

Abstract
Therapy-resistant microenvironments represent a major barrier toward effective elimination of disseminated cancer. Many hematologic and solid tumors are resistant to therapeutic antibodies in the bone marrow (BM), but not in the periphery (e.g., spleen). We previously showed that cyclophosphamide (CTX) sensitizes the BM niche to antibody therapeutics. Here, we showed that (i) BM resistance was not only induced by the tumor but also by the intrinsic BM microenvironment; (ii) CTX treatment overcame both resistance mechanisms by activating macrophage migration and phagocytosis, including significant upregulation of activating Fcγ receptors (FcγRIII and FcγRIV) and downregulating the inhibitory receptor, FcγRIIB; and (iii) CTX synergized with cetuximab (anti-EGFR) and trastuzumab (anti-Her2) in eliminating metastatic breast cancer in the BM of humanized mice. These findings provide insights into the mechanisms by which CTX synergizes with antibody therapeutics in resistant niche-specific organs and its applicability in treating BM-resident tumors.
Keywords: Cyclophosphamide, monoclonal antibody, macrophage, Fcγ receptors, bone marrow, resistant microenvironment, cancer
Introduction
Monoclonal antibodies (mAbs) are a major modality for cancer treatment because of their target specificity, efficacy, and low toxicity. Accumulating evidence suggests that mAb therapeutics can efficiently eliminate leukemia and lymphomas in peripheral organs, such as the spleen, but their efficacy is much lower in the bone marrow (BM)(1, 2). Similarly, metastasis of solid tumors, such as breast cancer, into the BM is often associated with poor treatment outcome and patient survival(3). These findings suggest that the BM is a resistant organ to antibody-mediated elimination of tumor cells (4). Overcoming BM resistance in cancer antibody immunotherapy is of significant clinical importance.
The therapeutic efficacy of mAbs often relies on immune effector cells, including macrophages and natural killer (NK) cells (5–8). Binding of a mAb to its target on malignant cells flags them for destruction by effector cells through FcγR-mediated antibody-dependent cellular cytotoxicity (ADCC) and phagocytosis (ADCP) (9). ADCC induces apoptosis/lysis of target cells by degranulation of effector cells such as NK cells. ADCP refers to the engulfment and digestion of mAb-opsonized targets by macrophages. Humans express up to six FcγRs (FcγRI [CD64], FcγRIIA [CD32A], FcγRIIB [CD32B], FcγRIIC [CD32C], FcγRIIIA [CD16A], and FcγRIIIB [CD16B]), with only FcγRIIB being a classical inhibitory immunoglobulin G (IgG) receptor. Mice have three activating FcγRs (FcγRI [CD64], FcγRIII [CD16], and FcγRIV [CD16–2]) and one inhibitory FcγR (FcγRIIB [CD32B])(6, 9, 10). Engagement of the activating FcγRs with the Fc portion of IgG results in cellular activation through immunoreceptor tyrosine-based activation motifs (ITAMs). In contrast, the intracellular domains of inhibitory FcγRIIB contains immunoreceptor tyrosine-based inhibitory motifs (ITIMs), and its ligation leads to inhibition of the effector cells (10, 11). Macrophages and NK cells express a number of activating and/or inhibitory FcγRs, depending on their tissue localization and microenvironmental cues (10, 11). We and others have shown that FcγR expression profiles on NK cells and macrophages are critical for therapeutic efficacy of mAbs, with almost all therapeutic mAbs relying on appropriate FcγR engagement for optimal activity (5, 7, 12, 13). Modulation of the FcγR expression profile and the activating to inhibitory (A:I) FcγR ratio can alter mAb efficacy (10, 14–16). A low A:I FcγR ratio, due to a reduction in activating FcγRs and/or elevation of inhibitory FcγRIIB, is a common feature of the immunosuppressive tumor microenvironment (14). In particular, expression of inhibitory FcγRIIB, which is known to suppress anti-cancer mAb immunotherapy (11, 17), is significantly increased on macrophages residing in the tumor microenvironment (14, 15, 18).
Cyclophosphamide (CTX) is an alkylating agent with potent effects on various components of the immune system and can synergize with mAb therapeutics (1, 19–23). Low-dose CTX therapy can transiently deplete suppressive regulatory T cells (Tregs) in both preclinical models and in patients, leading to clinical benefit (21, 23–25). This is because, compared to effector T cells, Tregs have metabolic adaptations that make them more susceptible to CTX-mediated cytotoxicity (26). We and others have shown that CTX treatment leads to recruitment and activation of myeloid effector cells, including macrophages, into the BM of tumor-bearing mice (1, 22, 27). As a result, CTX overcomes the BM resistant microenvironment and enhances the efficacy of mAb therapeutics in eliminating human ‘double-hit’ lymphoma in the BM (1). CTX and other chemotherapies are also known to combine effectively with rituximab (anti-CD20) [e.g., R-CHOP (rituximab plus CTX, doxorubicin, vincristine, and prednisone), R-CVP, etc.] for treating B-cell malignancies in humans (28–31). In agreement with preclinical data, high infiltration of macrophages into tumors has been shown to be associated with a favorable prognosis in follicular lymphoma (FL) patients undergoing R-CHOP, but not CHOP therapy (32). Despite these promising (pre)clinical effects, the underlying mechanisms by which CTX regulates macrophage function in different anatomical niches, such as the permissive lymphatic system organs and resistant BM, is not fully understood.
The BM resistance niche is not unique to tumor-bearing mice (1), and BM resistance to mAb-mediated cell depletion has also been observed in immunocompetent wild-type mice and ‘healthy’ humanized mice (2). These observations highlight the existence of underlying intrinsic mechanisms specific to the BM (i.e., in steady state), independent of tumor presence. Using immunodeficient NOD-SCID IL-2Rγ−/− (NSG) mice and NSG mice reconstituted with the human immune system (33), which allowed detailed analysis of both mouse and human macrophages in a niche-specific manner, we investigated the mechanisms by which CTX overcomes BM resistance. Our results revealed two mechanisms of BM resistance: one is intrinsic to the BM microenvironment and the other is induced by resident tumor cells. CTX overcame both resistance mechanisms by enhancing macrophage phagocytosis via downregulation of the inhibitory and upregulation of activating FcγRs, respectively, and therefore, synergized with therapeutic mAbs in eliminating target cells in the BM. We further show that the synergy extended to other direct-targeting therapeutic antibodies and solid tumors that often metastasize into the BM. Our findings shed light on the mechanisms and generality of CTX in overcoming BM resistance to antibody-mediated cancer immunotherapy.
Materials and Methods
Ethics Statement
All research with human samples and mice was performed in compliance with institutional guidelines, the World Medical Association’s Declaration of Helsinki, and the US Department of Health and Human Services Guide for the Care and use of Laboratory Animals. The MIT committee on the use of humans as experimental subjects (MIT COUHES) granted the research a waiver for the provision of human fetal liver tissues, as all human samples (normally disposed of as medical waste) were collected anonymously with parental consent by a 3rd party (Advanced Bioscience Resources, Inc., CA, USA) and purchased from that party for the purposes of research, as described below. Ethical approval for the use of lymphoma patient tissues, obtained as specified in the below sections, was granted under REC references 228/02/t (University of Southampton).
Cell line establishment and cultures
Human CD20+ lymphoma cell lines that also expressed GFP and luciferase were derived from a healthy donor as follows. Briefly, human CD34+ hematopoietic stem/progenitor cells (HSPCs) were transduced with a lentivirus expressing GFP and c-Myc and Bcl-2 oncogenes under the control of a B cell-specific Eμ enhancer/CD19 promoter (34). The transduced HSPCs were engrafted into NSG mice, where they rapidly gave rise to disseminated and aggressive human ‘double-hit’ lymphomas. The de novo-generated lymphoma cells grew readily in vitro and were transduced with lentivirus expressing luciferase and puromycin resistance genes. Puromycin-resistant lymphoma cells were selected and individual GFP+ and human CD20high cells were sorted into a 96-well plate using a BD FACS Aria (Beckton Dickinson, NJ) and expanded in vitro. Individual clones were evaluated for bioluminescence using a 96-well plate reader. Established GFP+ CD20high luciferase+ lymphoma cells (“GMB” cells) were cultured in RPMI 1640 containing 10% FCS, 2mM L-glutamine, 2-mercaptoethanol, non-essential amino acids, sodium pyruvate, penicillin/streptomycin (100 IU/mL) in an atmosphere of 5% CO2 at 37°C.
MDA-MA-231 cells (a gift from Richard Hynes in 2015) transduced with a lentivirus expressing GFP and luciferase were grown in glucose-rich DMEM medium supplemented with 10% FCS, l-glutamine (2mM), and penicillin/streptomycin (100 IU/mL) and incubated in an atmosphere of 5% CO2 at 37°C. All cell lines used were mycoplasma-free and passaged up to five times in culture, but were not reauthenticated in the past year.
Hematopoietic stem/progenitor cell (HSPC) isolation and generation of humanized mice
Human fetal livers were obtained from six aborted fetuses at 15–23 weeks of gestation, in accordance with the provider’s ethical guidelines (Advanced Bioscience Resources, Inc., CA, USA). All women gave written informed consent for the donation of their fetal tissue for research. Fetuses were collected within 2 hours of the termination of pregnancy. Fetal liver tissue was initially cut into small pieces and digested with collagenase VI (2 mg/mL in Dulbecco’s modified Eagle’s medium [DMEM]) for 30 minutes at 37°C with periodic mixing. Single-cell suspensions were prepared by passing the digested tissue through a 100 μm cell strainer (BD Biosciences, NJ, USA). CD34+ cells were purified with the use of a CD34+ selection kit (Stem Cell Technologies, Vancouver, BC, Canada), and the purity of CD34+ cells was 90%–99%. Viable cells were counted by trypan blue exclusion of dead cells. All cells were isolated under sterile conditions and cryopreserved in liquid nitrogen until further use.
NSG mice were purchased from the Jackson Laboratories (Bar Harbor, Maine, USA) and maintained under specific pathogen-free conditions in the animal facilities at MIT. To reconstitute mice, newborn pups (less than 2 days old) were irradiated with 100 cGy using a Gamma radiation source and injected intracardially with CD34+CD133+ cells (~2 × 105 cells/recipient). Engrafted mice were bled 12 weeks post HSPC engraftment and assessed for the presence of human CD45+ leukocyte subsets by flow cytometry. Typically mice with >40% human CD45+ leukocytes were used for the experiments.
