

Tidball et al. examine the effects of epilepsy-associated SCN8A variants on patient stem cell-derived neurons, and show that variant-specific changes in sodium currents lead to neuronal hyperexcitability. Drug application reduces the hyperexcitability in vitro, suggesting potential as a platform to identify precision therapies.
Keywords: developmental and epileptic encephalopathy, epileptogenesis, genetic epilepsy, voltage-gated sodium channel, induced pluripotent stem cell
Abstract
Missense variants in the SCN8A voltage-gated sodium channel gene are linked to early-infantile epileptic encephalopathy type 13, also known as SCN8A-related epilepsy. These patients exhibit a wide spectrum of intractable seizure types, severe developmental delay, movement disorders, and elevated risk of sudden unexpected death in epilepsy. The mechanisms by which SCN8A variants lead to epilepsy are poorly understood, although heterologous expression systems and mouse models have demonstrated altered sodium current properties. To investigate these mechanisms using a patient-specific model, we generated induced pluripotent stem cells from three patients with missense variants in SCN8A: p.R1872>L (Patient 1); p.V1592>L (Patient 2); and p.N1759>S (Patient 3). Using small molecule differentiation into excitatory neurons, induced pluripotent stem cell-derived neurons from all three patients displayed altered sodium currents. Patients 1 and 2 had elevated persistent current, while Patient 3 had increased resurgent current compared to controls. Neurons from all three patients displayed shorter axon initial segment lengths compared to controls. Further analyses focused on one of the patients with increased persistent sodium current (Patient 1) and the patient with increased resurgent current (Patient 3). Excitatory cortical neurons from both patients had prolonged action potential repolarization. Using doxycycline-inducible expression of the neuronal transcription factors neurogenin 1 and 2 to synchronize differentiation of induced excitatory cortical-like neurons, we investigated network activity and response to pharmacotherapies. Both small molecule differentiated and induced patient neurons displayed similar abnormalities in action potential repolarization. Patient induced neurons showed increased burstiness that was sensitive to phenytoin, currently a standard treatment for SCN8A-related epilepsy patients, or riluzole, an FDA-approved drug used in amyotrophic lateral sclerosis and known to block persistent and resurgent sodium currents, at pharmacologically relevant concentrations. Patch-clamp recordings showed that riluzole suppressed spontaneous firing and increased the action potential firing threshold of patient-derived neurons to more depolarized potentials. Two of the patients in this study were prescribed riluzole off-label. Patient 1 had a 50% reduction in seizure frequency. Patient 3 experienced an immediate and dramatic seizure reduction with months of seizure freedom. An additional patient with a SCN8A variant in domain IV of Nav1.6 (p.V1757>I) had a dramatic reduction in seizure frequency for several months after starting riluzole treatment, but then seizures recurred. Our results indicate that patient-specific neurons are useful for modelling SCN8A-related epilepsy and demonstrate SCN8A variant-specific mechanisms. Moreover, these findings suggest that patient-specific neuronal disease modelling offers a useful platform for discovering precision epilepsy therapies.
Introduction
During the recent explosion in exome sequencing of epilepsy patients, SCN8A was linked to early infantile epileptic encephalopathy type 13 (EIEE13) (Veeramah et al., 2012; Epi4K Investigators, 2013; Ohba et al., 2014; Larsen et al., 2015). EIEE13 patients have a wide spectrum of seizure types with seizure onset occurring on average at 5 months of age (Larsen et al., 2015). Developmental delays, lack of speech, and hypotonia are nearly universal. Seizures are typically severe in EIEE13 patients, refractory to antiseizure medications (ASMs), and lead to a high risk of sudden unexpected death in epilepsy (SUDEP), findings that have been recapitulated in mouse models (Veeramah et al., 2012; Wagnon et al., 2014; Kong et al., 2015; Larsen et al., 2015; Bunton-Stasyshyn et al., 2019).
SCN8A encodes the voltage-gated sodium channel (VGSC) α subunit, Nav1.6, which is the most abundant VGSC in the CNS, where it is primarily localized to axon initial segments (AIS) and nodes of Ranvier (Gasser et al., 2012). Nearly all SCN8A variants linked to EIEE13 thus far are de novo missense mutations resulting in single amino acid substitutions (Wagnon and Meisler, 2015), although one patient inherited the mutation from a mosaic parent (Larsen et al., 2015). Data from a knock-in mouse model and from heterologous expression of several mutations in cultured cells implicate a variety of potential sodium current (INa) abnormalities in disease pathogenesis, including slowed inactivation kinetics (Wagnon et al., 2016), increased persistent INa (Veeramah et al., 2012; Wagnon et al., 2016; Lopez-Santiago et al., 2017; Ottolini et al., 2017), and increased resurgent INa (Patel et al., 2016; Ottolini et al., 2017). The predicted outcomes of these changes in INa properties are delayed action potential repolarization and ectopic firing. Consistent with this, recordings from the Scn8aN1768D/+ EIEE13 mouse model showed early after depolarization (EAD)-like action potential waveforms in CA1 hippocampal neurons (Lopez-Santiago et al., 2017). A complete understanding of SCN8A mutation-specific mechanisms would necessitate the production of multiple mouse models. Moreover, because data from mice often do not translate to humans, it is desirable to test ASM efficacy in other models such as cultured human cortical neurons, which is now feasible in patient-derived cells using induced pluripotent stem cell (iPSC) technology.
While mouse and heterologous systems are useful, we sought to perform mechanistic studies in a human neuronal system that allows rapid testing of potential EIEE13 mutation-specific effects. To this end, we generated iPSC lines from three patients with EIEE13. Cortical excitatory patient neurons differentiated from the iPSCs exhibited elevated persistent or resurgent INa compared to control neurons, similar to findings in mouse and heterologous cell culture models. EIEE13 patient neurons also showed prolonged action potential repolarization and shorter AIS lengths. To reduce experimental variability inherent in small molecule cortical neuron differentiations, we generated doxycycline-inducible neurons (iNeurons) from the same iPSC lines. As assessed by burst duration and percentage of spikes in bursts, iNeurons displayed increased network bursting. Genotypic differences in network activity were rescued by administration of phenytoin or riluzole, the latter a drug with relative selectivity for persistent and resurgent INa. Notably, three EIEE13 patients with medically refractory epilepsy were prescribed off-label riluzole for compassionate use. All three patients had reductions in seizure frequency, suggesting the utility of patient-derived neuronal models for developing precision epilepsy therapies.
Materials and methods
Induced pluripotent stem cell reprogramming
Skin punch biopsies were obtained from three EIEE13 patients (Patients 1–3) and two healthy controls (Controls 1 and 4) without known genetic disorders with consent under a protocol approved by the Institutional Review Board of Michigan Medicine and reprogrammed by previously published methods (Tidball et al., 2017). Two additional control lines used in this study were previously reported as CC1 (herein Control 2) and CHD2 WT/WT2 (herein Control 3) from (Tidball et al., 2016, 2017), respectively, and were reprogrammed using identical methods. See the Supplementary material for further details of reprogramming and cell line validation.
CRISPR genome editing with simultaneous reprogramming
We have previously published methods combining CRISPR genome editing and iPSC reprogramming (Tidball et al., 2017, 2018). See the Supplementary material for further details describing the generation of the Patient 2 rescue iPSC line.
Excitatory neuron differentiation
We used a modification of the Shi et al. (2012) technique for differentiating iPSCs into excitatory cortical neurons. The neuronal cultures were transduced with lentivirus containing a mouse Camk2a promoter-driven GFP to identify mature neurons (Shcheglovitov et al., 2013; Nehme et al., 2018). See the Supplementary material for further details.
Generation and differentiation of stable doxycycline-inducible Ngn1/2 (iNeuron) iPSC lines
Established iPSC lines were transfected using Mirus LT-1 with 2 TALEN plasmids (pZT-C13-R1 and pZT-C13-L1, gifts of Jizhong Zou; addgene.org: #52638 and 52637, respectively) targeting the safe-harbour-like locus, CLYBL, and a targeting plasmid containing the doxycycline-inducible promoter system, which controls neurogenin 1 (Ngn1) and 2 (Ngn2) expression, and constitutively active mCherry (pUCM-CLYBL-Ngn1/2, a gift of Dr Michael Ward). Pure mCherry+ clonal lines were obtained by manual selection. To differentiate into iNeurons, cells were grown for 72 h in mTeSR1 media containing doxycycline. Cells were then replated onto polyethyleneimine (PEI)/laminin coated dishes in 3N medium containing doxycycline. On Day 8, the medium was replaced with BrainPhys medium containing N2 and SM1 supplements as well as brain and glial derived neurotrophic factors (BDNF and GDNF). See the Supplementary material for further details.
