

Abstract
The influence of dietary factors on brain health and mental function is becoming increasingly recognized. Similarly, mounting evidence supports a role for gut microbiota in modulating central nervous system function and behaviour. Still, the molecular mechanisms responsible for the impact of diet and associated microbiome in adult neurodegeneration are still largely unclear. In this study, we aimed to investigate whether and how changes in diet-associated microbiome and its metabolites impact on adult neurogenesis. Mice were fed a high-fat, choline-deficient diet, developing obesity and several features of the metabolic syndrome, including non-alcoholic steatohepatitis. Strikingly, our results showed, for the first time, that animals fed with this specific diet display premature increased neurogenesis, possibly exhausting the available neural stem cell pool for long-term neurogenesis processes. The high-fat, choline-deficient diet further induced neuroinflammation, oxidative stress, synaptic loss and cell death in different regions of the brain. Notably, this diet-favoured gut dysbiosis in the small intestine and cecum, up-regulating metabolic pathways of short-chain fatty acids, such as propionate and butyrate and significantly increasing propionate levels in the liver. By dissecting the effect of these two specific short-chain fatty acids in vitro, we were able to show that propionate and butyrate enhance mitochondrial biogenesis and promote early neurogenic differentiation of neural stem cells through reactive oxygen species- and extracellular signal-regulated kinases 1/2-dependent mechanism. More importantly, neurogenic niches of high-fat, choline-deficient-fed mice showed increased expression of mitochondrial biogenesis markers, and decreased mitochondrial reactive oxygen species scavengers, corroborating the involvement of this mitochondrial stress-dependent pathway in mediating changes of adult neurogenesis by diet. Altogether, our results highlight a mitochondria-dependent pathway as a novel mediator of the gut microbiota–brain axis upon dietary influences.
Keywords: gut microbiome, high-fat choline-deficient diet, mitochondrial oxidative stress, neurogenesis, short-chain fatty acids
Ribeiro et al. demonstrated a possible mechanism by which diet impairs adult neurogenesis in mice. A specific high caloric diet produces a gut microbiota signature that up-regulates the metabolism of short-chain fatty acids. These, in turn, are responsible to induce early neuronal differentiation by enhancing mitochondrial activity and oxidative stress in neural stem cells.
Graphical Abstract
Graphical Abstract.
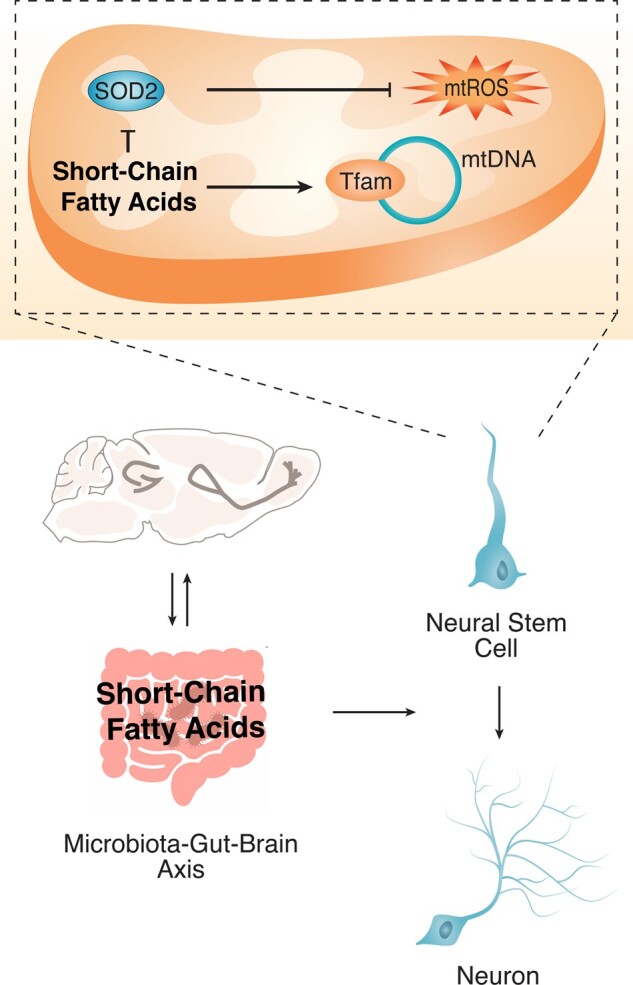
Introduction
Dietary factors are well known to influence neurological function and brain health. In fact, unbalanced dietary habits, particularly calorie-dense foods, are risk factors for impaired cognitive function and psychiatric disorders (Gomez-Pinilla, 2008). Similarly, mice fed a high-fat diet were shown to present cognitive deficits, anxiety and depressive-like behaviour (Almeida-Suhett et al., 2017). Diet also affects molecular events related to energy metabolism which, in turn, modulates neuronal signalling, synaptic plasticity and, ultimately, neuronal function (Gomez-Pinilla and Tyagi, 2013). As such, a better understanding of the signalling pathways connecting diet and brain function may allow for the development of novel non-invasive therapeutic strategies against neurological and psychiatric disorders.
Dietary nutrients are strong modulators of microbiota composition, function and metabolism, which in turn also impacts on host physiology (Gentile and Weir, 2018). The relevance of gut microbiota in the regulation of brain function has recently gained extensive interest (Khanna and Tosh, 2014). In fact, although the mechanisms that mediate this cross-talk remaining largely unknown, a microbiota–gut–brain axis has already been recognized. It is worth noting that gut microbiota is not only composed by bacteria but also by many other micro-organisms such as archaea, viruses, phages and fungus. However, although phages are known to exceed bacteria in both number and diversity, bacteria and their bacterial products are the most studied group of gut microbiota, with crucial roles in food processing, vitamin synthesis, inhibition of pathogens, host homeostasis (Ramakrishna, 2013) and even brain development (Heijtz et al., 2011; Rogers et al., 2016). Thus, disturbances in this symbiotic microbiota–host relationship might lead to pathological disorders in different organs, including the brain. In fact, gut dysbiosis has been shown to associate with neurological disorders, such as psychiatric disorders—depression and anxiety—autism and neurodegenerative diseases (Lurie et al., 2015; Rogers et al., 2016).
Curiously, microbiota composition is highly dynamic and sensitive to lifecycle, environmental, immune and nutritional factors. Along with changes in bacterial abundance, microbial metabolism also adjusts to surrounding fluctuations, leading to different bacterial products, which in turn modulate host physiology, metabolism, immune system and gene expression (Nicholson et al., 2012; Fung et al., 2017). Bacteria-derived molecules encompass lipopolysaccharides, peptidoglycans, short-chain fatty acids (SCFA), neurotransmitters and gaseous molecules (Cani and Knauf, 2016). Short-chain fatty acids, including acetate, propionate and butyrate, result from fermentation of non-digestible carbohydrates and some proteins in the cecum and large intestine (Koh et al., 2016), and embody powerful mitochondrial regulators. Apart from being used as bacterial energy substrates, SCFAs regulate fatty acid and glucose metabolism through an AMPK-PGC-1α-dependent mechanism, and are also capable of increasing mitochondrial bioenergetics and biogenesis (Rose et al., 2018; Uittenbogaard et al., 2018). Therefore, mitochondrial-sensitive processes are deeply prone to SCFA-mediated gut microbiota regulation.
The adult mammalian brain has the ability to generate new neurons through neurogenesis in a mitochondria-dependent process (Fang et al., 2016; Khacho et al., 2016). In the adult mammalian brain, neurogenesis occurs in two main neurogenic niches, including the sub-ventricular zone (SVZ) and the sub-granular zone (SGZ) in the dentate gyrus (DG) of the hippocampus. Neural stem cells (NSCs) self-renew, proliferate and differentiate into different neural cells (Zhao et al., 2008). In the case of SVZ, NSCs migrate until they reach the olfactory bulb (OB), where neural precursor cells mature and integrate.
Interestingly, reduction of adult hippocampal neurogenesis has been reported in animal models of memory loss (Donovan et al., 2006; Hamilton and Holscher, 2012) and psychiatric disorders, particularly depression and anxiety (Petrik et al., 2012; Fang et al., 2018), whereas an increased rate of SGZ neurogenesis induced by treatment with antidepressants improves psychiatric disorders (Perera et al., 2011), suggesting an important role of adult neurogenesis on neurological disorders. Importantly, mitochondrial metabolism was shown to finely tune NSC self-renewal and fate decision. In fact, mitochondrial mass, as well as oxidative phosphorylation and by-product reactive oxygen species (ROS) increase during neuronal differentiation in response to higher cellular energetic demands, and promoting transcription of neurogenic genes (Khacho et al., 2016). Besides mitochondrial biogenesis, alterations in mitochondrial dynamics—fusion and fission events—also regulate NSC cell-cycle progression and fate, namely by modulating mitochondrial bioenergetics and oxidative stress (Mitra, 2013). Although the impact of diet and gut microbiota on neuroplasticity has already been described, the underlying molecular mechanisms remain largely unknown.
Here, we demonstrate that gut microbiota-derived metabolites, produced upon dietary changes, modulate adult neurogenesis in a mitochondria-dependent manner. In particular, we show that a high-fat, choline-deficient (HFCD) diet induces gut dysbiosis to favour the production of propionate and butyrate from gut microbiota in the small intestine and in the cecum. Alterations in these specific SCFAs increase mitochondrial biogenesis in NSCs and induce premature neurogenic differentiation through a ROS- and ERK1/2-dependent manner, leading to rapid NSC pool depletion in both neurogenic niches.