Generation of humanized mice with metastasized breast cancer
Adult humanized NSG mice and NSG mice (~12 weeks old) were injected with 5×105 MDA-MA-231 human breast cancer cells carrying luciferase into the cardiac cavity (left ventricle) using ultrasound-guided imaging via a Vevo 770-ultrasound imaging system (VisualSonics) under general anesthesia, to model metastasis in vivo. Mice were treated, as indicated below, ten days post tumor cell engraftment. Tumor growth and spread was visualized using an IVIS Spectrum-bioluminescent imaging system.
Animal treatments
Mice were bred and maintained in local facilities. The MIT committee on animal care approved all the research components carried out in vivo. Animal experiments performed at the University of Southampton were approved by the local ethical committees and were performed under Home Office license P4D9C89EA. For generation of tumor models, 2×105 MDA-MB-231 breast cancer cells were engrafted via ultrasound-guided intracardiac injection into adult NSG or humanized mice; or 1×107 human CD20+ B lymphoma cell line derived from overexpression of Myc and Bcl-2 were intravenously injected into adult NSG mice, as previously described (34). Healthy or tumor-bearing mice were sex- and age-matched for experiments and intraperitoneally injected with CTX (100 mg/kg, Sigma-Aldrich, USA) and/or monoclonal antibody (10 mg/kg) as single agent or in combination. Tissues were harvested 1–7 days post injection and processed as detailed below.
Tissue preparation, antibodies, and flow cytometry
At various time points following treatment, mice were euthanized by CO2 asphyxiation and BM and spleen were harvested. The spleens were digested by collagenase D (Sigma-Aldrich, USA) at 37°C for 30 minutes, and the BM was flushed using syringes with a 27-gauge needle. All organs were then disrupted by grinding between frosted glass cover slips, and single-cell suspensions were prepared. Samples were lysed of red blood cells, and cells were counted in Trypan blue. FITC, PE, PerCP/cy5.5, APC, PE/Cy7, or APC/Cy7 conjugated antibodies directed to human CD3 (OKT3), CD14 (63D3), CD16 (3G8), CD19 (HIB19), CD20 (2H7), CD33 (WM53), CD34 (581), CD45 (2D1), CD56 (5.1H11), HLA-A2 (BB7.2), and mouse CD45.1 (A20), Ly6C (HK1.4), F4/80 (BM8), CD86 (GL-1) and MHCII (M5/114.15.2) were from Biolegend (USA). Human CD133 antibody was from Miltenyi Biotec (Germany). Fc-null–specific mouse FcγR mAb (F(ab’)2 (FcγRI, clone AT152–9; FcγRIIB, clone AT130–2; FcγRIII, clone AT154–2; FcγRIV, clone 9E9; all produced in-house) were produced following pepsin digestion (pH ~4.0) of monoclonal IgG at 37°C and subsequent HPLC purification and dialysis into PBS, as described (35); and labelled with FITC in-house, as previously reported (36). Cells were stained with appropriate combination of antibodies and then analyzed on LSR II flow cytometers (Beckton Dickinson, NJ, USA) in the MIT Koch Institute flow cytometry core facility and analyzed by FlowJo software. Cell sorting was performed on FACS Aria (Beckton Dickinson, NJ, USA) with a purity of 90%–99%. Sorted cells were subjected to lysis, and RNA (isolation indicated below) was kept frozen at −80°C until further use. Dead cells were excluded from analysis by DAPI (Sigma-Aldrich, USA) staining.
Transcriptome analysis
Splenic and BM mouse hCD45−F4/80+ and human hCD45+hCD14+ cells (macrophages) were sorted, as described above, from untreated control and CTX-treated non-tumor-bearing humanized mice on day 2 post CTX intraperitoneal injection. Cells were lysed in RLT lysis buffer containing β-mercaptoethanol, and total RNA was extracted with the RNeasy micro kit (Qiagen, USA) following the manufacturer’s instructions. Total RNA was assessed for quality and quantified using a total RNA 6000 Nano LabChip on a 2100 Bioanalyzer (Agilent Inc., USA). cDNA libraries were prepared according to the Illumina TruSeq RNA Sample Preparation Guide for SMARTer Universal Low Input RNA Kit (Clontech, USA). Gene expression was measured using HiSeq 2000 system (Illumina, USA). RNAseq data from mouse and human macrophages were aligned to mm10 and hg19, respectively, using Bowtie2 v2.2.3 (37). The number of mapped reads was quantified by RSEM v1.2.15 (38). Differential expression analysis between paired samples before and after treatment was performed using edgeR (39) with p <0.05 and >2 fold-change cut-offs. Differentially expressed genes were annotated using the online functional enrichment analysis tool DAVID (http://david.ncifcrf.gov/) (40). Gene set enrichment analysis was performed using Broad Institute Software (41), with the gene list pre-ranked according to logFC values from the edgeR output. The M1 and M2 macrophage gene sets (42) were obtained from the Molecular Signature Database (http://software.broadinstitute.org/gsea/msigdb/). The heatmap figure was visualized with MeV (43). Human and Mouse orthologues were matched based on MGI database (http://www.informatics.jax.org/). Raw sequences are deposited in the database of Gene Expression Omnibus (GEO) with accession ID: GSE135997.
Human lymphoma tissue immunohistochemistry (IHC)
Formalin-fixed, paraffin-embedded (FFPE) sections (4 µm) of consented human lymphoma patients’ lymph node (LN) and BM tissues were provided by Cellular Pathology, University Hospital Southampton NHS Foundation Trust. Sections were stained for immunohistochemistry (IHC) using the fully automated BOND-MAX IHC staining instrument (Leica Microsystems, Milton Keynes, UK), according to the manufacturer’s instructions and as previously reported (36). Briefly, sections were deparaffinized, pre-treated for heat-induced antigen retrieval (BOND ER2 protocol), and incubated with hydrogen peroxide followed by rabbit anti-human FcγRIIB (clone EP888Y; Abcam, Cambridge, UK), at a 1 in 2000 dilution. The antibody was subsequently bound to the poly-HRP IgG reagent, before incubation with 3,3’-diaminobenzidine (DAB). The sections were subsequently incubated with mouse anti-human CD68 (clone PG-M1; DAKO, Agilent Technologies LDA UK Limited, Stockport, UK) at a 1 in 250 dilution, which was then bound to the post primary IgG linker reagent (Leica Microsystems, Milton Keynes, UK). The substrate chromogen Fast Red (Leica Microsystems, Milton Keynes, UK) was then applied. All sections were counterstained using hematoxylin and mounted in CV Ultra mounting media (Leica Microsystems, Milton Keynes, UK). All images taken at original magnification x400, using an Olympus BX51 microscope and UplanApo 40x/0.85 objective and x10 eyepiece objective with a Leica DFC425 camera and Leica LAS V4.12 software.
Statistics
Mann-Whitney and one-way ANOVA statistical tests were performed throughout using GraphPadPrism (version 7), as indicated. For bar graphs, at least 3 experiments were performed, and error bars represent standard deviation. Mean of data were represented as bars in graphs.
Results
CTX potentiates antibody immunotherapy of human lymphoma in vivo
In our study, we used a human CD20+ B lymphoma cell line derived from overexpression of Myc and Bcl-2 (34) that also expressed the luciferase gene for easy in vivo monitoring of tumor burden by live mouse imaging. Lymphoma cells were engrafted into adult NSG mice by intravenous injection. Once tumors were established, mice were either left untreated or treated with rituximab (anti-CD20; human IgG1), CTX, or rituximab plus CTX. Treatment of lymphoma-bearing mice with rituximab alone resulted in the depletion of lymphoma cells in the spleen, but not the BM (Figure 1A). CTX treatment alone did not significantly affect tumor burden. The combination treatment of rituximab and CTX resulted in almost complete tumor elimination, not only in the spleen, but also in the BM (Figure 1A,B).
Figure 1. BM niche is resistant to mAb therapy and promotes immunosuppressive effector macrophages.