Immunostaining
Neurons grown on Nunc™ Lab-Tek™ III 8-well chamber slides (Thermo Scientific) were fixed in paraformaldehyde for 30 min at room temperature. After permeabilization with 0.2% TritonTM X-100 for 20 min at room temperature, cells were incubated in phosphate-buffered saline (PBS) containing 5% normal goat serum with 1% bovine serum albumin and 0.05% TritonTM X-100 for 1 h at room temperature. Cell were incubated in primary antibody overnight (see Supplementary Table 2 for antibodies and dilutions) in the same blocking buffer at 4°C, washed four times in PBS with 0.05% Tween-20 (PBST), and incubated for 90 min with secondary antibody. Cells were washed three times in PBST and incubated with bisbenzimide for 5 min. After additional PBS washes, coverslips were mounted on slides with Glycergel® mounting medium (Agilent Dako). Images were obtained on a Leica SP5 upright DMI 6000 confocal microscope.
Axon initial segment imaging and measurement
Neurons were differentiated and immunostained on Day 21 as described above with antibodies for ankyrin-G and microtubule associated protein 2ab-isoforms (MAP2ab). AIS were imaged on a Leica SP5 upright DMI 6000 confocal microscope using a water-immersion 63× objective. Images were taken at 1 Airy unit for the pinhole size with overlapped z-series steps covering the entire AIS thickness. Max projection images were obtained and exported as TIFF files, then imported into ImageJ. Segmented lines were overlaid by a blinded observer. The plot profiles (intensity of each pixel on the line) were exported and uploaded to a MATLAB program designed for determining AIS length (Dr Eugene Katrukha, Utrecht University). In short, this program performs a smoothening function, normalizes the intensity data, and applies a threshold of 40% to determine the ends of the AIS (Yau et al., 2014).
Electrophysiological recordings
Single GFP+ (labelled with a lentiviral Camk2a-GFP reporter) iPSC-derived neurons were analysed by whole-cell patch clamp recordings. Voltage clamp recordings were performed in the standard whole-cell configuration, using previously described conditions (Liu et al., 2013). Of note, voltage clamp of iPSC-derived neurons can become unstable, making it sometimes difficult to complete long voltage-step protocols. All experiments were carried out at room temperature (21–22°C). See the Supplementary material for further details.
Multielectrode array recordings
On Day 3 of doxycycline treatment, differentiating iNeurons were plated onto PEI/laminin coated Axion 96-well multielectrode array (MEA) recording plates at 1.5 × 105 cells in a 5 µl drop on the electrode grid (8 electrodes/well) of each well. Cells were grown as described above with half-volume media changes three times a week. Every 2–4 days a 5-min recording was performed at 37°C after a 5-min equilibration time on the MEA. At 30 days after doxycycline treatment, the MEA plates were used for ASM testing. Because of differences in the overall activity of individual plates, the definition of a network burst was changed from default settings to reduce false positive and false negative network burst detections (Matsuda et al., 2018). Therefore, we used an iterative process of altering the minimum percentage of electrodes participating (from 25% to 50%, or two to four electrodes) and the minimum number of spikes (from 10 to 100 spikes) for each plate on the most active recording day (typically Day 30). A maximum interspike interval of 75 ms was used to define bursts for all analyses.
Between Days 30 and 33, drug testing was performed on MEA plates that did not show signs of peeling or excessive cell death (shown by loss of overall activity or lack of network bursting in all wells). Concentrated (10×) ASMs were added to the plate in a 10% volume (20 µl) with 1% dimethyl sulfoxide (DMSO), or DMSO alone (control). Thus, final concentrations were 1× of the expected concentration with 0.1% DMSO. Plates were allowed to equilibrate for 5 min on the Maestro MEA followed by a 10-min baseline measurement. The plate was immediately removed, and the drugs were added. Each drug/concentration was added to one row yielding six wells used per line. The plate was placed back on the MEA recording device and allowed to equilibrate for 5 min followed by a 10-min experimental recording. When performing drug-testing analyses, the percentage of spikes in network bursts values were only used from wells that had a pretreatment level of at least 5% spikes in network bursts. For all other measures, wells with <0.01 Hz mean firing rate were removed from analyses. Each well of the drug-testing mean firing rate data was normalized to the pretreatment level to account for large plate-to-plate variability in overall activity.
Statistical analyses
Results are expressed as mean ± standard error of the mean (SEM) except for the iNeuron action potential dynamics, which are displayed as 95% confidence intervals (CI). For each line within a dataset, 3–10 independent cultures of neurons were used, except for resurgent current measurements for Patient 2 (two independent cultures). Data were analysed and organized using Excel (Microsoft). Prism (GraphPad) was used for generating graphs and statistical analyses. Sodium current, AIS length, and action potential dynamic data were analysed by the non-parametric Kruskal-Wallis test with uncorrected Dunn’s post-test. For MEA data analyses, when no data-points were missing, we used repeated measures two-way ANOVA. Post hoc analyses were performed by two-stage linear step-up procedure for multiple comparison controlling for false discovery rate (Benjamini et al., 2006).
Data availability
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
Results
EIEE13 patient-derived neurons show variant-specific differences in INa properties
To understand the disease mechanisms of individual EIEE13 patient SCN8A variants in a human neuronal model, we generated integration-free iPSCs by episomal transfection of fibroblasts from three patients with EIEE13 (Fig. 1A and Table 1) and two non-epileptic controls, Controls 1 and 4 (Table 1). Patient variants were confirmed in each iPSC line by PCR of genomic DNA followed by Sanger sequencing (Supplementary Fig. 1A). We used two additional control lines, Controls 2 and 3 (Table 1) who have been previously published (Tidball et al., 2016, 2017). Each line used for subsequent experiments was negative for episomal reprogramming vector integration, had no observable genomic abnormalities as tested by either g-band karyotype or single-nucleotide polymorphism chip microarray analyses, and expressed markers of pluripotency (Supplementary Fig. 1B, C and data not shown). A subset of the iPSC lines (two patients and one control) were differentiated to embryoid bodies, resulting in the formation of three germ layers as assessed by immunofluorescence microscopy (Supplementary Fig. 1D).
Figure 1.
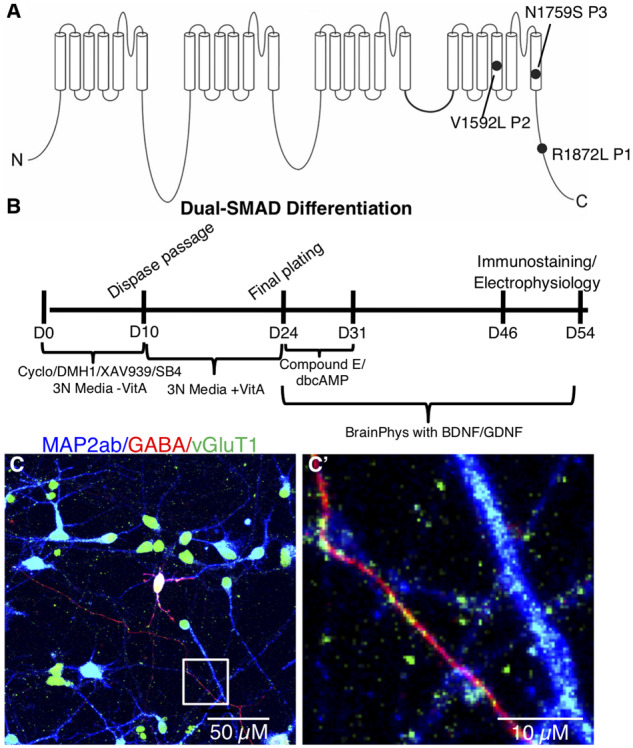
Patient variants and glutamatergic neuronal differentiation. (A) Diagram of the SCN8A gene with de novo missense variants from the three patients included in this study. (B) Differentiation protocol for generating excitatory cortical neurons from iPSCs using dual-SMAD inhibition combined with WNT and SHH inhibition. (C) Mature iPSC-derived neuronal cultures labelled with MAP2ab (blue), GABA (red), and vGluT1 (green). (C’) Enlarged image of the boxed area in C highlighting vGLUT1-labelled (green) puncta apposed to GABA- (red) and MAP2ab-positive (blue) neuronal processes.
Table 1.