Materials and methods
Animals, diets and sample collection
We used mice fed a HFCD diet, a well-established progressive liver disease—non-alcoholic steatohepatitis (NASH)—model, with several features of the metabolic syndrome, including obesity and insulin resistance (Gans, 1976; Kishida et al., 2016). Three-week-old C57BL/6N male non-littermate mice were acquired at the same time from Charles River Laboratories International, Inc. (Wilmington, MA, USA) and were randomly distributed three to four mice/cage. One week before starting HFCD diet feeding, the animals were again randomly distributed three to four mice/cage to dilute the possible effect of housing in mediating microbiome composition alterations and fed either a normal diet (control) (n = 14) or a HFCD diet (n = 14) (TestDiet, St. Louis, MO, USA) for 14 and 24 weeks. Additionally, mice received a one-time intra-peritoneal injection of 25 mg/kg diethylnitrosamine at the start of the feeding protocol to accelerate disease progression (Travis et al., 1991; Kishida et al., 2016). At the indicated time points, animals were killed by exsanguination under isoflurane anaesthesia. Luminal samples from small intestine and caecum were collected for DNA extraction and next-generating sequencing (NGS) analysis of 16S rRNA gene. The liver was collected for propionate and butyrate analysis via LC-MS/MS and the brain was removed for region-specific isolation and biochemical studies. Neurogenic niches (SVZ, DG and OB), as well as the hippocampus and cortex, were isolated from one hemisphere and flash-frozen in liquid nitrogen for RNA and protein extraction. The contralateral brain hemisphere was fixed with 4% paraformaldehyde (PFA) at 4°C overnight for free-floating immunohistochemistry.
The protocol procedures were prepared before and reviewed and approved by the Institution ethics committee and national competent authorities for animal protection. Animals received humane care in a temperature-controlled environment with a 12-h light–dark cycle. All animal experiments were performed in accordance with Portuguese laws (DL 113/2013, 2880/2015, 260/2016) on Animal Care and with the EU Directive (86/609/EEC; 2010/63/EU) on the protection of animals used for experimental and other scientific purposes. In addition, animal welfare fulfilled the recommendation from the ‘Guide for the Care and Use of Laboratory Animals’ prepared by the National Academy of Sciences and published by the National Institutes of Health (NIH publication 86-23 revised 1985). Importantly, throughout the conducted experiments, group allocations and data analysis were randomized by different researchers for blinded outcome assessments.
Microbial community analysis
Microbial community analysis was performed by bioinformatics analysis. DNA from intestinal and cecum luminal samples was extracted using QIAamp DNA Stool Mini Kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions. Next, metagenomic content was predicted using the 16S rRNA profile. Briefly, the variable 4 (V4) region of the bacterial 16S rRNA gene was amplified and sequenced through NGS using Illumina MiSeq Benchtop Sequencer (San Diego, CA, USA) and following Illumina recommendations. Polymerase chain reaction of the 16S rRNA gene was performed using the primer pair F515/R806, with the following cycling conditions: 94°C for 3 min, then 35 cycles of 94°C for 60 s and 72°C for 105 s, with a final extension step of 72°C for 10 min. The QIIME pipeline was used to de-multiplex and quality filter the samples for taxonomic assignment (Caporaso et al., 2010). Reads were truncated at a stretch of four or more consecutive low-quality bases (phred q score, <20) with script split_libraries_fastq, and only reads at ≥75% of the original length were retained. Operational taxonomic units were assigned with 97% similarity against the Greengenes database (McDonald et al., 2012). Principal co-ordinates analysis (PCoA) plots were generated using QIIME. Linear discriminant analysis effect size (LEfSe) was used to determine significant alterations in relative abundance of microbial taxa and predicted Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways function between mice fed the control and the HFCD diet (Segata et al., 2011). The KEGG pathway functions were categorized at level 3 using the phylogenetic investigation of communities by reconstruction of unobserved state (PICRUSt) tool (Langille et al., 2013). When applicable, the classes were defined as treatment groups (control or HFCD). Significance thresholds were performed at the default settings: α ≤ 0.05 for the Kruskal–Wallis test among classes, and ≥2.0 for the logarithmic linear discriminant analysis (LDA) score.
Liver propionate and butyrate analysis
The levels of both SCFAs, propionate and butyrate, were measured in the liver of the ND- and HFCD-fed animals (Metabolon Inc., Durham, NC, USA). Mouse tissue samples were analysed by LC-MS/MS (Metabolon Method TAM135: ‘LC-MS/MS Method for the Quantitation of Short-Chain Fatty Acid [C2–C6] in Human Feces’). After sample derivatization, the samples were injected onto an Agilent 1290/AB Sciex QTrap 5500 LC MS/MS system equipped with a C18 reversed-phase UHPLC column. The mass spectrometer was operated in negative mode using electrospray ionization. The peak area of the individual analyte product ions was measured against the peak area of the product ions of the corresponding internal standards. Quantitation was performed using a weighted linear least squares regression analysis generated from fortified calibration standards prepared immediately prior to each run. LC-MS/MS raw data were collected and processed using SCIEX OS-MQ software v1.7. Titre was normalized by the amount of starting material.
Analysis of ROS and caspase-3/-7 activity
The levels of ROS were detected using 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA; Sigma-Aldrich Corp., St. Louis, MO, USA), a cell-permeant non-fluorescent molecule that forms the fluorescent compound dichlorofluorescein when oxidized by ROS. Briefly, 10 mg of brain tissue was homogenized using a motor-driven grinder on 100 μl of ice-cold phosphate-buffered saline (PBS). Insoluble particles were removed by centrifugation at 10 200g for 5 min at 4°C. Ten micromolar H2DCFDA were incubated with 50 μl of the lysate at room temperature (RT) for 30 min. In addition, using the same lysate, caspase-3/-7 activity was measured using the Caspase-Glo 3/7 Assay (Promega Corp., Madison, WI, USA). The reagent was incubated for 30 min at RT releasing a luminescent signal. Fluorescence and luminescence were measured using GloMax-Multi+ Detection System (Promega Corp.). Both caspase-3/-7 activity and total ROS were normalized with total protein amount.
Free-floating immunohistochemistry and cell counting
Brain hemispheres were sectioned from the OB to the occipital lobe, using a cryostat Leica CM 3050S (Leica Biosystems, Wetzlar, Germany), to produce 40 μm-thick, free-floating coronal sections. Sections were divided into 10 series.
Sections were degelatinized with PBS at 37°C, then blocked at RT for 1 h with PBS containing 3% bovine serum albumin (Sigma-Aldrich Corp.) and 2% Triton-X-100 (Sigma-Aldrich Corp.). Next, slices were incubated overnight at 4°C with primary antibodies goat anti-doublecortin (DCX) (1-500; sc-8066; Santa Cruz Biotechnology Inc., Dallas, TX, USA) and anti-PGC-1α (1:500, ST1202, Calbiochem, San Diego, CA, USA); and rabbit anti-Ki67 (1:200; ab166667; Abcam Plc, Cambridge, UK) and anti-mitochondrial transcription factor A (Tfam) (1:200; ab131607, Abcam); and mouse anti-Synaptophysin (1:1000; MAB5258, Merck Millipore) diluted in blocking solution. After rinsing, the secondary antibodies anti-goat Alexa Fluor 488, anti-mouse Alexa 568 and anti-rabbit Alexa Fluor 568 (all 1:500; Thermo Fisher Scientific Inc., Waltham, MA, USA) were incubated with Hoechst 33258 (50 μg/ml; Sigma-Aldrich Corp.) in PBS at RT for 2 h. Finally, samples were mounted using Mowiol 4-88 (Calbiochem). Negative controls were obtained by omitting the primary antibody to validate the results.
Sections were viewed with a Leica DMi8 confocal fluorescence microscope (Leica Microsystems, Germany). Images were acquired with a 20× objective. For synaptophysin detection, samples were single-layer scanned with a Zeiss LSM 880 with Airyscan, a confocal point-scanning microscope (Carl Zeiss, Jena, Germany) equipped with a 63×/1.4 oil differential interference contrast plan-apochromat objective. All imaging analysis was performed using ImageJ Fiji 1.52e software. DCX+, Ki67+ and DCX/Ki67+ cells were counted in the SVZ and DG regions in one-in-ten series, representing the whole neurogenic niches. Using Hoechst staining, areas of SVZ and DG were manually traced and calculated. The volume of SVZ and DG from each animal was estimated by multiplying the total area of each neurogenic niche with the distance between each section (400 μm). Finally, the number of DCX+, Ki67+ and DCX/Ki67+ cells was normalized by the volume of each neurogenic niche per brain hemisphere.
Total RNA extraction and quantitative RT-PCR (qRT-PCR)
Sub-ventricular zone and the DG regions were dissected out and flash-frozen separately. The remaining hippocampus and the cortex tissues were also collected. RNA was extracted from frozen neurogenic-enriched tissues and cells using Ribozol™ (AMRESCO, LLC, Solon, OH, USA), as described previously (Simoes et al., 2013).
cDNA was prepared from 500 to 1000 ng total RNA using NZY Reverse Transcriptase (NZYTech, Lisbon, Portugal), according to the manufacturer’s instructions. Real-time RT-PCR was performed using SensiFast™ SYBR® Hi-ROX Kit (Bioline USA Inc., Taunton, MA, USA) in an Applied Biosystems QuantStudio 7 Flex Real-Time PCR System (Thermo Fisher Scientific Inc.). Primers used for qRT-PCR are listed in Table 1. Relative gene expression was calculated based on the standard curve and normalized to the level of Hprt housekeeping gene and expressed as fold change from controls.
Table 1.
