

Scriba et al. report a novel RFC1 pentanucleotide motif (ACAGG)exp associated with CANVAS in two families. The three patients all display fasciculations and elevated serum creatine kinase levels, suggesting that (ACAGG)exp causes a slight variation on the characteristic CANVAS phenotype.
Keywords: sensory neuropathy; cerebellar ataxia, neuropathy, vestibular areflexia syndrome; CANVAS; RFC1; repeat expansion
Abstract
Cerebellar ataxia, neuropathy and vestibular areflexia syndrome (CANVAS) is a progressive late-onset, neurological disease. Recently, a pentanucleotide expansion in intron 2 of RFC1 was identified as the genetic cause of CANVAS. We screened an Asian-Pacific cohort for CANVAS and identified a novel RFC1 repeat expansion motif, (ACAGG)exp, in three affected individuals. This motif was associated with additional clinical features including fasciculations and elevated serum creatine kinase. These features have not previously been described in individuals with genetically-confirmed CANVAS. Haplotype analysis showed our patients shared the same core haplotype as previously published, supporting the possibility of a single origin of the RFC1 disease allele. We analysed data from >26 000 genetically diverse individuals in gnomAD to show enrichment of (ACAGG) in non-European populations.
Introduction
Cerebellar ataxia, neuropathy and vestibular areflexia syndrome (CANVAS) (OMIM: 614575) is a recessive late-onset neurological disease with slow progression (Migliaccio et al., 2004; Szmulewicz et al., 2011a, b). CANVAS was first characterized as a distinct syndrome by Szmulewicz et al. in 2011, who determined that non-length dependent sensory deficit is secondary to neuronopathy and is an integral part of the syndrome (Szmulewicz et al., 2011a). Prominent clinical features associated with CANVAS include imbalance, saccadic smooth pursuit, downbeat nystagmus and autonomic dysfunction (Wu et al., 2014), gait ataxia, dysarthria, sensory symptoms (Cortese et al., 2020) and an impaired visually enhanced vestibulo-ocular reflex (Migliaccio et al., 2004; Szmulewicz et al., 2011a). Chronic cough has been reported to arise years before neurological symptoms (Wu et al., 2014). Pathologically, patients typically display cerebellar atrophy particularly affecting the vermis and hemisphere crus I, and marked atrophy of dorsal root ganglia. Nerve conduction studies show absent sensory nerve action potentials, often in conjunction with normal motor conduction (Szmulewicz et al., 2011a, b; Umeh et al., 2016). Nerve ultrasound shows characteristically small peripheral nerves (Pelosi et al., 2018).
A biallelic pentanucleotide expansion in the second intron of the replication factor C subunit 1 (RFC1) gene was recently identified as a genetic basis for CANVAS (Cortese et al., 2019). This locus displays considerable heterogeneity. The pathogenic RFC1 expansion (AAGGG)exp differs from the reference allele (AAAAG)11 in both motif sequence and the number of repeats (Cortese et al., 2019; Rafehi et al., 2019). The initial report also described two other non-pathogenic expansions observed within healthy individuals: (AAAAG)exp and (AAAGG)exp (Cortese et al., 2019). Two additional motifs were subsequently described: (AAGAG)exp and (AGAGG)exp (Akçimen et al., 2019). However, no homozygous individuals were observed for either configuration, so pathogenicity of these motifs remains uncertain. Further adding to the complexity of the locus is an alternate pathogenic allele configuration (AAAGG)10-25(AAGGG)exp(AAAGG)exp, which appears to be specific to the Māori population (Beecroft et al., 2020).
By screening an Asian Pacific cohort for CANVAS, we discovered three patients with a novel, likely pathogenic RFC1 repeat motif (ACAGG)exp. These patients show additional clinical features that have not been previously described in genetically defined CANVAS, including fasciculations and elevated creatine kinase levels. We also show these patients share the same core haplotype as previously described, which further supports a single origin of the CANVAS disease allele.
