

Abstract
Massively parallel sequencing technologies such as exome sequencing are increasingly applied across medicine. Connaughton et al. report a high diagnostic yield of exome sequencing among adults with hereditary nephropathy or nephropathy of unknown cause. Their findings support broader use of genomic sequencing in nephrology and highlight key associated questions, including how to identify those patients for whom testing is indicated, pinpoint pathogenic variants, and balance the resultant health care benefits and clinical follow-up burden.
In recent years, massively parallel sequencing has enabled high-throughput assessment of individual human genomes for causal variants for a variety of clinical disorders, supporting specific diagnosis and, in many cases, guiding management.
In adult medicine, these advances have been most spectacular in oncology, an area in which genome-wide sequence analysis of tumor and germline DNA have enabled selection of therapies targeted to the underlying molecular defects. To date, applications of massively parallel sequencing in nephrology have been limited largely to patients with pediatric-onset and/or familial disease, with use of multigene panels targeting solely the clinically suspected nephropathy.1 However, the fact that Mendelian causes currently are thought to account for approximately 10% of cases of end-stage kidney failure, and Mendelian nephropathies show high genetic and phenotypic heterogeneity,2 provides strong rationale for expanding both the scope of genes and clinical indications tested. In this issue of Kidney International, Connaughton et al.3 contribute significantly to a growing body of work demonstrating the value and challenges of performing such expanded genetic testing in nephrology.4,5
Connaughton et al.3 evaluated the diagnostic yield of exome sequencing (ES) in a cohort of 114 Irish adult probands with chronic kidney disease (CKD) of diverse etiologies, with nonfamilial as well as familial disease. In ES, the coding portion of the genome is captured and sequenced, enabling assessment of the majority of human genes, including the ~3600 already known to be associated with Mendelian disorders. This technique thus substantially expands the diagnostic search space as compared to phenotype-driven panels, which typically feature several dozen genes. Thus, ES eliminates the need for sequential gene panel testing for patients with nondiagnostic CKD—etiology unknown, and can clarify the clinical impression for those who may have been initially misdiagnosed. The genome-wide approach of ES also has uncovered genotype–phenotype relationships that were not previously appreciated. For example, ES studies have demonstrated that COL4A3 and COL4A4 mutations can manifest as focal segmental glomerulosclerosis, expanding the phenotypic spectrum of COL4A-associated nephropathy.6 Finally, use of ES has revealed that a subset of patients harbor diagnostic mutations for 2 distinct Mendelian disorders,7 helping clinicians to resolve complex clinical presentations.
The patients assessed by Connaughton et al.3 span the major categories of hereditary nephropathy, such as cystic kidney disease, congenital anomalies of the kidney and urinary tract, steroid-resistant nephrotic syndrome, and tubulointerstitial disease, as well as CKD—etiology unknown. Of these 114 probands, 94 (81%) harbored other features consistent with a hereditary form of disease, with 78 (68%) reporting a positive family history of CKD and 16 (14%) having extra-renal features. Connaughton et al.3 report an overall diagnostic yield of 37% (42 of 114), encompassing 29 distinct single-gene etiologies of CKD. Of these 29 monogenic disorders, 21 (72%) were detected only once, in a single proband, highlighting the genetic heterogeneity of Mendelian nephropathies. Among the 42 positive cases, ES reclassified the clinically suspected disease in 9 (22%), and for 16 (38%), it provided a diagnosis for CKD—etiology unknown. Diagnostic yield was higher among cases with other clinical features supportive of a Mendelian etiology of nephropathy, with detection rates of 69% (11 of 16) and 36% (28 of 78) among individuals with a positive family history for CKD and extra-renal features, respectively, versus 15% (3 of 20) in those without either, supporting them as indications for genetic testing. However, yield did not significantly differ between cases with versus without early-onset disease (prior to age 18 years), or according to median age of ESRD. Finally, consistent with prior reports,7 dual diagnoses were detected in 4.7% of positive cases, who harbored diagnostic mutations in 2 different Mendelian nephropathy-associated genes.