List of primers used for PCR
| Gene | Sequence (5′-3′) |
|---|---|
| Il-1β | 5′-TGCCACCTTTTGACAGTGATG-3′ (fwd) |
| 5′-TGATGTGCTGCTGCGAGATT-3′ (rev) | |
| Hprt | 5′-GGTGAAAAGGACCTCTCGAAGTG-3′ (fwd) |
| 5′-ATAGTCAAGGGCATATCCAACAACA-3′ (rev) | |
| Mash1 | 5′-AGATGAGCAAGGTGGAGACG-3′ (fwd) |
| 5′-TGGAGTAGTTGGGGGAGATG-3′ (rev) | |
| Mki67 (for Ki67) | 5′-CCTTTGCTGTCCCCGAAGA-3′ (fwd) |
| 5′-GGCTTCTCATCTGTTGCTTCCT-3′ (rev) | |
| Rbfox3 (for NeuN) | 5′-CCAGGCACTGAGGCCAGCACACAGC-3′ (fwd) |
| 5′-CTCCGTGGGGTCGGAAGGGTGG-3′ (rev) | |
| Neurod1 (for NeuroD1) | 5′-CGCAGAAGGCAAGGTGTC-3′ (fwd) |
| 5′-TTTGGTCATGTTTCCACTTCC-3′ (rev) | |
| Nlrp3 | 5′-AGAGCCTACAGTTGGGTGAAATG-3′ (fwd) |
| 5′-CCACGCTACCAGGAAATCTC-3′ (rev) | |
| Ppargc1a (for PGC-1α) | 5′-GGACATGTGCAGCCAAGACTCT-3′ (fwd) |
| 5′-CACTTCAATCCACCCAGAAAGCT-3′ (rev) | |
| Sirt3 | 5′-TGCTACTCATTCTTGGGACCT-3′ (fwd) |
| 5′-CACCAGCCTTTCCACACC-3′ (rev) | |
| Sox2 | 5′-′GGGTTCTTGCTGGGTTTTGATTCT-3′ (fwd) |
| 5′-CGGTCTTGCCAGTACTTGCTCTCA-3′ (rev) | |
| Tfam | 5′-CACCCAGATGCAAAACTTTCAG-3′ (fwd) |
| 5′-CTGCTCTTTATACTTGCTCACAG-3′ (rev) | |
| Tlr4 | 5′-TCCCTGCATAGAGGTAGTTCCTA-3′ (fwd) |
| 5′-CTTCAAGGGGTTGAAGCTCAG-3′ (rev) | |
| Tnf-α | 5′-AGGCACTCCCCCAAAAGATG-3′ (fwd) |
| 5′-TGAGGGTCTGGGCCATAGAA-3′ (rev) | |
| Tubb3 (for βIII-tubulin) | 5′-GCGCCTTTGGACACCTATTCA-3′ (fwd) |
| 5′-TTCCGCACGACATCTAGGACTG-3′ (rev) | |
| mt-Co1 (for CO1) | 5′-ATCTGTTCTGATTCTTTGGGCAC-3′ (fwd) |
| 5′-AGCCTAGAAAGCCAATAGACATTA-3′ (rev) | |
| Rn18s (for 18S) | 5′-TAGAGGGACAAGTGGCGTTC-3′ (fwd) |
| 5′-CGCTGAGCCAGTCAGTGT-3′ (rev) |
Neural stem cell cultures and cell treatments
Neural stem cells were derived from 14.5-days post-coitum (dpc) mouse fetal forebrain, as described previously (Conti et al., 2005; Pollard et al., 2006). Forebrain progenitor cells in the embryo are clonally related to post-natal NSCs and are quiescent until they are activated in adulthood (Fuentealba et al., 2015), thus modelling the quiescent pool of adult NSCs. These NSCs continuously expand by symmetrical division and are capable of tri-potential differentiation (Conti et al., 2005; Fonseca et al., 2013). Briefly, NSCs were maintained in a monolayer and in self-renewal conditions in Euromed-N medium (EuroClone®S.p.A., Pavia, Italy), at 37°C, in a humidified atmosphere of 5% CO2. The self-renewal medium was supplemented with 1% penicillin–streptomycin, 1% N-2 supplement, 20 ng/ml epidermal growth factor (EGF) and 20 ng/ml basic fibroblast growth factor (βFGF). Penicillin–streptomycin and N-2 supplements were purchased from Gibco™, Life Technologies Corp. (Carlsbad, CA, USA). Both EGF and βFGF were purchased from PeproTech® EC (London, UK). In self-renewal medium, NSCs were plated with a density of around 1 × 105 cells/cm2. In brief, 24 h after platting, NSCs were incubated with SCFAs sodium propionate (P1880; Sigma-Aldrich Corp.) and sodium butyrate (B5887; Sigma-Aldrich Corp.). Short-chain fatty acids were incubated for 24 h to assess cell viability, proliferation, neurogenic differentiation, mRNA and protein levels of mitochondrial regulators; 2 h to measure mitochondrial ROS; or 5, 15, 60 and 120 min to determine the protein levels of p-ERK1/2. In addition, 0.5 μM N-acetyl-l-cysteine (NAC, T7250; Sigma-Aldrich Corp.), a well-established anti-oxidant, was incubated 2 h previously SCFA treatment to assess the ROS involvement in SCFA effect. Cells were collected and processed for flow cytometry, immunocytochemistry, immunoblotting and qRT-PCR assays. For the proliferation assay, 10 μM BrdU (BD Pharmingen, BD Biosciences) was added to the culture medium for the last 4 h of 24 h-SCFA treatment.
Evaluation of cell death and viability
Cell death was assessed by the Guava ViaCount® assay (Guava Technologies, Millipore Corp.), according to the manufacturer’s instructions. After incubation of SCFAs in self-renewal conditions, the cell-culture medium and adherent cells previously detached with StemPro Accutase (A11105-01; Gibco™) were collected and centrifuged for 5 min at 600g and re-suspended in PBS with 2% foetal bovine serum. Cell suspension was mixed with Guava ViaCount reagent and incubated for 20 min at RT. Sample acquisition was performed using Guava easyCyte 5HT flow cytometer (Guava Technologies).
Measurement of mitochondrial ROS and DNA copy number
After 2 h of propionate and butyrate treatment, mitochondrial superoxide anion was measured using MitoSOX™ Red mitochondrial superoxide indicator (M36008; Molecular Probes™, Life Technologies Corp.), as described previously (Ribeiro et al., 2019). In addition, mitochondrial DNA (mtDNA) copy number was determined as described previously (Ribeiro et al., 2019). Primer sequences are listed in Table 1.
Immunocytochemistry
Cells were fixed with PFA in PBS (4%, w/v), then blocked for 1 h at RT in PBS, containing 0.1% Triton X-100, 1% FBS and 10% normal donkey serum, and further incubated with primary rabbit anti-MAP2 (1:100, AB5622, Merck Millipore, Billerica, MA, USA) and mouse anti-BrdU (1:100, M0744, Dako) overnight at 4°C. The appropriate secondary antibody anti-rabbit Alexa Fluor 568 and anti-mouse Alexa Fluor 568 antibodies diluted 1:200 were then incubated for 2 h at RT. Nuclei were stained with Hoechst 33342 dye, and the preparations were mounted using Mowiol. Image acquisition was performed with an AxioScope.A1 microscope (Carl Zeiss Microscopy GmbH), equipped AxioCam HRm camera. Images were processed using ImageJ Fiji 1.52e software.
Transfection assays
Neural stem cells were transfected with a plasmid encoding a constitutively active form of mitogen-activated protein kinase (MEK1*) (pCMV-deltaN3-MEK1), kindly provided by Dr. Roger J. Davis (Howard Hughes Medical Institute). Empty plasmid pcDNA3.1(−) harbouring a CMV promoter (pCMV) was used as control. We used Lipofectamine® 3000 (Invitrogen, Thermo Fischer), according to the manufacturer’s instructions. Briefly, Opti-MEM® (Gibco™) containing the mixture of Lipofectamine® 3000 and DNA (2 μl:1 μg) was added and cells were incubated for 24 h. After 24 h of transfection, cells were treated with propionate and butyrate and collected after 24 h to assess cell fate by PCR, as described above.
Statistical analysis
Statistical significance was assessed using unpaired two-tailed Student’s t-test when two groups were compared, one-way ANOVA followed by Dunnett’s post-test for multiple comparisons to one control or two-way ANOVA followed by Tukey post-test for multiple comparisons. The analysis was performed using GraphPad Prism version 5.0, GraphPad Software (San Diego, CA, USA). The values of P < 0.05 were considered statistically significant.
Data availability
The data supporting the findings in this study are available from the corresponding author upon request.
Results
Dietary changes induce neural damage and alterations in NSC dynamics
Dietary factors impact on brain function (Gomez-Pinilla, 2008; Almeida-Suhett et al., 2017). In addition, diet is an important modulator of neurogenesis, which contributes to cognitive performance and mood (Lindqvist et al., 2006; Stangl and Thuret, 2009). To dissect the role of diet and gut microbiome in neurogenic niches, mice were fed a HFCD diet for 14 and 24 weeks, mimicking short- and long-term malnutrition, respectively. Mice on this diet typically develop features of the metabolic syndrome, including obesity and insulin resistance, as well as different degrees of non-alcoholic fatty liver disease. In fact, our results confirmed the occurrence of liver steatosis and lipid accumulation (Supplementary Fig. 1) (Kishida et al., 2016). As NSC fate determination is deeply affected by neighbouring cells and surrounding regions, we began to assess brain alterations in mice on this diet, specifically general brain damage. Cell death and oxidative stress were evaluated by measuring caspase-3 activity and ROS levels, respectively, in whole brain lysate. In long-term HFCD-fed mice, caspase-3 was markedly activated in the brain, as assessed by the enzymatic assay (P < 0.05; Fig. 1A). In addition, increased ROS levels were detected (P < 0.05; Fig. 1B). We evaluated protein expression of two members of the ROS scavenging system, namely superoxide dismutase 2 (Sod2), and its positive activator sirtuin-3 (Sirt3), a NAD+-dependent mitochondrial deacetylase (Sundaresan et al., 2009). Short-term HFCD diet increased Sod2 in hippocampus and cortex regions, whereas Sirt3 protein was increased in the cortex alone (at least P < 0.01; Fig. 1C), suggesting engagement of endogenous protective mechanisms against oxidative stress. Curiously, prolonged HFCD feeding induced a significant decrease in Sirt3 levels in the hippocampus (P < 0.01; Fig. 1C), suggestive of opposite events, namely augmented Sod2 acetylation and activation, and subsequently increased oxidative stress. As oxidative stress is particularly harmful to dendritic spines (Mast et al., 2008) and synaptic loss precedes neuronal death, we also analysed the relative abundance of pre-synaptic and post-synaptic proteins synaptophysin and PSD-95, respectively, in the hippocampus and cortex regions of HFCD-fed mice. As expected, synaptophysin markedly decreased in the hippocampus of long-term HFCD-fed mice, comparing to controls, whereas in the cortex, loss of both synaptic markers was soon observed in short-term HFCD diet-fed animals (at least P < 0.05; Fig. 1D and E). Interestingly, the statistical difference of Sirt3, Sod2 and synaptic proteins between the two control groups (Fig. 1C and D) clearly reflected expected changes associated with aging.