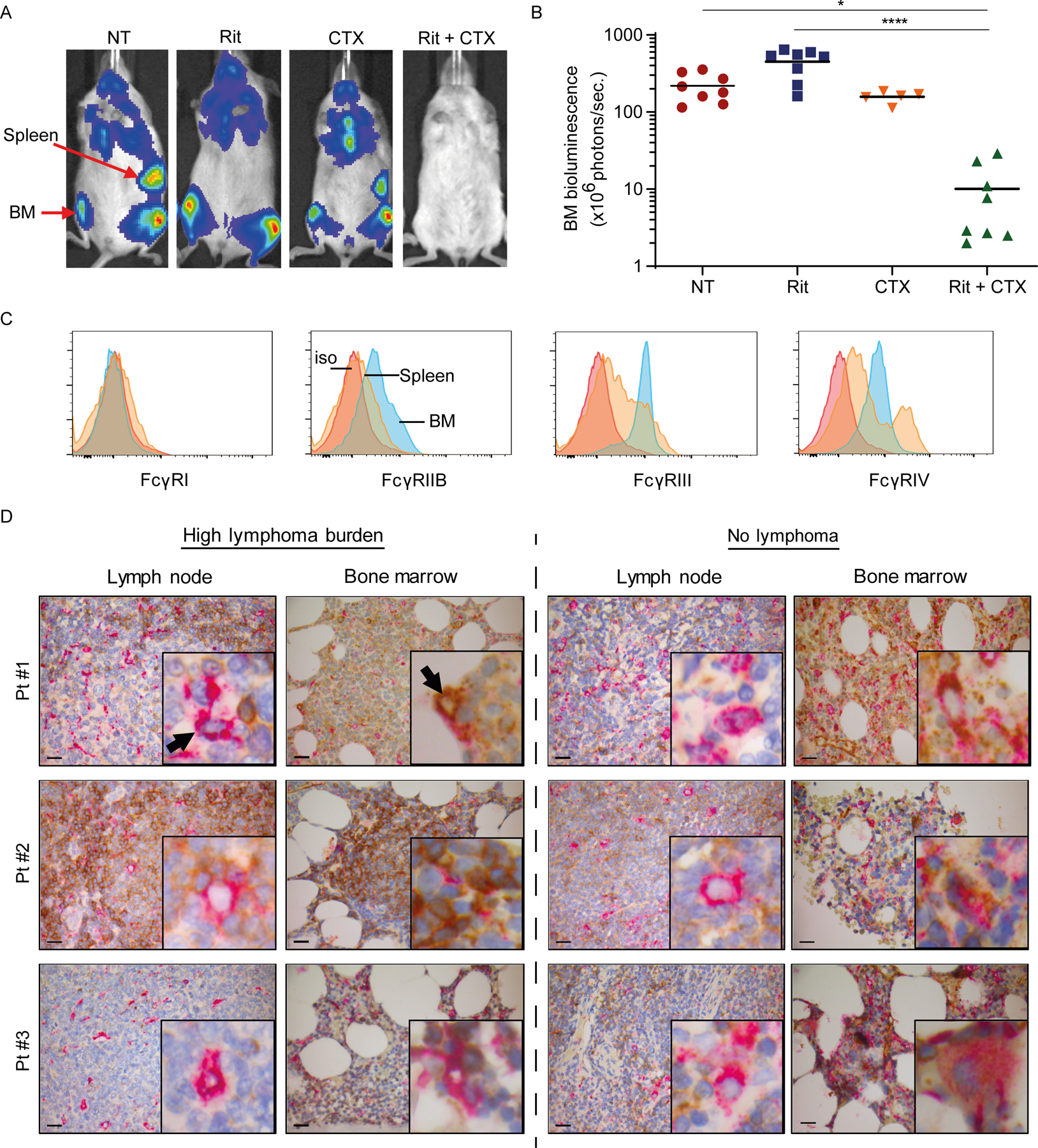
Ten million luciferase+GFP+ CD20+ human B-cell lymphoma cells were intravenously injected into adult NSG mice (5–8 mice/group from 1 experiment) and were left untreated (NT) or treated with either rituximab (Rit, 10 mg/kg) or CTX (100 mg/kg) as single agents or in combination (Rit+CTX) on day 21 after tumor injection. Tumor burden, indicated by bioluminescence intensity, was monitored on day 21 (before treatment) and 7 days after the indicted treatments. (A) Representative mouse images and (B) bioluminescence signal intensity in the BM. The positions of the spleen and BM are indicated. Each dot represents one mouse.; *p<0.05, ****p<0.0001 by one-way ANOVA. Means are shown. (C) Representative flow cytometry histograms of FcγR expression on F4/80+ macrophages from spleen and BM of a NSG mouse (5–8 mice/group). Red trace: isotype control (iso); orange trace: spleen; blue trace: BM. (D) FFPE diagnostic LN biopsies and staging BM trephines from three consented patients with follicular lymphoma were double-stained with antibodies to human CD68 (pink) and FcγRIIB (brown). Paired LN and BM samples from each patient were mounted on the same slide and stained. Representative IHC images are shown (original magnification x400; scale bar is 20µm) from areas with high lymphoma burden (left panel) and areas with no lymphoma involvement (right panel). The insert in each image represents 10x higher magnification of a representative macrophage taken from the same section. Black arrows point to CD68+ macrophages in the LN and BM.
Therapeutic antibodies rely on the presence of innate immune effector cells, such as macrophages, via binding to their activating FcγRs in order to eliminate tumor cells in vivo. Our previous observation that rituximab and alemtuzumab alone were only capable of depleting lymphoma cells from the spleen, but not BM, suggests that either there is not a sufficient number of macrophages in the BM or BM-resident macrophages are less efficient in eliminating opsonized tumor cells than splenic macrophages(1). To test the latter possibility, we phenotyped and compared activating and inhibitory FcγR expression on F4/80+ macrophages in the spleen and BM of non-tumor–bearing healthy NSG mice. BM-resident F4/80+ macrophages had higher expression of inhibitory FcγRIIB and lower expression of activating FcγRIII and FcγRIV vs. the spleen (Figure 1C). This confirmed that the BM can be a suppressive niche and that BM-resident macrophages are likely less efficient in FcγR-mediated phagocytosis than those in the spleen.
To corroborate these findings, next we investigated whether human BM-resident macrophages also expressed higher FcγRIIB (inhibitory receptor) in lymphoma patients. Diagnostic lymph node (LN) biopsies and staging BM trephines were obtained from four consented patients, one with diffuse large B cell lymphoma (DLBCL) and three with follicular lymphoma (FL). Paired LN and BM samples from each patient were mounted on the same slide and were double-stained with antibodies to human CD68 (macrophage marker) and FcγRIIB. Very few FcγRIIB+ macrophages were found in LN sections, regardless of their proximity to lymphoma cells. FcγRIIB staining was weak, indicting low expression (Figure 1D, Supplementary Fig. S1). In contrast, the majority of CD68+ macrophages in the BM stained for FcγRIIB, indicating high expression, both in areas with high lymphoma burden (left panel) and areas with no or low lymphoma burden (right panel)(Figure 1D, Supplementary Figure S2). Consistent with our previous report (44), here FL cells and normal B cells from patient sections had low to modest expression of FcγRIIB, which varied from patient to patient (Figure 1D). On the other hand, DLBCL cells had high expression of FcγRIIB, evident in both LN and BM (Supplementary Fig. S1). These results showed that the expression of FcγRIIB is significantly higher in macrophages in the BM than in the LN in lymphoma patients.
CTX reprograms BM macrophages by increasing A:I FcγR ratio
To investigate the effect of CTX on macrophage FcγR expression, we used non-tumor–bearing NSG mice because they allow us to investigate the effect of CTX on macrophages in the absence of T, B, NK cells, and tumor cells. NSG mice were dosed with 100 mg/kg CTX and FcγR expression on F4/80+ macrophages in the BM was analyzed 1, 2, and 3 days after treatment. Compared to untreated mice, the inhibitory FcγRIIB showed the greatest decrease on day 1 and day 2, whereas the activating FcγRIII and FcγRIV showed the biggest increase on day 2 following CTX treatment (Fig. 2A–B). We, therefore, focused on day 2 in subsequent experiments. Compared to FcγR expression on macrophages from BM of untreated mice, CTX treatment increased FcγRI by ~1.1-fold (not significant), FcγRIII by ~1.2-fold (p< 0.0001), and FcγRIV by ~1.45-fold (p <0.001)(Fig. 2C). In contrast, CTX treatment decreased FcγRIIB expression by ~1.4-fold (p <0.0001). CTX treatment had no significant effect on the expression of FcγRs on macrophages in the spleen of the same mice (Fig. 2D). Consistently, CTX significantly increased the A:I FcγR ratio on macrophages in the BM (Fig. 2E), but not in the spleen (Fig. 2F). CTX treatment also increased expression of MHC class II and the costimulatory molecule CD86 on macrophages in the BM (Fig. 2G), but not in the spleen. In contrast to macrophages, CTX treatment did not have the same influence on Ly6C+ monocyte activation status, and resulted in a modest increase in the expression of FcγRIIB in the BM (Supplementary Fig. S2). These observations demonstrated that CTX treatment reprogrammed macrophages, but not monocytes, to express higher activating FcγRs and other immune-stimulatory factors, while reducing inhibitory FcγRIIB expression in a BM-specific manner.
Figure 2. CTX treatment induces a higher A:I FcγR ratio in BM-resident, but not splenic, macrophages in non-tumor-bearing mice.
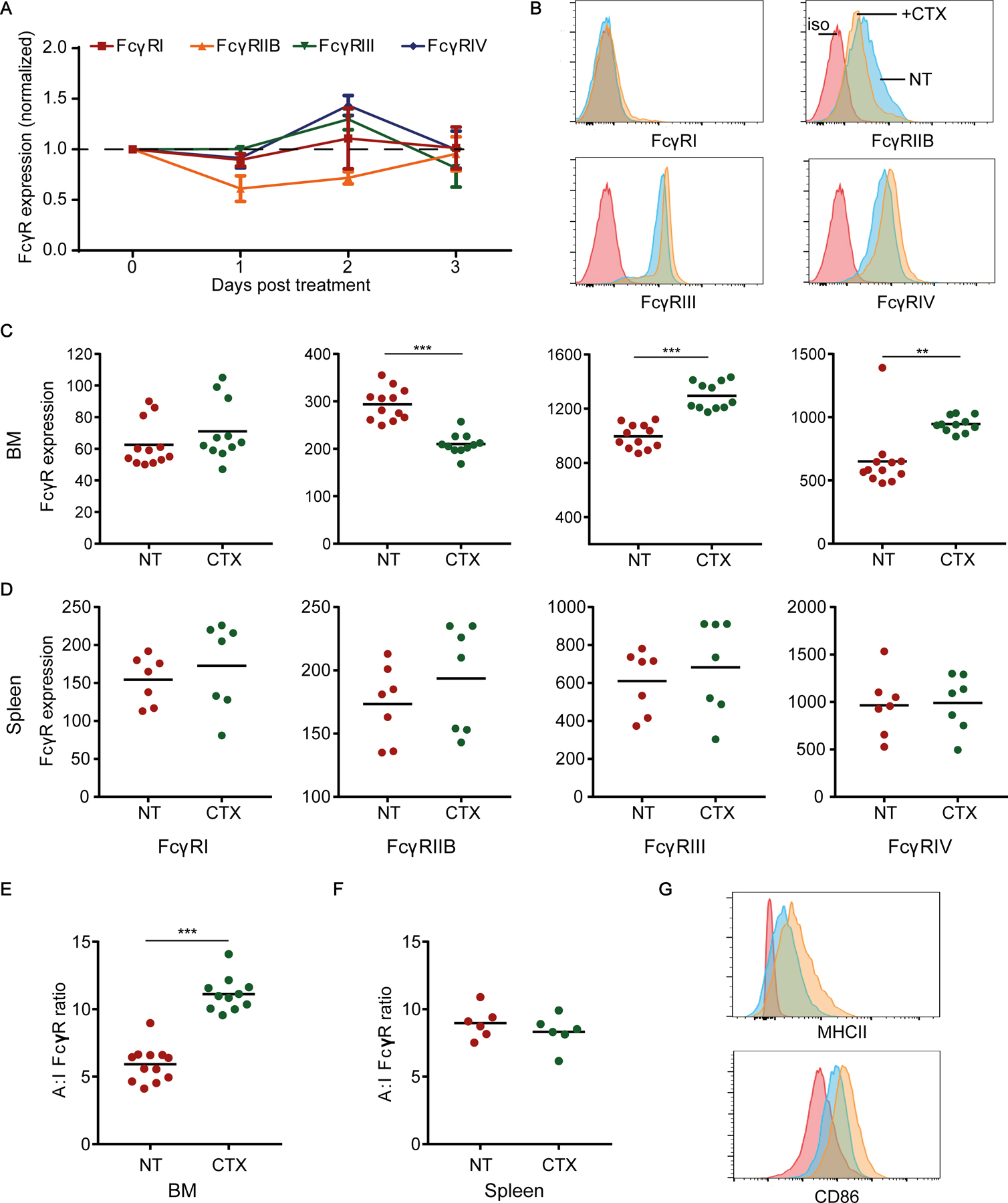
(A) FcγR expression on F4/80+ macrophages in the BM 1, 2, and 3 days after CTX treatment normalized to corresponding FcγR expression on untreated F4/80+ macrophages in the BM of control non-treated (NT) mice. Mean ± SD shown; 5 mice/group of 1 experiment. (B-D) Comparison of FcγR expression by F4/80+ macrophages in the BM and spleen of NT and CTX-treated NSG mice (2 days post-treatment). (B) Representative flow cytometry histograms of various FcγR expression by BM F4/80+ macrophages (4–5 mice/group, combined from at least 2 independent experiments). Red trace: isotype (iso) control; blue trace: NT; orange trace: CTX treated. Comparison of FcγR expression by F4/80+ macrophages in the (C) BM and (D) spleen of NT and CTX-treated NSG mice (2 days after CTX treatment). (E-F) Comparison of A:I FcγR ratio on F4/80+ macrophages from (E) BM and (F) spleen of NT and CTX-treated mice. (G) Representative flow cytometry histograms showing MHC class II and CD86 expression by BM F4/80+ macrophages. Red trace: isotype control; blue trace: NT; orange trace: CTX-treated (day 2). Median figures are shown for flow cytometry graphs. Each dot in C-F represents one mouse (combined from at least 2 independent experiments); **p< 0.001, ***p< 0.0001 by Mann-Whitney test.