Altogether, these findings provide valuable insight regarding the diagnostic yield of ES and associated genetic and phenotypic spectrum among patients clinically diagnosed with various hereditary nephropathies. The high detection rate, particularly among patients with CKD—etiology unknown, supports prior reports of the merits of more-comprehensive genetic analysis, helping end the “diagnostic odyssey” experienced by patients and their clinicians. On case-level evaluation, Connaughton et al.3 find that in many cases, the genetic findings have important implications for clinical management, including reclassification of disease, initiation of targeted workup, and changes in therapy. The clinical implications of their findings are remarkably concordant with another recently published study of ES in adults with CKD,4 highlighting the promise of genetic diagnostics to support targeted interventions, and if performed early enough, even presymptomatic disease screening and preventive care.
Connaughton et al.3 perform a targeted analysis of ES data, filtering for rare (defined here using control minor allele frequency cutoffs of 0.1% for dominant disorders and 1% for recessive disorders) protein- or splice-altering variants occurring in a set of 478 Mendelian nephropathy-associated genes. The curated gene list represents a valuable contribution to ongoing efforts to comprehensively catalog and characterize genes associated with Mendelian nephropathy,2,4,5 and the phenotype-driven approach supports efficient identification of variants explicative for the primary renal disease indication. The frequency-based filtering applied has been demonstrated to greatly reduce the burden of misclassified variants emerging from ES,5 reflecting the fact that the majority of causal variants for Mendelian disorders are ultra-rare. However, this approach holds the challenge of determining the appropriate minor allele frequency thresholds. Certain established causal variants for Mendelian nephropathies, such as the NPHS2 p.R229Q allele, associated with steroid-resistant nephrotic syndrome, and the COL4A5 p.G624D allele, associated with X-linked Alport syndrome, are present at appreciable control frequencies, such that they could be removed with indiscriminate use of ultra-stringent minor allele frequency–-based filtering.5
As in prior diagnostic massively parallel sequencing studies in nephrology,4,8 Connaughton et al.3 surveyed a selected population comprised of patients harboring clinical diagnoses consistent with inherited forms of renal disease, 78% of whom also had a positive family history for nephropathy. In contrast, most individuals with CKD are clinically diagnosed with acquired causes, such as diabetes- and hypertension-attributed nephropathy, and across etiologies, approximately 25% of cases report a positive family history. Moreover, 99% of the cases assessed in this study self-identified as white Europeans, further limiting its generalizability to the greater, ethnically diverse CKD population. The yield of genetic testing can vary widely among clinical categories of renal disease,8 as can the frequencies of candidate pathogenic variants by ethnicity. Thus, assessment of an ethnically diverse cohort including patients representing all major etiologic categories is needed to provide insight regarding the genetic and phenotypic spectrum of Mendelian nephropathies, resolve the clinical relevance of more common alleles, and ultimately develop evidence-based guidelines for genetic testing in nephrology.
These findings also point to future challenges for nephrologists. Should ES be deployed as a predictive test, as may occur with broader use of genetic testing across clinical medicine? Generally, patients undergoing clinical ES receive variants classified as pathogenic, likely pathogenic, or of uncertain significance in genes related to the primary disease indication for testing; however, many laboratories now offer the option of also receiving otherwise medically relevant genetic findings unrelated to this primary disease indication, including those for other Mendelian disorders.9 Both how to interpret a candidate pathogenic variant for nephropathy emerging from diagnostic ES that is also found in adults not known to have CKD and how rare a variant must be in the general population to be causal are unclear at present, contributing to the high burden of variants of uncertain significance. Conversely, the genome-wide scope of ES also can result in the discovery of medically actionable mutations for Mendelian disorders other than nephropathy. Such secondary findings represent both an advantage and challenge of genomic sequencing, because although they are not explicative for a patient’s CKD, they not only can initiate extra-nephrologic subspecialty referrals, but also may have meaningful implications for its management. For example, secondary findings for hereditary cancer predisposition could not only trigger oncologic workup, but also inform usage of immunosuppressive therapy, in the context of the patient’s primary renal disease and/or renal transplantation. Given the abundance of rare, putatively damaging variation present in each human genome, and the fact that many Mendelian disorders have multi-organ involvement, broader usage of genomic sequencing could lead to patients and their families undergoing considerable clinical workup involving multiple subspecialty referrals and substantial health care spending.5 Thus, how to balance the opportunity for early detection of medically actionable conditions with the potential financial and psychosocial burden of the associated medical follow-up remains unclear.