Figure 1.

HFCD diet induces brain damage, synaptic loss and oxidative stress. Mice were fed a control (n = 4–7 for each time point) or HFCD diet (n = 4–7 for each time point) for 14 and 24 weeks. The wholebrain extract, as well as the cortex and hippocampus regions were collected for enzymatic and ROS assays and Western blot, as described in Materials and methods section. Effect of HFCD diet on caspase-3/7 enzymatic activity (A) and ROS generation (B) in wholebrain extracts after 24 weeks of feeding. Immunoblotting (top) and densitometry (bottom) of Sirt3 and Sod2 (C) and synaptophysin and PSD-95 (D) in the hippocampus and cortex. β-Actin was used as loading control. (E) Representative images of immunofluorescence detection of synaptophysin in brains of control and HFCD diet-fed mice. Nuclei were counterstained with Hoechst 33258 (blue). Scale bar, 5 μm. Data are expressed as fold change over control. Representative immunoblots are shown. Data represent mean values ± SEM for at least five individual mice. *P < 0.05, **P < 0.01 and ***P < 0.001 compared to control. Syn: synaptophysin.
As inflammation is a hallmark of neurological impairment, and also impacts neurogenesis through several cytokines and intracellular mediators (Cai, 2013), we next evaluated the expression of pro-inflammatory cytokines in both hippocampus and cortex regions, through qRT-PCR. Expression levels of interleukin (IL)-1β, tumour necrosis factor α (TNF-α) and pro-inflammatory markers nod-like receptor protein-3 (NLRP3) and toll-like receptor 4 (TLR4) increased in the hippocampus of short-term HFCD-fed mice, compared to control (Fig. 2A). In the cortex of short-term HFCD-fed animals, only IL-1β was found increased (at least P < 0.05; Fig. 2B). No changes were found in long-term HFCD-fed mice, comparing with control group. Because neurological disorders are known to associate with malnutrition and since neurogenesis is deregulated in neurodegenerative diseases and depression-like behaviours (Perera et al., 2007; Hamilton and Holscher, 2012), we investigated alterations in NSC fate in our model, uniquely present in neurogenic niches. Cell proliferation was assessed by the number of Ki67-positive NSCs in both DG and SVZ neurogenic regions of HFCD-fed mice. Curiously, Ki67-positive cells and its co-localization with the early neuronal differentiation marker DCX increased in the SVZ neurogenic region in short-term HFCD diet-fed mice (P < 0.05; Fig. 3A and B). In contrast, a significant decrease in the number of Ki67/DCX-positive cells in the SVZ region was observed in HFCD diet-fed animals for 24 weeks (P < 0.05; Fig. 3A and B). The short-term HFCD diet also increased the expression of stemness marker SRY (sex determining region Y)-box 2 (Sox2), corroborating the increased NSC pool in this neurogenic niche (P < 0.05; Fig. 3C).
Figure 2.

HFCD diet induces neuroinflammation at short-term. Mice were fed a control (n = 4–7 for each time point) or HFCD diet (n = 3–7 for each time point) for 14 and 24 weeks. The total RNA from cortex and hippocampus regions were extracted for qRT-PCR analysis, as described in Materials and methods section. Expression levels of pro-inflammatory markers, including IL-1β, TNF-α, Nlrp3 and Tlr4 in the hippocampus (A) and cortex (B). Hprt was used as loading control. Data are expressed as fold change over control. Data represent mean values ± SEM for at least five individual mice. *P < 0.05 and **P < 0.01 compared to control.
Figure 3.


HFCD diet exhausts NSC pool by increasing adult neurogenesis. Mice were fed a control (n = 3–7 for each time point) or HFCD diet (n = 3–7 for each time point) for 14 and 24 weeks. Brain slices and total RNA from SVZ-, OB- and DG-enriched extracts were processed for immunohistochemistry and qRT-PCR, respectively, as described in Materials and methods section. (A) Representative images of immunofluorescence detection and (B) quantitative analysis of neuronal differentiation and proliferation markers DCX+, Ki67+ and DCX+/Ki67+ from SVZ of control and HFCD diet-fed mice. (C) Expression levels of Sox2 in the SVZ-enriched extracts. (D) Representative images of immunofluorescence detection and (E) quantitative analysis of neuronal differentiation and proliferation markers DCX+, Ki67+ and DCX+/Ki67+ from DG of control and HFCD diet-fed mice. Nuclei were counterstained with Hoechst 33258 (blue). Scale bar, 5 μm. Quantitative analysis of neuronal markers βIII-tubulin (F), NeuroD1 (G) and Mash1 (H) in OB- (top) and DG- (bottom) enriched extracts. Hprt was used as loading control. Data are expressed as fold change over control. Data represent mean values ± SEM for at least five individual mice. *P < 0.05, **P < 0.01 and ***P < 0.001 compared to control.
Notably, immunohistochemistry analysis revealed a significant increase of newly born cells in both SVZ and DG neurogenic regions in short-term HFCD-fed mice, as indicated by the increased number of DCX-positive cells (at least P < 0.01; Fig. 3A, B, C, and E). In agreement, these animals also displayed augmented βIII-tubulin expression, a marker of early neuronal differentiation, in both DG and OB neurogenic regions (P < 0.05; Fig. 3F), whereas NeuroD1 and Mash1, two transcription factors required for the differentiation and maturation of NSCs (Gao et al., 2009; Raposo et al., 2015), increased in the OB and DG regions, respectively (P < 0.05; Fig. 3G and H). Strikingly, the cumulative feed intake of this high caloric diet induced a marked reduction in adult neurogenesis. In fact, although no changes in DCX-positive cells were observed in DG, a significant decrease in DCX-positive cells was evident in the SVZ neurogenic region (P < 0.001; Fig. 3A, B, D, and E). Furthermore, mice also displayed reduced βIII-tubulin levels in hippocampal neurogenic niche (P < 0.05; Fig. 3F), and reduced expression levels of Mash1 and NeuroD1 in OB regions (at least P < 0.05; Fig. 3G and H).
Taken together, our data demonstrate that short-term HFCD diet triggers proliferation of SVZ-derived NSCs and premature neuronal differentiation in both adult neurogenic niches. However, sustained dietary changes may lead to exhaustion of the NSC pool and impairment of neurogenesis.
Diet-induced gut dysbiosis associates with increased SCFA metabolic pathways
As diet strongly impacts on gut microbiota (Gomez-Pinilla and Tyagi, 2013; Almeida-Suhett et al., 2017), and gut dysbiosis is a putative causal factor of neurological disorders (Rogers et al., 2016; Slattery et al., 2016), we hypothesized that altered gut microbiota could play a role in altering neurogenesis induced by the HFCD diet. To test our hypothesis, we analysed the gut microbiome in luminal samples from the small intestine and cecum of HFCD-fed mice. V4 variable region of the 16S rRNA gene from intestinal microbiota was sequenced. By performing a PCoA on gut microbiota composition, a clear separation was found between either short- or long-term HFCD diet and corresponding control groups clusters, in both small intestine (Fig. 4A) and cecum (Fig. 4B). These results indicate that HFCD diet induced gut dysbiosis in both digestive compartments. To depict our analysis in terms of microbiota composition relative abundance between HFCD-fed mice and control, we performed LEfSe analysis. At the phylum level, Proteobacteria was one of the most highly enriched microbiota populations in the small intestine of long-term HFCD diet-fed mice, as well as in the cecum of short-term HFCD-fed mice (Supplementary Fig. 2). Firmicutes were significantly decreased in the small intestine and cecum of long-term HFCD-fed mice (Supplementary Fig. 2). Additionally, these dietary changes induced more discriminating taxa in small intestine than in cecum, possibly due to the fact that diversity of microbiota composition is usually much lower in small intestine than in cecum (Supplementary Fig. 2).
Figure 4.

HFCD diet exerts gut dysbiosis. Mice were fed a control (n = 5 for each time point) or HFCD diet (n = 8–10 for each time point) for 14 and 24 weeks. Luminal samples of small intestine and cecum were collected as described in Materials and methods section. The influence of HFCD diet on the composition of the small intestine (A) and cecum (B) gut microbiota was assessed by PCoA. Data represent mean values ± SEM for four individual mice.
The cross-talk between the brain and the microbiota can be supported by different pathways, such as the neural pathway, the immune system and microbiota metabolites (Fung et al., 2017). Thus, variation of metabolites from gut microbiota can strongly influence neurological function. To evaluate the potential impact of these gut microbiota differences on the systemic circulation and the brain, we used a LEfSe approach, to determine KEGG pathways that were enriched or under-represented in HFCD diet groups. In short-term HFCD-fed mice, we found that 53 and 32 third-level classification KEGG pathways were markedly different in the small intestine and cecum, respectively (Fig. 5A and Supplementary Fig. 3). Long-term HFCD diet significantly altered more third-level classification KEGG pathways, namely 141 and 54 in the small intestine and cecum, respectively (Fig. 5B and Supplementary Fig. 3). Again, the small intestine presented with the largest number of changes in microbial function, comparing with the cecum. Among these pathways, HFCD diet increased SCFA metabolic pathways, such as propionate and butyrate. In fact, HFCD diet increased propionate metabolism in the small intestine of all animals (Fig. 5A and Supplementary Fig. 3A), and in the cecum of long-term feeding animals (Fig. 5B), suggesting a shift of propionate-producing bacteria from the cecum to the small intestine. Long-term HFCD diet also increased butyrate metabolism in the small intestine (Supplementary Fig. 3A).
Figure 5.