Patient information
| Subject | Seizure onset | Age at biopsy/sex | Mutations | Seizure type | Other findings | Previous ASMs | Current medications |
|---|---|---|---|---|---|---|---|
| Patient 1 | 1 month | 16 years/female | p.R1872>L | Myoclonic at first, followed by gelastic | Intellectual disability, pancerebellar atrophy and ataxia | Keto, PB, CZP, VPA, LTG, PRM, GBP, OXC, LEV, ZON, FBM | PHT, CLB, TPM, vagal nerve stimulator |
| Patient 2 | 5 months | 9 months/female | p.V1592>L | Focal onset with eye deviation | Developmental delays, hypotonia, apnea | – | OXC, infrequent seizures |
| Patient 3 | 10 months | 7 years/female | p.N1759>S | Myoclonic jerks, altered mental status | Encephalopathy, developmental delays, ataxia | LAC, PHT, OXC, CLB | LEV |
| Patient 4a | Neonatal | 1 years/male | p.V1757>I | Tonic | Hyperekplexia, bradycardia, apnea | ZON, LOR | PHT, OXC, GBP, CZP |
| Control 1 | – | 36 years/female | – | – | – | – | – |
| Control 2 | – | 17 years/female | – | – | – | – | – |
| Control 3 | – | Newborn/male | – | – | – | – | – |
| Control 4 | Mother of Patient 1 | 47 years/female | – | – | – | – | – |
Information for patients and controls from which the iPSC lines were derived are presented including, age at biopsy, sex, SCN8A de novo variant, seizure type, medications, and other findings. CLB = clobazam; CZP = clonazepam; FBM = felbamate; GBP = gabapentin; Keto = ketogenic diet; LAC = lacosamide; LEV = levetiracetam; LOR = lorazepam; LTG = lamotrigine; OXC = oxcarbazepine; PB = phenobarbital; PHT = phenytoin; PRM = primidone; TPM = topiramate; VPA = valproic Acid; ZON = zonisamide.
Neuronal data for this patient are not presented in this report.
We differentiated the iPSCs into cortical excitatory neurons using a dual-SMAD inhibition protocol for specifying PAX6+ neuroepithelium (Fig. 1B). Our use of DMH1 for brain morphogenic protein (BMP) inhibition instead of dorsomorphin or noggin was based on its more specific inhibition of activin receptor-like kinase-2 (ALK2) (Neely et al., 2012). We also added the Wnt-antagonist XAV939, which is known to increase the forebrain identity of dual-SMAD differentiated neurons (Maroof et al., 2013), as well as cyclopamine to inhibit any endogenous sonic hedgehog (SHH) signalling and, thereby, decrease the number of subpallial GABAergic neurons generated. Using immunocytochemistry, nearly all patient and control neurons were positive for vesicular glutamate transporter 1 (vGLUT1) with ∼10% GABAergic neurons, which often double-labelled with both vGLUT1 and GABA (Fig. 1C and C’), consistent with recent studies that reported several cell types in the rodent brain that co-express these two markers (Perreault et al., 2012; Root et al., 2018). About 30% of cells expressed CTIP2 at Day 43 of differentiation (data not shown), as was reported in the original publication of the protocol we adapted (Shi et al., 2012). We also performed qRT-PCR for SCN8A, SCN1A, SCN1B, and SCN4B and found no differences between the patient and control groups (Supplementary Fig. 2). The mRNA expression for SCN4B was not detectable in our cultures.
We next measured INa properties of EIEE13 patient and control iPSC-derived neurons. Whole-cell voltage clamp recordings were performed on isolated neurons labelled with GFP driven by the Camk2a promoter, a marker of excitatory cortical neurons that has been shown to identify neurons with more mature electrophysiological characteristics (Fig. 2F) (Shcheglovitov et al., 2013; Nehme et al., 2018). Representative traces are shown in Fig. 2A–D. INa density measurements are shown at −30 mV, the peak voltage for most neurons tested. Patient 1 neurons had decreased transient INa density that approached significance [233 ± 39 pA/pF (n = 24) for controls versus 140 ± 12 pA/pF (n = 24) for Patient 1, P = 0.058; Fig. 2I]. Persistent INa was measured at 50 ms after onset of a depolarizing step to −30 mV and normalized to peak INa. Neurons from Patients 1 and 2 had significantly higher percentages of persistent INa compared to controls [control: 3.6 ± 0.5% (n = 24); Patient 1: 5.7 ± 0.6% (n = 24), P = 0.012; Patient 2: 6.81 ± 1.33% (n = 10), P = 0.014; Fig. 2J]. In contrast, persistent INa in Patient 3 neurons were not significantly different from controls [Patient 3: 3.61 ± 0.44 (n = 17), P = 0.985], suggesting an alternative mechanism.
Figure 2.

Increased persistent or resurgent INa in EIEE13 patient neurons. (A–D) Representative INa traces for control and patient neurons. (E) Persistent INa traces for Control 2 (C2), Patient 1 (P1) and Patient 2 (P2) are overlaid to demonstrate increased currents in patient neurons. (F) Representative epifluorescent image of a Camk2a-GFP labelled neuron from which whole-cell patch clamp recordings were made. (G and H) Resurgent INa recordings from Control 2 and Patient 3, respectively. (I and J) Quantification of peak INa density at −30 mV membrane potential and persistent INa density 50 ms after depolarization divided by peak INa, respectively. (K) Resurgent INa quantified after a 30 mV depolarization followed by −30 mV repolarization voltage step. Error bars are SEM. *P < 0.05, ** P < 0.01,*** P < 0.001, and **** P < 0.0001 by Kruskal-Wallis test with uncorrected Dunn’s post-test. For I–J controls n = 24 (C1 = 15, C2 = 9), P1 = 24 (Clone1 = 12, Clone2 = 12), P2 = 10 (Clone1 = 4, Clone2 = 6), P2r = 6, and P3 = 17 (Clone1 = 8, Clone2 = 5, Clone3 = 4). For K controls n = 10 (C1 = 6, C2 = 4), P1 = 8 (Clone1 = 3, Clone2 = 5), P2 = 4 (Clone1 = 1, Clone2 = 3), and P3 = 11 (Clone1 = 4, Clone2 = 4, Clone3 = 3).
Resurgent INa density was recorded using the voltage-step protocol shown in Fig. 2H. Representative traces are shown in Fig. 2G and H. Patient 3 neurons had significantly higher resurgent INa density [7.3 pA/pF (n = 10)] compared to controls [0.5 pA/pF (n = 11), P < 0.0001] while Patient 1 [1.3 pA/pF (n = 8), P = 0.375] and Patient 2 neurons [3.3 pA/pF (n = 4), P = 0.0706] did not (Fig. 2K).
Voltage-dependent properties across the full I-V curve range for a subset of the recorded neurons were not significantly different between genotypes; however, a trend for decreased peak INa (Gmax) for Patients 1 and 3 fits with the significant results found at −30 mV (Supplementary Table 4). Additionally, there are no differences in inactivation kinetics between controls and patient cells (Supplementary Table 4).
Deletion of the pathogenic allele in Patient 2 cells rescues persistent INa
An edited Patient 2 iPSC line was generated using simultaneous CRISPR gene-editing and iPSC reprogramming. The ‘nickase’ Cas9 variant, D10A, was used with two guide RNA sequences that flanked the patient variant. A silent mutation was engineered in the single stranded oligo donor (ssODN) template to alter one of the PAM sequences to block further nickase breaks. One line with only the wild-type variant was identified; however, the silent mutation was not present (Supplementary Fig. 1A). Further long-range PCR amplified a ∼500-bp deletion of the pathogenic allele (Supplementary Fig. 3).
This Patient 2 rescue line, which we call P2r, was differentiated using the same protocol as control and patient lines. Transient INa density at −30 mV was larger in the P2r line than in Patient 2 neurons [P2r: 386 ± 75 pA/pF (n = 6); Patient 2: 171 ± 26 pA/pF (n = 10), P = 0.040; Fig. 2I], consistent with the observed trend for larger transient INa density values in neurons from controls compared to those from Patients 1 and 2 (Fig. 2I). Because transient INa density in the CRISPR gene-edited line was similar to control levels, we concluded that hemizygous SCN8A expression was not deleterious. Importantly, the elevated percent persistent INa observed in Patient 2 neurons was rescued in the gene edited line, P2r, with values similar to controls but significantly different from Patient 2 [P2r: 2.9 ± 0.9% (n = 6); Patient 2: 6.8 ± 1.3% (n = 10), P = 0.015; controls: 3.6 ± 0.5% (n = 24), P = 0.486, Fig. 2J].