Massively parallel sequencing–based genetic testing promises to revolutionize the workup of suspected hereditary nephropathy, enabling specific diagnosis and personalized care. The findings of Connaughton et al.3 not only support this promise, but also point to key gaps in our knowledge that must be filled to realize it (Figure 1). Future studies that assess the frequency of pathogenic variants for Mendelian nephropathy in well-phenotyped cases and controls, compare the diagnostic yield of ES across broader clinical indications and relative to targeted panels, and examine the long-term health benefits and costs of genetic testing, will provide much-needed insight regarding these questions, and help to achieve genomic medicine for patients with kidney disease.
Figure 1 |. Massively parallel sequencing (MPS) in nephrology: current practices, potential future usage, and associated questions.
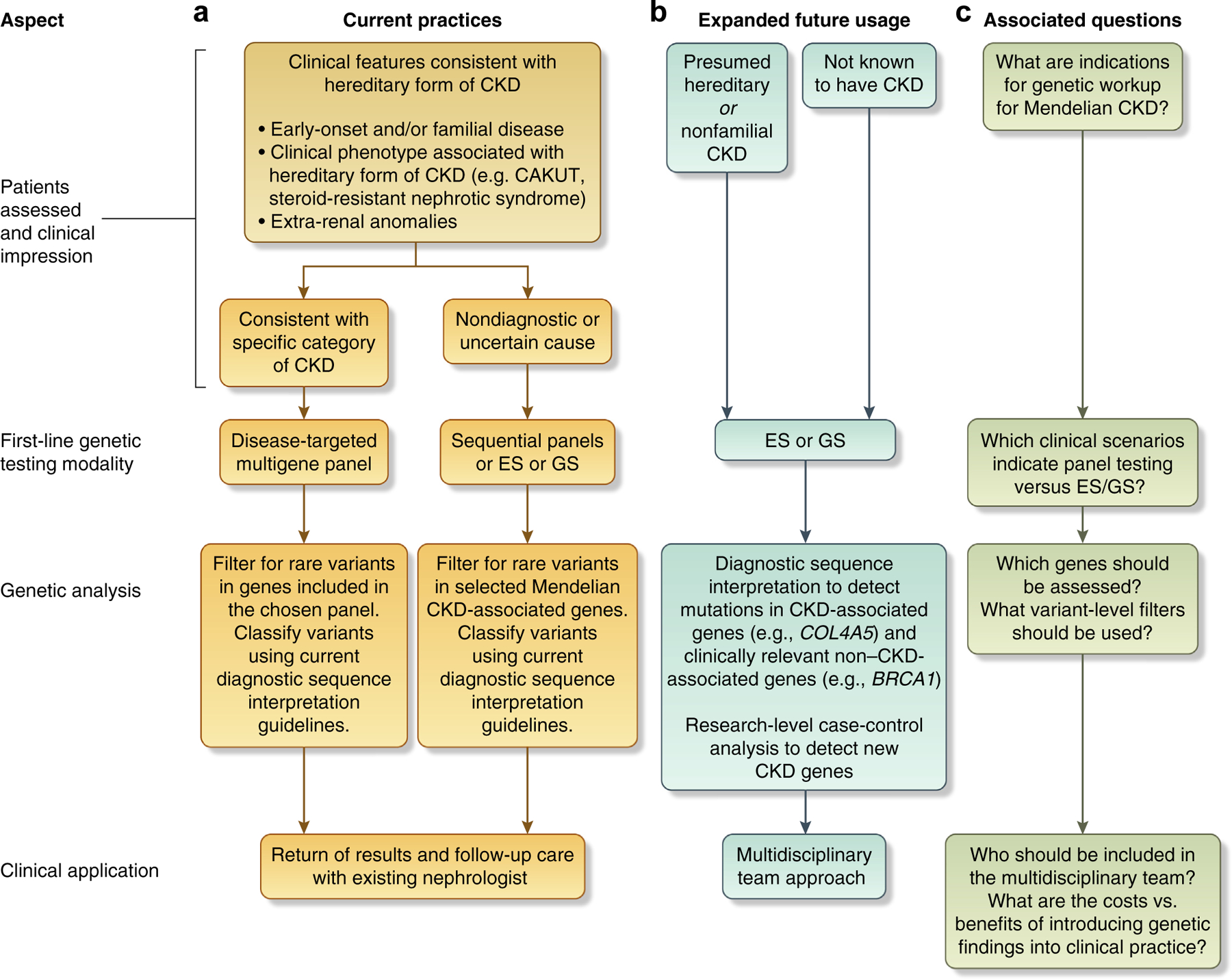
The key aspects of MPS in nephrology include (i) patient populations assessed and determination of the first-line genetic testing modality; (ii) genetic analysis procedures; and (iii) clinical application of the genetic findings detected. (a) Current use of MPS consists largely of applying disease-targeted multigene panels as a first-line test to probands clinically diagnosed with the associated disease. If the clinical impression is uncertain or the presentation wholly nondiagnostic, sequential gene panel testing or exome sequencing (ES) or genome sequencing (GS) may be applied instead. Genetic analysis assesses rare variants occurring in the genes included on the panel; for ES or GS, rare variants in known Mendelian nephropathy-associated genes are assessed. Return of results and follow-up clinical care are generally conducted as part of the patient’s routine nephrologic care, with his or her existing nephrologist. (b) Potential future use of MPS involves applying genome-wide testing (exome sequencing or genome sequencing) to a broader population, which may include individuals not known to have chronic kidney disease (CKD; e.g., for purposes of disease screening and presymptomatic genetic diagnosis, or, among probands undergoing genomic sequencing for other disease indications, as secondary genetic findings) as well as patients with CKD of diverse etiologies, including those presumed to have acquired causes of disease (e.g., diabetic nephropathy). The scope of genetic analysis may expand to all genes (genome-wide), integrating genetic diagnostics with case-control analyses for novel gene discovery. As the disorders detected often involve multiple organ systems, and the patients sequenced may not have previously been under nephrologic care, return of results and clinical follow-up will involve a multidisciplinary