HFCD diet promotes SCFA metabolic pathways. Mice were fed a control (n = 5 for each time point) or HFCD diet (n = 8–10 for each time point) for 14 and 24 weeks. Luminal samples of small intestine and cecum were collected as described in Materials and methods section. The impact of HFCD diet on the metabolism of gut microbiota was predicted by LDA analysis in both feeding time points. The pathway of KEGG was predicted by LDA coupled with effect size measurements in the small intestine after 14 weeks (A) and in the cecum after 24 weeks (B). The most differentially abundant pathways enriched by HFCD diet and ND were indicated in red and green, respectively. Arrows mark enriched SCFA metabolic pathway. Only pathways meeting an LDA significant threshold of >2 were shown. Validation of propionate pathway and measurement of propionate concentration in the liver of the ND- and HFCD-fed mice by LC-MS/MS (C). Data represent mean values ± SEM for four individual mice. *P < 0.05 compared to control.
In order to corroborate the predicted bioinformatic SCFA data, we performed direct measurements of butyrate and propionate levels in mice liver. In fact, because the portal system drains directly into the liver, this organ is trusted for assessing changes in systemic circulation. Interestingly, our results corroborated KEGG analysis and revealed that 24 weeks of HFCD diet significantly increased propionate levels in liver (Fig. 5C and Supplementary Fig. 3A). Butyrate remained below the detection limit at 24 weeks of diet (data not shown).
Diet-associated gut metabolites induce alterations in NSC fate, mitochondrial biogenesis and oxidative stress
To determine whether metabolites from HFCD diet-associated microbiota, namely propionate and butyrate, modulate neurogenesis, we dissected the effect of these two specific SCFAs on NSC mitochondrial activity, viability, proliferation and differentiation potential, in vitro (Conti et al., 2005). Our results showed that propionate did not significantly reduce cell viability of NSCs up to 1 mM concentration. However, both SCFAs significantly increased cell death to 10–20% at 1 mM, comparing to controls (at least P < 0.05; Fig. 6A). To avoid massive cell death, we then performed subsequent experiments with 1 mM of each compound. The effects of SCFAs on NSC proliferation and differentiation were assessed through the expression of proliferation (Ki67), stemness (Sox2), early (βIII-tubulin) and late (NeuN) neuronal markers. Interestingly, propionate and butyrate significantly increased the expression of neuronal markers βIII-tubulin and NeuN (at least P < 0.01; Fig. 6B). These results were also corroborated by immunocytochemistry of MAP2 (Fig. 6B). Concerning proliferation, butyrate decreased Ki67 mRNA levels, whereas propionate increased Ki67 levels (P < 0.001; Fig. 6C), possibly indicating different proportions of symmetric and asymmetric divisions triggered by these two SCFAs. Indeed, propionate increased BrdU incorporation while butyrate decreased, as shown by fluorescence microscopy (Fig. 6C). These specific SCFAs also increased the expression of stemness marker Sox2 (P < 0.001; Fig. 6D). More importantly, and in agreement with the augmented adult neurogenesis observed in short-term HFCD-fed mice, these results reveal that both SCFAs, propionate and butyrate, enhance early neurogenic differentiation.
Figure 6.

Short-chain fatty acids stimulate neuronal differentiation, mitochondrial biogenesis and oxidative stress of NSCs. Mouse NSCs were treated with 1, 5, 10, 100, 250, 500 or 1000 μM of propionate (n = 3–9) and butyrate (n = 3–8) in self-renewal conditions. After 24 h of propionate and butyrate treatment, cells were collected for viability, mitochondrial mass, qRT-PCR, immunocytochemistry and Western blot analysis, whereas cells treated with only 2 h of these SCFAs were collected for mtROS, as described in Materials and methods section. (A) Effect of SCFAs on NSC viability, assessed by Guava ViaCount flow cytometry. Data are expressed by percentages of viable cells. Effect of SCFAs on (B, left) βIII-tubulin and NeuN, and (C, left) Ki67 and (D) Sox2 mRNA levels in NSCs. Hprt was used as loading control. Data are expressed as fold change over control (untreated cells). Representative images of immunofluorescence detection of cells labelled with anti-MAP2 (B, right) and anti-BrdU (C, right) antibodies. Nuclei were stained with Hoechst 33258. Scale bar, 5 μm. (E) Effect of SCFAs on mitochondrial biogenesis markers in NSCs. Quantification of relative mtDNA copy number (left) assessed by qPCR analysis of mitochondria-encoded gene mt-Co1. Nuclear Rn18s was used as loading control. Quantification of Tfam expression levels (right) assessed by qRT-PCR. Hprt was used as loading control. (F) Effect of SCFAs on mtROS levels in NSCs, assessed by flow cytometry. Data are expressed by percentages of cells stained with MitoSOX™ Red reagent. (G) Representative immunoblots (top) of Sirt3, total Sod2 and Ac-Sod2 protein levels and respective densitometry analysis (bottom). β-Actin was used as loading control. Data are expressed as fold change over control. Data represent mean values ± SEM for at least three independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001 compared to control cells.
To dissect the role of mitochondria in SCFA-induced changes in neurogenesis, we next investigated the effect of propionate and butyrate on NSC mitochondria. In fact, SCFAs have been described to regulate mitochondrial bioenergy, biogenesis and dynamics in the small intestine, liver, muscle and adipose tissues (Rose et al., 2018; Uittenbogaard et al., 2018). On the other hand, mitochondria are known to regulate NSC self-renewal, differentiation and viability (Sena and Chandel, 2012; Khacho et al., 2016). Our results showed that propionate and butyrate increased mtDNA copy number and mitochondrial transcription factor A (Tfam) mRNA expression in NSCs (at least P < 0.05; Fig. 6E), suggesting that both propionate and butyrate increase the number of mitochondria in NSCs. Curiously, SCFAs also increased mitochondrial ROS (mtROS) (at least P < 0.05; Fig. 6F), possibly by increasing mitochondrial mass but also by preventing the elimination of NSC mtROS. In fact, propionate and butyrate had no effect on total levels of the major ROS mitochondrial scavenger Sod2, or its activator protein, Sirt3 (Kong et al., 2010; Chen et al., 2011), but increased acetyl-Sod2 levels, the inactive form of Sod2 (P < 0.05; Fig. 6G). This increase in acetyl-Sod2 form may rely on the loss of Sirt3 activity rather than changes in Sod2 and Sirt3 total levels, as observed in vivo. These results highlight the effect of propionate and butyrate on neuronal differentiation, mitochondrial biogenesis and oxidative stress of NSCs.
Diet-associated gut metabolites trigger premature differentiation of NSCs in a ROS- and p-ERK1/2-dependent manner
Apart from inducing cellular toxicity, elevated levels of ROS have also been demonstrated to act as key intracellular signals for cell-cycle progression and NSC fate (Khacho et al., 2016). Therefore, we investigated whether ROS induced by HFCD-associated gut metabolites were crucial for NSC differentiation, by blocking ROS accumulation through the use of antioxidant NAC, in NSCs treated with propionate and butyrate (Sun, 2010). Our results show that NAC significantly decreased SCFA-induced βIII-tubulin and NeuN expression (at least P < 0.05; Fig. 7A), whereas SCFA-induced Sox2 was further enhanced in the presence of NAC (at least P < 0.05; Fig. 7B).
Figure 7.

Short-chain fatty acids trigger neuronal differentiation of NSCs in a ROS- and p-ERK1/2-dependent manner. Mouse NSCs were treated with 1 mM of propionate (n = 3–7) and butyrate (n = 3–7) and 0.5 μM NAC for 24 h in self-renewal conditions. Cells were collected for qRT-PCR and Western blot analysis, as described in Materials and methods section. Effect of SCFAs on βIII-tubulin and NeuN (A), and Sox2 (B) mRNA levels. Hprt was used as loading control. A time-dependent assay was performed to evaluate the effect of propionate and butyrate on ERK1/2 phosphorylation. As described in Materials and methods section, NSCs were transfected with a MEK over-expression plasmid to evaluate the impact of ERK1/2 activation on SCFA-mediated changes in NSC fate. (C) Representative immunoblots (top) of p-ERK1/2 protein levels and respective densitometry analysis (bottom). β-Actin was used as loading control. (D) Representative immunoblots showing the efficiency of MEK transfection in NSCs. (E) Effect of ERK1/2 activation on SCFA-mediated changes in βIII-tubulin and NeuN, and Ki67 mRNA levels (F). Hprt was used as loading control. Data are expressed as fold change over control. Data represent mean values ± SEM for at least three independent experiments. *P < 0.05, **P < 0.01 and ***P < 0.001 compared to untreated cells.
Increased levels of ROS have also been associated with modulation of redox signalling pathways, including mitogen-activated protein kinase (MAPK) cascades and, specifically, extracellular signal-related protein kinase1/2 (ERK1/2) (Zhang et al., 2016; Choi et al., 2019). In addition, several studies have already correlated the repression of ERK1/2 pathway with stem cell differentiation (Na et al., 2010). To test the role of ERK1/2 in either SCFA- or ROS-mediated effects on neurogenesis, we evaluated p-ERK1/2 levels in NSCs incubated with propionate and butyrate. Curiously, propionate and butyrate were shown to significantly inhibit ERK1/2 phosphorylation in NSCs (at least P < 0.05; Fig. 7C). To elucidate the significance SCFA-mediated inhibition of ERK1/2 phosphorylation in neuronal differentiation, NSCs were transfected with a plasmid coding for a constitutively active form of MEK1 (MEK1*). This form constitutively phosphorylates, and consequently activates, ERK1/2 (Fig. 7D). Importantly, MEK1* prevented propionate- and butyrate-induced neurogenesis, blocking SCFA-induced βIII-tubulin (at least P < 0.05; Fig. 7E), as well as NSC proliferation (P < 0.001; Fig. 7F). Altogether, these data suggest that HFCD-associated gut metabolites, propionate and butyrate, induce neuronal differentiation of NSCs, at least in part, through a ROS- and ERK1/2-dependent signalling.