Materials and methods
Cohort
This project was approved by the Human Research Ethics Committee of the University of Western Australia (RA/4/20/1008) with reciprocal ethics approval from Curtin University (HRE2019-0566). DNA samples from whole blood were obtained from the Diagnostic Genomics Department (PathWest) for each of the probands and additional family members, where available. Clinical data were collected by the corresponding clinicians.
The patients described here represent a subset of a larger CANVAS cohort that was genetically screened for the RFC1 pathogenic repeat expansion. Twenty-nine of these patients have been described elsewhere (Beecroft et al., 2020; Cortese et al., 2020).
Family Indo1
This family consisted of two affected brothers and five unaffected siblings (Fig. 1A). The parents were second cousins. The family resides in Indonesia, but are of Chinese descent. Further information about their ancestry was not available.
Figure 1.
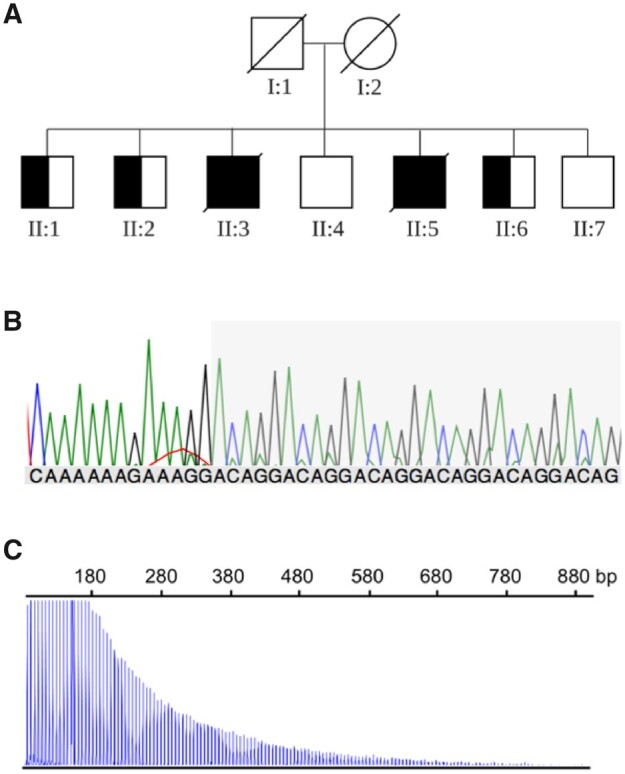
Novel configuration of the RFC1 expansion in an Asian Pacific family. (A) Pedigree of Family Indo1. Half-shaded symbols represent individuals identified as (ACAGG) repeat expansion carriers by flanking PCR and RP-PCR. (B) Representative Sanger chromatogram of long-range, nested PCR products containing the novel (ACAGG) expansion at the RFC1 locus. The grey shaded region indicates the (ACAGG) repeat sequence. (C) Representative positive RP-PCR fragment analysis result for the (ACAGG) repeat expansion.
Patient N1
This female patient is an isolated proband from Niue for whom no familial DNA was available. Although there was no recorded consanguinity, the population of Niue is ∼1500 individuals (Australian Department of Foreign Affairs and Trade, 2019). Her mother was of Niuean descent, and her father had one Niuean and one English parent.
Preliminary genetic screening
A combination of flanking PCR and repeat primed-PCR (RP-PCR) was used to genotype each individual, described by Cortese et al. (2019). In brief, flanking PCR is performed with primers that bind just outside the RFC1 repeat region. Absence of a PCR product suggests the presence of a biallelic expansion that is too large to be amplified with the standard PCR extension time. Individuals that did not show a product by flanking PCR were subsequently tested by RP-PCR for the three previously known RFC1 repeat expansion motifs: (AAAAG), (AAAGG), and (AAGGG). Reaction composition, thermocycling conditions and primers are provided in Supplementary Table 1.