EIEE13 neurons have early after depolarizations and slowed action potential repolarization
To investigate changes in neuronal excitability, we recorded spontaneous activity from Camk2a-GFP-positive iPSC-derived neurons in current-clamp mode. We observed EADs in Patient 3 neurons, similar to findings in the Scn8aN1768D/+ EIEE13 mouse model (Lopez-Santiago et al., 2017), but not in neurons from controls, Patient 1 or Patient 2 (Fig. 3A, B and data not shown). We evoked single action potentials with 1-ms current injections in Patient 1 and Patient 3 neurons. We focused primarily on these two patients because they had the most severe clinical phenotypes and represented both classes of INa abnormalities. Representative traces are depicted in Fig. 3C. Patient 1 neurons showed prolonged repolarization and Patient 3 neurons showed EADs (arrows). Many Patient 1 and 3 neurons did not return to resting membrane potential levels within the duration of the action potential recording. Because of this, standard ADP50 and ADP80 measurements could not be accurately determined. Instead, we used three time points following the action potential peak (5, 10, and 40 ms) to compare the three groups (Fig. 3E–G). The resulting peak amplitude values were similar between groups, demonstrating that these measures were appropriate (Fig. 3D). Patient 1 neuronal action potentials were more depolarized than controls at 5 and 10 ms, respectively (Fig. 3E and F). Action potentials in Patient 3 neurons were more depolarized than controls at the 10 and 40 ms time points (Fig. 3F and G). Passive membrane properties, including resting membrane potential, input resistance, and capacitance, were similar between patients and controls (Supplementary Table 3).
Figure 3.

Impaired action potential repolarization in EIEE13 patient iPSC-derived neurons. (A) Representative traces of spontaneous activity in Patient 3 (P3) neurons. Arrows indicate instances of EAD-like waveforms. (B) Single spontaneous action potentials displayed with a higher resolution timescale show EAD-like waveforms in Patient 3 neurons (arrows). (C) Examples of evoked action potential firing for Control 2 (C2), Control 3 (C3) Patient 1 (P1) and Patient 3 (P3). Dotted lines denote the resting membrane potential (VMR). Arrows denote abnormalities in patient neuron action potential repolarization. (D) The peak amplitude in millivolts as measured from the resting membrane potential to the peak potential. (E–G) Measurements of voltage difference from the resting membrane potential at 5, 10, and 40 ms after the initial evoked action potential. Error bars represent standard error of the mean (SEM). *P < 0.05, **P < 0.01 by Kruskal-Wallis test with uncorrected Dunn’s post-test.
EIEE13 patient iPSC-derived neurons have reduced axon initial segment length
Nav1.6, the protein encoded by SCN8A, primarily localizes to the neuronal AIS and nodes of Ranvier via interactions with its binding partner ankyrin-G (Gasser et al., 2012). To determine whether neurons with EIEE13 variants display AIS alterations, we immunostained for ankyrin-G (Gutzmann et al., 2014) as a proxy for the AIS in iPSC neurons. Nearly all neurons tested contained a single, well-defined AIS (Fig. 4A–C). We observed a significant decrease in AIS length in neurons derived from all three patient lines compared to controls (Fig. 4D and E). On average, the AIS length in patient neurons was decreased by 32% (38% for Patient 1, 25% for Patient 2, and 33% for Patient 3) compared to the three controls.
Figure 4.

Decreased AIS length in EIEE13 patient-derived excitatory neurons. (A–C) Excitatory neurons from Control 1 (C1), Patient 1 (P1) and Patient 3 (P3) respectively, immunostained for ankyrin-G (red) and MAP2ab (green), with nuclear Hoechst labelling in blue. Arrowheads point to the start and end of each AIS from different neurons. (D and E) Quantification of AIS lengths for excitatory neurons from three controls (C2–4) and three patients (P1–3) plotted as box and whisker plots in D depicting the mean, minimum, maximum, and upper and lower quartile. The same quantification is plotted as cumulative distributions in E with 5 µM bin size. *P < 0.05 by Kruskal-Wallis test with uncorrected Dunn’s post-test.
EIEE13 patient-derived iNeurons show differences in action potential dynamics
We next used the iPSC neuron model for population analyses of neuronal activity and drug screening by MEA recordings. While the dual-SMAD differentiation technique paired with the Camk2a promoter-driven GFP reporter was useful for assessing individual neuron electrophysiology and AIS length, network activity assessed by MEA recordings was highly variable, possibly due to heterogeneous neuronal maturation and variability in the percentage of GABAergic neurons between cultures (data not shown). To reduce variability, we engineered iPSC lines to rapidly and homogeneously generate iNeurons by stably expressing doxycycline-inducible Ngn1 and Ngn2. As previously described, these cells quickly and efficiently differentiate into neurons after doxycycline addition (Zhang et al., 2013; Busskamp et al., 2014; Lam et al., 2017). Ngn1/2 induction produces neurons expressing upper cortical layer markers, including CUX1, CUX2, and BRN2, but not deeper layer markers such as CTIP2 (Zhang et al., 2013; Busskamp et al., 2014; Nehme et al., 2018). Stable insertion was successfully achieved for Patient 1, Patient 3, Control 2, and Control 3 lines using TALENs and homology arms targeting the CLYBL gene that is considered a ‘safe-harbour-like’ locus in the human genome (Cerbini et al., 2015). We first differentiated iNeurons (Fig. 5B) and measured evoked action potentials in single cells by patch clamp methods. Action potentials from patient iNeurons showed reduced repolarization rates, similar to observations in the dual-SMAD neurons [controls: −34.1 ± 1.7 mV/ms (n = 47); Patient 1: −28.7 ± 1.8 mV/ms (n = 46), P = 0.010; Patient 2: −26.1 ± 1.6 mV/ms (n = 29), P = 0.003] (Fig. 5A and F). In addition, Patient 1 and Patient 3 cells showed increased action potential amplitude half-widths compared to controls [controls: 3.03 ± 0.12 ms (n = 47); Patient 1: 3.65 ± 0.18 ms (n = 46), P = 0.004; Patient 3: 3.51 ± 0.16 mV/ms (n = 29), P = 0.014] (Fig. 5D). Finally, Patient 3 neurons exhibited reduced action potential peak amplitude and rate of depolarization compared with controls (Fig. 5C and E). While the individual metrics presented for iNeurons are different from the dual-SMAD neurons presented in Fig. 3, the action potential traces are strikingly similar between the dual-SMAD and iNeurons for each individual line (Supplementary Fig. 4).
Figure 5.

Altered action potential repolarization in EIEE13 patient-derived iNeurons. (A) Representative traces of evoked action potential firing for controls (C2 and 3), Patient 1, and Patient 3 (P1 and P3). Dotted line denotes the resting membrane potential (VMR). Arrows denote abnormalities in patient neuron action potential repolarization. (B) Schematic of iNeuron differentiation protocol from iPSC lines with stable cassette insertion in CLYBL safe-harbour-like locus. (C–F) Measurement of key action potential characteristics including the peak amplitude in millivolts as measured from the resting membrane potential to the peak potential (C), action potential width at half the peak height (D), and the rates of depolarization and repolarization (E and F, respectively). Error bars represent SEM. *P < 0.05, **P < 0.01, ***P < 0.001 by Kruskal-Wallis test with uncorrected Dunn’s post-test.
Increased burstiness of EIEE13 patient iNeurons
After differentiation, iNeurons were plated onto 96-well MEA plates (detailed in the ‘Materials and methods’ section and Fig. 5B) and activity was measured for over a month. Individual raster plots and instantaneous firing frequency plots of neuronal activity on Day 33 revealed that EIEE13 network activity, particularly that of Patient 1 neurons, displayed periods of rapid network bursts that become shorter and more frequent with time, followed by a brief period of low activity (Fig. 6A–C). Both the increased burst activity and pattern of burst discharges in EIEE13 iNeurons are consistent with an epileptiform-like phenotype. After more than 4 weeks in culture (Days 29–33), we found significant increases in measures of bursting activity in patient iNeurons compared to controls, as assessed by burst duration and the percentage of total spikes that were in network bursts for both Patient 1 and Patient 3 (Fig. 6F, G, J and K) (see ‘Materials and methods’ section for burst and network burst criteria). Another measure of burstiness, the coefficient of variation of interspike interval was elevated in Patient 1 only (Fig. 6E and I) (Nawrot et al., 2008). These measures of burstiness were not due to overall increases in firing, given that Patient 1 neurons tended to have lower overall activity compared with controls as measured by the weighted mean firing rate and Patient 3 was only slightly elevated (Fig. 6D and H).