Dietary changes induce mitochondrial biogenesis and oxidative state alterations in adult neurogenic niches
Our results have thus far suggested that propionate and butyrate, produced upon HFCD diet-associated gut microbial changes, are capable of regulating NSC mitochondrial biogenesis and oxidative stress. Therefore, we finally assessed the expression of different mitochondrial-associated proteins in neurogenic niches of HFCD-fed mice. Tfam, a key activator of mitochondrial transcription and mitochondrial genome replication, was increased in DG of short-term HFCD-fed mice as shown by qRT-PCR and immunohistochemistry (P < 0.05; Fig. 8A). In fact, this is in agreement with the pattern of neurogenesis found in DG neurogenic niche (Fig. 3). Notably, dietary changes also significantly decreased Sirt3 in DG- and OB-enriched extracts (at least P < 0.05; Fig. 8B), suggesting that NSCs may be further exposed to elevated ROS levels in these contexts. Particularly, long-term HFCD diet-mediated inhibition of Sirt3 in OB-enriched extracts may result from decreased expression of peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α), a positive upstream regulator of Sirt3 (Chen et al., 2005) as we observed by qRT-PCR and immunostaining in the SVZ (P < 0.05; Fig. 8C). All these in vivo molecular alterations, together with the SCFA-induced signature in NSCs, strongly support the idea that mitochondria are, in part, responsible for the impact of diet-associated gut microbiome metabolites on adult NSC fate.
Figure 8.

Dietary changes induce mitochondrial biogenesis and oxidative state in adult neurogenic niches. Mice were fed a control (n = 4–5 for each time point) or HFCD diet (n = 4–7 for each time point) for 14 and 24 weeks. Total RNA from DG- and OB-enriched extracts were processed for qRT-PCR, and brain slices were immunostained, as described in Materials and methods section. Effect of HFCD diet on Tfam (A, left), Sirt3 (B) and PGC-1α (C, left) mRNA levels in enriched extracts derived from the DG and OB. Hprt was used as loading control. Data are expressed as fold change over control. Representative images of immunofluorescence detection of DG and SVZ labelled with anti-Tfam (A, right) and anti-PGC-1α (C, right) antibodies. Nuclei were stained with Hoechst 33258. Scale bar, 5 μm. Data represent mean values ± SEM for at least five individual mice. *P < 0.05, **P < 0.01 and ***P < 0.001 compared to control.
Discussion
Changes in host dietary patterns alter gut microbial composition and bacterial metabolism, ultimately favouring the species that can better adapt to new fuel sources. Importantly, diet-induced changes in microbiota potential impact on host physiology and disease processes (Gomez-Pinilla and Tyagi, 2013). By manipulating various dietary factors, microbiota plasticity embodies a therapeutic opportunity to improve host health.
Gut microbiota has recently emerged as a novel regulatory factor of brain function, namely through the action of microbiota metabolites (Ogbonnaya et al., 2015; Sarkar et al., 2018). In fact, gut microbiota imbalances associate with a variety of neurological and psychiatric disorders, some with impaired neurogenesis, including cognitive decline, depression and anxiety (Ghaisas et al., 2016; Rogers et al., 2016). Furthermore, diet-induced whole-body metabolic disturbances, particularly features of the metabolic syndrome like obesity, insulin resistance and type II diabetes, are becoming increasingly recognized as risk factors for many neurological disorders (Etchegoyen et al., 2018; Gasparova et al., 2018). In particular, non-alcoholic fatty liver disease, considered as the hepatic manifestation of the metabolic syndrome, correlates with gut dysbiosis and has also been associated with neurological comorbidities (Seyan et al., 2010; Patel et al., 2017). In fact, patients with NASH may present hepatic encephalopathy (Seyan et al., 2010) and other neurological alterations, such as cognitive impairment, depression and anxiety (Elwing et al., 2006; Youssef et al., 2013). A major challenge resides in understanding the molecular mechanisms associated with this inter-organ communication to ameliorate neural regeneration and brain function after abnormal dietary habits or gut dysbiosis. Our data clearly demonstrate a molecular link between diet-associated whole-body metabolic disturbances, including NASH, gut dysbiosis, and adult neurogenesis, highlighting mitochondrial stress as the culprit for gut microbiota metabolite-mediated modulation of NSCs.
Mice fed a high-fat diet (HFD), including NASH-inducing diets, have already been shown to present anxiety- and depressive-like behaviour as well as cognitive impairment (Almeida-Suhett et al., 2017). Here, we show that mice on a HFCD diet display increased cell death and oxidative stress in several areas of the brain, namely the cortex and the hippocampus, in parallel with loss of synaptic markers. Notably, apart from brain damage, neurological disorders have also been associated with deregulated adult neurogenesis (Fang et al., 2018). Indeed, several studies have reported that HFD induces lipid peroxidation in the brain, while also decreasing brain-derived neurotrophic factor, a pro-neurogenic factor, as well as its receptor (Park et al., 2010; Arcego et al., 2016). On the other hand, as inflammation is a hallmark of neurological impairment, and based on the fact that inflammation might also impact neurogenesis through several intracellular mediators, such as NLRP3 (Cai, 2013), we also evaluated the pro-inflammatory responses and neurogenesis alterations in HFCD-fed mice. In line with HFD activating immune response in the brain and brain periphery (Baufeld et al., 2016), we found increased levels of several pro-inflammatory cytokines in the brains of HFCD-fed mice. Regarding adult neurogenesis, short-term diet-associated changes stimulated premature adult neuronal differentiation in the DG and SVZ regions of mouse brain, whereas long-term feeding led to an exhaustion of this process. In agreement, HFD has been already reported to induce memory impairment, depressive-like behaviour and stress-induced depression by inhibiting hippocampal neurogenesis (Lindqvist et al., 2006; Arcego et al., 2018). Furthermore, HFD has also been shown to induce apoptosis of newborn neurons, resulting in reduced neurogenesis in the hypothalamus, a non-canonical neurogenic niche (McNay et al., 2012). Therefore, neurogenesis decline may result from direct brain damage and inflammation (Gould and Tanapat, 1997; Parent et al., 1997), but also from premature neuronal differentiation processes, followed by depletion of the active NSC pool in the brain, as our results suggest. In fact, neurogenesis has already been shown to be triggered during metabolic disorders and/or upon HFD, where the switch of quiescent NSCs into activated NSCs leads to premature exhaustion of NSC (Cavallucci et al., 2016).
Gut microbiota has recently arisen as a new regulator of neurogenesis, emotion and stress, learning and memory and social behaviour (Ogbonnaya et al., 2015; Sarkar et al., 2018). Here, we show that shifting mice from a control to a HFCD diet promotes gut dysbiosis and, particularly, enhanced microbial metabolism of two specific SCFAs, namely propionate and butyrate. In fact, although SCFAs are mainly produced from fibre-enriched diets (Koh et al., 2016), a high abundance of genes involved in propionate and butyrate metabolism have been reported in faeces of HFD-fed mice (Xiao et al., 2017). Furthermore, and in agreement with our data, faeces from NASH-affected patients present high abundance of SCFAs, as well as enzymes involved in butyrate and propionate metabolism, and domination of SCFA-producing bacteria (Loomba et al., 2017; Rau et al., 2018). More importantly, our metabolite analysis showed that HFCD diet significantly increases propionate levels in the liver, whereas butyrate remained undetectable. This may also suggest that propionate might be the major responsible for the neurogenesis differences in vivo.
Short-chain fatty acids are also important mitochondrial modulators (Clark and Mach, 2017). Because proliferation and differentiation of NSCs are highly sensitive to mitochondrial function (Antico Arciuch et al., 2012), we hypothesized that both propionate and butyrate could constitute a dominant link between gut microbiota and neurogenesis. In this regard, we demonstrated that propionate and butyrate modulate mitochondrial biogenesis and mass, increasing mtDNA copy number and Tfam expression in NSCs. In fact, others have also reported that SCFAs are capable of stimulating mitochondrial biogenesis (Tang et al., 2011; Uittenbogaard et al., 2018). Accordingly, we also showed that short-term HFCD diet increased Tfam expression in mice neurogenic niches, possibly due to increased SCFA metabolism. In parallel, we found that propionate and butyrate induced early neurogenic differentiation of NSCs through a ROS-dependent mechanism. Indeed, in vitro and mouse model studies have reported increased differentiation of NSCs by butyrate (Uittenbogaard et al., 2018; Val-Laillet et al., 2018). However, neurogenesis enhancement by propionate has never been assessed. Interestingly, ROS have recently gained a new perspective as physiological signalling molecules in cell differentiation, promoting a transcriptional program of pro-neuronal genes (Sena and Chandel, 2012). Non-toxic, high levels of ROS induce expression of genes that promote commitment and differentiation of NSCs (Khacho et al., 2016). Here, we demonstrate that mtROS accumulation by SCFAs results from an unbalanced radical scavenger system in the brain of HFCD-fed mice. In parallel, neurogenic niches of HFCD-fed mice presented decreased Sirt3 expression, the positive regulator of Sod2, further contributing to the increased susceptibility of NSCs to the effects of ROS. In fact, the increase of total Sod2 levels observed in brain of HFCD-fed mice might represent an adaptative cellular mechanism to counteract the decrease of Sod2 activation under an oxidative stress scenario.
Several studies have reported the involvement of ERK1/2 in neurogenesis (Pereira et al., 2015; Vithayathil et al., 2015; Tang et al., 2017), while propionate and butyrate decrease ERK1/2 phosphorylation in colon carcinoma cells (Davido et al., 2001). Interestingly, maintenance of quiescent NSCs in neurogenic niches was also shown to be assured by ERK1/2 phosphorylation (Pereira et al., 2015), with loss of ERK1/2 inhibiting maintenance of neural progenitors as they migrate, resulting in premature depletion of neural progenitor cells (Vithayathil et al., 2015). Here, we show that ERK1/2 phosphorylation, and subsequently its activation, is decreased in NSCs upon exposure of SCFAs, supporting the idea that decreased p-ERK1/2 plays a role in premature NSC differentiation. In fact, the role of both mitochondrial ROS and inactive form of ERK on cell-cycle arrest and differentiation progression has been well established (Pereira et al., 2015; Khacho et al., 2016).
Conclusion
Overall, we have shown that HFCD diet produces a specific gut microbiota signature in the small intestine and cecum, increases propionate and butyrate, changes NSC fate and promotes mitochondrial biogenesis and oxidative stress. More importantly, we identified the mitochondrial by-product ROS as a key mediator of SCFA-induced NSC alterations, leading to a premature differentiation and depletion of NSC pool in adult neurogenic niches of HFCD-fed mice. Although other host feedback mechanisms triggered by diet-associated gut microbiome must be further investigated, our findings add novel understandings of gut–microbiota–brain interactions in regulating adult neurogenesis, propelling mitochondrial stress into the centre of this cross-talk.