PCR-based sequencing of the RFC1 expansion sequence
A multi-step PCR protocol was developed for amplification and subsequent Sanger sequencing of the novel RFC1 repeat motif. Long-range PCR was used to amplify the repeat locus, followed by nested PCR to provide clearer sequencing downstream. PCR products of the expected size (∼355 bp) were purified by gel extraction or band stab PCR (Wilton et al., 1997) to enrich for desired target amplicons. Reaction composition, thermocycling conditions and primers are provided in Supplementary Table 1. Purified products were Sanger sequenced at the Australian Genome Research Facility (AGRF).
ACAGG repeat primed PCR
Following the discovery of the novel (ACAGG) motif by Sanger sequencing, we designed an ACAGG-specific RP-PCR. This allowed confirmation of the motif sequence and visualization of any large interruptions or alternate motifs present in the expansion. Primer sequences, and reaction and thermocycling conditions are provided in Supplementary Table 1. Fragment length separation was performed by AGRF.
Southern blot
Southern blot was performed as described in Cortese et al. (2019).
Next-generation sequencing
Illumina whole exome sequencing (WES) was performed on both affected brothers from Family Indo1 (Patients II:3 and II:5). Whole genome sequencing (WGS) was performed on Patient N1. Paired-end sequencing reads (150 bp) were generated using Novaseq 6000 sequencing (Illumina) with a 30-fold average read depth. The BAM file was viewed with Integrative Genomics Viewer software (IGV) (version 2.7.0, Broad Institute) with soft-clipping to facilitate visualization of reads that contained part of the repeat configuration. All sequencing was performed by AGRF following GATK4 best-practices (Poplin et al., 2017).
Haplotype analysis
For Patient N1, we compared the WGS against the selected markers used for the haplotype analysis by Cortese et al. (2019) (spanning chr4: 38157510-40712481, hg19). For Individuals Indo1 II:3 and II:5, informative HapMap2 markers were extracted from exome sequencing data using Linkdatagen (Bahlo and Bromhead, 2009), and prepared for analysis with Merlin (Abecasis et al., 2002). Markers were excluded if they were covered to a read depth of <20-fold. Merlin was used to generate the most likely haplotypes for these individuals.
Frequency of the (ACAGG) motif in the gnomAD dataset
Analysis of 26 745 ethnically diverse samples from gnomAD v3 (Karczewski et al., 2020) was performed with ExpansionHunter Denovo software (Dolzhenko et al., 2020). Carrier frequency estimates were performed for populations with >1000 individuals (Richards et al., 2015) that had not been through a significant genetic bottleneck.
Data availability
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
Results
Cohort and clinical features
Family Indo1
Individuals II:3 and II:5 presented with weakness about the ankles, poor balance and chronic cough at 55 and 54 years of age, respectively. Muscle wasting was initially restricted to the calf muscles but progressed to the anterior muscles below the knee and the intrinsic muscles of the hand and forearm. Fasciculations also became visible. Creatine kinase levels were elevated, ranging between 580 and 1020 U/l (normal < 195). Deep tendon reflexes were initially globally reduced but deteriorated over time until they were absent. Cerebellar signs included saccadic interruption of eye movements and a wide-based gait, and later in life, dysarthria. Both individuals had an abnormal vestibulo-ocular reflex. Nerve conduction studies showed severe sensorimotor peripheral neuropathy. Muscle biopsy showed atrophic myofibres and group atrophy, consistent with a neurogenic cause. Electron microscopy of the muscle showed subsarcolemmal accumulation of pleomorphic mitochondria with large electron-dense inclusions. MRI of the head showed progressive atrophy of the cerebellum, principally of the vermis, and thinning of the brainstem. The family returned to Indonesia and were lost to follow-up; the affected individuals are now deceased.
