

Abstract
Cellular replacement in the heart is restricted to postnatal stages with the adult heart largely postmitotic. Studies show that loss of regenerative properties in cardiac cells seems to coincide with alterations in metabolism during postnatal development and maturation. Nevertheless, whether changes in cellular metabolism are linked to functional alternations in cardiac cells is not well studied. We report here a novel role for uncoupling protein 2 (UCP2) in regulation of functional properties in cardiac tissue derived stem‐like cells (CTSCs). CTSC were isolated from C57BL/6 mice aged 2 days (nCTSC), 2 month (CTSC), and 2 years old (aCTSC), subjected to bulk‐RNA sequencing that identifies unique transcriptome significantly different between CTSC populations from young and old heart. Moreover, results show that UCP2 is highly expressed in CTSCs from the neonatal heart and is linked to maintenance of glycolysis, proliferation, and survival. With age, UCP2 is reduced shifting energy metabolism to oxidative phosphorylation inversely affecting cellular proliferation and survival in aged CTSCs. Loss of UCP2 in neonatal CTSCs reduces extracellular acidification rate and glycolysis together with reduced cellular proliferation and survival. Mechanistically, UCP2 silencing is linked to significant alteration of mitochondrial genes together with cell cycle and survival signaling pathways as identified by RNA‐sequencing and STRING bioinformatic analysis. Hence, our study shows UCP2‐mediated metabolic profile regulates functional properties of cardiac cells during transition from neonatal to aging cardiac states.
Keywords: age, cardiac cells, heart, neonatal, stem cells, UCP2
Loss in cardiac regeneration coincides with metabolic alterations. Whether cellular metabolism regulates cardiac cells function from young to old age remains unknown. Here we demonstrate that uncoupling protein 2 (UCP2) promotes increased glycolysis, proliferation, and survival in cardiac tissue derived stem‐like cells in neonatal heart. In aging, UCP2 is reduced increasing oxidative phosphorylation, reducing glycolysis parallel with reduced proliferation and survival.
Significance statement.
This study found that cardiac tissue derived stem‐like cells (CTSCs) from neonatal (2‐day‐old) and aged (2‐year‐old) mouse heart exhibit unique expression of genes as determined by bulk sequencing analysis. Neonatal CTSCs express genes that promote metabolism designed to increase functional properties of the cells. However, CTSC aging is accompanied by adverse changes in expression of genes that alters metabolism associated with decreased function. Additionally, this study reports a novel role for mitochondrial protein UCP2 in mediating salutary effects on neonatal CTSC whereas loss of UCP2 with age or in neonatal CTSCs blocks beneficial effects of metabolism on cellular function.
1. INTRODUCTION
Cardiovascular disease continues to be the leading cause of death in both men and women. Adult heart exhibits low cellular replacement 1 , 2 and is unable to replace dead cardiomyocytes (CMs) due to injury 3 initiating a downward spiral of adverse ventricular remodeling that ends up in heart failure. In contrast, developmental cardiac tissue is a proliferative organ capable of regenerating lost myocardium after injury. 4 , 5 , 6 Interestingly, cardiac cells exhibit a specialized metabolic profile during development that alters as the heart undergoes maturation and coincides with loss of regenerative processes. 5 Whether cellular metabolism regulates regenerative processes in the heart is not well studied.
Aging and disease are known to adversely affect cellular function in the heart with implications for cardiac tissue homeostasis and cellular turnover. 1 , 2 Recently, cell metabolism and regulation of cell fate decisions during normal tissue homeostasis and aging has gained importance. 7 Developmental tissue harbors pluripotent stem cells (PSCs) with specialized metabolism that adapts to changes in pluripotency, self‐renewal, proliferation, and differentiation. 8 , 9 Adult tissue stem cells such as hematopoietic stem cells largely exist in quiescence favoring glycolysis that preserves stemness. 10 Cellular differentiation requires adult stem cells to undergo a metabolic shift toward oxidative phosphorylation (OXPHOS) increasing reactive oxygen species (ROS) generation. 11 Accumulating ROS levels overtime combined with enhanced organismal age ultimately pushes adult stem cells toward exhaustion and stem cell aging. In the context of the heart, evidence suggests that cardiac progenitor cells (CPCs) reside in hypoxic niches 12 , 13 and their emergence from the niche is characterized by metabolic changes. Recently, Zhang et al document a critical role played by a mitochondrial gene called uncoupling protein 2 (UCP2) for regulation of PSC metabolism and differentiation. 14 UCP2 regulates energy generation and redox homeostasis, 15 is known to be expressed in the heart 16 and promotes cardioprotection in response to injury. 17 Nevertheless, whether cellular metabolism and in particular UCP2 is linked to enhanced functional properties of cardiac cells in the heart during the regenerative postnatal period 18 or toward regulation of adverse changes accrued in cardiac cells with age remains untested.
In this article, we show that transcriptomic analysis and functional characterization of cardiac tissue derived stem‐like cells (CTSCs) populations in the neonatal, adult, and aged heart demonstrate unique transcriptome and increased functional properties of neonatal CTSC. Our data suggest that neonatal CTSCs possess a cellular metabolic profile linked to enhanced expression of mitochondrial protein UCP2 that alters with age, potentially opening cellular metabolism as a new avenue for enhancement of cell‐based therapies for cardiac repair.
2. MATERIALS AND METHODS
2.1. Cell isolation and culture
All CTSCs were isolated from the hearts of C57BL/6 Mice (Jackson). Hearts were subjected to novel tissue digestion method, and separation of various cellular populations by plating, and replating of supernatant to remove fast adhering cells. CTSC Growth Media composition was similar to previously published 19 and included DMEM‐F12 (Gibco), 10% Embryonic Stem Cell Fetal Bovine Serum (Gibco), 1% Penicillin‐Streptomycin‐Glutamine (PSG) (Gibco), 1x Insulin‐Transferrin‐Selenium (Gibco), Recombinant Human‐EGF 10 ng/mL (Peprotech), Recombinant Human‐FGF 10 ng/mL (Peprotech), and Leukemia Inhibitory Factor (Millipore Sigma), and incubated in a 37°C 5% CO2 incubator. Routine cell culturing passaging included dissociation of cells using 0.25% Trypsin EDTA (Gibco) and centrifugation for 5 minutes at 1500 RPM. Pelleted cells were resuspended in CTSC Growth media and counted using a hemocytometer and plated. For all subsequent experiments, all three CTSCs types were kept between passages 10 and 15.
2.2. CyQuant, MTT, PDT, resazurin, viability assays
CyQuant NF Cell Proliferation (Invitrogen) assay, Resazurin (CST), and MTT (ATCC) assay were conducted by plating CTSCs in quadruplicate (2000 cells/well) in a 96‐well flat bottom plate. Assays were performed based on manufacturer's instructions. Population doubling time (PDT) and viability assays were performed by plating 50 000 cells/well in a 6‐well plate. Trypan blue stain was added to calculate viability and an online population doubling time calculator was used (http://www.doubling-time.com/compute.php). Assays were conducted on days 1, 3, and 5 postplating.
2.3. RNA‐sequencing
CTSCs were prepared for bulk‐RNA sequencing with three replicates of each CTSC line consisting of 1 million cells per replicate cultured at passage 10 for 48 hours prior to sequencing. Samples were sent out to GeneWiz for RNA isolation, cDNA library construction, and sequencing. In brief, RNA was isolated from cells by GeneWiz including mRNA sequencing via PolyA selection. Library preparation involved fragmentation and random priming, strand cDNA synthesis, end repair, and adapter ligation with polymerase chain reaction (PCR) enrichment for sequencing. Sequencing was carried out on Illumina HiSeq System with 2x 150 bp PE HO configuration.
2.4. Bioinformatics analysis
Raw data quality was evaluated with FastQC, reads were trimmed using Trimmomatric v.0.36 to remove adapter sequences and poor‐quality nucleotides. Reads were then mapped onto Mus musculus GRCm38 reference genome using STAR aligner v.2.5.2b. Genome was available on ENSEMBL. Generated BAM files were used to extract unique gene hit counts using FeatureCounts from Subread package v.1.5.2. Genes were identified on mapped reads followed by downstream differential expression analysis using R package DESeq2. Genes with less than 5 reads per sample were removed. Wald test was used to statistically identify genes with P value <.05. Using regularized logarithm (rlog) function, count data was transformed for visualization on a log2 scale.
2.5. Immunocytochemistry
Immunocytochemistry and TUNEL assays were performed as described previously 20 , 21 with additional detail in Supporting Information including a list of antibodies in Table S2.
2.6. Measurement of oxygen consumption rate, extracellular acidification rate, and fuel flexibility
A Seahorse Bioscience XF96 Extracellular Flux Analyzer was utilized to measure oxygen consumption rates (OCRs), extracellular acidification rate (ECAR), and fuel flexibility in CTSCs using a protocol similar to that was previously reported. 20 At the conclusion of each experiment, cells were lysed in RIPA buffer and protein concentration was determined for each well using a standard Bradford assay. All calculations for assessment of OCR/ECAR were reported as mean ± SD (pmol O2/[min mg]).