Supplementary Material
Acknowledgements
The authors are also thankful to Dr. Roger J. Davis (Howard Hughes Medical Institute, USA) for kindly providing constitutively active form of MEK1 overexpression vector. Finally, the authors thank all members of the laboratory, especially Beatriz Estremores, for technical assistance.
Funding
This work was supported by PTDC/MED-NEU/29650/2017 and SFRH/BD/100674/2014 from Fundação para a Ciência e Tecnologia, Portugal.
Competing Interests
The authors report no competing interests.
Glossary
- DCX =
doublecortin X
- DG =
dentate gyrus
- ERK1/2 =
extracellular signal-related protein kinase1/2
- HFCD =
high-fat, choline-deficient
- HFD =
high-fat diet
- KEGG =
Kyoto encyclopedia of genes and genomes
- LEfSe =
linear discriminant analysis of effect size
- MEK =
mitogen-activated protein kinase
- mtDNA =
mitochondrial DNA
- mtROS =
mitochondrial ROS
- NASH =
non-alcoholic steatohepatitis
- NSC =
neural stem cell
- OB =
olfactory bulb
- PGC-1α =
peroxisome proliferator-activated receptor γ coactivator-1 α
- ROS =
reactive oxygen species
- SCFA =
short-chain fatty acids
- Sirt3 =
sirtuin-3
- Sod2 =
superoxide dismutase 2
- Sox2 =
SRY (sex determining region Y)-box 2
- SVZ =
sub-ventricular zone
- Tfam =
mitochondrial transcription factor A
Contributor Information
Maria F Ribeiro, Research Institute for Medicines (iMed.ULisboa), Faculty of Pharmacy, Universidade de Lisboa, Lisbon, Portugal.
André A Santos, Research Institute for Medicines (iMed.ULisboa), Faculty of Pharmacy, Universidade de Lisboa, Lisbon, Portugal.
Marta B Afonso, Research Institute for Medicines (iMed.ULisboa), Faculty of Pharmacy, Universidade de Lisboa, Lisbon, Portugal.
Pedro M Rodrigues, Research Institute for Medicines (iMed.ULisboa), Faculty of Pharmacy, Universidade de Lisboa, Lisbon, Portugal.
Sónia Sá Santos, Research Institute for Medicines (iMed.ULisboa), Faculty of Pharmacy, Universidade de Lisboa, Lisbon, Portugal.
Rui E Castro, Research Institute for Medicines (iMed.ULisboa), Faculty of Pharmacy, Universidade de Lisboa, Lisbon, Portugal.
Cecília M P Rodrigues, Research Institute for Medicines (iMed.ULisboa), Faculty of Pharmacy, Universidade de Lisboa, Lisbon, Portugal.
Susana Solá, Research Institute for Medicines (iMed.ULisboa), Faculty of Pharmacy, Universidade de Lisboa, Lisbon, Portugal.
REFERENCES
- Almeida-Suhett CP, Graham A, Chen Y, Deuster P. Behavioral changes in male mice fed a high-fat diet are associated with IL-1beta expression in specific brain regions. Physiol Behav 2017; 169: 130–40. [DOI] [PubMed] [Google Scholar]
- Antico Arciuch VG, Elguero ME, Poderoso JJ, Carreras MC. Mitochondrial regulation of cell cycle and proliferation. Antioxid Redox Signal 2012; 16: 1150–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcego DM, Krolow R, Lampert C, Toniazzo AP, Berlitz C, Lazzaretti C, et al. Early life adversities or high fat diet intake reduce cognitive function and alter BDNF signaling in adult rats: interplay of these factors changes these effects. Int J Dev Neurosci 2016; 50: 16–25. [DOI] [PubMed] [Google Scholar]
- Arcego DM, Toniazzo AP, Krolow R, Lampert C, Berlitz C, Dos Santos Garcia E, et al. Impact of high-fat diet and early stress on depressive-like behavior and hippocampal plasticity in adult male rats. Mol Neurobiol 2018; 55: 2740–53. [DOI] [PubMed] [Google Scholar]
- Baufeld C, Osterloh A, Prokop S, Miller KR, Heppner FL. High-fat diet-induced brain region-specific phenotypic spectrum of CNS resident microglia. Acta Neuropathol 2016; 132: 361–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D. Neuroinflammation and neurodegeneration in overnutrition-induced diseases. Trends Endocrinol Metab 2013; 24: 40–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cani PD, Knauf C. How gut microbes talk to organs: the role of endocrine and nervous routes. Mol Metab 2016; 5: 743–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 2010; 7: 335–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallucci V, Fidaleo M, Pani G. Neural stem cells and nutrients: poised between quiescence and exhaustion. Trends Endocrinol Metab 2016; 27: 756–69. [DOI] [PubMed] [Google Scholar]
- Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem 2005; 280: 26185–92. [DOI] [PubMed] [Google Scholar]
- Chen Y, Zhang J, Lin Y, Lei Q, Guan KL, Zhao S, et al. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep 2011; 12: 534–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SA, Kim YH, Park YH, Yang HJ, Jeong PS, Cha JJ, et al. Novel crosstalk between Vps26a and Nox4 signaling during neurogenesis. Cell Death Differ 2019; 26: 1582–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark A, Mach N. The crosstalk between the gut microbiota and mitochondria during exercise. Front Physiol 2017; 8: 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti L, Pollard SM, Gorba T, Reitano E, Toselli M, Biella G, et al. Niche-independent symmetrical self-renewal of a mammalian tissue stem cell. PLoS Biol 2005; 3: e283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davido DJ, Richter F, Boxberger F, Stahl A, Menzel T, Luhrs H, et al. Butyrate and propionate downregulate ERK phosphorylation in HT-29 colon carcinoma cells prior to differentiation. Eur J Cancer Prev 2001; 10: 313–21. [DOI] [PubMed] [Google Scholar]
- Donovan MH, Yazdani U, Norris RD, Games D, German DC, Eisch AJ. Decreased adult hippocampal neurogenesis in the PDAPP mouse model of Alzheimer’s disease. J Comp Neurol 2006; 495: 70–83. [DOI] [PubMed] [Google Scholar]
- Elwing JE, Lustman PJ, Wang HL, Clouse RE. Depression, anxiety, and nonalcoholic steatohepatitis. Psychosom Med 2006; 68: 563–9. [DOI] [PubMed] [Google Scholar]
- Etchegoyen M, Nobile MH, Baez F, Posesorski B, Gonzalez J, Lago N, et al. Metabolic syndrome and neuroprotection. Front Neurosci 2018; 12: 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang D, Yan S, Yu Q, Chen D, Yan SS. Mfn2 is required for mitochondrial development and synapse formation in human induced pluripotent stem cells/hiPSC derived cortical neurons. Sci Rep 2016; 6: 31462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J, Demic S, Cheng S. The reduction of adult neurogenesis in depression impairs the retrieval of new as well as remote episodic memory. PLoS One 2018; 13: e0198406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca MB, Sola S, Xavier JM, Dionisio PA, Rodrigues CM. Amyloid beta peptides promote autophagy-dependent differentiation of mouse neural stem cells: a beta-mediated neural differentiation. Mol Neurobiol 2013; 48: 829–40. [DOI] [PubMed] [Google Scholar]
- Fuentealba LC, Rompani SB, Parraguez JI, Obernier K, Romero R, Cepko CL, et al. Embryonic origin of postnatal neural stem cells. Cell 2015; 161: 1644–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung TC, Olson CA, Hsiao EY. Interactions between the microbiota, immune and nervous systems in health and disease. Nat Neurosci 2017; 20: 145–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gans JH. Diethylnitrosamine-induced changes in mouse liver morphology and function. Proc Soc Exp Biol Med 1976; 153: 116–20. [DOI] [PubMed] [Google Scholar]
- Gao Z, Ure K, Ables JL, Lagace DC, Nave KA, Goebbels S, et al. Neurod1 is essential for the survival and maturation of adult-born neurons. Nat Neurosci 2009; 12: 1090–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparova Z, Janega P, Weismann P, Falougy HE, Kaprinay BT, Liptak B, et al. Effect of metabolic syndrome on neural plasticity and morphology of the hippocampus: correlations of neurological deficits with physiological status of the rat. Gen Physiol Biophys 2018; 37: 619–32. [DOI] [PubMed] [Google Scholar]
- Gentile CL, Weir TL. The gut microbiota at the intersection of diet and human health. Science 2018; 362: 776–80. [DOI] [PubMed] [Google Scholar]
- Ghaisas S, Maher J, Kanthasamy A. Gut microbiome in health and disease: linking the microbiome-gut-brain axis and environmental factors in the pathogenesis of systemic and neurodegenerative diseases. Pharmacol Ther 2016; 158: 52–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Pinilla F. Brain foods: the effects of nutrients on brain function. Nat Rev Neurosci 2008; 9: 568–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Pinilla F, Tyagi E. Diet and cognition: interplay between cell metabolism and neuronal plasticity. Curr Opin Clin Nutr Metab Care 2013; 16: 726–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould E, Tanapat P. Lesion-induced proliferation of neuronal progenitors in the dentate gyrus of the adult rat. Neuroscience 1997; 80: 427–36. [DOI] [PubMed] [Google Scholar]
- Hamilton A, Holscher C. The effect of ageing on neurogenesis and oxidative stress in the APP(swe)/PS1(deltaE9) mouse model of Alzheimer’s disease. Brain Res 2012; 1449: 83–93. [DOI] [PubMed] [Google Scholar]
- Heijtz RD, Wang S, Anuar F, Qian Y, Bjorkholm B, Samuelsson A, et al. Normal gut microbiota modulates brain development and behavior. Proc Natl Acad Sci USA 2011; 108: 3047–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khacho M, Clark A, Svoboda DS, Azzi J, MacLaurin JG, Meghaizel C, et al. Mitochondrial dynamics impacts stem cell identity and fate decisions by regulating a nuclear transcriptional program. Cell Stem Cell 2016; 19: 232–47. [DOI] [PubMed] [Google Scholar]
- Khanna S, Tosh PK. A clinician's primer on the role of the microbiome in human health and disease. Mayo Clin Proc 2014; 89: 107–14. [DOI] [PubMed] [Google Scholar]