Patient N1
The patient first presented aged 57 when a CT scan performed for syncope found evidence of cerebellar atrophy. Prior to this, the patient had experienced severe leg cramps and poor balance for ∼10 years. She had difficulties with speaking quickly and experienced frequent choking. Examination showed gaze-evoked nystagmus with some downbeat component, broken smooth pursuit, and positive head impulse test. Limb examination showed widespread fasciculations, absent reflexes, loss of vibration sense to the costal margin, loss of position sense distally with pseudoathetosis, and mild intention tremor. The patient could not stand with feet together or perform a tandem gait. Creatine kinase level was elevated at 502 U/l (normal <195). MRI scan showed vermal, crus1, and crus2 atrophy. Nerve conduction studies showed absent sensory potentials but normal motor studies. Electromyography showed widespread, mild, chronic motor unit loss consistent with the patient's fasciculations and elevated creatine kinase. Autonomic testing confirmed impairment of parasympathetic function. The patient experienced a slow inexorable progression of neurological symptoms until death at 71 years of age. Further information is available in Supplementary Table 2.
Genetic investigations identified a novel RFC1 repeat configuration
The three affected individuals showed no product by flanking PCR, suggesting a biallelic expansion at the RFC1 locus. All unaffected siblings in Family Indo1 generated a robust PCR product, suggesting at least one allele was small enough to be amplified by PCR (data not shown). These individuals were negative for the (AAAAG), (AAAGG), and (AAGGG) motifs by Sanger sequencing and RP-PCR. Our multistep amplification and sequencing protocol allowed clean Sanger sequencing of the RFC1 repeat locus, which showed an apparently homozygous (ACAGG)exp repeat motif in all three affected individuals (Fig. 1B and C). Three unaffected siblings (Individuals II:1, II:2 and II:6) in Family Indo1 were heterozygous for the (ACAGG) motif, while Individuals II:4 and II:7 had two normal alleles. Southern blot was performed for Patient Indo1 II:5, showing a band of ∼10 000 kb (homozygous). Given that control DNA gives a band of ∼5000 kb, the expansion in our patient would be ∼5000 kb. This corresponds to ∼1015 repeated units (Supplementary Fig. 1).
For Patient N1, the RFC1 locus configuration was also visible with WGS data. A number of soft-clipped reads spanned the repeat expansion and the region flanking the RFC1 repeat locus (Fig 2). No reads contained alternative repeat configurations, indicating that Patient N1 is homozygous for the ACAGG motif.
Figure 2.

Visualization of the (ACAGG) repeat expansion from whole genome sequencing of Patient N1. Reads containing the RFC1 intron 2 (ACAGG) motif are visible with soft-clipping enabled on IGV. This shows the patient is homozygous for the (ACAGG) motif.
Haplotype analysis
Comparison of the Patient N1 haplotype with that of Cortese et al. (2019) indicated that these individuals share the same core haplotype, as well as a subset of the ‘Māori’ haplotype we recently described (Beecroft et al., 2020) (Supplementary Table 3). Similarly, the affected individuals in Family Indo1 shared the same ‘Māori’ haplotype, as extracted from WES markers (Supplementary Table 4) (Beecroft et al., 2020). The reference and minor allele frequencies were extracted from the whole 1000 Genomes dataset (1000 Genomes Project Consortium et al., 2015) using LDlink (Machiela and Chanock, 2015).
gnomAD analysis
Analysis of 26 745 samples from gnomAD v3 (Karczewski et al., 2020) identified the RFC1 (ACAGG) motif in seven individuals. Of these, two were African, four were South Asian, and one was East Asian. The (ACAGG) motif appears to be enriched in non-European populations, since of the 26 745 individuals analysed, 9954 were non-Finnish Europeans, 7281 were African, 4919 were admixed American/Latino, 1841 were Finnish, 1510 were South Asian, 894 were East Asian, and 346 were Ashkenazi Jewish. Haplotyping was not possible. Carrier frequency ranged from 0% in Europeans to 0.03% in Africans and 0.26% in the South Asians (Supplementary Table 5).