2.7. ATP content determination
CTSCs were isolated by trypsinization and the pellet was used to determine ATP according to manufacturer's instruction (Invitrogen).
2.8. Proteome profiler array
Mouse angiogenesis array assay was conducted according to manufacturer's instruction (R&D Systems). Cell culture supernatant was collected to perform assay. Quantification of dot‐blot intensity was carried out on ImageJ.
2.9. Lactate, pyruvate, PK activity assays
CTSCs were grown to confluence followed by preparation for measurement of lactate (Biovision), pyruvate (Abcam), and pyruvate kinase activity (Biovision) for all three cell types according to manufacturer's protocol.
2.10. Immunoblot
Immunoblot analysis was performed as described previously 19 , 21 with additional detail in the Supporting Information.
2.11. RNA interference
Small interfering RNA (siRNA; 20 μmol/L) for UCP2 (Dharmacon) transfection was performed with CTSC plated in 6‐well dishes and transfected with 3 μL of siRNA and 12 μL of Hi Perfect transfection reagent (Qiagen) in 85 μL of DMEM/F12 without serum.
2.12. Statistical analysis
Statistical analysis is performed using unpaired Student's t test for data comparing two groups and one‐way or two‐way analysis of variance (ANOVA) with Bonferroni post hoc test for comparing more than two groups for data exhibiting normal distribution. For data that do not exhibit normal distribution, Mann‐Whitney test was used. All data sets were assessed for normality using Shapiro‐Wilk test. P < .05 is considered statistically significant. Error bars represent ±SD. Statistical analysis is performed using Graph Pad prism v 8.0 software.
3. RESULTS
3.1. Isolation and characterization of CTSCs
In this study, we describe isolation of CTSCs from the heart during cardiac tissue maturation and aging. For this purpose, hearts from 2‐day‐old, 2‐month‐old, and 2‐year‐old C57BL/6 mice were subjected to enzymatic digestion followed by plating, replating supernatant and passaging in culture media that led to emergence of cells that were designated as CTSCs (Figure 1A). Morphological analysis of three CTSC lines demonstrated a spindle like shape in CTSCs from 2‐month‐old mice, spindle‐flattened shape for aCTSC from 2‐year‐old mice, and increased roundness in nCTSC from 2‐day‐old mice (Figure 1B,C). Analysis of cell area showed smaller cell body in nCTSC (1437.7 μm2) compared to aCTSC that were more flattened (Figure 1D,E). Analysis of cell surface markers by flow cytometry demonstrated three CTSCs lines to be negative for hematopoietic markers CD45, CD11b, CD105, and c‐kit. CTSCs lines were low Isl1+ (Table S2E) VCAM+, and high Sca‐1+, Integrin B1+ and ecto‐5‐nucleotidase+ (Figure S1A), ABCG1+ (Table S2E) in conjunction with quantitative real time polymerase chain reaction (qRT‐PCR) expression analysis (Figure S1B) and immunocytochemistry‐based detection (Figure S1C‐F). To determine whether CTSC media promotes stem cell characteristics, CMs and cardiac fibroblasts (CFs) were isolated from neonatal and adult mouse hearts and plated in CTSC medium. No morphological changes were observed in both cell types after 10 days of culture from neonatal and adult mouse that indicate a stem cell phenotype, in fact, there was significant mortality in adult CF and CM cultures (Figure S1G). The main question was whether CTSCs represent an unknown cell population and for this purpose, isolated CTSCs lines were subjected to bulk‐RNA sequencing followed by comparison to transcriptome of cardiac cell types and stem cells using previously published databases. 22 , 23 , 24 Principal component analysis (PCA) demonstrated CTSC lines transcriptome to show high degree of variance in comparison to CMs, CF, endothelial cells (ECs), CPCs, and stem cell types such as ESCs and mesenchymal stem cells (MSCs) (Figure 1F). Additionally, comparison of putative genes for CM shows 36.3%, CF 41.1%, ECs 60.2%, CPCs 57.1%, and MSCs 59.3% homology with nCTSCs (Figure 1G) (Tables S2A‐F). Interestingly, nCTSCs transcriptome showed least variance to ESCs and shared 31.7% homology (Figure 1F,G) with expression of pluripotent markers such as Oct‐4, Sox‐2, Nanog (Figure S2A), and LIN28a as measured by qRT‐PCR, Western blot, and immunocytochemistry (Figure S2B‐D). Expression of pluripotent markers was significantly increased in nCTSC compared to CTSCs while aCTSC had no or low expression for pluripotent genes (Figure S2B‐D). Finally, we compared nCTSCs to recently described nine CPC populations in the embryonic heart 25 with the idea to conduct a qualitative assessment of gene expression between nCTSC as the most unaltered cell type in the postnatal heart, to the published database. Results showed that nCTSC share the highest percentage of top markers expressed by the progenitor cell populations of the anterior heart field (AHF) suggesting possible origin and requires further validation by lineage tracing experiments (Table S3). Collectively, the results suggest CTSCs lines to be a novel cell population with stem cell like characteristics that persists in the adult heart of normal and aged animals.
FIGURE 1.
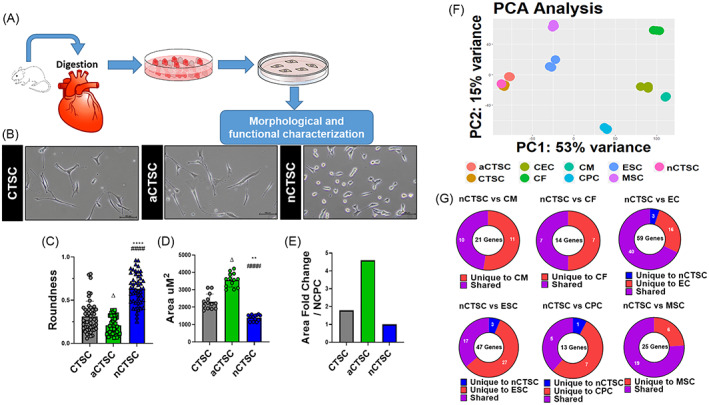
RNA‐sequencing identifies CTSCs in the heart. A, Illustration describes isolation strategy for CTSCs based upon digestion of whole heart from 2‐day‐old, 2‐month‐old, and 2‐year‐old mice followed by plating in growth medium. At day 1 postplating, adherent cells were discarded, and supernatant was replated and the cell were grown for 2 to 3 weeks that led to emergence of CTSCs followed by analysis molecular and functional analyses. B, Morphological analysis of the CTSC populations isolated from 2‐day‐old (nCTSC), 2‐month‐old (CTSC), and 2‐year‐old (aCTSC) mice. C, Roundness, D, cell area, and, E, Area fold change was calculated using ImageJ. Scale bar = 100 μm. n = 20 number of cell counted/three independent experiments for all three cell lines. F, Principal component analysis details clustering of biological replicates of the same condition between CTSCs population from different ages and other cardiac cell and stem cells types (n = 3 replicates/cell lines used for RNA sequencing). G, Comparison of CTSC transcriptome to putative markers for CMs, CFs, ECs, ESCs, MSCs, and CPCs using R&D systems database. nCTSC vs CTSCs *P < .05, **P < .01, ***P < .001, nCTSC vs aCTSC # P < .05, ## P < .01, ### P < .001, and nCTSC vs aCTSCs Δ P < .05, ΔΔ P < .01, ΔΔΔ P < .001. See also Figures S1 and S2, and Tables S2 and S3. CFs, cardiac fibroblasts; CMs, cardiomyocytes; CPCs, cardiac progenitor cells; CTSCs, cardiac tissue derived stem‐like cells; ECs, endothelial cells; MSCs, mesenchymal stem cells
3.2. Proliferation, survival, migration, and senescence alter with age in three CTSCs lines
The next set of studies aimed to characterize proliferation and survival in three CTSC lines and alteration with age. Increased cell counts (Figure 2A) in conjunction with significant decrease in population doubling time (PDT) (Figure 2B) for nCTSC compared to CTSC while aCTSC were the slowest. Similarly, nCTSC showed increased proliferation (Figure 2C), metabolic activity (Figure 2D), and viability (Figure 2E) compared to CTSC, while aCTSC showed lowest proliferation potential. Cell cycle in three CTSC lines showed increased percentage of nCTSC in S/G2 phases of the cell cycle with corresponding decrease in G0/G1 phase compared to CTSCs while aCTSC show inverse cell cycle phasing (Figure 2F). To assess survival, all three CTSC types were exposed to 18 hours H2O2 induced oxidative stress followed by TUNEL staining. nCTSC showed significantly reduced while aCTSC the highest TUNEL+ nuclei compared to CTSC (Figure S3A). Next, a wound healing assay for all three cell types was performed to determine age‐associated changes in cell migration. Results showed that all three cell lines possess the ability to migrate but nCTSC have the most area covered while aCTSC have the least, 24 hours postinjury (Figure S3B). Finally, senescence associated β‐galactosidase staining showed nCTSC exhibit significantly lower β‐gal+ cells (1.4%) compared to CTSCs (11.8%) while aCTSC had the highest number of β‐gal+ cells (21.5%) (Figure S3C). Collectively, nCTSC represent a highly proliferative cell type within the cardiac tissue possibly due to minimal adverse effects of age and environment while aCTSC exhibit adverse changes that are hallmarks of stem cell aging.
