholar]
- 8. Ferris SP, Velazquez Vega J, Aboian M, Lee JC, Van Ziffle J, Onodera C et al (2020) High‐grade neuroepithelial tumor with BCOR exon 15 internal tandem duplication‐a comprehensive clinical, radiographic, pathologic, and genomic analysis. Brain Pathol 30:46–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gessi M, Hattingen E, Dörner E, Goschzik T, Dreschmann V, Waha A et al (2016) Dysembryoplastic neuroepithelial tumor of the septum pellucidum and the supratentorial midline: histopathologic, neuroradiologic, and molecular features of 7 cases. Am J Surg Pathol 40:806–811. [DOI] [PubMed] [Google Scholar]
- 10. Gessi M, Moneim YA, Hammes J, Goschzik T, Scholz M, Denkhaus D et al (2014) FGFR1 mutations in Rosette‐forming glioneuronal tumors of the fourth ventricle. J Neuropathol Exp Neurol 73:580–584. [DOI] [PubMed] [Google Scholar]
- 11. Goode B, Mondal G, Hyun M, Ruiz DG, Lin YH, Van Ziffle J et al (2018) A recurrent kinase domain mutation in PRKCA defines chordoid glioma of the third ventricle. Nat Commun 9:810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Harter DH, Omeis I, Forman S, Braun A (2006) Endoscopic resection of an intraventricular dysembryoplastic neuroepithelial tumor of the septum pellucidum. Pediatr Neurosurg 42:105–107. [DOI] [PubMed] [Google Scholar]
- 13. Iorgulescu JB, Van Ziffle J, Stevers M, Grenert JP, Bastian BC, Chavez L et al (2018) Deep sequencing of WNT‐activated medulloblastomas reveals secondary SHH pathway activation. Acta Neuropathol 135:635–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kline CN, Joseph NM, Grenert JP, van Ziffle J, Talevich E, Onodera C et al (2017) Targeted next‐generation sequencing of pediatric neuro‐oncology patients improves diagnosis, identifies pathogenic germline mutations, and directs targeted therapy. Neuro‐Oncol 19:699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee J, Putnam AR, Chesier SH, Banerjee A, Raffel C, Van Ziffle J et al (2018) Oligodendrogliomas, IDH‐mutant and 1p/19q‐codeleted, arising during teenage years often lack TERT promoter mutation that is typical of their adult counterparts. Acta Neuropathol Commun 6:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lopez GY, Van Ziffle J, Onodera C, Grenert JP, Yeh I, Bastian BC et al (2019) The genetic landscape of gliomas arising after therapeutic radiation. Acta Neuropathol 137:139–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mackay A, Burford A, Carvalho D, Izquierdo E, Fazal‐Salom J, Taylor KR et al (2017) Integrated molecular meta‐analysis of 1,000 pediatric high‐grade and diffuse intrinsic pontine glioma. Cancer Cell 32:520–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Onguru O, Deveci S, Sirin S, Timurkaynak E, Gunhan O (2003) Dysembryoplastic neuroepithelial tumor in the left lateral ventricle. Minim Invasive Neurosurg 46:306–309. [DOI] [PubMed] [Google Scholar]
- 19. Ozawa T, Brennan CW, Wang L, Squatrito M, Sasayama T, Nakada M et al (2010) PDGFRA gene rearrangements are frequent genetic events in PDGFRA‐amplified glioblastomas. Genes Dev 24:2205–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Paugh BS, Zhu X, Qu C, Endersby R, Diaz AK, Zhang J et al (2013) Novel oncogenic PDGFRA mutations in pediatric high‐grade gliomas. Cancer Res 73:6219–6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pekmezci M, Stevers M, Phillips JJ, Van Ziffle J, Bastian BC, Tsankova NM et al (2018) Multinodular and vacuolating neuronal tumor of the cerebrum is a clonal neoplasm defined by genetic alterations that activate the MAP kinase signaling pathway. Acta Neuropathol 135:485–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pekmezci M, Villanueva‐Meyer JE, Goode B, Van Ziffle J, Onodera C, Grenert JP et al (2018) The genetic landscape of ganglioglioma. Acta Neuropathol Commun 6:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Phillips JJ, Gong H, Chen K, Joseph NM, van Ziffle J, Bastian BC et al (2019) The genetic landscape of anaplastic pleomorphic xanthoastrocytoma. Brain Pathol 29:85–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Qaddoumi I, Orisme W, Wen J, Santiago T, Gupta K, Dalton JD et al (2016) Genetic alterations in uncommon low‐grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol 131:833–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rivera B, Gayden T, Carrot‐Zhang J, Nadaf J, Boshari T, Faury D et al (2016) Germline and somatic FGFR1 abnormalities in dysembryoplastic neuroepithelial tumors. Acta Neuropathol 131:847–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sievers P, Appay R, Schrimpf D, Stichel D, Reuss DE, Wefers AK et al (2019) Rosette‐forming glioneuronal tumors share a distinct DNA methylation profile and mutations in FGFR1, with recurrent co‐mutation of PIK3CA and NF1. Acta Neuropathol 138:497–504. [DOI] [PubMed] [Google Scholar]
- 27. Solomon DA, Korshunov A, Sill M, Jones DTW, Kool M, Pfister SM et al (2018) Myxoid glioneuronal tumor of the septum pellucidum and lateral ventricle is defined by a recurrent PDGFRA p. K385 mutation and DNT‐like methylation profile. Acta Neuropathol 136:339–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang F, Qiao G, Li X, Gui Q (2007) A dysembryoplastic neuroepithelial tumor in the area of the caudate nucleus in a 57‐year‐old woman: case report. Neurosurgery 61:E420. [DOI] [PubMed] [Google Scholar]
- 29. Wood MD, Tihan T, Perry A, Chacko G, Turner C, Pu C et al (2018) Multimodal molecular analysis of astroblastoma enables reclassification of most cases into more specific molecular entities. Brain Pathol 28:192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xiong J, Liu Y, Chu SG, Chen H, Chen HX, Mao Y et al (2012) Dysembryoplastic neuroepithelial tumor‐like neoplasm of the septum pellucidum: review of 2 cases with chromosome 1p/19q and IDH1 analysis. Clin Neuropathol 31:31–38. [DOI] [PubMed] [Google Scholar]
- 31. Yuan J, Sharma N, Choudhri H, Figueroa R, Sharma S (2011) Intraventricular dysembryoplastic neuroepithelial tumor in a pediatric patient: is it the most common extracortical location for DNT? Childs Nerv Syst 27:485–490. [DOI] [PubMed] [Google Scholar]
- 32. Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B et al (2013) Whole‐genome sequencing identifies genetic alterations in pediatric low‐grade gliomas. Nat Genet 45:602–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of the 479 genes targeted for sequencing on the UCSF500 Cancer Panel.
Table S2. PDGFRA mutations identified in the eight myxoid glioneuronal tumors, PDGFRA p.K385‐mutant.
Table S3. Chromosomal copy number alterations identified in the eight myxoid glioneuronal tumors, PDGFRA p.K385‐mutant.
Data Availability Statement
Scanned image files of H&E‐stained sections from the eight tumors from which representative images are presented are available for downloading and viewing at the following link: https://figshare.com/projects/Myxoid_glioneuronal_tumor_PDGFRA_p_K385-mutant/35651. Sequencing data files are available from the authors upon request.