Discussion
Extending the heterogeneity of the RFC1 repeat locus
Previous studies identified five different configurations of the RFC1 intron 2 repeat expansion. Here, we identified three CANVAS patients from two families that tested negative for motifs (AAAAG), (AAAGG) and (AAGGG) by flanking PCR and RP-PCR. We therefore suspected these individuals instead harboured large biallelic expansions of an unknown motif. All three individuals were shown to carry biallelic expansions of a novel repeat unit, (ACAGG), by three independent methods. The identification of expanded repeats of the same configuration in CANVAS patients from two unrelated families suggests that this configuration is likely to be pathogenic. The ACAGG allele segregates with disease in Family Indo1.
Genotype-phenotype correlation: an extended CANVAS phenotype
The affected individuals displayed the classical CANVAS triad of ataxia, neuronopathy and vestibular areflexia (Migliaccio et al., 2004; Szmulewicz et al., 2011a, b, 2014; Wu et al., 2014). However, they also shared phenotypic features that may be specific to this genotype, notably, fasciculations, elevated serum creatine kinase levels, and denervation on EMG/muscle biopsy. In contrast, subtle denervation changes were found on EMG without fasciculation or raised creatine kinase in other CANVAS patients. This suggests CANVAS may involve anterior horn cells in addition to the previously described dorsal root ganglia and cranial nerve ganglia involvement (Szmulewicz et al., 2011a, 2014) and that this aspect of the disease may be prominent amongst patients with this genetic variant.
A single origin of a permissive haplotype?
The (ACAGG) motif shares the same core haplotype as the (AAGGG) configuration described in the Caucasian cohort from Cortese et al. (2019). This core haplotype is also shared by other non-Caucasian CANVAS cohorts (Beecroft et al., 2020; Nakamura et al., 2020). The haplotype for Patient N1 not only shared the core haplotype associated with the pathogenic (AAGGG)exp previously described by Cortese et al. (2019), but also matches a small section of the recent extended Māori haplotype (Beecroft et al., 2020) (Supplementary Table 3). This suggests the ‘Niue haplotype’ may predate the ‘Māori haplotype’. This would align with the ‘out of Taiwan’ theory of Austronesian migration, which postulates migration from mainland Asia through Niue and then to New Zealand (Chambers and Edinur, 2016). Together, these findings support the theory of a single origin of the disease (Rafehi et al., 2019). This raises the question as to whether this common CANVAS haplotype is a permissive haplotype. It is also unclear if there is some intrinsic aspect of this haplotype that allows motif change to (AAGGG) or (ACAGG), or promotes repeat expansion into the pathogenic range. The gnomAD analysis shows that the (ACAGG) is not unique to our patients, and is enriched in non-European cohorts. CANVAS may underlie a significant proportion of undiagnosed ataxia in non-European populations.
GC content and pathogenicity
Cortese et al. (2019) showed that the wild-type (AAAAG)exp expanded to 15–200 repeats, the benign variant (AAAGG)exp to 40–1000 repeats, and the pathogenic motif (AAGGG)exp to 400–2000 repeats. These motifs contain 20%, 40% and 60% GC content, respectively. This implies a positive correlation between GC content and RFC1 repeat expansion size. This is supported by findings from Kiktev et al. (2018), who showed that alteration of a yeast gene to 63% GC content raised mutation rates >6-fold compared to 43% or 31% GC content (Kiktev et al., 2018). The discovery of a third, expanded RFC1 allele with 60% GC content supports a link between GC content of the repeat, its size, and its pathogenicity.
Conclusions
We have identified a novel pathogenic RFC1 repeat expansion motif (ACAGG)exp for CANVAS that appears to be associated with the additional phenotypic features of fasciculations and elevated serum creatine kinase. Our analysis of gnomAD data shows this motif is enriched in non-Europeans, and may be a significant cause of ataxia, particularly in South and East Asian populations. All three affected individuals included in this study showed the same core haplotype that had previously been noted by Cortese et al. (2019). This supports the possibility of a single, ancient origin of the CANVAS allele (Rafehi et al., 2019). Further work is required to determine if this is a permissive haplotype.