FIGURE 2.
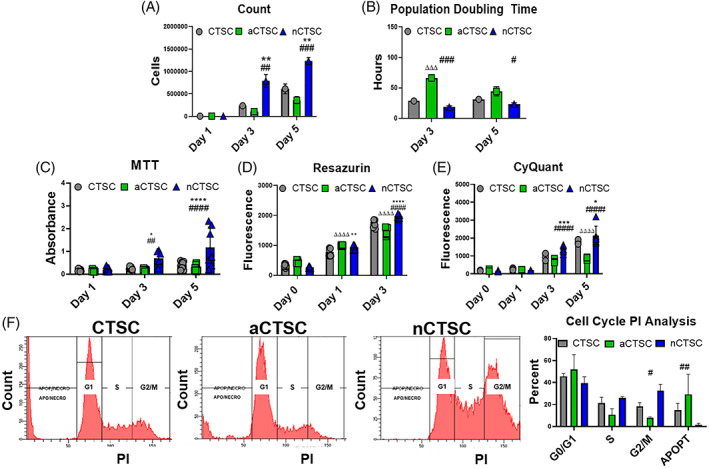
Increased viability, metabolic activity, and proliferation rate in nCTSC compared to other CTSC populations. A, Viability cell counts show increased cell number in nCTSCs compared to CTSC and aCTSC over the course of 5 days in culture (n = 6). B, Decreased population doubling time in nCTSC compared to CTSCs and aCTSC (n = 6). C, Increased metabolic activity in nCTSC compared to CTSCs and aCTSC at day 3 and 5 after culture as measured by MTT assay (n = 5) as well as Resazurin assay (n = 5) in (D). E, nCTSC exhibit increased cell proliferation at day 3 and day 5 compared to CTSCs and aCTSC as assessed by CyQuant assay (n = 5). F, Flow cytometry‐based measurement of cell cycle shows decreased percentage of cells in G1 and a corresponding increase in percentage of cells in S/G2 phases in nCTSC compared to CTSC and aCTSC (n = 3). nCTSC vs CTSCs *P < .05, **P < .01, ***P < .001, nCTSC vs aCTSC # P < .05, ## P < .01, ### P < .001, and nCTSC vs aCTSCs Δ P < .05, ΔΔ P < .01, ΔΔΔ P < .001. See also Figures S3 and S4. CTSCs, cardiac tissue derived stem‐like cells
3.3. Age‐induced changes in secretome from three CTSC lines
Cell‐based therapy for cardiac repair shows increased benefit for cardiac structure and function even though less few cells survive post transplantation. 26 Salutary effects of the transplanted stem cells have been attributed to the ability to secrete pro‐regenerative factors at the site of injury. Therefore, secretome of three CTSCs was assessed for secreted cytokines and chemokines and to determine whether differences between secretome from neonatal and aged heart. Proteome profiler array‐based detection showed differential expression of various growth factors, cytokines, and chemokines categorized as having a role in inflammation, cell migration, cellular growth, cellular development, or being antiangiogenic (Figure S4A). Interestingly, growth factors such as Amphiregulin, PDGF‐AA, and IGFBP3 were either exclusively or highly present in nCTSC while increased expression of pro‐inflammatory factors was found in aCTSC secretome. Additionally, expression of growth factors such as SDF‐1, HGF, VEGF, and IGF‐1 was validated by Western blot analysis. Results showed significantly high expression in nCTSC compared to CTSCs and aCTSC for all the growth factors assessed (Figure S4B,C). Taken together, secretome in the three CTSC lines is diverse and unique and changes substantially with increasing age that may have influence on the therapeutic efficacy of CTSCs lines for cardiac repair.
3.4. Bulk‐RNA sequencing identifies unique metabolic properties for CTSCs lines
Analysis of morphological and functional properties showed significant differences in CTSC lines that become pronounced with age. In order to identify mechanistic basis for functional differences, transcriptome from three CTSC lines was assessed by RNA‐sequencing followed by bioinformatics analyses (Figure S5A). Bulk RNA‐sequencing demonstrated a total of 19 303 genes altered between the three cell populations. In comparison to CTSCs, nCTSC showed 24% significantly upregulated and 27% significantly downregulated genes, in contrast to 27% significantly upregulated genes and 29% significantly downregulated genes observed in aCTSC transcriptome (Figure 3A). Volcano plot representation detailed genes that were not only significant but also distinguished by a log fold‐change threshold of 1.5 (Figure 3B). Next, a PCA for all three CTSC lines was performed that showed consistency within each sample transcriptome together with identification of variance between each cell line (Figure 3C). Using differentially expressed genes (DEGs) data sets, gene ontology (GO) and gene set enrichment (GSEA) analyses were conducted. GO analysis identified the top over‐represented terms for biological processes including significant alteration in metabolic processes within nCTSC and aCTSC compared to CTSCs (Figure 3D). Moreover, comparison of nCTSCs with aCTSC identified GO terms significantly altered with age together with GSEA analysis for changes in cell cycle and fatty‐acid metabolism signaling pathways (Figure S5H,I). Interestingly, significant GO terms for nCTSC in comparison to CTSCs included cell proliferation and cell migration while in addition to the terms found in nCTSC, aCTSCs demonstrated significant representation of apoptotic processes and response to oxidative stress. Since GO analysis showed metabolic gene alterations in CTSCs lines therefore expression of genes involved in various metabolic signaling pathways was visualized using rlog‐based expression values normalized via row‐z score. Genes involved in glycolysis, oxidative phosphorylation, fatty‐acid metabolism, amino‐acid metabolism, glutamine metabolism, and glutathione metabolism were expressed as heatmaps that detail their expression in each sample. Increased expression of glycolytic genes was observed in nCTSCs compared to CTSCs while aCTSC had greater expression of metabolic genes involved in oxidative phosphorylation and fatty‐acid metabolism (Figure S5B‐G). Using ClusterProfiler package, a KEGG enrichment analysis was conducted to identify over‐represented signaling pathways present amongst the CTSC lines. KEGG analysis revealed terms over‐represented such as fatty acid biosynthesis and cell cycle. Finally, GSEA was conducted to identify gene sets present in nCTSC vs aCTSC. GSEA noted enriched KEGG‐based GSEA terms such as cell cycle, glycolysis, and fatty acid metabolism (Figure 3E). Collectively, GO analysis and GSEA reveal a unique profile present in both nCTSC and aCTSC when compared to CTSCs and a significant enrichment of genes involved in regulation of metabolic processes that were altered with age.
FIGURE 3.