Figure 6.

Increased burstiness in Patient 1 and Patient 3 iNeurons . (A) Control and patient iNeuron networks recorded by MEA 33 days after plating. Instantaneous firing frequencies over a 5-min recording show a unique bursting patterns in patient lines compared to control. (B and C) Raster plots for the eight electrodes in an individual well of a 96-well MEA plate for Control 2 (C2) and Patient 1 (P1). (D–K) Eight paired MEA experiments were performed on 96-well MEA plates with 48 wells per line. Mean values were recorded for each line for each experiment. For some independent experiments, two identical plates were used. In this case the mean of the two plates was used for the individual data-point. (D and H) The measurement of the mean firing frequency normalized to the number of active electrodes in a well. (E and I) The coefficient of variation for all interspike intervals (ISI). (F and J) Burst duration as defined by Axion software. (G and K) The per cent of total spikes in network bursts. Network bursts identification is defined in the ‘Materials and methods’ section. (D–G) include both controls and P1: n = 8 controls (C2 = 6, C3 = 2), n = 8 P1. (H–K) include both controls and P3: n = 8 controls (C2 = 2, C3 = 6), n = 8 P3. Data are depicted as box and whisker plots of mean, maximum, minimum, and upper and lower quartiles. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 by two-way ANOVA with two-stage linear step-up procedure post-test.
Riluzole and phenytoin attenuate bursting phenotypes in EIEE13 patient iNeurons
We next tested the effects of the ASM phenytoin, which has been used with some success in EIEE13 patients to reduce seizures, as well as riluzole, a drug that has been shown to inhibit persistent and resurgent INa (Urbani and Belluzzi, 2000; Theile and Cummins, 2011). Riluzole also blocks EAD-like action potential wave forms in the Scn8aN1768D/+ EIEE13 mouse model without significant reduction of the rising phase of the action potential (Lopez-Santiago et al., 2017). Drug concentrations were chosen based on patient CSF data from amyotrophic lateral sclerosis (ALS, riluzole) or epilepsy (phenytoin) patients (Groeneveld et al., 2001; Rambeck et al., 2006). Experiments included drug concentrations at a half-log above and a half-log below those values, respectively. In whole-cell patch clamp recordings, 3 µM riluzole completely and reversibly inhibited spontaneous action potential firing of Patient 3 neurons (Fig. 7A). In evoked firing experiments, riluzole did not inhibit the first action potential but blocked all subsequent repetitive firing at nearly all current injections (data not shown). Riluzole also increased the voltage threshold for initiating a single action potential (Fig. 7B) of Patient 3 and control neurons.
Figure 7.

Phenytoin and riluzole suppress activity of EIEE13 patient iNeurons. (A) Patch-clamp recording of a spontaneously active Patient 3 (P3) neuron before, during, and after washout of 3 µM riluzole. Riluzole inhibited all spontaneous activity but was completely reversible with washout. (B) The threshold voltage for single evoked action potentials was measured before and after riluzole exposure. For both controls and Patient 3, riluzole raised the action potential threshold to more depolarized potentials. (C and D) Changes in per cent spikes in network bursts in patient and control iNeurons in response to increasing concentrations of riluzole (C; 0, 0.3, 1, and 3 µM) and phenytoin (D; 0, 8, 24, and 80 µM). n = 8 independent experiments for C2 and P1, respectively; n = 5 independent experiments for C3 and P3, respectively. Within group comparisons for drug dosage effect compared to vehicle (DMSO) were performed using a one-way ANOVA. (E and F) Grouped control and patient data from C and D, respectively. n = 13 for each group. Data are depicted as box and whisker plots of mean, maximum, minimum, and upper and lower quartiles. Two-way unpaired ANOVA (due to missing values) was performed. All post hoc analyses used the two-stage linear step-up procedure post-test. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Acute effects of the drugs were determined by MEA recordings following a 5-min equilibration time of the drugs in the culture wells. Since the percentage of spikes in network bursts was the most reliable excitability difference for the two patient lines compared to controls (Fig. 6G and K), we compared the effects of riluzole and phenytoin on this parameter as well as on the mean firing rate. We found that 1 µM riluzole significantly decreased the percentage of spikes in network bursts in patient, but not control, iNeuron cultures (Fig. 7C and E). A similar, patient-specific effect was seen with 24 µM phenytoin (Fig. 7D and F). Two-way ANOVA analysis from vehicle to therapeutic concentration (1 µM for riluzole and 24 µM for phenytoin) had a significant genotype by treatment interaction (riluzole: P = 0.036, phenytoin: P = 0.035) meaning that the cells responded to both drugs in a genotype-dependent manner. When we plotted the mean firing rate normalized to the pretreatment mean firing rate, we observed a steady decrease in activity with increasing dosage but no differences in the effect of either drug between patient and control (Supplementary Fig. 5). These results indicate an increased sensitivity of bursting activity in patient iNeurons for both drugs compared to controls, with no genotype-related differential drug effects on overall activity. Notably, over the concentration range tested, riluzole had a greater inhibitory effect on both percentage of spikes in network bursts (Fig. 7) and overall mean firing rate (Supplementary Fig. 5) than phenytoin.
Riluzole reduced seizures in three patients with medically refractory EIEE13
During this study, Patients 1 and 3 (from which Patient 1 and Patient 3 lines were derived, respectively) deteriorated clinically despite treatment with VGSC and non-VGSC ASMs (see Table 1 for previous medications). Given the possibility of riluzole as a potential therapy based on our data, its FDA-approval for ALS, its prior use in children with spinal muscular atrophy and obsessive-compulsive disorder (Russman et al., 2003; Grant et al., 2014) and the severity of the patients’ epilepsy, the families, in consultation with their clinicians, elected to begin off-label riluzole treatment. Patient 1 was administered 25 mg/day of riluzole as an add-on therapy to phenytoin, clobazam, and topiramate. This dose was increased over 4 weeks to 50 mg twice daily. During the 20 weeks prior to the riluzole trial, the patient had 164 recorded seizures (8.2 seizures/week), with one seizure-free week, and a maximum of 24 seizures in 1 week. During the first 20 weeks of riluzole treatment, the patient had 83 recorded seizures (4.2 seizures/week), with two separate seizure free weeks, and a maximum of 11 seizures in 1 week. Thus, this patient had a ∼50% decrease in seizure frequency during riluzole treatment. Despite this apparent benefit, riluzole treatment was discontinued during recurrent hospitalizations with urinary tract infections associated with seizure clusters.
Patient 3, who previously failed trials of lacosamide, phenytoin, oxcarbazepine, and clobazam (Table 1), was taking high dose levetiracetam and having daily seizures with altered mental status and ataxia. The patient was administered 50 mg/day of riluzole as add-on therapy. A dramatic reduction in seizures was reported at 1-month follow-up, with no episodes of altered mental status, no myoclonic jerks, and improved EEG background. Riluzole was then increased to 75 mg/day; however, the patient became excessively sleepy at this dosage. Returning the patient to 50 mg/day coincided with an increase in seizures. After 1 year of treatment and up to 3 months seizure-free on riluzole, the patient’s family elected to stop riluzole treatment. Since discontinuation, the patient has had 11 months of seizure freedom on levetiracetam monotherapy.
We generated iPSCs from an additional patient (Patient 4) with a missense variant (p.V1757>I) altering an amino acid located two residues upstream from Patient 3 (p.N1759>S). We have yet to begin characterizing neurons from this patient; however, given the results of riluzole treatment in Patient 3, the subject’s paediatric epileptologist and the family elected to pursue treatment with riluzole. At the start of treatment at 11 months old, the patient was taking phenytoin, oxcarbazepine, gabapentin, and clonazepam. After initiation of riluzole treatment, he experienced a significant reduction in seizures: initially going from multiple per day to just a few per day with some of the first seizure free days in his lifetime (longest run was ∼2 weeks). Within 4 months of initiation, seizure frequency increased again to pretreatment baseline. The riluzole dose was increased without any improvement. Rufinamide was subsequently added while titrating his other ASMs, ultimately weaning him off riluzole and rufinamide at age 20 months in favour of lacosamide. A comorbid movement disorder was not changed by riluzole.