[Note added at proof stage: Recently, Tsuchiya et al. (2020) identified a single Japanese CANVAS patient with bi-allelic expansions at the RFC1 locus of the (ACAGG) motif, within a cohort of 37 late onset ataxia cases. This study provides further evidence of the pathogenicity of the (ACAGG) repeat expansion and supports that it is present in East Asian populations.]
Supplementary Material
Acknowledgements
We sincerely thank the patients and their families for their participation in this study.
Funding
We extend our sincere thanks to the families involved in this study. S.J.B. is funded by The Fred Liuzzi Foundation (TFLF) (Melbourne, Australia). N.G.L. (APP1117510) and G.R. (APP1122952) are supported by the Australian National Health and Medical Research Council (NHMRC). G.R. is also supported by a Western Australian Department of Health Future Health’s WA Merit Award. This work is funded by an NHMRC Project Grant (APP1080587) and the Margaret and Terry Orr Memorial Fund. A.C. thanks Medical Research Council (MR/179744) and Fondazione CARIPLO (2019-1836) for grant support. H.H. and M.M.R. are grateful to the Medical Research Council (MRC), MRC Centre grant (G0601943), and M.M.R. is also grateful to the National Institutes of Neurological Diseases and Stroke and office of Rare Diseases (U54NS065712) for their support. H.H. is also supported by Ataxia UK, The MSA Trust, MDUK and The Muscular Dystrophy Association (MDA). M.M.R. is supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. B.W. is funded by the National Human Genome Research Institute, the National Eye Institute, and the National Heart, Lung and Blood Institute grant UM1 HG008900 and in part by National Human Genome Research Institute grant R01 HG009141. The funding agencies were not involved in the design, completion, or writing of this study.
Competing interests
The authors report no competing interests.
Supplementary material
Supplementary material is available at Brain online.
Glossary
- CANVAS =
cerebellar ataxia with neuropathy and bilateral vestibular areflexia syndrome
References
- 1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature 2015; 526: 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR.. Merlin-rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 2002; 30: 97–101. [DOI] [PubMed] [Google Scholar]
- Akçimen F, Ross JP, Bourassa CV, Liao C, Rochefort D, Gama MTD, et al. Investigation of the RFC1 repeat expansion in a Canadian and a Brazilian Ataxia Cohort: identification of novel conformations. Front Genet 2019; 10: 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Australian Department of Foreign Affairs and Trade. Niue country brief. 2019. Available from: https://www.dfat.gov.au/geo/niue/Pages/niue-country-brief (9 March 2020, date last accessed).
- Bahlo M, Bromhead CJ.. Generating linkage mapping files from Affymetrix SNP chip data. Bioinformatics 2009; 25: 1961–2. [DOI] [PubMed] [Google Scholar]
- Beecroft SJ, Cortese A, Sullivan R, Yau W, Dyer Z, Wu TY, et al. A Māori specific RFC1 pathogenic repeat configuration in CANVAS, likely due to a founder allele. Brain 2020; 143: awaa203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers G, Edinur H.. The Austronesian diaspora: a synthetic total evidence model. Glob J Anthropol Res 2016; 2: 53–65. [Google Scholar]