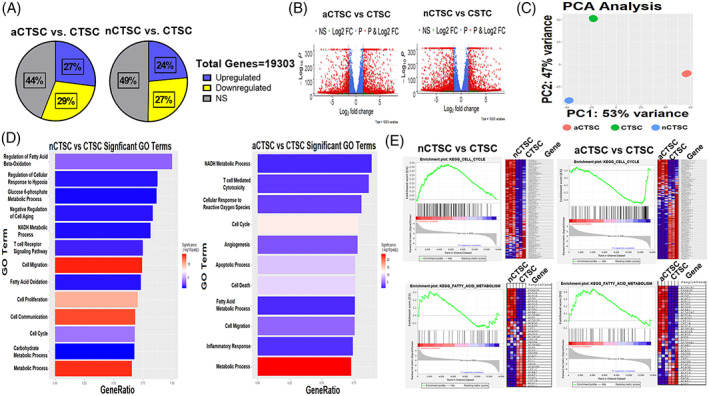
Alterations in metabolic processes underlies CTSC transition from neonatal to aged animals. A, Global transcriptome visualization of significantly upregulated, downregulated, or unchanged genes between nCTSC, aCTSC in comparison to CTSCs. A total of 19 303 genes were identified between cell lines. B, Volcano Plot is generated as visualization after differential expression analysis. Plot details fold difference between log2 normalized expressions vs −log10(adjusted P value). Each dot represents a gene and is colored based on the threshold set for log2 FC (±1.5) and −log10(adjusted P value) (>5). C, PCA plot details clustering of biological replicates of the same condition. Plot confirms biological samples (n = 3) are of great similarity with differences greatest compared to aCTSC. D, GO plot identifies top significant biological processes‐based GO terms. Plot details −log10(adjusted P value) based in intensity of each bar fill and are ranked based on number of DEG/total genes present in GO term (GeneRatio). E, GSEA‐enrichment plots derived from the Broad Institute software identifies top enriched gene sets based on MSigDB's KEGG curated gene sets, Cell Cycle, and Fatty Acid Metabolism. Plot is described in multiple portions. The running enrichment score (top portion) for the gene set represents ranked list of genes. The leading‐edge subset (middle) of the gene set are the genes that contribute most to the ES. The bottom portion of each plot shows the value of ranking matrices as it moves down the list of ranked genes. The colored horizontal bar changes from red to blue to detail a change from positively correlated genes (red) to negatively correlated genes (blue). Positive ESs indicate enrichment at the top of the ranked list while negative ESs point out enrichment at the bottom of the ranked list. See also Figure S5 and Tables S4 to S6. CTSCs, cardiac tissue derived stem‐like cells; DEGs, differentially expressed genes; ES, enrichment score; GO, gene ontology; GSEA, gene set enrichment analysis; PCA, principal component analysis
3.5. Increased glycolytic flux and decreased mitochondrial respiration for energy generation in nCTSC
PSCs possess unique cellular metabolism that enables high levels of proliferation and is altered by cellular differentiation. 8 , 9 Therefore, it was hypothesized that transition of CTSC from neonatal to aged heart leads to significant metabolic alterations. To further validate metabolic changes within CTSCs lines, OCRs and ECARs were measured for all three lines. Results demonstrated increased ECAR and glycolysis in nCTSC compared to CTSCs while aCTSC were the least glycolytic (Figure 4A). In conjunction, OCR was increased in aCTSC and the lowest in nCTSC compared to CTSCs (Figure 4B). Interestingly, nCTSCs exhibit increased proton leak despite enhanced ATP generation suggesting lack of mitochondrial respiration for energy production. Further validation showed highest ATP production in nCTSC compared to CTSCs and aCTSC (Figure 4C). nCTSC demonstrated significantly reduced pyruvate levels (Figure 4D) but higher pyruvate kinase activity (Figure 4E) and lactate production (Figure 4F) together with increased expression of PDK1 and reduced PDH compared to CTSCs and aCTSC (Figure 5A,B) in concurrence with cells demonstrating high glycolytic drive and cellular proliferation. Furthermore, nCTSCs exhibit lower citrate synthase activity (Figure 5C) and mtDNA (Figure 5D) together with lower mitochondrial membrane potential (Figure 5E) indicating inefficient mitochondrial respiration in nCTSC compared to aCTSC. Additionally, nCTSCs show increased glucose dependence, capacity, and inability to utilize fatty acids as a substrate (Figure 5F). Interestingly, a distinct mitochondrial morphology was observed by TOMM20 immunostaining (Figure 5G) together with changes in OPA1 expression (Figure S6D) in all three CTSC lines possibly linking changes in mitochondria to bioenergetics. Taken together, nCTSCs primarily generate energy through activation of glycolytic pathways in contrast to aCTSC that rely on oxidative phosphorylation, thereby providing a possible explanation for higher proliferative rates in nCTSC compared to aCTSC.
FIGURE 4.
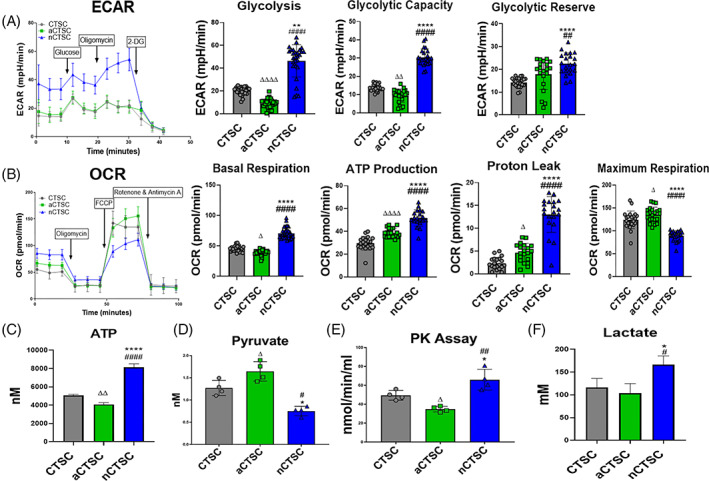
Assessment of cellular energetics shows increased glycolysis in nCTSC. A, Measurement of ECARs in nCTSC, CTSCs, aCTSC by seahorse bioanalyzer show increased ECARs, glycolysis, glycolytic capacity, and glycolytic reserve in nCTSC compared to CTSCs and aCTSC (n = 15 replicates/line/three independent experiments). B, Analysis of oxidative phosphorylation (OCR) shows significant reduction of OCR in nCTSC while aCTSC have the highest OCR compared to CTSCs together with measurement of basal respiration, ATP production, proton leak, and maximal respiration (n = 15 replicates/line/three independent experiments). Data for ECAR and OCR were normalized to cell number and protein content. C, ATP quantification assay shows increased ATP production in nCTSC compared to CTSCs and aCTSC (n = 4). D, nCTSC exhibit increased intracellular pyruvate levels as compared to CTSCs and aCTSC as measured by pyruvate measurement kit (n = 4). E, Increased activity of pyruvate kinase in nCTSC compared to CTSCs and aCTSC determine by pyruvate kinase assay kit (n = 4). F, Enhanced intracellular lactate production in nCTSC compared to CTSCs and aCTSC as measured by lactate detection kit (n = 4). nCTSC vs CTSCs *P < .05, **P < .01, ***P < .001, nCTSC vs aCTSC # P < .05, ## P < .01, ### P < .001, and nCTSC vs aCTSCs Δ P < .05, ΔΔ P < .01, ΔΔΔ P < .001. See also Figure S5. CTSCs, cardiac tissue derived stem‐like cells; ECARs, extracellular acidification rates; OCR, oxygen consumption rate
FIGURE 5.
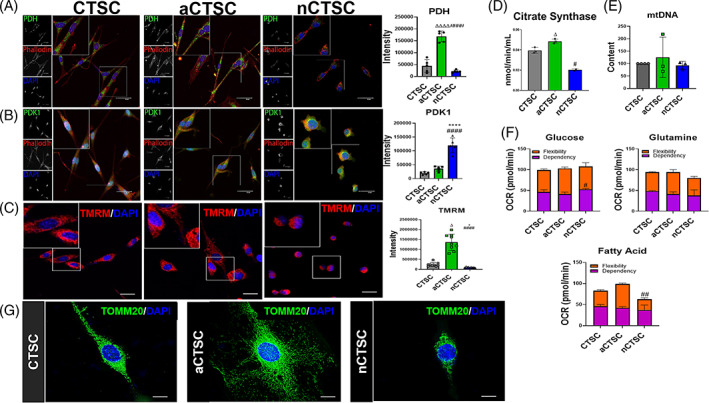
Decreased mitochondrial respiration in nCTSCs that alters with age. A, Decreased expression of PDH in nCTSC while aCTSC exhibit the highest PDH expression compared to CTSCs along with quantification. B, nCTSC show increased expression of PDK1 while aCTSC have the lowest as compared to CTSCs along with quantification. PDH/PDK1 (green), Phalloidin (red), nuclei (blue). Scale bar = 20 μm. C, Citrate synthase activity assay shows decreased activity in nCTSCs while highest in aCTSC as compared to CTSCs. D, Decreased mtDNA in nCTSC while aCTSC have the highest compared to CTSCs as measured by mtDNA content assessment (n = 4). E, nCTSC demonstrate low mitochondrial membrane potential visualized by TMRM staining while aCTSC have the highest TMRM intensity. TMRM (red), nuclei (blue), Scale bar = 20 μm (n = 4). F, Substrate flexibility, capacity, and dependence measurements in CTSCs using fuel flex assay (n = 6 replicates/line/three independent experiments). G, Unique mitochondrial morphology in three CTSC types analyzed for TOMM20 (green), nuclei (DAPI). Scale bar = 10 μm. nCTSC vs CTSCs *P < .05, **P < .01, ***P < .001, nCTSC vs aCTSC # P < .05, ## P < .01, ### P < .001, and nCTSC vs aCTSCs Δ P < .05, ΔΔ P < .01, ΔΔΔ P < .001. CTSCs, cardiac tissue derived stem‐like cells; DAPI, 4′,6‐diamidino‐2‐phenylindole; PDH, pyruvate dehydrogenase; TMRM, tetramethylrhodamine, methyl ester, perchlorate
3.6. UCP2 is essential for maintaining nCTSC metabolism and proliferation
nCTSC utilize glycolysis for energy generation together with increased proliferation rates, so the next question was to delineate the molecular signaling responsible for maintaining unique nCTSC metabolic profile. For this purpose, a screen for known metabolic targets was conducted on nCTSC using qRT‐PCR. As expected, increased expression of glycolytic enzymes was observed in nCTSC but interestingly, increased expression of mitochondrial genes such as uncoupling proteins 2 and 3 were observed compared to CTSCs and aCTSC (Figure 6A). Given the recent report highlighting the role of UCP2 in maintenance of pluripotency and energy dynamics, 14 it was hypothesized that nCTSC glycolytic phenotype and proliferative state may be regulated by UCP2. Expression of UCP2 was validated by western blot analysis confirming significantly high expression of UCP2 in nCTSC compared to CTSC and aCTSC (Figure 6B). To validate the UCP2 for maintenance of cell characteristics, nCTSC were subjected to siRNA knockdown of UCP2 (Figure 6C) while aCTSC were lentivirally modified to overexpress UCP2 followed by assessment of ECAR by seahorse analysis. Results showed loss of ECAR activity and glycolysis rather than a shift to oxidative phosphorylation (Figure 6D) in conjunction with significantly reduced expression of glycolytic enzymes and genes (Figure 6E) in siUCP2‐nCTSC compared to scrambled controls. Conversely, increased ECAR and glycolysis was observed in aCTSCs overexpressing UCP2 (Figure S6C). Knockdown of UCP2 in nCTSC impaired glucose flexibility thereby increasing fatty acid dependence for energy generation (Figure S6A), suggesting a role for UCP2 in maintaining glucose metabolism in nCTSC. The main question was whether loss of glycolytic metabolism impairs nCTSC ability to proliferate and survive and we observed a robust morphological change in nCTSC that exhibited a rounded apoptotic morphology after UCP2 knockdown compared to control cells (Figure S6B), making measurement of additional functional parameters challenging. Concurrently, loss of cell cycle progression and increased apoptosis after UCP2 blunting was observed compared to scrambled cells (Figure 6F) together with reduced metabolic activity (Figure 6G) and viability (Figure 6H) in nCTSC. Collectively, data suggest a critical role for UCP2 in regulating nCTSC energy dynamics that in turn affects nCTSC ability to proliferate and survive.