CTX activates BM macrophages in lymphoma-bearing mice in vivo
NSG mice were injected intravenously with human CD20+ B lymphoma cells and 21 days later single cell suspensions were prepared from the BM and assayed for FcγR expression by myeloid cells. Compared to FcγR expression on BM macrophages in non-tumor-bearing NSG mice, BM macrophages in tumor-bearing mice expressed higher FcγRIIB, but lower FcγRIII and FcγRIV (Fig. 3A–B). These results are consistent with previous observations of regulatory effects of tumor cells on FcγR expression (10, 11, 14).
Figure 3. CTX treatment enhances A:I FcγR ratio in lymphoma-bearing mouse BM macrophages.
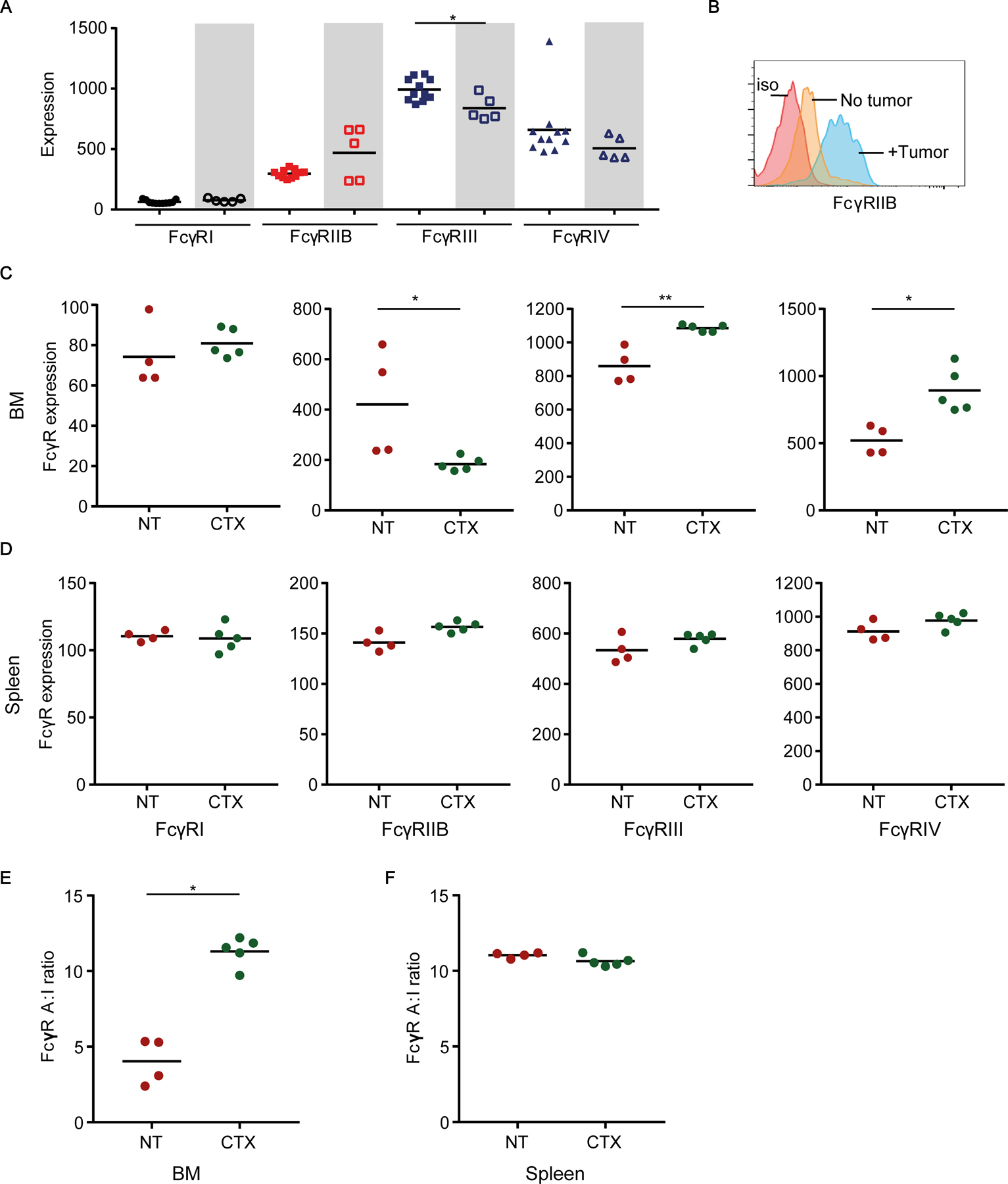
(A) Comparison of FcγR expression on BM F4/80+ macrophages from non-tumor-bearing (filled symbols) and lymphoma-bearing (open symbols; shaded boxes) NSG mice. FcγR expression was assayed 21 days following intravenous injection of 1×107 lymphoma cells. (B) Representative flow cytometry histograms of the inhibitory FcγRIIB expression on BM F4/80+ macrophages. Red trace: isotype (iso) control; orange trace: non-tumor-bearing mouse; blue trace: tumor-bearing mouse. (C-D) Comparison of median fluorescence intensity of activating and inhibitory FcγRs on F4/80+ macrophages from the (C) BM and (D) spleen of lymphoma-bearing mice with or without CTX treatment. (E-F) Comparison of A:I FcγR ratio on F4/80+ macrophages from the (E) BM and (F) spleen of tumor-bearing mice with or without CTX treatment (day 2). Median figures are shown for flow cytometry graphs. Each dot represents one mouse (3–5 mice/group combined from 1–3 independent experiments); *p<0.05, **p<0.001 by Mann-Whitney test.
Similar to non-tumor-bearing mice, treatment of tumor-bearing NSG mice with CTX for 2 days resulted in a higher expression of the activating FcγRIII and FcγRIV, but lower expression of the inhibitory FcγRIIB on BM macrophages (Fig. 3C). Similarly, no significant change was observed in the FcγR expression by macrophages in the spleen of treated tumor-bearing mice (Fig. 3D). Consequently, the A:I FcγR ratio was significantly increased by more than two-fold on BM macrophages (Fig. 3E), but not on splenic macrophages in CTX-treated lymphoma-bearing mice versus untreated mice (Fig. 3F). This increase in A:I FcγR ratio on BM macrophages following CTX treatment was equivalent to that seen in the permissive spleen niche. Thus, CTX also enhanced the A:I FcγR ratio on the BM-resident macrophages in tumor-bearing mice.
CTX treatment potentiates antibody-mediated depletion of human B cells from the BM
In addition to the resistant nature of BM in tumor-bearing mice we previously observed (1), Nimmerjahn and colleagues also report that the BM-resistance niche exists in tumor-free, immunocompetent wild-type mice, as well as ‘healthy’ humanized mice (2). To investigate whether CTX treatment potentiated depletion of normal human B cells, humanized mice were generated by engrafting human CD34+ hematopoietic stem/progenitor cells (HSPCs) into NSG pups. Administration of rituximab alone in adult humanized mice resulted in efficient depletion of human CD45+CD19+ B cells from the circulation and spleen, but not BM (Fig. 4A–B), as reported previously (2). We then treated humanized mice with rituximab and CTX as single agents or in combination and measured the frequency of human B cells in the spleen and BM 7 days post treatment. Although CTX treatment resulted in a modest depletion of B cells from the spleen and the BM (Fig. 4C–D), combination of rituximab and CTX resulted in a further significant depletion of B cells from both spleen and the BM. These results showed that the poor depletion of B-lymphoma cells from the BM by rituximab was not only due to the immunosuppressive tumor microenvironment, but also because of other intrinsic properties of the BM niche in general, which could be overcome by concomitant CTX treatment.
Figure 4. BM-resident human B cells are resistant to rituximab-mediated depletion, but are sensitized by CTX treatment.