team, including other medical subspecialists for the organ systems affected by the given disease, and genetic counselors. (c) The key questions associated with this expanded, future use include (i) the clinical indications for genetic workup for Mendelian forms of CKD, (ii) determining which genes should be assessed and which filtering parameters most effectively identify bonafide disease-causal variants in the context of the abundance of putatively damaging variation present in any human genome, and (iii) determining which health care professionals should be involved and how to balance the opportunity to identify potentially medically actionable conditions with the financial and psychosocial burdens of the often considerable clinical follow-up.
ACKNOWLEDGMENTS
This work is supported by grants from the National Institutes of Health (grant numbers U01-HG008680 [AAG] and F30-DK116473 [EEG]).
Footnotes
DISCLOSURE
All the authors declared no competing interests.
REFERENCES
- 1.Vivante A, Hildebrandt F. Exploring the genetic basis of early-onset chronic kidney disease. Nat Rev Nephrol. 2016;12: 133–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Devuyst O, Knoers NV, Remuzzi G, et al. Rare inherited kidney diseases: challenges, opportunities, and perspectives. Lancet. 2014;383:1844–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Connaughton DM, Kennedy C, Shril S, et al. Monogenic causes of chronic kidney disease in adults. Kidney Int. 2019;95:914–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lata S, Marasa M, Li Y, et al. Whole-exome sequencing in adults with chronic kidney disease: a pilot study. Ann Intern Med. 2018;168:100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rasouly HM, Groopman EE, Heyman-Kantor R, et al. The burden of candidate pathogenic variants for kidney and genitourinary disorders emerging from exome sequencing. Ann Intern Med. 2018. 10.7326/M18-1241. [DOI] [PubMed]
- 6.Malone AF, Phelan PJ, Hall G, et al. Rare hereditary COL4A3/COL4A4 variants may be mistaken for familial focal segmental glomerulosclerosis. Kidney Int. 2014;86:1253–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Posey JE, Harel T, Liu P, et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med. 2017;376: 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mallett AJ, McCarthy HJ, Ho G, et al. Massively parallel sequencing and targeted exomes in familial kidney disease can diagnose underlying genetic disorders. Kidney Int. 2017;92:1493–1506. [DOI] [PubMed] [Google Scholar]
- 9.O’Daniel JM, McLaughlin HM, Amendola LM, et al. A survey of current practices for genomic sequencing test interpretation and reporting processes in US laboratories. Genet Med. 2017;19:575–582. [DOI] [PMC free article] [PubMed] [Google Scholar]