FIGURE 6.
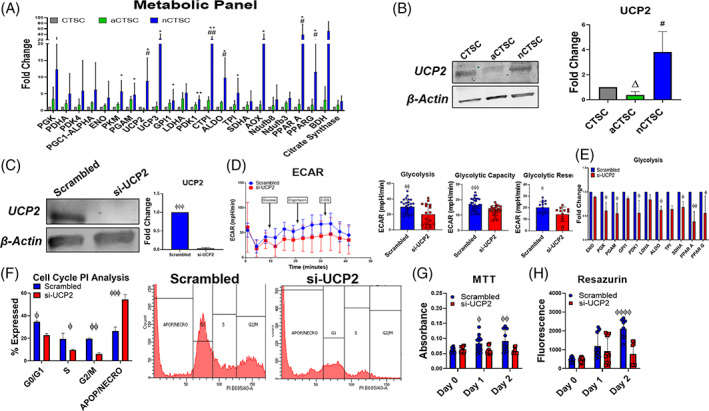
Identification of UCP2 as a regulator of nCTSC metabolic function and proliferation. A, Assessment of metabolic genes in CTSCs populations by qRT‐PCR analysis (n = 4). B, Increased expression of UCP2 expression in nCTSC while aCTSC have the lowest expression compared to CTSCs as confirmed by immunoblot (n = 4). C, Validation of UCP2 knockdown by siRNA in nCTSC compared to scrambled treated nCTSC by immunoblot (n = 4). D, Reduced ECAR, glycolysis, glycolytic capacity, and glycolytic reserve in siRNA treated nCTSC compared to scrambled controls as measured by seahorse assay (n = 4 replicates/line/three independent experiments). E, Reduced expression of glycolytic genes and enzymes in si‐UCP2 nCTSC compared to scrambled nCTSC as measured by qRT‐PCR analysis (n = 4). F, Flow cytometry cell cycle assessment shows increased apoptosis and reduced cell cycle activity in si‐UCP2 treated nCTSC compared to control nCTSC (n = 4). nCTSC vs siUCP2‐nCTSC *P < .05, **P < .01, ***P < .001. CTSCs, cardiac tissue derived stem‐like cells; ECAR, extracellular acidification rate; qRT‐PCR, quantitative real time polymerase chain reaction; siRNA, small interfering RNA; UCP2, uncoupling protein 2
3.7. Loss of UCP2 alters metabolic and proliferative signaling pathways in nCTSCs
In order to provide a mechanistic connection with UCP2 knockdown and impaired proliferation and survival, nCTSC with or without UCP2 silencing were subjected to RNA‐sequencing. Results showed that UCP2 silencing in nCTSC caused 26% genes to be downregulated and 24% upregulated while 50% remain unchanged (Figure 7A), further represented in a volcano plot showing significantly altered genes in siUCP2‐nCTSC compared to control (Figure 7B). Principle component analysis identified significant variance between siUCP2‐nCSTC and scrambled and UCP2 overexpressing nCTSCs (UCP2‐GFP) control cells (Figure 7C) together with GO analysis for over‐representation of biological processes using significantly altered DEGs present between si‐UCP2 and nCTSC. Over‐represented GO terms were filtered out for redundancy and visualized as clusters representing various biological processes using the online software REVIGO (Figure 7D). Additionally, top DEGs for cell cycle and survival pathways were represented as heat map showing a significant alteration of cell cycle/survival pathways in siUCP2‐nCTSC compared to scrambled cells (Figure 7E). Finally, to identify protein‐protein interactions, DEGs of si‐UCP2 and nCTSC were uploaded onto STRING software to identify protein‐protein interactions, functional enrichment analysis and network visualization through Cytoscape. With STRING's database and our DEG list, we were able to generate a network of proteins involved with UCP2 and the effect of UCP2 silencing on protein targets. Interestingly, several mitochondrial proteins belonging to inner and outer membrane were altered together with cell membrane receptors and survival/proliferation genes in nCTSC after UCP2 silencing indicating a critical role for UCP2 in maintenance of energy dynamics and cell proliferation and survival (Figure 7F). In summary, UCP2 silencing in nCTSC leads to impaired regulation of genes involved in energy dynamics, proliferation, and survival suggesting a role for UCP2 in maintenance of cellular function by modulating energy balance in the nCTSC.
FIGURE 7.
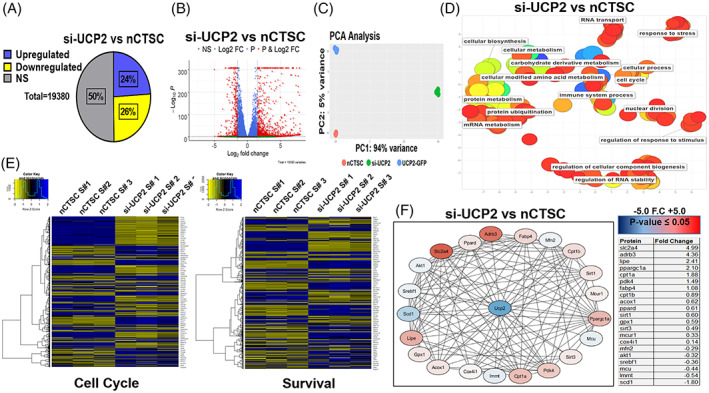
UCP2 silencing impairs nCTSC proliferation, survival, and metabolic signaling. A, Bulk‐RNA sequencing identifies significantly upregulated and downregulated genes in nCTSC after UCP2 silencing. B, Volcano plot representation of siUCP2 treated nCTSC compared to control nCTSC. C, Principal component analysis defines variance between nCTSCs and si‐UCP2‐nCTSCs and clusters biological replicates of the same condition while nCTSC overexpressing UCP2 were employed as a positive control condition. D, REVIGO analysis details significant GO terms clustered together. Color of each dot (GO Term) is the log10 (P value) generated from the GO analysis with blue as low and red as high. Size of the the dot is based on the frequency of the GO term. Broad based GO terms will have a larger dot size. E, Cell signaling pathways for cell cycle and survival are significantly altered between siUCP2‐nCTSC compared to nCTSC. F, Protein‐protein interactions analyzed by STRING software and visualized through Cytoscape shows the most highly upregulated, downregulated proteins in relation to UCP2 after knockdown in nCTSC. See also Figure S6 and Tables S7 and S8. CTSCs, cardiac tissue derived stem‐like cells; GO, gene ontology; UCP2, uncoupling protein 2
4. DISCUSSION
Our findings here link cellular metabolic profile to functional properties of a novel stem‐like cell population in the heart termed as CTSCs. Additionally, our data identifies a role for UCP2 in maintenance of enhanced cellular proliferation and survival exhibited by neonatal CTSCs. Loss of UCP2 with age leads to compensatory changes in metabolism and attenuation of cellular proliferation and survival in aged CTSCs.