Discussion
Our results demonstrate the utility of iPSC-derived neurons to identify variant-specific INa abnormalities in patients with EIEE13. Of three patients with distinct SCN8A variants studied, cortical-like neurons from two showed increased proportions of persistent INa and one showed increased resurgent INa. All EIEE13 patient neurons had prolonged action potential repolarization and decreased AIS lengths. The iNeurons generated using the doxycycline-inducible Ngn1/2 system recapitulated the prolonged action potential repolarization observed in patient neurons and demonstrated increased network bursting measured by MEA recordings. These recordings provided a platform for testing the VGSC-selective drugs, phenytoin and riluzole. Riluzole administration produced similar effects as phenytoin, one of the most widely used therapies for EIEE13 (Boerma et al., 2016). Both drugs reduced network bursting activity at pharmacologically relevant concentrations with some specificity for the disease variant neuronal networks. Positive responses to off-label riluzole were seen in all three patients that received treatment, suggesting that further study of riluzole or riluzole derivatives as an EIEE13 therapy is warranted.
The increase in persistent INa that we identified in human iPSC-derived neurons is consistent with previous studies in other model systems. Elevated persistent INa was reported in the first identified EIEE13 disease variant (p.N1768D) modelled in transduced hippocampal neurons (Veeramah et al., 2012). A mouse model of the p.N1768D variant displayed increased persistent INa, slowed repolarization, and EAD-like action potential waveforms in CA1 hippocampal neurons (Lopez-Santiago et al., 2017). Immortalized cells transiently expressing SCN8A cDNA with the p.N1768D variant exhibited both increased persistent and resurgent INa (Patel et al., 2016), suggesting that the effect of this mutation on INa may be cell-type-dependent. Here, we found increased persistent INa in patient-derived neuronal cultures expressing two different SCN8A variants. However, unlike Patient 1 (p.R1872L) and Patient 2 (p.V1592L) neurons, Patient 3 neurons (p.N1759S) showed unaltered levels of persistent INa but, instead, showed elevated resurgent INa density. Neurons from Patient 3, but not from Patients 1 or 2, also exhibited EAD-like waveforms during spontaneous firing. A recent study of variants in SCN9A linked to small fibre neuropathy described increased resurgent INa, broader action potentials, and EADs (Xiao et al., 2019), similar changes as described here for Patient 3 neurons. These data suggest that elevated resurgent INa may be an underlying mechanism for the development of EAD-like waveforms in some types of mutant neurons.
In addition to the clear differences in INa recorded between patient and control neurons, our hemizygous P2r rescue experiment provides an isogenic control in the absence of the disease variant to demonstrate that the observed increase in persistent INa in Patient 2 neurons is due to the SCN8A de novo variant, rather than to other genetic differences. While haploinsufficiency of some VGSC genes can lead to epilepsy (e.g. SCN1A in Dravet syndrome), heterozygous Scn8a loss-of-function in mice results in no overt phenotype, except subtle absence seizures (Papale et al., 2009). In fact, the Scn8amed-jo/+ mouse crossed with a Dravet model (Scn1a+/−) resulted in rescue of induced seizure threshold to wild-type levels (Martin et al., 2007). Taken together, this information and the observation that transient INa was not reduced in P2r neurons suggest that this line is an appropriate rescue for Patient 2 neurons.
Both dual-SMAD neurons and iNeurons were used for measuring action potential dynamics. We found similar prolonged repolarization for both Patient 1 and Patient 3 neurons compared with controls. For dual-SMAD neurons, this was apparent by the failure to return to the resting membrane potential at various time points after the action potential peak while iNeurons showed a large difference in the maximum repolarization rate and half width. It is important to note that early dual-SMAD differentiated neurons are most similar to deep cortical layer neurons by gene expression (Shi et al., 2012) while iNeurons express markers of upper cortical layers (Busskamp et al., 2014). In previous mouse studies, electrophysiological disease phenotypes from SCN8A disease variants have been shown to be region specific (Lopez-Santiago et al., 2017). Therefore, it is not surprising that these two neuron types have slight differences in phenotypic manifestations.
The observed reduction in AIS length in EIEE13 patient neurons was unexpected. Others have proposed that AIS shortening is a compensatory mechanism to counter hyperexcitability, and animal models have shown AIS shortening in cortical neurons after TBI or stroke (Baalman et al., 2013; Hinman et al., 2013; Evans et al., 2015). AIS length is also known to vary with developmental timing and input in the visual cortex (Gutzmann et al., 2014). More work is necessary to determine whether AIS shortening in EIEE13 neurons reflects a compensatory attempt to decrease hyperexcitability or instead relates to altered development. Interestingly, in terms of the latter possibility, we did not observe AIS shortening in iNeurons (data not shown), which may lose developmental phenotypes observed with small molecule differentiation protocols that more faithfully recapitulate developmental events (Schafer et al., 2019).
MEA recordings demonstrated increased burstiness in Patient 1 and Patient 3 iNeuron cultures. We used iNeurons to decrease variability by forcing synchronized differentiation into pure excitatory cortical-like neurons. While this approach decreased the variability in our experiments considerably, purely excitatory cultures exhibit spontaneous network burst firing, potentially an epileptiform-like event, under basal conditions (Matsuda et al., 2018). While both control and patient iNeuron networks exhibited these events, the EIEE13 patient iNeuron networks had greater ‘burstiness’ than controls as measured by longer burst durations and greater percentage of spikes in network bursts. Patient 1 neurons also displayed a greater interspike interval coefficient of variation than controls (Fig. 6F). These measures suggest increased activity within network bursts, likely due to the cell-intrinsic SCN8A gain-of-function mechanisms we identified using single cell electrophysiological analyses. These differences are similar to changes in network bursts observed following the administration of chemoconvulsants such as bicuculline, pentylentetrazole, and 4-aminopyridine (Odawara et al., 2016). A recent publication investigating Kleefstra syndrome using iNeurons on MEAs identified disease specific alterations using similar outcomes measures, including burst duration, per cent of spikes out of bursts, and interburst interval coefficient of variation (Frega et al., 2019). Importantly, EIEE13 patient cultures demonstrated ‘burst of bursts’ events with high-frequency network bursting that did not occur in controls. These events were often followed by reduced activity, resulting in no significant differences in burst frequency calculated over the 5-min recording period. The reproducible differences in bursting parameters we observed between EIEE13 patient and control neuronal networks allowed us to begin assessing the effects of anti-epileptic drugs.
Currently, the VGSC blocking ASMs, including high-dose phenytoin, are the most effective and commonly used therapies for patients with SCN8A EIEE13 variants (Boerma et al., 2016; Gardella et al., 2018). However, these drugs frequently cause adverse effects and most EIEE13 patients continue to have frequent seizures despite the use of multiple ASMs (Braakman et al., 2017). We chose to examine a VGSC-selective inhibitor not currently used for epilepsy, riluzole, for its potential to more specifically attenuate persistent and resurgent INa than phenytoin. Riluzole is used for the treatment of ALS and is known to inhibit INa by binding to and stabilizing the inactive state of VGSC α subunits (Song et al., 1997). Riluzole has also been shown to preferentially inhibit both persistent and resurgent INa (Urbani and Belluzzi, 2000; Spadoni et al., 2002; Theile and Cummins, 2011; Xie et al., 2011). Even though riluzole has been used to effectively inhibit seizures in mice, rats, and baboons, it has not been used to treat epilepsy patients (Mizoule et al., 1985; Romettino et al., 1991; Doble, 1996; De Sarro et al., 2000; Yoshida et al., 2001). Although riluzole affects a wide array of channels and receptors, at the concentrations found in patients and tested in our MEA recordings (<10 µM), riluzole is thought to selectively inhibit persistent INa and potentiate calcium-dependent potassium currents (Bellingham, 2011). Application of either phenytoin or riluzole in concentrations reflecting patient CSF concentrations identified in the literature resulted in a variant-specific reduction in burstiness parameters to control levels (Groeneveld et al., 2001; Rambeck et al., 2006). Thus, our drug-testing platform is informative at treatment-relevant concentrations.