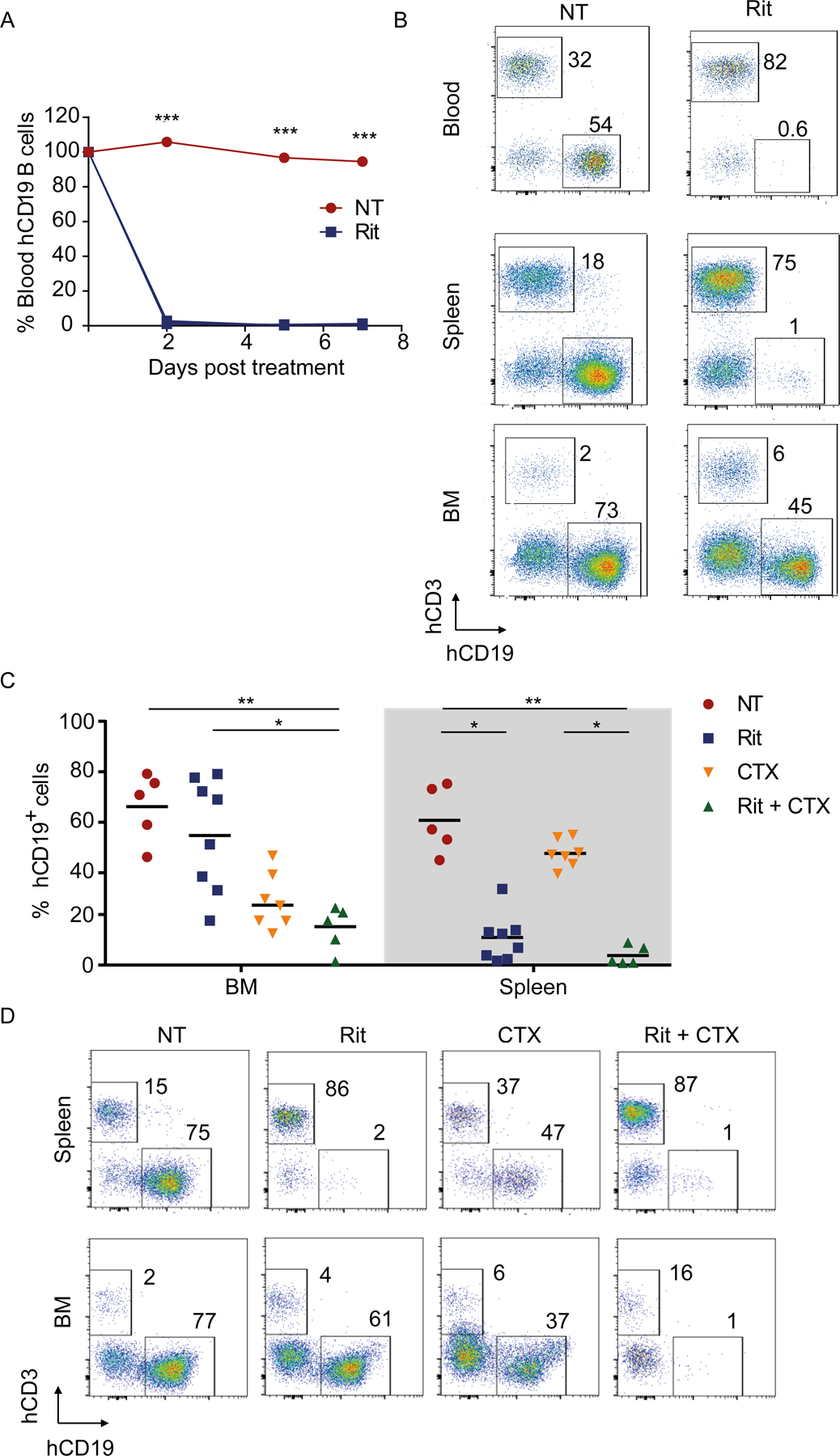
Adult humanized mice were injected with rituximab (Rit, 10 mg/kg). Mice were bled on day 2, 5, and 7 and the percentage of human (h) CD19+ B cells was quantified (3 mice/group). (A) Shown are percentages of human CD19+ B cells normalized to the percentage of B cells in the same mouse prior to rituximab treatment; ***p<0.001 by one-way ANOVA. (B) Seven days after rituximab treatment, single-cell suspensions were prepared from harvested organs and assayed for human CD45+CD19+ B cells by flow cytometry. Shown are hCD19 versus hCD3 staining profiles from the blood, spleen, and the BM of a representative mouse. The numbers indicate percentages of cells in the respective gate. (C-D) Humanized mice were intraperitoneally injected with either Rit (10 mg/kg) or CTX (100 mg/kg) as single agents or in combination. (C) Organs were harvested 7 days post treatment and percentage of live hCD45+hCD19+ B cells were quantified in the BM and spleen. Each dot represents one mouse; *p <0.05, **p <0.001 by one-way ANOVA test. (D) Representative flow cytometry plots showing hCD19 versus hCD3 staining profiles from spleen and the BM of representative mice (up to 8 mice/group from 3 independent experiments).
CTX treatment potentiates antibody-mediated depletion of human breast cancer cells in the BM
BM metastasis by breast, lung, and liver cancers is often associated with treatment resistance (3). To investigate whether CTX synergized with antibody therapeutics in eliminating BM metastasis of solid tumors, we generated metastatic human breast cancer-bearing mice by ultrasound-guided intracardiac injection of luciferase-expressing MDA-MB-231 breast cancer cells into NSG or humanized mice. Although allogeneic human lymphoma cells were readily rejected in humanized mice, but not NSG mice following intravenous injection (Supplementary Fig. S3A–B), breast cancer cells engrafted in unrelated humanized mice were not rejected (Fig. 5). When breast cancer cells were readily detected in the liver and/or BM of NSG mice at around 10 days post-engraftment, mice were either left untreated or treated with cetuximab, CTX, or the combination of cetuximab and CTX once every 5 days. Tumor burden increased from day 10 to day 25 in untreated, cetuximab-treated, and CTX-treated mice, as indicated by bioluminescence intensity (Fig. 5A–B), although the increase in tumor burden was slower in CTX-treated mice. In contrast, tumor burden was stabilized from day 10 to 25 in mice treated with combination CTX and cetuximab (Fig. 5A–B). Consistently, BM was filled with tumor cells in untreated, cetuximab-treated, and CTX-treated mice, whereas, BM was mostly cleared of tumor cells in CTX plus cetuximab-treated mice (Fig. 5C–D). Untreated and cetuximab-treated mice exhibited earlier and bigger lesions of the bones, whereas CTX plus cetuximab-treated mice did not show any apparent bone lesions (Fig. 5C–D). Survival of CTX plus cetuximab-treated mice was significantly longer than untreated and single-agent–treated mice. Similarly, treatment with CTX plus trastuzumab also significantly extended the survival (Fig. 5E). In humanized mice engrafted with MDA-MB-231 breast cancer cells, CTX, but not cetuximab, treatment delayed tumor progression, whereas tumor burden was significantly decreased following the combination therapy of CTX and cetuximab (Fig. 5F–G). These results showed that CTX synergized with anti-EGFR therapeutics to eliminate breast cancer cells in the BM and liver, indicating efficacy in multiple tumor locations using different therapeutic antibodies.
Figure 5. CTX potentiates mAb-mediated depletion of human breast cancer cells in the BM.
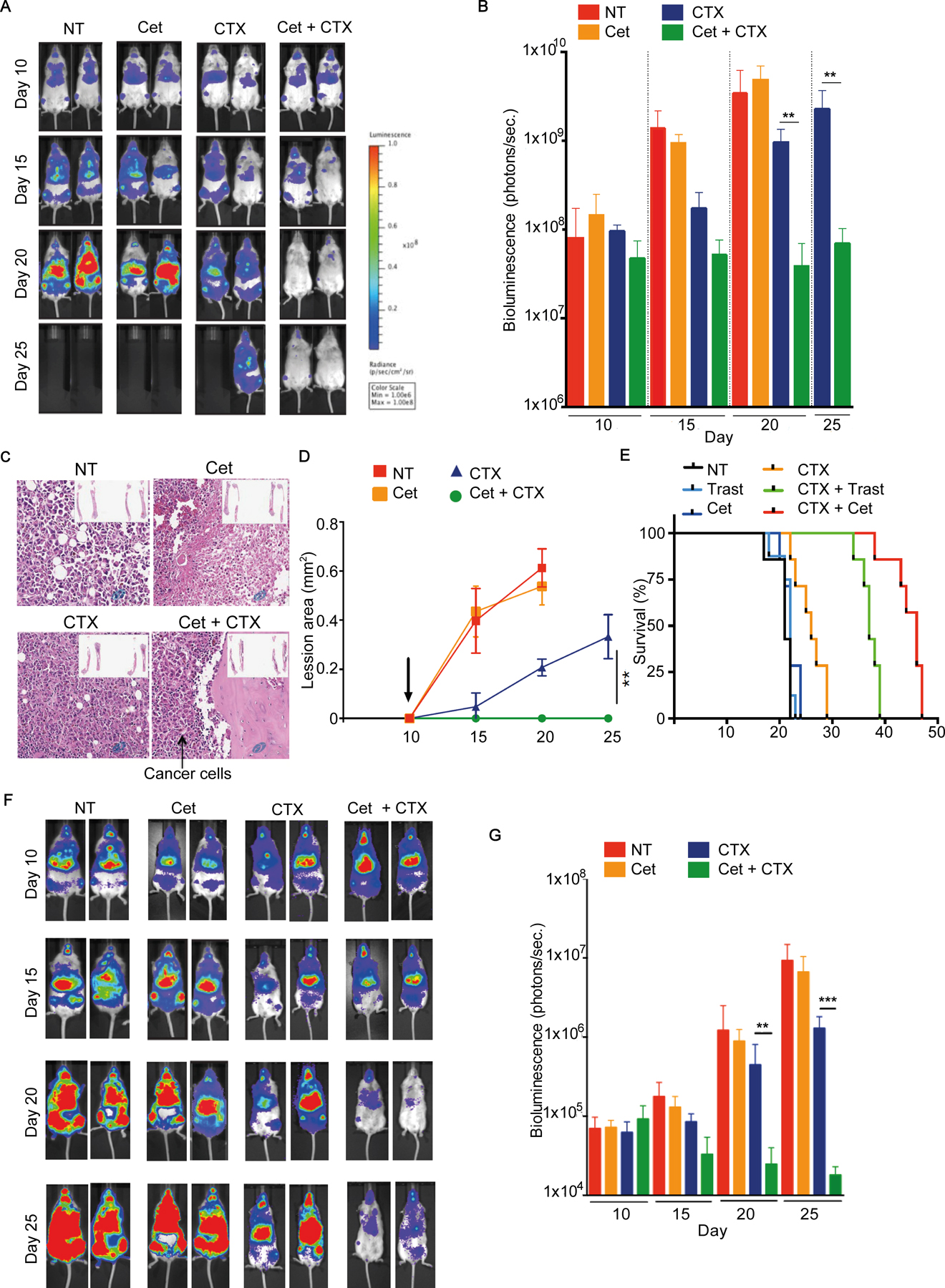
(A-E) Adult NSG or (F-G) humanized mice were engrafted with 5×104 MDA-MB-231 breast cancer cells by intracardiac injection. Randomized tumor-bearing mice and treated with either 10 mg/kg of the indicted therapeutic mAb or CTX (100 mg/kg) as single agents or in combination on day 10, 15, and 20 post tumor engraftment. Bioluminescence intensities were assessed by IVIS imaging on the indicated days. (A) Representative mouse images and (B) quantifications of bioluminescence intensity shown (mean+SD). (C) Representative H&E stains of femur sections (40x) of mice at day 20 after tumor engraftment. Inserts: tibia and femur from representative mice. (D) Bone lesions were scanned by micro CT at the indicated days; sizes of lesions (mean ±SD) are shown overtime. (E) Survival of cohorts of mice with different treatments. Trastuzumab and cetuximab alone or plus CTX were used, as indicated. (F) Representative bioluminescent images and (G) quantifications of tumor-bearing humanized mice (mean+SD). 3–5 mice/group, repeated at least 3 times for each experiment; **p<0.001, **p<0.001 by Mann-Whitney test.