The neonatal heart possesses ability to repair and regenerate after injury which is lost as the heart matures. 4 Stem/progenitor cells isolated from neonatal animals and young humans demonstrate strong regenerative and secretory properties able to promote myocardial repair after injury. 18 , 27 Recent studies indicate that proliferative capacity of neonatal cardiac tissue coincides with CMs undergoing metabolic changes to support increased demands of cellular proliferation. 5 Similarly, CPCs in the epicardial regions of the heart are known to possess increased levels of glycolysis for energy generation preserving stemness. 13 CPCs activation leads to a shift toward oxidative phosphorylation accompanied by reduced proliferation and differentiation 13 suggesting a role for cellular metabolism in regulating cardiac cell function. Contrary to neonatal heart, aging cardiac tissue exhibits impaired metabolic flexibility that increases sensitivity to stress. 28 Nevertheless, whether metabolic signaling plays a role in proliferation and survival of cardiac cells in the neonatal heart and is altered with cardiac age is not well studied. In this study, we have identified a novel CTSCs in the neonatal, adult, and aged heart that exhibits stem cell‐like properties and expresses unique transcriptional profile when compared to other cardiac cell types or embryonic, mesenchymal or c‐kit+ cardiac stem cells as determined by RNA‐sequencing. Interestingly, CTSCs express typical stem cell markers along with several embryonic markers such as LIN28a highest expressed in the neonatal CTSCs with low or no expression in aged CTSCs in accordance with a previous study that provides evidence for an uncommitted progenitor cells population in the adult heart expressing embryonic marker SSEA‐1 and able to repair the heart after myocardial infarction. 29 Our cell isolation strategy is novel and unique, and we chose to isolate CTSCs from a 2‐day‐old heart with the rationale to identify a regenerative cell in the postnatal heart that is largely unaltered. Our finding that the heart harbors a stem cell‐like population is in conjunction with numerous studies over the years describing multiple cell populations in the adult heart expressing stem cell markers such as c‐kit, 30 Sca‐1, 31 Isl1, 32 ABCG2 33 together with ability to enhance cardiac function after ex vivo expansion and transplantation in myocardial injury model. Next, we compared neonatal CTSCs transcriptome to a published database for six different CPC populations identified in the embryonic heart. 25 Our results identified similarities with one of the populations from AHF in accordance with the published database, suggesting possible origin of the cells. The next set of studies were designed to extensively characterize functional properties of CTSCs isolated from neonatal and aged heart. Cellular features such as proliferation and survival were increased in CTSCs from the neonatal cardiac tissue and were significantly decreased in aCTSC with the opposite effect on cellular senescence, in concordance with numerous studies that report a similar change in other stem cell types that have been isolated from young and old hearts. 34 , 35 , 36 Interestingly, nCTSC have a unique secretome comprising of various growth factors, cytokines, and anti‐inflammatory factors that are reduced in aCTSC suggesting alteration of paracrine secretion in the CTSCs with age.
A unique aspect of the study is the finding that CTSC cellular metabolism regulated neonatal cell proliferation and survival but is largely altered with age adversely affecting aged CTSC functional properties. Recent studies show that energy metabolism is uniquely adapted in PSCs to support high proliferation rates and self‐renewing ability and transitions from glycolysis to oxidative phosphorylation upon differentiation and commitment of PSCs, invariably reducing cellular proliferation. 8 , 9 Similar studies on tissue resident stem cells show that cardiac stem/progenitor cells in the heart reside in hypoxic niches operating under glycolytic metabolism but as the cells undergo differentiation, oxidative phosphorylation becomes the predominant energy generating pathway. 12 , 13 In accordance, our data using RNA‐sequencing and molecular assays for cellular metabolism shows CTSCs from the neonatal heart to predominately operate under glycolytic metabolism compared to CTSCs from adult heart. In contrast, CTSCs from aged heart exhibit increased oxidative phosphorylation, mitochondrial content, and activity (Figures 3 and 4). Interestingly, nCTSCs show increased proton leak together with lower mitochondrial membrane potential (ΔΨ) suggesting immature mitochondria and unique metabolic adaptations in nCTSC regulating energy dynamics (Figures 4 and 5). Recently, it has been shown that PSCs possess underdeveloped and inactive mitochondria together with increased expression of UCP2 that primarily drives glycolytic metabolism by preventing mitochondrial glucose oxidation and increased substrate shunting, whereas, loss of UCP2 leads to PSC differentiation. 14 In concordance, our results identify increased expression of UCP2 in nCTSC that progressively declines as CTSCs age and is found to be the lowest in aCTSC. UCPs are members of the anion carrier protein family and are present in the inner membrane of the mitochondria. 15 UCP1 was first discovered in brown tissue to act as regulators of respiration couplings and are involved in thermogenesis. 15 Other isoforms such as UCP2 is more specific to the heart and skeletal muscle and recently has been shown to be highly expressed in the murine heart during embryonic stages together with reduction in the postnatal heart. 16 UCP2 overexpression in the adult heart in known to promote cardioprotection by reducing ROS generation and preventing mitochondrial Ca+ overload. 17 Collectively, glycolytic energy metabolism in nCTSC and high UCP2 expression together with the published role for UCP2 in maintaining glycolysis, our data link metabolic signaling to cellular features such as proliferation and survival in CTSCs and concurs with recent studies that implicate cellular metabolism as prelude and critical for various cellular processes. 37 , 38 , 39
5. CONCLUSION
In conclusion, we report here novel transcriptomic differences between CTSCs from neonatal, adult, and aged heart. CTSCs possess a specialized metabolic profile when isolated from neonatal heart due to high expression of UCP2 that results in increased glycolytic flux and reduced oxidative phosphorylation, immature mitochondrial phenotype. However, with age UCP2 expression declines altering metabolism from glycolysis to oxidative phosphorylation. Additionally, we identify a critical role for UCP2 in maintenance of neonatal CTSC characteristics that are altered with loss of UCP2 potentially affecting mitochondrial proteins together with cellular proliferation and survival signaling pathways.
CONFLICT OF INTEREST
The authors declared no potential conflicts of interest.
AUTHOR CONTRIBUTIONS
J.K., V.O.C.R.: performed and analyzed experiments, data analysis and interpretation, wrote the manuscript with contributions from F.A.R., H.W., and S.R.H.; A.E.Y., N.K., K.B., D.H.: performed and analyzed experiments, data analysis and interpretation; M.W.: performed and analyzed experiments; M.K.: data analysis and interpretation, conception, design, and financial support; S.M.: wrote the manuscript with contributions from F.A.R., H.W., and S.R.H.
Supporting information
Figure S1 Characterization of CTSC populations. A) Flow cytometry‐based characterization for CTSC, nCTSC and aCTSC for detection of Sca‐1, Integrin‐β1, VCAM, ecto‐5 nucleotidase, Endoglin, CD45 and Integrin αM. Donkey anti‐rabbit Alexa flour 488 antibody was used as the secondary antibody. n = 3/line. B) Analysis of cell surface markers on all 3 CTSC lines by q‐RT‐PCR. (n = 4/line). Immunocytochemistry on 3 CTSC lines for (C) CD45, (D) c‐kit, (E) Nkx2‐5 and (F) GATA‐4. CD45/c‐kit/Nkx2‐5/GATA‐4 (green), Phalloidin (red) and nuclei (blue). Scale bar = 20 μm. n = 4/line. G) Morphological analysis of mouse neonatal/adult cardiomyocytes and adult cardiac fibroblasts (CF) cultured in CTSC media for 10 days. Scale bar = 100 μm.nCTSC vs CTSC *P < 0.05, **P < 0.01, ***P < 0.001 and nCTSC vs aCTSC # P < 0.05, ## P < 0.01, ### P < 0.001.
Figure S2: Analysis of pluripotent marker expression. A) Increased expression of pluripotent markers in nCTSC compared to CTSCs and aCTSC by qRT‐PCR analysis (n = 4). B) Increased expression of LIN28a in nCTSC compared to CTSCs and aCTSC. LIN28a (green), Phalloidin (red), Nuclei (blue). Scale bar = 40 μm. (n = 4). C) Immunoblot analysis for LIN28a with quantification. (n = 5). D) mRNA expression of LIN28a in CTSCs. (n = 5). nCTSC vs CTSC *P < 0.05, **P < 0.01, ***P < 0.001 and nCTSC vs aCTSC # P < 0.05, ## P < 0.01, ### P < 0.001.