Patients 1, 3, and 4 experienced medically refractory seizures despite using standard VGSC blocking (including phenytoin, carbamazepine, oxcarbazepine, lamotrigine, etc) and other ASMs. The mutation position in the Nav1.6 channel may be, in part, why VGSC-blocking drugs were ineffective in Patients 3 and 4 (Table 1). Previous work showed that the SCN2A/Nav1.2 variants N1769>A and V1767>A (among others on segment IVS6) altered the effectiveness of etidocaine on INa (Ragsdale et al., 1994). The equivalent residues in Nav1.6 are N1759 (mutated in Patient 3) and V1757 (mutated in Patient 4). Neither patient had adequate seizure relief with typical ASMs—phenytoin, lamotrigine, and carbamazepine—that are thought to interact at this same site (Lipkind and Fozzard, 2010). Riluzole, on the other hand, has a theoretical binding pocket interacting with residues 1787, 1801, and 1845 of Nav1.6 (Sierra Bello et al., 2012). Part of the impetus for Patients 3 and 4 to begin riluzole treatment was the indication in the literature that riluzole binds to a different site than traditional VGSC blockers and that patients with variants located near the local anaesthetic binding site may not respond to typical drugs such as phenytoin. Both Patients 3 and 4 had remarkable responses to initial riluzole treatment; however, Patient 3 now remains seizure-free without riluzole while the apparent effect in Patient 4 was transient. These observations demonstrate that further studies are needed to assess the efficacy and safety of riluzole treatment in EIEE13 before considering randomized trials. Importantly, the findings by our group in patient-derived neurons and others in heterologous systems/cultured neurons (Barker et al., 2016; Wagnon et al., 2016) of mutation-specific gain-of-function effects in EIEE13 suggests that the ASM choice may be variant-specific. The patient-derived neuronal models that we describe offer a useful approach for this type of precision epilepsy therapy.
Supplementary Material
Acknowledgements
We thank the parents and patients who provided the skin biopsies and clinical information for this study. We also thank Dr Michael Ward and the Inherited Neurodegenerative Diseases Unit at NINDS for the gift of the pUCM-CLYBL-Ngn1&2 plasmid.
Funding
This work was supported by an American Epilepsy Society/Wishes for Elliott Fellowship (A.M.T) and grant funding from the National Institutes of Health/National Institute of Neurological Disorders and Stroke: NS090364 (J.M.P.) and NS088571 (L.L.I. and J.M.P.). This work was supported by the Michigan Institute for Clinical & Health Research Drug Repurposing Initiative.
Competing interests
The authors report no competing interests.
Glossary
- AIS =
axon initial segments
- ASM =
antiseisure medication
- EIEE =
early infantile epileptic encephalopathy
- iNeuron =
doxycycline-inducible neuron
- iPSC =
induced pluripotent stem cell
- MEA =
multielectrode array
- VGSC =
voltage-gated sodium channel
References
- Baalman KL, Cotton RJ, Rasband SN, Rasband MN.. Blast wave exposure impairs memory and decreases axon initial segment length. J Neurotrauma 2013; 30: 741–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker BS, Ottolini M, Wagnon JL, Hollander RM, Meisler MH, Patel MK.. The SCN8A encephalopathy mutation p.Ile1327Val displays elevated sensitivity to the anticonvulsant phenytoin. Epilepsia 2016; 57: 1458–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellingham MC. A review of the neural mechanisms of action and clinical efficiency of riluzole in treating amyotrophic lateral sclerosis: what have we learned in the last decade? CNS Neurosci Ther 2011; 17: 4–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Krieger AM, Yekutieli D.. Adaptive linear step-up procedures that control the false discovery rate. Biometrika 2006; 93: 491–507. [Google Scholar]
- Boerma RS, Braun KP, van de Broek MP, van Berkestijn FM, Swinkels ME, Hagebeuk EO, et al. Remarkable phenytoin sensitivity in 4 children with SCN8A-related epilepsy: a molecular neuropharmacological approach. Neurotherapeutics 2016; 13: 192–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braakman HM, Verhoeven JS, Erasmus CE, Haaxma CA, Willemsen MH, Schelhaas HJ.. Phenytoin as a last‐resort treatment in SCN 8A encephalopathy. Epilepsia Open 2017; 2: 343–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunton-Stasyshyn RKA, Wagnon JL, Wengert ER, Barker BS, Faulkner A, Wagley PK, et al. Prominent role of forebrain excitatory neurons in SCN8A encephalopathy. Brain 2019; 142: 362–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busskamp V, Lewis NE, Guye P, Ng AH, Shipman SL, Byrne SM, et al. Rapid neurogenesis through transcriptional activation in human stem cells. Mol Syst Biol 2014; 10: 760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerbini T, Funahashi R, Luo Y, Liu C, Park K, Rao M, et al. Transcription activator-like effector nuclease (TALEN)-mediated CLYBL targeting enables enhanced transgene expression and one-step generation of dual reporter human induced pluripotent stem cell (iPSC) and neural stem cell (NSC) lines. PLoS One 2015; 10: e0116032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sarro G, Siniscalchi A, Ferreri G, Gallelli L, De Sarro A.. Nmda and AMPA/kainate receptors are involved in the anticonvulsant activity of riluzole in DBA/2 mice. Eur J Pharmacol 2000; 408: 25–34. [DOI] [PubMed] [Google Scholar]
- Doble A. The pharmacology and mechanism of action of riluzole. Neurology 1996; 47(6 Suppl 4): 233S–41S. [DOI] [PubMed] [Google Scholar]
- Epi4K Investigators. De novo mutations in the classic epileptic encephalopathies. Nature 2013; 501: 217–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans MD, Dumitrescu AS, Kruijssen DL, Taylor SE, Grubb MS.. Rapid modulation of axon initial segment length influences repetitive spike firing. Cell Rep 2015; 13: 1233–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frega M, Linda K, Keller JM, Gümüş-Akay G, Mossink B, van Rhijn J-R, et al. Neuronal network dysfunction in a model for Kleefstra syndrome mediated by enhanced NMDAR signaling. Nat Commun 2019; 10: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardella E, Marini C, Trivisano M, Fitzgerald MP, Alber M, Howell KB, et al. The phenotype of SCN8A developmental and epileptic encephalopathy. Neurology 2018; 91: e1112–24. [DOI] [PubMed] [Google Scholar]
- Gasser A, Ho T-Y, Cheng X, Chang K-J, Waxman SG, Rasband MN, et al. An ankyrinG-binding motif is necessary and sufficient for targeting Nav1.6 sodium channels to axon initial segments and nodes of Ranvier. J Neurosci 2012; 32: 7232–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant PJ, Joseph LA, Farmer CA, Luckenbaugh DA, Lougee LC, Zarate CA Jr, et al. 12-week, placebo-controlled trial of add-on riluzole in the treatment of childhood-onset obsessive–compulsive disorder. Neuropsychopharmacol 2014; 39: 1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groeneveld G, Van Kan H, Toraño JS, Veldink J, Guchelaar H-J, Wokke J, et al. Inter-and intraindividual variability of riluzole serum concentrations in patients with ALS. J Neurol Sci 2001; 191: 121–5. [DOI] [PubMed] [Google Scholar]
- Gutzmann A, Ergül N, Grossmann R, Schultz C, Wahle P, Engelhardt M.. A period of structural plasticity at the axon initial segment in developing visual cortex. Front Neuroanat 2014; 8: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinman JD, Rasband MN, Carmichael ST.. Remodeling of the axon initial segment after focal cortical and white matter stroke. Stroke 2013; 44: 182–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong W, Zhang Y, Gao Y, Liu X, Gao K, Xie H, et al. SCN8A mutations in Chinese children with early onset epilepsy and intellectual disability. Epilepsia 2015; 56: 431–8. [DOI] [PubMed] [Google Scholar]
- Lam RS, Töpfer FM, Wood PG, Busskamp V, Bamberg E.. Functional maturation of human stem cell-derived neurons in long-term cultures. PLoS One 2017; 12: e0169506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen J, Carvill GL, Gardella E, Kluger G, Schmiedel G, Barisic N, et al. The phenotypic spectrum of SCN8A encephalopathy. Neurology 2015; 84: 480–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipkind GM, Fozzard HA.. Molecular model of anticonvulsant drug binding to the voltage-gated sodium channel inner pore. Mol Pharmacol 2010; 78: 631–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Lopez‐Santiago LF, Yuan Y, Jones JM, Zhang H, O'malley HA, et al. Dravet syndrome patient‐derived neurons suggest a novel epilepsy mechanism. Ann Neurol 2013; 74: 128–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Santiago LF, Yuan Y, Wagnon JL, Hull JM, Frasier CR, O’Malley HA, et al. Neuronal hyperexcitability in a mouse model of SCN8A epileptic encephalopathy. Proc Natl Acad Sci USA 2017; 114: 2383–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroof AM, Keros S, Tyson JA, Ying S-W, Ganat YM, Merkle FT, et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell 2013; 12: 559–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MS, Tang B, Papale LA, Yu FH, Catterall WA, Escayg A.. The voltage-gated sodium channel Scn8a is a genetic modifier of severe myoclonic epilepsy of infancy. Hum Mol Genet 2007; 16: 2892–9. [DOI] [PubMed] [Google Scholar]