- Cortese A, Simone R, Sullivan R, Vandrovcova J, Tariq H, Yan YW, et al. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat Genet 2019; 51: 649–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortese A, Tozza S, Yau WY, Rossi S, Beecroft SJ, Jaunmuktane Z, et al. Cerebellar ataxia, neuropathy, vestibular areflexia syndrome due to RFC1 repeat expansion. Brain 2020; 143: 480–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolzhenko E, Bennett MF, Richmond PA, Trost B, Chen S, van Vugt J, et al. ExpansionHunter Denovo: a computational method for locating known and novel repeat expansions in short-read sequencing data. Genome Biol 2020; 21: 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJFrancioli LCTiao GCummings BBAlföldi JWang Q, . et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020; 581: 434–43. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiktev DA, Sheng Z, Lobachev KS, Petes TD.. GC content elevates mutation and recombination rates in the yeast Saccharomyces cerevisiae. Proc Natl Acad Sci USA 2018; 115: E7109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machiela MJ, Chanock SJ.. LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 2015; 31: 3555–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliaccio AA, Halmagyi GM, McGarvie LA, Cremer PD.. Cerebellar ataxia with bilateral vestibulopathy: description of a syndrome and its characteristic clinical sign. Brain 2004; 127: 280–93. [DOI] [PubMed] [Google Scholar]
- Nakamura H, Doi H, Mitsuhashi S, Miyatake S, Katoh K, Frith MC, et al. Long-read sequencing identifies the pathogenic nucleotide repeat expansion in RFC1 in a Japanese case of CANVAS. J Hum Genet 2020; 65: 475–80. [DOI] [PubMed] [Google Scholar]
- Pelosi L, Mulroy E, Leadbetter R, Kilfoyle D, Chancellor AM, Mossman S, et al. Peripheral nerves are pathologically small in cerebellar ataxia neuropathy vestibular areflexia syndrome: a controlled ultrasound study. Eur J Neurol 2018; 25: 659–65. [DOI] [PubMed] [Google Scholar]
- Poplin R, Ruano-Rubio V, DePristo MA, Fennell TJ, Carneiro MO, Van der Auwera GA, et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 2017: 201178. doi: 10.1101/201178. [Google Scholar]
- Rafehi H, Szmulewicz DJ, Bennett MF, Sobreira NL, Pope K, Smith KR, et al. Bioinformatics-based identification of expanded repeats: a non-reference intronic pentamer expansion in RFC1 causes CANVAS. Am J Hum Genet 2019; 105: 151–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szmulewicz DJ, McLean CA, Rodriguez ML, Chancellor AM, Mossman S, Lamont D, et al. Dorsal root ganglionopathy is responsible for the sensory impairment in CANVAS. Neurology 2014; 82: 1410–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szmulewicz DJ, Waterston JA, Halmagyi GM, Mossman S, Chancellor AM, McLean CA, et al. Sensory neuropathy as part of the cerebellar ataxia neuropathy vestibular areflexia syndrome. Neurology 2011; 76: 1903–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szmulewicz DJ, Waterston JA, MacDougall HG, Mossman S, Chancellor AM, McLean CA, et al. Cerebellar ataxia, neuropathy, vestibular areflexia syndrome (CANVAS): a review of the clinical features and video-oculographic diagnosis. Ann N Y Acad Sci 2011; 1233: 139–47. [DOI] [PubMed] [Google Scholar]
- Tsuchiya M, Nan H, Koh K, Ichinose Y, Gao L, Shimozono K, et al. RFC1 repeat expansion in Japanese patientswith late-onset cerebellar ataxia. J Hum Genet 2020; doi: 10.1038/s10038-020-0807-x. [DOI] [PubMed] [Google Scholar]
- Umeh CC, Polydefkis M, Chaudhry V, Zee DS.. Sweat gland denervation in cerebellar ataxia with neuropathy and vestibular areflexia syndrome (CANVAS). Mov Disord Clin Pract 2016; 46–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilton SD, Lim L, Dye D, Laing N.. Bandstab: a PCR-based alternative to cloning PCR products. Biotechniques 1997; 22: 642–5. [DOI] [PubMed] [Google Scholar]
- Wu TY, Taylor JM, Kilfoyle DH, Smith AD, McGuinness BJ, Simpson MP, et al. Autonomic dysfunction is a major feature of cerebellar ataxia, neuropathy, vestibular areflexia ‘CANVAS’ syndrome. Brain 2014; 137: 2649–56. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, upon reasonable request.