CTX treatment reprograms human and mouse BM macrophages in vivo
To further investigate the immunomodulating effects of CTX in vivo, we performed transcriptional profiling of macrophages from the spleen and BM before and after CTX treatment. ‘Healthy’ (non–tumor-bearing) humanized mice were treated with 100 mg/kg CTX for 2 days. Both human CD45+CD14+ and mouse F4/80+ macrophages, from untreated control (naïve) and CTX-treated mice were isolated by cell sorting from the BM and spleen of the same mice and used for RNA sequencing (RNAseq). Principal component analysis (PCA) showed that both mouse and human macrophages grouped according to the tissue of origin independently of treatment (Fig. 6A), suggesting that macrophage gene expression signatures were regulated by tissue microenvironment, as previously reported (45, 46). CTX induced significant changes in gene expression in mouse macrophages: 247 genes were upregulated and 624 genes were downregulated in the spleen; and 346 genes were upregulated and 197 were downregulated in the BM. Among the significantly modulated genes, 59 were shared between the macrophages in the spleen and BM (Fig. 6B). Functional pathway enrichment analysis revealed that most of the genes affected by CTX in the BM macrophages were involved in phagocytosis, antigen processing and presentation, apoptotic cell clearance, migration, and immune defense (Fig. 6C), all of which are hallmarks of an activated inflammatory phenotype involving TNFα and IFNγ signaling, as confirmed by gene set enrichment analysis (GSEA)(Fig. 6D). In contrast, genes upregulated in splenic macrophages were mostly involved in transcription, cell proliferation, migration, and immune/inflammatory responses. Consistent with the flow cytometry immunophenotyping (Fig. 2), in untreated mice, FcγRIIB transcript expression was higher in the BM macrophages than splenic macrophages, whereas FcγRIII and FcγRIV transcript expression was lower in the BM macrophages than splenic macrophages (Fig. 6E). Following CTX treatment, the expression of the FcγRIIB transcript was decreased, whereas expression of FcγRIII and FcγRIV transcripts were increased in BM macrophages (Fig. 6E). As a result, CTX treatment induced a significantly higher A:I FcγR ratio in BM-resident macrophages, but not the splenic macrophages. We also examined the expression of macrophage proinflammatory (M1) and anti-inflammatory (M2) genes by BM and splenic macrophages isolated from naïve untreated mice (42). Splenic macrophages were significantly enriched for expression of M1-polarizing genes, whereas BM-resident macrophages were enriched for expression of M2-polarizing genes (Supplementary Fig. S4A). CTX treatment induced a significant M2-to-M1 shift in the BM macrophages (Supplementary Fig. S4B).
Figure 6. Transcriptome analysis of human and mouse macrophages from spleen and BM of CTX-treated humanized mice.
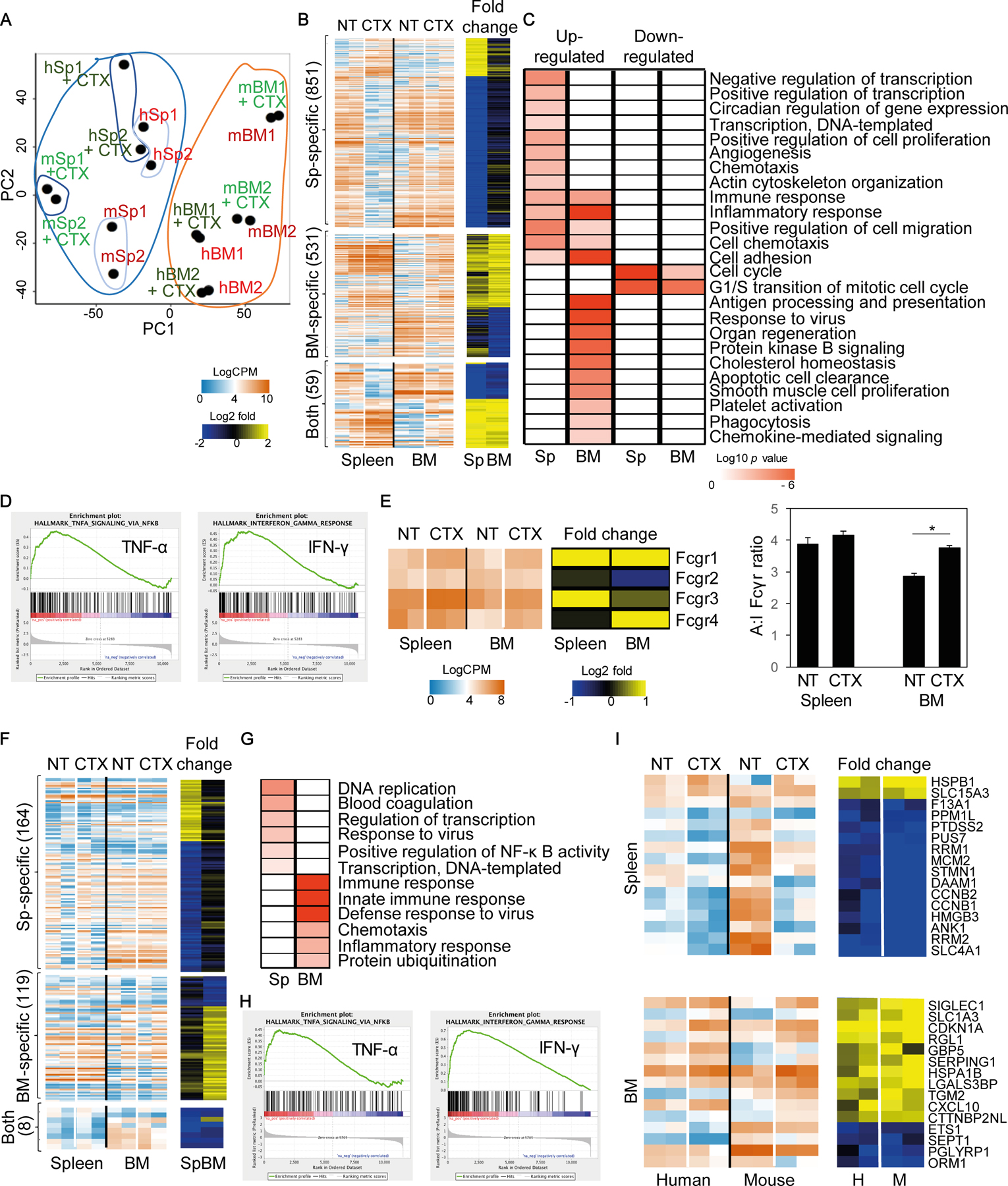
Adult humanized mice were injected with CTX (100 mg/kg) and 2 days later spleen and BM were harvested (pooled from 2 mice; data combined from 2 independent experiments). Both mouse and human macrophages were purified by cell sorting and subjected to RNAseq analysis (m: mouse; h: human). (A) PCA analysis of spleen (Sp) and BM CD45+CD14+ human and F4/80+ mouse macrophages. Blue area indicates splenic cells and orange line indicates BM cells. (B) Heatmap showing differentially expressed genes specifically induced by CTX in the mouse macrophages in the spleen, BM, or both, along with gene fold-changes in each organ following CTX treatment. (C) Functional pathway enrichment analysis of differentially expressed genes induced by CTX in the mouse macrophages. (D) GSEA analysis showing positive coloration with mouse TNFα (NES=1.89; FWER p=0.003) and IFNγ (NES=1.95; FWER p < 0.001) signaling elements. (E) Heatmap representation of FcγR genes in spleen and BM of mouse macrophages, along with their respective fold changes following CTX treatment and a graph showing calculated A:I FcγR ratios for macrophages isolated from each organ pre- and post-treatment (*p<0.05). (F) Heatmap showing differentially expressed genes specifically induced by CTX in the human macrophages in the spleen, BM, or both tissues, along with gene fold-changes in each organ following CTX treatment. (G) Functional pathway enrichment analysis of differentially expressed genes induced by CTX in the human macrophages in the spleen and BM. (H) GSEA analysis showing positive coloration with human TNFα (NES=1.86; FWER p<0.001) and IFNγ (NES=3.01; FWER p<0.001) signaling elements. (I) Heatmap showing differentially expressed genes induced by CTX in both mouse and human macrophages in the spleen and the BM, respectively. NES; normalized enrichment score; FWER; familywise-error rate.