- Kishida N, Matsuda S, Itano O, Shinoda M, Kitago M, Yagi H, et al. Development of a novel mouse model of hepatocellular carcinoma with nonalcoholic steatohepatitis using a high-fat, choline-deficient diet and intraperitoneal injection of diethylnitrosamine. BMC Gastroenterol 2016; 16: 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh A, De Vadder F, Kovatcheva-Datchary P, Backhed F. From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell 2016; 165: 1332–45. [DOI] [PubMed] [Google Scholar]
- Kong X, Wang R, Xue Y, Liu X, Zhang H, Chen Y, et al. Sirtuin 3, a new target of PGC-1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS One 2010; 5: e11707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 2013; 31: 814–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindqvist A, Mohapel P, Bouter B, Frielingsdorf H, Pizzo D, Brundin P, et al. High-fat diet impairs hippocampal neurogenesis in male rats. Eur J Neurol 2006; 13: 1385–8. [DOI] [PubMed] [Google Scholar]
- Loomba R, Seguritan V, Li W, Long T, Klitgord N, Bhatt A, et al. Gut microbiome-based metagenomic signature for non-invasive detection of advanced fibrosis in human nonalcoholic fatty liver disease. Cell Metab 2017; 25: 1054–62.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lurie I, Yang YX, Haynes K, Mamtani R, Boursi B. Antibiotic exposure and the risk for depression, anxiety, or psychosis: a nested case-control study. J Clin Psychiatry 2015; 76: 1522–8. [DOI] [PubMed] [Google Scholar]
- Mast JD, Tomalty KM, Vogel H, Clandinin TR. Reactive oxygen species act remotely to cause synapse loss in a Drosophila model of developmental mitochondrial encephalopathy. Development 2008; 135: 2669–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 2012; 6: 610–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNay DE, Briancon N, Kokoeva MV, Maratos-Flier E, Flier JS. Remodeling of the arcuate nucleus energy-balance circuit is inhibited in obese mice. J Clin Invest 2012; 122: 142–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra K. Mitochondrial fission-fusion as an emerging key regulator of cell proliferation and differentiation. Bioessays 2013; 35: 955–64. [DOI] [PubMed] [Google Scholar]
- Na J, Furue MK, Andrews PW. Inhibition of ERK1/2 prevents neural and mesendodermal differentiation and promotes human embryonic stem cell self-renewal. Stem Cell Res 2010; 5: 157–69. [DOI] [PubMed] [Google Scholar]
- Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, et al. Host-gut microbiota metabolic interactions. Science 2012; 336: 1262–7. [DOI] [PubMed] [Google Scholar]
- Ogbonnaya ES, Clarke G, Shanahan F, Dinan TG, Cryan JF, O’Leary OF. Adult hippocampal neurogenesis is regulated by the microbiome. Biol Psychiatry 2015; 78: e7–9–e9. [DOI] [PubMed] [Google Scholar]
- Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci 1997; 17: 3727–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HR, Park M, Choi J, Park KY, Chung HY, Lee J. A high-fat diet impairs neurogenesis: involvement of lipid peroxidation and brain-derived neurotrophic factor. Neurosci Lett 2010; 482: 235–9. [DOI] [PubMed] [Google Scholar]
- Patel PJ, Hayward KL, Rudra R, Horsfall LU, Hossain F, Williams S, et al. Multimorbidity and polypharmacy in diabetic patients with NAFLD: implications for disease severity and management. Medicine 2017; 96: e6761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira L, Font-Nieves M, Van den Haute C, Baekelandt V, Planas AM, Pozas E. IL-10 regulates adult neurogenesis by modulating ERK and STAT3 activity. Front Cell Neurosci 2015; 9: 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera TD, Coplan JD, Lisanby SH, Lipira CM, Arif M, Carpio C, et al. Antidepressant-induced neurogenesis in the hippocampus of adult nonhuman primates. J Neurosci 2007; 27: 4894–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera TD, Dwork AJ, Keegan KA, Thirumangalakudi L, Lipira CM, Joyce N, et al. Necessity of hippocampal neurogenesis for the therapeutic action of antidepressants in adult nonhuman primates. PLoS One 2011; 6: e17600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrik D, Lagace DC, Eisch AJ. The neurogenesis hypothesis of affective and anxiety disorders: are we mistaking the scaffolding for the building? Neuropharmacology 2012; 62: 21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard SM, Conti L, Sun Y, Goffredo D, Smith A. Adherent neural stem (NS) cells from fetal and adult forebrain. Cereb Cortex 2006; 16: i112–20. [DOI] [PubMed] [Google Scholar]
- Ramakrishna BS. Role of the gut microbiota in human nutrition and metabolism. J Gastroenterol Hepatol 2013; 28: 9–17. [DOI] [PubMed] [Google Scholar]
- Raposo A, Vasconcelos FF, Drechsel D, Marie C, Johnston C, Dolle D, et al. Ascl1 coordinately regulates gene expression and the chromatin landscape during neurogenesis. Cell Rep 2015; 10: 1544–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau M, Rehman A, Dittrich M, Groen AK, Hermanns HM, Seyfried F, et al. Fecal SCFAs and SCFA-producing bacteria in gut microbiome of human NAFLD as a putative link to systemic T-cell activation and advanced disease. United European Gastroenterol J 2018; 6: 1496–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro MF, Genebra T, Rego AC, Rodrigues CMP, Sola S. Amyloid beta peptide compromises neural stem cell fate by irreversibly disturbing mitochondrial oxidative state and blocking mitochondrial biogenesis and dynamics. Mol Neurobiol 2019; 56: 3922–36. [DOI] [PubMed] [Google Scholar]
- Rogers GB, Keating DJ, Young RL, Wong ML, Licinio J, Wesselingh S. From gut dysbiosis to altered brain function and mental illness: mechanisms and pathways. Mol Psychiatry 2016; 21: 738–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose S, Bennuri SC, Davis JE, Wynne R, Slattery JC, Tippett M, et al. Butyrate enhances mitochondrial function during oxidative stress in cell lines from boys with autism. Transl Psychiatry 2018; 8: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar A, Harty S, Lehto SM, Moeller AH, Dinan TG, Dunbar RIM, et al. The microbiome in psychology and cognitive neuroscience. Trends Cogn Sci 2018; 22: 611–36. [DOI] [PubMed] [Google Scholar]
- Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol 2011; 12: R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell 2012; 48: 158–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyan AS, Hughes RD, Shawcross DL. Changing face of hepatic encephalopathy: role of inflammation and oxidative stress. World J Gastroenterol 2010; 16: 3347–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoes AE, Pereira DM, Amaral JD, Nunes AF, Gomes SE, Rodrigues PM, et al. Efficient recovery of proteins from multiple source samples after TRIzol((R)) or TRIzol((R))LS RNA extraction and long-term storage. BMC Genomics 2013; 14: 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slattery J, MacFabe DF, Kahler SG, Frye RE. Enteric ecosystem disruption in autism spectrum disorder: can the microbiota and macrobiota be restored? Curr Pharm Des2016; 22: 6107–21. [DOI] [PubMed] [Google Scholar]
- Stangl D, Thuret S. Impact of diet on adult hippocampal neurogenesis. Genes Nutr 2009; 4: 271–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun SY. N-acetylcysteine, reactive oxygen species and beyond. Cancer Biol Ther 2010; 9: 109–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Invest 2009; 119: 2758–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang MM, Lin WJ, Zhang JT, Zhao YW, Li YC. Exogenous FGF2 reverses depressive-like behaviors and restores the suppressed FGF2-ERK1/2 signaling and the impaired hippocampal neurogenesis induced by neuroinflammation. Brain Behav Immun 2017; 66: 322–31. [DOI] [PubMed] [Google Scholar]
- Tang Y, Chen Y, Jiang H, Nie D. Short-chain fatty acids induced autophagy serves as an adaptive strategy for retarding mitochondria-mediated apoptotic cell death. Cell Death Differ 2011; 18: 602–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travis CC, McClain TW, Birkner PD. Diethylnitrosamine-induced hepatocarcinogenesis in rats: a theoretical study. Toxicol Appl Pharmacol 1991; 109: 289–304. [DOI] [PubMed] [Google Scholar]
- Uittenbogaard M, Brantner CA, Chiaramello A. Epigenetic modifiers promote mitochondrial biogenesis and oxidative metabolism leading to enhanced differentiation of neuroprogenitor cells. Cell Death Dis 2018; 9: 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Val-Laillet D, Guerin S, Coquery N, Nogret I, Formal M, Rome V, et al. Oral sodium butyrate impacts brain metabolism and hippocampal neurogenesis, with limited effects on gut anatomy and function in pigs. FASEB J 2018; 32: 2160–71. [DOI] [PubMed] [Google Scholar]
- Vithayathil J, Pucilowska J, Goodnough LH, Atit RP, Landreth GE. Dentate gyrus development requires ERK activity to maintain progenitor population and MAPK pathway feedback regulation. J Neurosci 2015; 35: 6836–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Sonne SB, Feng Q, Chen N, Xia Z, Li X, et al. High-fat feeding rather than obesity drives taxonomical and functional changes in the gut microbiota in mice. Microbiome 2017; 5: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youssef NA, Abdelmalek MF, Binks M, Guy CD, Omenetti A, Smith AD, et al. Associations of depression, anxiety and antidepressants with histological severity of nonalcoholic fatty liver disease. Liver Int 2013; 33: 1062–70. [DOI] [PubMed] [Google Scholar]
- Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu Y, et al. ROS and ROS-mediated cellular signaling. Oxid Med Cell Longev 2016; 2016: 4350965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Deng W, Gage FH. Mechanisms and functional implications of adult neurogenesis. Cell 2008; 132: 645–60. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings in this study are available from the corresponding author upon request.