Figure S3: Measurement of survival, migration and cellular senescence. A) Decreased TUNEL+ cells in nCTSC in response to H2O2 challenge compared to CTSCs and aCTSC. TUNEL (green), Phalloidin (red), Nuclei (Blue). Scale bar = 40 μm. (n = 3). B) Wound assay shows increased migration ability of nCTSC to populate the scratch wound after 24 hours compared to CTSCs and aCTSC. C) Decreased β‐gal+ cells in nCTSC compared to CTSCs and aCTSC as assessed by β‐galactosidase senescence. n = 500 cells counted/3 independent experiments. nCTSC vs CTSCs *P < 0.05, **P < 0.01, ***P < 0.001, nCTSC vs aCTSC # P < 0.05, ## P < 0.01, ### P < 0.001 and nCTSC vs aCTSCs Δ p < 0.05, ΔΔ p < 0.01, ΔΔΔ p < 0.001.
Figure S4: Analysis of changes in CTSC secretome with age. A) Proteome profiler array for the assessment of secreted growth factors, cytokines, chemokines from CTSC populations along with dot plot image and corresponding quantification. B) Immnoblot analysis for growth factors such as SDF‐1, IGF‐1, HGF and VEGF in CTSC populations along with corresponding quantification (C) (n = 4). nCTSC vs CTSCs *P < 0.05, **P < 0.01, ***P < 0.001, nCTSC vs aCTSC # P < 0.05, ## P < 0.01, ### P < 0.001 and nCTSC vs aCTSCs Δ p < 0.05, ΔΔ p < 0.01, ΔΔΔ p < 0.001.
Figure S5: Schematic representation of RNA‐sequencing and bioinformatics analyses of CTSCs populations. A) CTSCs from all 3 time points together with or without UCP2 siRNA treatment were cultured followed by isolation of 1 million cells per vials with 3 replicates of each line. Samples were sent out to GeneWiz for RNA isolation, cDNA library construction, sequencing, processing and generation of a raw count files. Using files generated from sequencing, differential expression analysis was performed using DESeq2. Gene Ontology analysis using goseq, ClusterProfiler was used to conduct KEGG Enrichment analysis, GSEA for gene set enrichment analysis, and heatmaps were generated by Gplots using the processed files. Raw count data was transformed on the log2 scale for visualization using the rld function in DESeq2. Using these values, genes present within GO terms for each metabolic pathways such as B) Glycolysis, C) OXPHOS, D) Fatty‐acid metabolism, E) Amino Acid metabolism, F) Glutamine metabolism and G) Glutathione metabolism, were extracted and visualized into heatmaps using the R package gplots. Comparison of gene expression profiles between nCTSCs and aCTSC identified top significant GO terms (H) and GSEA analysis for fatty‐acid metabolism and cell cycle signaling pathways (I).
Figure S6: Morphological and molecular changes in nCTSC following UCP2 knockdown.
A) Flexibility, dependency and capacity of nCTSC to utilize glucose, glutamine and fatty acids for energy generation with or without UCP2 knockdown as measured by fuel flex assay (n = 4 replicates/line/3 independent experiments). B) UCP2 silencing in nCTSC leads to adverse morphological changes and reduced cell numbers as compared to scrambled nCTSC. nCTSC vs siUCP2‐nCTSC *P < 0.05, **P < 0.01, ***P < 0.001. C) Increased ECAR and glycolysis in aCTSC lentivirally modified to expression UCP2 as measured by Seahorse bioanalyzer. n = 10 replicates/sample for 3 independent experiments. D). Analysis of mitochondrial protein OPA1 (inner) in all 3 CTSC lines. aCTSC vs aCTSCs‐UCP2 σ p < 0.05, σσ p < 0.01, σσσ p < 0.001.
Table S4 Supporting information
Table S5 Supporting information
Table S6 Supporting information
Table S7 Supporting information
Table S8 Supporting information
ACKNOWLEDGMENTS
We thank all members of the Khan laboratory for their valuable discussions as well Temple Flow Cytometry core. This work was supported by National Institute of Health Grant HL135177, American Heart Association Scientific Development Grant 15SDG22680018, and H1801 WW Smith Charitable Trust to M.K. and HL137850 and 15SDG25550038 to S.M.
Kurian J, Yuko AE, Kasatkin N, et al. Uncoupling protein 2‐mediated metabolic adaptations define cardiac cell function in the heart during transition from young to old age. STEM CELLS Transl Med. 2021;10:144–156. 10.1002/sctm.20-0123
Funding information WW Smith Charitable Trust, Grant/Award Numbers: 15SDG25550038, HL137850, H1801; American Heart Association Scientific Development, Grant/Award Number: 15SDG22680018; National Institute of Health, Grant/Award Number: HL135177
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from corresponding author upon a reasonable request.
REFERENCES
- 1. Bergmann O, Bhardwaj RD, Bernard S, et al. Evidence for cardiomyocyte renewal in humans. Science. 2009;324(5923):98‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ahuja P, Sdek P, MacLellan WR. Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol Rev. 2007;87(2):521‐544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maillet M, van Berlo JH, Molkentin JD. Molecular basis of physiological heart growth: fundamental concepts and new players. Nat Rev Mol Cell Biol. 2013;14(1):38‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Porrello ER, Mahmoud AI, Simpson E, et al. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331(6020):1078‐1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. de Carvalho A, Bassaneze V, Forni MF, et al. Early postnatal cardiomyocyte proliferation requires high oxidative energy metabolism. Sci Rep. 2017;7(1):15434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mollova M, Bersell K, Walsh S, et al. Cardiomyocyte proliferation contributes to heart growth in young humans. Proc Natl Acad Sci USA. 2013;110(4):1446‐1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ito K, Ito K. Metabolism and the control of cell fate decisions and stem cell renewal. Annu Rev Cell Dev Biol. 2016;32:399‐409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ito K, Suda T. Metabolic requirements for the maintenance of self‐renewing stem cells. Nat Rev Mol Cell Biol. 2014;15(4):243‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Teslaa T, Teitell MA. Pluripotent stem cell energy metabolism: an update. EMBO J. 2015;34(2):138‐153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Simsek T, Kocabas F, Zheng J, et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell. 2010;7(3):380‐390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Takubo K, Goda N, Yamada W, et al. Regulation of the HIF‐1alpha level is essential for hematopoietic stem cells. Cell Stem Cell. 2010;7(3):391‐402. [DOI] [PubMed] [Google Scholar]
- 12. Sanada F, Kim J, Czarna A, et al. C‐kit‐positive cardiac stem cells nested in hypoxic niches are activated by stem cell factor reversing the aging myopathy. Circ Res. 2014;114(1):41‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kimura W, Sadek HA. The cardiac hypoxic niche: emerging role of hypoxic microenvironment in cardiac progenitors. Cardiovasc Diagn Ther. 2012;2(4):278‐289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang J, Khvorostov I, Hong JS, et al. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 2011;30(24):4860‐4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tian XY, Ma S, Tse G, Wong WT, Huang Y. Uncoupling protein 2 in cardiovascular health and disease. Front Physiol. 2018;9:1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hilse KE, Rupprecht A, Egerbacher M, et al. The expression of uncoupling protein 3 coincides with the fatty acid oxidation type of metabolism in adult murine heart. Front Physiol. 2018;9:747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Teshima Y, Akao M, Jones SP, Marbán E. Uncoupling protein‐2 overexpression inhibits mitochondrial death pathway in cardiomyocytes. Circ Res. 2003;93(3):192‐200. [DOI] [PubMed] [Google Scholar]
- 18. Simpson DL, Mishra R, Sharma S, Goh SK, Deshmukh S, Kaushal S. A strong regenerative ability of cardiac stem cells derived from neonatal hearts. Circulation. 2012;126(11 suppl 1):S46‐S53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Khan M, Mohsin S, Avitabile D, et al. Beta‐adrenergic regulation of cardiac progenitor cell death versus survival and proliferation. Circ Res. 2013;112(3):476‐486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Borden A, Kurian J, Nickoloff E, et al. Transient introduction of miR‐294 in the heart promotes cardiomyocyte cell cycle reentry after injury. Circ Res. 2019;125(1):14‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Khan M, Nickoloff E, Abramova T, et al. Embryonic stem cell‐derived exosomes promote endogenous repair mechanisms and enhance cardiac function following myocardial infarction [in English]. Circ Res. 2015;117(1):52‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lighthouse JK, Burke RM, Velasquez LS, et al. Exercise promotes a cardioprotective gene program in resident cardiac fibroblasts. JCI Insight. 2019;4(1):e92098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lother A, Bergemann S, Deng L, Moser M, Bode C, Hein L. Cardiac endothelial cell transcriptome. Arterioscler Thromb Vasc Biol. 2018;38(3):566‐574. [DOI] [PubMed] [Google Scholar]