Similarly, CTX induced significant changes in gene expression in human macrophages in vivo: 37 genes were upregulated and 168 genes were downregulated in the spleen; and 61 genes were upregulated and 47 genes were downregulated in the BM (Fig. 6F). Among the significantly affected genes, 8 were shared between the two niches. Functional pathway enrichment analysis revealed that differentially expressed genes in the splenic macrophages were involved in transcription, DNA replication, immune responses, and NF-κB signaling, whereas genes in BM macrophages were involved in inflammatory/immune responses, chemotaxis, and protein ubiquitination (Fig. 6G). Similarly to mouse BM-resident macrophages, GSEA analysis revealed that TNFα and IFNγ signaling pathways were upregulated in BM-resident human macrophages (Fig. 6H). We further compared genes shared between mouse and human macrophages from the spleen and BM of humanized mice. Several genes were similarly regulated by CTX treatment in both species, such as the upregulation of HSPB1 and SLC15A3 and downregulation of F13A1, PPM1L, and PTDSS2 in splenic macrophages; and upregulation of SIGLEC1, SLC1A3, CDKN1A, and CXCL10 and downregulation of ETS1, SEPT1, PGLYPR1, and ORM1 in BM-resident macrophages (Fig. 6I).
Discussion
We have previously shown that alemtuzumab (anti-CD52) is effective in eliminating human B lymphoma in the peripheral organs, such as spleen and liver, but not the BM. BM resistance can be overcome by combination chemoimmunotherapy of low-dose CTX and alemtuzumab, partly through recruitment of macrophages into the BM (1). In this study, we further investigated the nature of BM resistance, mechanism of action of CTX in overcoming the resistance, and generality of CTX in synergizing with other therapeutic antibodies for effective elimination of solid tumors from the BM. We showed that the BM resistance results partly from the intrinsic resistant property of the BM and partly from the tumor-induced suppressive microenvironment. In “healthy” humanized mice in the absence of tumor engraftment, administration of rituximab efficiently depleted human B cells from the peripheral blood and spleen, but not the BM, suggesting that the BM could be a resistant niche in the absence of the tumor. The presence of tumor in the BM likely makes the BM more resistant, indicated by the increased expression of the inhibitory FcγRIIB and decreased expression of activating FcγRIII and FcγRIV by BM macrophages. Our findings are consistent with previous observations showing the intrinsic resistance of BM to antibody-mediated depletion of human B cells (1, 2) and induction of FcγRIIB expression on BM macrophages in the presence of tumor (14, 18). Consistent with the preclinical observations, we also demonstrated that in matched LN and BM samples from the same lymphoma patient, FcγRIIB was expressed by BM-resident CD68+ macrophages, but not by macrophages in LN, regardless of their proximity to the tumor. Our study extends the previous studies by showing that CTX chemotherapy could transiently overcome both the intrinsic resistant property of the BM and the resistance induced by tumor cells. We showed that healthy human B cells are efficiently depleted by rituximab in humanized mice in the presence of CTX.
We further demonstrated that CTX overcame BM resistance in the presence or the absence of tumors through the same mechanisms. In the BM of both non-tumor-bearing NSG mice and NSG mice with engrafted tumor, CTX activated BM-resident macrophages to increase the expression of activating FcγRIII and FcγRIV, but significantly decreased the expression of the inhibitory FcγRIIB at both the transcript and protein levels. No significant changes were observed in macrophage FcγR expression in the spleen, suggesting that CTX had less profound effects on these cells. As such, splenic macrophages were efficient at phagocytosing opsonized tumor cells in vivo (in the absence of CTX stimulation), implying their activation threshold was lower than BM-resident macrophages. The organ-specific activation of macrophages was in agreement with our previous report, whereby a synergy was observed in tumor-bearing mice dosed with alemtuzumab and CTX within one day of each other (1). CTX treatment did not have the same influence on Ly6C+ monocyte FcγR expression, suggesting that CTX activation of myeloid cells was both organ- and myeloid cell subset (macrophage)-specific.
We showed that CTX synergized with antibody-mediated elimination of tumor cells in the BM could be extended to solid tumors, such as metastatic breast cancer and other antibodies, such as cetuximab and trastuzumab. In both NSG mice and humanized mice with breast cancer engraftment, the combination treatment of CTX and cetuximab (or trastuzumab) efficiently eliminated tumor cells, whereas CTX or cetuximab alone did not. A limited number of studies have investigated the clinical benefits of chemoimmunotherapy in treating metastatic breast cancer to date (47, 48). However, the full mechanism of action of such combination therapies is not yet clear. The observation that CTX was also capable of synergizing with cetuximab and trastuzumab to treat BM-metastasized human breast cancer cells in NSG mice and fully reconstituted humanized NSG mice is clinically relevant. First, it provides mechanistic insights in relevant in vivo models and emphasizes that CTX-induced activation of macrophages was independent of the cancer type in the resistant niche. Second, it highlights that CTX chemotherapy was able to potentiate the efficacy of different therapeutic antibodies, irrespective of the tumor origin and the target antigen. These encouraging results suggest that CTX may have a broader application in combination therapy with therapeutic antibodies and warrant further investigation. This combination therapy may also enhance the therapeutic activity of certain immune checkpoint inhibitors, such as anti–CTLA-4, which have been suggested to exert their effect by preferentially depleting tumor-resident Tregs via ADCP (49, 50).
Our detailed transcriptional analysis of both mouse and human macrophages from CTX-treated humanized mice further shed light on the mechanism by which CTX activates macrophages in the BM. CTX treatment differentially activated both spleen- and BM-resident mouse and human macrophages. Gene signatures modulated in BM-resident macrophages represented immune- and inflammatory-related genes involved in phagocytosis, antigen processing and presentation, apoptotic cell clearance, migration, and immune defense, consistent with increased activities. In agreement, GSEA analysis of murine and human macrophages suggested that IFNγ and TNFα signaling pathways were significantly induced by CTX treatment. In contrast to splenic macrophages which had an M1-polarizing gene signature, BM-resident macrophages were more enriched for M2-polarizing genes in naïve, untreated mice, which significantly shifted towards an M1 phenotype upon short-term CTX treatment. In addition to confirming a significantly higher A:I FcγR ratio in the mouse BM-resident macrophages, but not in splenic macrophages, at the transcriptome level, we identified several genes that were similarly affected by CTX in mouse and human macrophages from spleen and BM. The commonly upregulated genes identified in BM-resident macrophages included SIGLEC1 (CD169, sialoadhesin), HSPA1B (HSP70), and CXCL10. Both SIGLEC1 and HSPA1B are key genes implicated in macrophage phagocytosis. SIGLEC1, a member of the I-type lectin family, is induced by Toll-like receptor (TLR) agonists (TLR-7 and -9) and type I IFNs (51). SIGLEC1 contributes to host defense via scavenging functions and promoting macrophage activation and phagocytosis (52). HSPA1B encodes a 70-kDa heat shock protein and has been reported to activate macrophages and enhance phagocytosis (53, 54). Similarly, CXCL10 is an IFN-inducible chemokine, which is a chemotactic factor for T cells and myeloid cells (55). On the other hand, four genes (ETS1, SEPT1, PGLYPR1, and ORM1) were commonly downregulated in BM-resident mouse and human macrophages following CTX treatment. ETS1 is a transcription factor, induced by a variety of factors including TNFα, which is responsible for vascular inflammation and remodeling (56). Nevertheless, CTX did not reprogram BM macrophages to have a similar transcriptome profile as splenic macrophages, indicated by spleen and BM macrophages clustering separately regardless of their activation status, consistent with effects of the microenvironment on macrophage phenotype and activity (45, 46).
Our findings suggest modulating FcγRIIB as an effective cancer immunotherapeutic target. In the case of cancer therapy, this was originally demonstrated by Clynes and colleagues, who show that antibody therapy is significantly improved in FcγRIIB-deficient mice (17). Because this approach is not viable in the clinic, modulating FcγR expression and the A:I FcγR ratio is an alternative way to augment antibody efficacy. FcγRIIB can be targeted by a specific blocking antibody (57, 58) or its expression regulated by modulating the tumor microenvironment (11, 14). Our current data indicated that CTX treatment was able to selectively target BM-resident macrophages by lowering inhibitory FcγRIIB expression and increasing activating FcγR expression. This transient increase of A:I FcγR ratio on BM-resident macrophages, together with activation of macrophage for phagocytosis, migration, and apoptotic cell clearance could result in more effective macrophages for eliminating antibody-bound tumor cells in the BM. These results are consistent and complementary to data where patient-derived xenograft models were used to study this phenomenon in ‘double-hit’ lymphoma therapy(59).
In conclusion, our study further elucidated the nature of BM resistance, the mechanisms by which CTX synergized with antibody therapeutics, and generality of this synergy for other antibodies and human solid tumors. Despite major therapeutic advances, most hematological (e.g., mature B-cell malignancies) and metastatic solid cancers (e.g., breast cancer) remain incurable (60, 61). One potential facet underpinning treatment resistance is the powerful pro-survival microenvironment in the BM (29, 62–65). Our current study adds to the growing evidence that CTX could be used to overcome the suppressive BM microenvironment and, therefore, synergize with antibody-mediated elimination of tumor cells in the BM.
Supplementary Material
Acknowledgements and Grant Support
We would like to thank the Koch Institute Flow Cytometry (Glenn Paradis), Bioinformatics and Computing (Stuart Levin) and Animal Imaging and Preclinical Testing cores (Scott Malstrom) for their technical assistance, Starsha Kolodziej and Megan Kaiser for their support with generating and maintaining the humanized mice, Adam Drake for the parental lymphoma cell lines and Feng Shi for lentiviral vector expressing luciferase and puromycin. We would also like to thank the Antibody and Vaccine Group antibody production team and Alison Tutt for the provision of FITC-labelled FcγR antibodies, Ruth French for advice on statistical analysis. This work was supported in part by a Bloodwise Visiting Fellowship grant (14043) to AR, the Southampton CRUK Experimental Cancer Medicine Centre, the Koch Institute Support (core) Grant P30-CA14051 from the National Cancer Institute, and NIH grants 1R01NS104315 and 1R35CA197605.
Footnotes
Conflicts of Interest: A.R., S.A.B., M.J.G., S.H.L., and M.S.C. receive institutional support from BioInvent International for grants and patents. M.S.C. is a retained consultant for BioInvent International.