- Matsuda N, Odawara A, Katoh H, Okuyama N, Yokoi R, Suzuki I.. Detection of synchronized burst firing in cultured human induced pluripotent stem cell-derived neurons using a 4-step method. Biochem Biophys Res Commun 2018; 497: 612–8. [DOI] [PubMed] [Google Scholar]
- Mizoule J, Meldrum B, Mazadier M, Croucher M, Ollat C, Uzan A, et al. 2-Amino-6-trifluoromethoxy benzothiazole, a possible antagonist of excitatory amino acid neurotransmission—I: anticonvulsant properties. Neuropharmacology 1985; 24: 767–73. [DOI] [PubMed] [Google Scholar]
- Nawrot MP, Boucsein C, Molina VR, Riehle A, Aertsen A, Rotter S.. Measurement of variability dynamics in cortical spike trains. J Neurosci Methods 2008; 169: 374–90. [DOI] [PubMed] [Google Scholar]
- Neely MD, Litt MJ, Tidball AM, Li GG, Aboud AA, Hopkins CR, et al. DMH1, a highly selective small molecule BMP inhibitor promotes neurogenesis of hiPSCs: comparison of PAX6 and SOX1 expression during neural induction. ACS Chem Neurosci 2012; 3: 482–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehme R, Zuccaro E, Ghosh SD, Li C, Sherwood JL, Pietilainen O, et al. Combining NGN2 programming with developmental patterning generates human excitatory neurons with NMDAR-mediated synaptic transmission. Cell Rep 2018; 23: 2509–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odawara A, Katoh H, Matsuda N, Suzuki I.. Physiological maturation and drug responses of human induced pluripotent stem cell-derived cortical neuronal networks in long-term culture. Sci Rep 2016; 6: 26181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohba C, Kato M, Takahashi S, Lerman‐Sagie T, Lev D, Terashima H, et al. Early onset epileptic encephalopathy caused by de novo SCN8A mutations. Epilepsia 2014; 55: 994–1000. [DOI] [PubMed] [Google Scholar]
- Ottolini M, Barker BS, Gaykema RP, Meisler MH, Patel MK.. Aberrant sodium channel currents and hyperexcitability of medial entorhinal cortex neurons in a mouse model of SCN8A encephalopathy. J Neurosci 2017; 2709–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papale LA, Beyer B, Jones JM, Sharkey LM, Tufik S, Epstein M, et al. Heterozygous mutations of the voltage-gated sodium channel SCN8A are associated with spike-wave discharges and absence epilepsy in mice. Hum Mol Genet 2009; 18: 1633–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel RR, Barbosa C, Brustovetsky T, Brustovetsky N, Cummins TR.. Aberrant epilepsy-associated mutant Nav1. 6 sodium channel activity can be targeted with cannabidiol. Brain 2016; 139: 2164–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perreault ML, Fan T, Alijaniaram M, O'Dowd BF, George SR.. Dopamine D1–D2 receptor heteromer in dual phenotype GABA/glutamate-coexpressing striatal medium spiny neurons: regulation of BDNF, GAD67 and VGLUT1/2. PLoS One 2012; 7: e33348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragsdale DS, McPhee JC, Scheuer T, Catterall WA.. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science 1994; 265: 1724–8. [DOI] [PubMed] [Google Scholar]
- Rambeck B, Jürgens UH, May TW, Pannek HW, Behne F, Ebner A, et al. Comparison of brain extracellular fluid, brain tissue, cerebrospinal fluid, and serum concentrations of antiepileptic drugs measured intraoperatively in patients with intractable epilepsy. Epilepsia 2006; 47: 681–94. [DOI] [PubMed] [Google Scholar]
- Romettino S, Lazdunski M, Gottesmann C.. Anticonvulsant and sleep-waking influences of riluzole in a rat model of absence epilepsy. Eur J Pharmacol 1991; 199: 371–3. [DOI] [PubMed] [Google Scholar]
- Root DH, Zhang S, Barker DJ, Miranda-Barrientos J, Liu B, Wang H-L, et al. Selective brain distribution and distinctive synaptic architecture of dual glutamatergic-GABAergic neurons. Cell Rep 2018; 23: 3465–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russman BS, Iannaccone ST, Samaha FJ.. A phase 1 trial of riluzole in spinal muscular atrophy. Arch Neurol 2003; 60: 1601–3. [DOI] [PubMed] [Google Scholar]
- Schafer ST, Paquola AC, Stern S, Gosselin D, Ku M, Pena M, et al. Pathological priming causes developmental gene network heterochronicity in autistic subject-derived neurons. Nat Neurosci 2019; 22: 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcheglovitov A, Shcheglovitova O, Yazawa M, Portmann T, Shu R, Sebastiano V, et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature 2013; 503: 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Kirwan P, Smith J, Robinson HP, Livesey FJ.. Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat Neurosci 2012; 15: 477–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra Bello O, Gonzalez J, Capani F, Barreto GE. In silico docking reveals possible Riluzole binding sites on Nav1.6 sodium channel: Implications for amyotrophic lateral sclerosis therapy. J Theor Biol 2012; 315: 53–63. [DOI] [PubMed] [Google Scholar]
- Song J-H, Huang C-S, Nagata K, Yeh JZ, Narahashi T.. Differential action of riluzole on tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels. J Pharmacol Exp Ther 1997; 282: 707–14. [PubMed] [Google Scholar]
- Spadoni F, Hainsworth AH, Mercuri NB, Caputi L, Martella G, Lavaroni F, et al. Lamotrigine derivatives and riluzole inhibit INa, P in cortical neurons. Neuroreport 2002; 13: 1167–70. [DOI] [PubMed] [Google Scholar]
- Theile JW, Cummins TR.. Inhibition of Navβ4 peptide-mediated resurgent sodium currents in Nav1.7 channels by carbamazepine, riluzole, and anandamide. Mol Pharmacol 2011; 80: 724–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidball AM, Dang LT, Glenn TW, Kilbane EG, Klarr DJ, Margolis JL, et al. Rapid generation of human genetic loss-of-function iPSC lines by simultaneous reprogramming and gene editing. Stem Cell Rep 2017; 9: 725–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidball AM, Neely MD, Chamberlin R, Aboud AA, Kumar KK, Han B, et al. Genomic instability associated with p53 knockdown in the generation of Huntington’s disease human induced pluripotent stem cells. PLoS One 2016; 11: e0150372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidball AM, Swaminathan P, Dang LT, Parent J.. Generating loss-of-function iPSC lines with combined CRISPR indel formation and reprogramming from human fibroblasts. Bio Protoc 2018; 8: e2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbani A, Belluzzi O.. Riluzole inhibits the persistent sodium current in mammalian CNS neurons. Eur J Neurosci 2000; 12: 3567–74. [DOI] [PubMed] [Google Scholar]
- Veeramah KR, O'Brien JE, Meisler MH, Cheng X, Dib-Hajj SD, Waxman SG, et al. De novo pathogenic SCN8A mutation identified by whole-genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet 2012; 90: 502–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagnon JL, Barker BS, Hounshell JA, Haaxma CA, Shealy A, Moss T, et al. Pathogenic mechanism of recurrent mutations of SCN8A in epileptic encephalopathy. Ann Clin Transl Neurol 2016; 3: 114–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagnon JL, Korn MJ, Parent R, Tarpey TA, Jones JM, Hammer MF, et al. Convulsive seizures and SUDEP in a mouse model of SCN8A epileptic encephalopathy. Hum Mol Genet 2014; 24: 506–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagnon JL, Meisler MH.. Recurrent and non-recurrent mutations of SCN8A in epileptic encephalopathy. Front Neurol 2015; 6: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Barbosa C, Pei Z, Xie W, Strong JA, Zhang J-M, et al. Increased resurgent sodium currents in Nav1. 8 contribute to nociceptive sensory neuron hyperexcitability associated with peripheral neuropathies. J Neurosci 2019; 39: 1539–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie R-G, Zheng D-W, Xing J-L, Zhang X-J, Song Y, Xie Y-B, et al. Blockade of persistent sodium currents contributes to the riluzole-induced inhibition of spontaneous activity and oscillations in injured DRG neurons. PLoS One 2011; 6: e18681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau KW, van Beuningen SF, Cunha-Ferreira I, Cloin BM, van Battum EY, Will L, et al. Microtubule minus-end binding protein CAMSAP2 controls axon specification and dendrite development. Neuron 2014; 82: 1058–73. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Noguchi E, Tsuru N, Ohkoshi N.. Effect of riluzole on the acquisition and expression of amygdala kindling. Epilepsy Res 2001; 46: 101–9. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 2013; 78: 785–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, upon reasonable request.