- 24. Nothjunge S, Nuhrenberg TG, Gruning BA, et al. DNA methylation signatures follow preformed chromatin compartments in cardiac myocytes. Nat Commun. 2017;8(1):1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. de Soysa TY, Ranade SS, Okawa S, et al. Single‐cell analysis of cardiogenesis reveals basis for organ‐level developmental defects. Nature. 2019;572(7767):120‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tang XL, Li Q, Rokosh G, et al. Long‐term outcome of administration of c‐kit(POS) cardiac progenitor cells after acute myocardial infarction: transplanted cells do not become cardiomyocytes, but structural and functional improvement and proliferation of endogenous cells persist for at least one year. Circ Res. 2016;118(7):1091‐1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sharma S, Mishra R, Bigham GE, et al. A deep proteome analysis identifies the complete secretome as the functional unit of human cardiac progenitor cells. Circ Res. 2017;120(5):816‐834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lesnefsky EJ, Chen Q, Hoppel CL. Mitochondrial metabolism in aging heart. Circ Res. 2016;118(10):1593‐1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ott HC, Matthiesen TS, Brechtken J, et al. The adult human heart as a source for stem cells: repair strategies with embryonic‐like progenitor cells. Nat Clin Pract Cardiovasc Med. 2007;4(suppl 1):S27‐S39. [DOI] [PubMed] [Google Scholar]
- 30. Mohsin S, Khan M, Toko H, et al. Human cardiac progenitor cells engineered with Pim‐I kinase enhance myocardial repair [in English]. J Am Coll Cardiol. 2012;60(14):1278‐1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Noseda M, Harada M, McSweeney S, et al. PDGFRalpha demarcates the cardiogenic clonogenic Sca1+ stem/progenitor cell in adult murine myocardium. Nat Commun. 2015;6:6930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Laugwitz KL, Moretti A, Lam J, et al. Postnatal isl1+ cardioblasts enter fully differentiated cardiomyocyte lineages. Nature. 2005;433(7026):647‐653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Martin CM, Meeson AP, Robertson SM, et al. Persistent expression of the ATP‐binding cassette transporter, Abcg2, identifies cardiac SP cells in the developing and adult heart. Dev Biol. 2004;265(1):262‐275. [DOI] [PubMed] [Google Scholar]
- 34. Castaldi A, Dodia RM, Orogo AM, et al. Decline in cellular function of aged mouse c‐kit(+) cardiac progenitor cells. J Physiol. 2017;595(19):6249‐6262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Walravens AS, Vanhaverbeke M, Ottaviani L, et al. Molecular signature of progenitor cells isolated from young and adult human hearts. Sci Rep. 2018;8(1):9266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mohsin S, Khan M, Nguyen J, et al. Rejuvenation of human cardiac progenitor cells with Pim‐1 kinase. Circ Res. 2013;113(10):1169‐1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Derlet A, Rasper T, Roy Choudhury A, et al. Metabolism regulates cellular functions of bone marrow‐derived cells used for cardiac therapy. Stem Cells. 2016;34(8):2236‐2248. [DOI] [PubMed] [Google Scholar]
- 38. Moussaieff A, Rouleau M, Kitsberg D, et al. Glycolysis‐mediated changes in acetyl‐CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab. 2015;21(3):392‐402. [DOI] [PubMed] [Google Scholar]
- 39. Takubo K, Nagamatsu G, Kobayashi CI, et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell. 2013;12(1):49‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Characterization of CTSC populations. A) Flow cytometry‐based characterization for CTSC, nCTSC and aCTSC for detection of Sca‐1, Integrin‐β1, VCAM, ecto‐5 nucleotidase, Endoglin, CD45 and Integrin αM. Donkey anti‐rabbit Alexa flour 488 antibody was used as the secondary antibody. n = 3/line. B) Analysis of cell surface markers on all 3 CTSC lines by q‐RT‐PCR. (n = 4/line). Immunocytochemistry on 3 CTSC lines for (C) CD45, (D) c‐kit, (E) Nkx2‐5 and (F) GATA‐4. CD45/c‐kit/Nkx2‐5/GATA‐4 (green), Phalloidin (red) and nuclei (blue). Scale bar = 20 μm. n = 4/line. G) Morphological analysis of mouse neonatal/adult cardiomyocytes and adult cardiac fibroblasts (CF) cultured in CTSC media for 10 days. Scale bar = 100 μm.nCTSC vs CTSC *P < 0.05, **P < 0.01, ***P < 0.001 and nCTSC vs aCTSC # P < 0.05, ## P < 0.01, ### P < 0.001.
Figure S2: Analysis of pluripotent marker expression. A) Increased expression of pluripotent markers in nCTSC compared to CTSCs and aCTSC by qRT‐PCR analysis (n = 4). B) Increased expression of LIN28a in nCTSC compared to CTSCs and aCTSC. LIN28a (green), Phalloidin (red), Nuclei (blue). Scale bar = 40 μm. (n = 4). C) Immunoblot analysis for LIN28a with quantification. (n = 5). D) mRNA expression of LIN28a in CTSCs. (n = 5). nCTSC vs CTSC *P < 0.05, **P < 0.01, ***P < 0.001 and nCTSC vs aCTSC # P < 0.05, ## P < 0.01, ### P < 0.001.
Figure S3: Measurement of survival, migration and cellular senescence. A) Decreased TUNEL+ cells in nCTSC in response to H2O2 challenge compared to CTSCs and aCTSC. TUNEL (green), Phalloidin (red), Nuclei (Blue). Scale bar = 40 μm. (n = 3). B) Wound assay shows increased migration ability of nCTSC to populate the scratch wound after 24 hours compared to CTSCs and aCTSC. C) Decreased β‐gal+ cells in nCTSC compared to CTSCs and aCTSC as assessed by β‐galactosidase senescence. n = 500 cells counted/3 independent experiments. nCTSC vs CTSCs *P < 0.05, **P < 0.01, ***P < 0.001, nCTSC vs aCTSC # P < 0.05, ## P < 0.01, ### P < 0.001 and nCTSC vs aCTSCs Δ p < 0.05, ΔΔ p < 0.01, ΔΔΔ p < 0.001.
Figure S4: Analysis of changes in CTSC secretome with age. A) Proteome profiler array for the assessment of secreted growth factors, cytokines, chemokines from CTSC populations along with dot plot image and corresponding quantification. B) Immnoblot analysis for growth factors such as SDF‐1, IGF‐1, HGF and VEGF in CTSC populations along with corresponding quantification (C) (n = 4). nCTSC vs CTSCs *P < 0.05, **P < 0.01, ***P < 0.001, nCTSC vs aCTSC # P < 0.05, ## P < 0.01, ### P < 0.001 and nCTSC vs aCTSCs Δ p < 0.05, ΔΔ p < 0.01, ΔΔΔ p < 0.001.
Figure S5: Schematic representation of RNA‐sequencing and bioinformatics analyses of CTSCs populations. A) CTSCs from all 3 time points together with or without UCP2 siRNA treatment were cultured followed by isolation of 1 million cells per vials with 3 replicates of each line. Samples were sent out to GeneWiz for RNA isolation, cDNA library construction, sequencing, processing and generation of a raw count files. Using files generated from sequencing, differential expression analysis was performed using DESeq2. Gene Ontology analysis using goseq, ClusterProfiler was used to conduct KEGG Enrichment analysis, GSEA for gene set enrichment analysis, and heatmaps were generated by Gplots using the processed files. Raw count data was transformed on the log2 scale for visualization using the rld function in DESeq2. Using these values, genes present within GO terms for each metabolic pathways such as B) Glycolysis, C) OXPHOS, D) Fatty‐acid metabolism, E) Amino Acid metabolism, F) Glutamine metabolism and G) Glutathione metabolism, were extracted and visualized into heatmaps using the R package gplots. Comparison of gene expression profiles between nCTSCs and aCTSC identified top significant GO terms (H) and GSEA analysis for fatty‐acid metabolism and cell cycle signaling pathways (I).
Figure S6: Morphological and molecular changes in nCTSC following UCP2 knockdown.
A) Flexibility, dependency and capacity of nCTSC to utilize glucose, glutamine and fatty acids for energy generation with or without UCP2 knockdown as measured by fuel flex assay (n = 4 replicates/line/3 independent experiments). B) UCP2 silencing in nCTSC leads to adverse morphological changes and reduced cell numbers as compared to scrambled nCTSC. nCTSC vs siUCP2‐nCTSC *P < 0.05, **P < 0.01, ***P < 0.001. C) Increased ECAR and glycolysis in aCTSC lentivirally modified to expression UCP2 as measured by Seahorse bioanalyzer. n = 10 replicates/sample for 3 independent experiments. D). Analysis of mitochondrial protein OPA1 (inner) in all 3 CTSC lines. aCTSC vs aCTSCs‐UCP2 σ p < 0.05, σσ p < 0.01, σσσ p < 0.001.
Table S4 Supporting information
Table S5 Supporting information
Table S6 Supporting information
Table S7 Supporting information
Table S8 Supporting information
Data Availability Statement
The data that support the findings of this study are available from corresponding author upon a reasonable request.