References
- 1.Pallasch CP et al. , Sensitizing protective tumor microenvironments to antibody-mediated therapy. Cell 156, 590–602 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lux A et al. , A humanized mouse identifies the bone marrow as a niche with low therapeutic IgG activity. Cell reports 7, 236–248 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Macedo F et al. , Bone Metastases: An Overview. Oncol Rev 11, 321 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Croucher PI, McDonald MM, Martin TJ, Bone metastasis: the importance of the neighbourhood. Nat Rev Cancer 16, 373–386 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Gul N et al. , Macrophages eliminate circulating tumor cells after monoclonal antibody therapy. The Journal of clinical investigation 124, 812–823 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gul N, van Egmond M, Antibody-Dependent Phagocytosis of Tumor Cells by Macrophages: A Potent Effector Mechanism of Monoclonal Antibody Therapy of Cancer. Cancer research 75, 5008–5013 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Beers SA et al. , Antigenic modulation limits the efficacy of anti-CD20 antibodies: implications for antibody selection. Blood 115, 5191–5201 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P, Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol 14, 399–416 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nimmerjahn F, Gordan S, Lux A, FcgammaR dependent mechanisms of cytotoxic, agonistic, and neutralizing antibody activities. Trends in immunology 36, 325–336 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Dahal LN, Roghanian A, Beers SA, Cragg MS, FcgammaR requirements leading to successful immunotherapy. Immunological reviews 268, 104–122 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Roghanian A, Stopforth RJ, Dahal LN, Cragg MS, New revelations from an old receptor: Immunoregulatory functions of the inhibitory Fc gamma receptor, FcgammaRIIB (CD32B). Journal of leukocyte biology, (2018). [DOI] [PubMed]
- 12.Nimmerjahn F, Ravetch JV, Antibodies, Fc receptors and cancer. Current opinion in immunology 19, 239–245 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Clynes R, Antitumor antibodies in the treatment of cancer: Fc receptors link opsonic antibody with cellular immunity. Hematology/oncology clinics of North America 20, 585–612 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Dahal LN et al. , STING Activation Reverses Lymphoma-Mediated Resistance to Antibody Immunotherapy. Cancer research 77, 3619–3631 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bournazos S, Wang TT, Ravetch JV, The Role and Function of Fcgamma Receptors on Myeloid Cells. Microbiology spectrum 4, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benonisson H et al. , High FcgammaR Expression on Intratumoral Macrophages Enhances Tumor-Targeting Antibody Therapy. J Immunol 201, 3741–3749 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Clynes RA, Towers TL, Presta LG, Ravetch JV, Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nature medicine 6, 443–446 (2000). [DOI] [PubMed] [Google Scholar]
- 18.Arce Vargas F et al. , Fc-Optimized Anti-CD25 Depletes Tumor-Infiltrating Regulatory T Cells and Synergizes with PD-1 Blockade to Eradicate Established Tumors. Immunity 46, 577–586 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Proietti E et al. , Importance of cyclophosphamide-induced bystander effect on T cells for a successful tumor eradication in response to adoptive immunotherapy in mice. The Journal of clinical investigation 101, 429–441 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sistigu A et al. , Immunomodulatory effects of cyclophosphamide and implementations for vaccine design. Seminars in immunopathology 33, 369–383 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Scurr M et al. , Low-Dose Cyclophosphamide Induces Antitumor T-Cell Responses, which Associate with Survival in Metastatic Colorectal Cancer. Clin Cancer Res 23, 6771–6780 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Radojcic V et al. , Cyclophosphamide resets dendritic cell homeostasis and enhances antitumor immunity through effects that extend beyond regulatory T cell elimination. Cancer immunology, immunotherapy : CII 59, 137–148 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moschella F et al. , Unraveling cancer chemoimmunotherapy mechanisms by gene and protein expression profiling of responses to cyclophosphamide. Cancer research 71, 3528–3539 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Ahlmann M, Hempel G, The effect of cyclophosphamide on the immune system: implications for clinical cancer therapy. Cancer Chemother Pharmacol 78, 661–671 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Ghiringhelli F et al. , CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol 34, 336–344 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Madondo MT, Quinn M, Plebanski M, Low dose cyclophosphamide: Mechanisms of T cell modulation. Cancer Treat Rev 42, 3–9 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Doloff JC, Waxman DJ, VEGF receptor inhibitors block the ability of metronomically dosed cyclophosphamide to activate innate immunity-induced tumor regression. Cancer research 72, 1103–1115 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nickenig C et al. , Combined cyclophosphamide, vincristine, doxorubicin, and prednisone (CHOP) improves response rates but not survival and has lower hematologic toxicity compared with combined mitoxantrone, chlorambucil, and prednisone (MCP) in follicular and mantle cell lymphomas: results of a prospective randomized trial of the German Low-Grade Lymphoma Study Group. Cancer 107, 1014–1022 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Scott DW, Gascoyne RD, The tumour microenvironment in B cell lymphomas. Nat Rev Cancer 14, 517–534 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Habermann TM et al. , Rituximab-CHOP versus CHOP alone or with maintenance rituximab in older patients with diffuse large B-cell lymphoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 24, 3121–3127 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Keating MJ et al. , Early results of a chemoimmunotherapy regimen of fludarabine, cyclophosphamide, and rituximab as initial therapy for chronic lymphocytic leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 23, 4079–4088 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Taskinen M, Karjalainen-Lindsberg ML, Nyman H, Eerola LM, Leppa S, A high tumor-associated macrophage content predicts favorable outcome in follicular lymphoma patients treated with rituximab and cyclophosphamide-doxorubicin-vincristine-prednisone. Clin Cancer Res 13, 5784–5789 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Drake AC, Chen Q, Chen J, Engineering humanized mice for improved hematopoietic reconstitution. Cellular & molecular immunology 9, 215–224 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leskov I et al. , Rapid generation of human B-cell lymphomas via combined expression of Myc and Bcl2 and their use as a preclinical model for biological therapies. Oncogene 32, 1066–1072 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Elliott TJ, Glennie MJ, McBride HM, Stevenson GT, Analysis of the interaction of antibodies with immunoglobulin idiotypes on neoplastic B lymphocytes: implications for immunotherapy. J Immunol 138, 981–988 (1987). [PubMed] [Google Scholar]
- 36.Tutt AL et al. , Development and Characterization of Monoclonal Antibodies Specific for Mouse and Human Fcgamma Receptors. J Immunol 195, 5503–5516 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Langmead B, Trapnell C, Pop M, Salzberg SL, Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li B, Dewey CN, RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robinson MD, McCarthy DJ, Smyth GK, edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang DW et al. , DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res 35, W169–175 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Subramanian A et al. , Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martinez FO, Gordon S, Locati M, Mantovani A, Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol 177, 7303–7311 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Saeed AI et al. , TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34, 374–378 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Lim SH et al. , Fc gamma receptor IIb on target B cells promotes rituximab internalization and reduces clinical efficacy. Blood 118, 2530–2540 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Aschenbrenner AC, Schultze JL, New “programmers” in tissue macrophage activation. Pflugers Archiv : European journal of physiology 469, 375–383 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Xue J et al. , Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 40, 274–288 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Slamon DJ et al. , Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 344, 783–792 (2001). [DOI] [PubMed] [Google Scholar]
- 48.Chen G et al. , A feasibility study of cyclophosphamide, trastuzumab, and an allogeneic GM-CSF-secreting breast tumor vaccine for HER2+ metastatic breast cancer. Cancer Immunol Res 2, 949–961 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arce Vargas F et al. , Fc Effector Function Contributes to the Activity of Human Anti-CTLA-4 Antibodies. Cancer Cell 33, 649–663 e644 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bulliard Y et al. , Activating Fc gamma receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J Exp Med 210, 1685–1693 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.York MR et al. , A macrophage marker, Siglec-1, is increased on circulating monocytes in patients with systemic sclerosis and induced by type I interferons and toll-like receptor agonists. Arthritis Rheum 56, 1010–1020 (2007). [DOI] [PubMed] [Google Scholar]
- 52.Macauley MS, Crocker PR, Paulson JC, Siglec-mediated regulation of immune cell function in disease. Nat Rev Immunol 14, 653–666 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang R, Town T, Gokarn V, Flavell RA, Chandawarkar RY, HSP70 enhances macrophage phagocytosis by interaction with lipid raft-associated TLR-7 and upregulating p38 MAPK and PI3K pathways. J Surg Res 136, 58–69 (2006). [DOI] [PubMed] [Google Scholar]
- 54.Asea A et al. , HSP70 stimulates cytokine production through a CD14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nature medicine 6, 435–442 (2000). [DOI] [PubMed] [Google Scholar]
- 55.Petrovic-Djergovic D et al. , CXCL10 induces the recruitment of monocyte-derived macrophages into kidney, which aggravate puromycin aminonucleoside nephrosis. Clin Exp Immunol 180, 305–315 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhan Y et al. , Ets-1 is a critical regulator of Ang II-mediated vascular inflammation and remodeling. The Journal of clinical investigation 115, 2508–2516 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rankin CT et al. , CD32B, the human inhibitory Fc-gamma receptor IIB, as a target for monoclonal antibody therapy of B-cell lymphoma. Blood 108, 2384–2391 (2006). [DOI] [PubMed] [Google Scholar]
- 58.Roghanian A et al. , Antagonistic human FcgammaRIIB (CD32B) antibodies have anti-tumor activity and overcome resistance to antibody therapy in vivo. Cancer Cell 27, 473–488 (2015). [DOI] [PubMed] [Google Scholar]
- 59.Lossos C et al. , Mechanisms of Lymphoma Clearance Induced by High-Dose Alkylating Agents. Cancer Discov 9, 944–961 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Burger JA, Ghia P, Rosenwald A, Caligaris-Cappio F, The microenvironment in mature B-cell malignancies: a target for new treatment strategies. Blood 114, 3367–3375 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nahta R, Yu D, Hung MC, Hortobagyi GN, Esteva FJ, Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nat Clin Pract Oncol 3, 269–280 (2006). [DOI] [PubMed] [Google Scholar]
- 62.Liotta LA, Kohn EC, The microenvironment of the tumour-host interface. Nature 411, 375–379 (2001). [DOI] [PubMed] [Google Scholar]
- 63.Bakker E, Qattan M, Mutti L, Demonacos C, Krstic-Demonacos M, The role of microenvironment and immunity in drug response in leukemia. Biochimica et biophysica acta 1863, 414–426 (2016). [DOI] [PubMed] [Google Scholar]
- 64.Meads MB, Hazlehurst LA, Dalton WS, The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin Cancer Res 14, 2519–2526 (2008). [DOI] [PubMed] [Google Scholar]
- 65.Tikhonova AN et al. , The bone marrow microenvironment at single-cell resolution. Nature, (2019). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.