

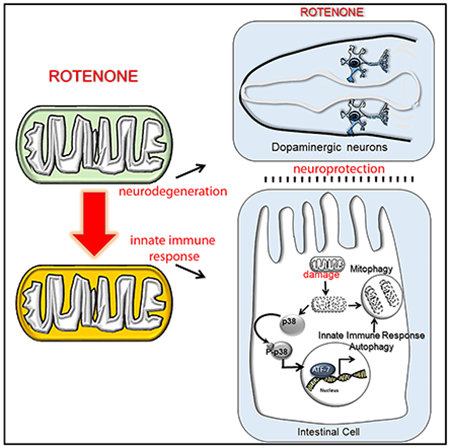
SUMMARY
Immunological mediators that originate outside the nervous system can affect neuronal health. However, their roles in neurodegeneration remain largely unknown. Here, we show that the p38MAPK-mediated immune pathway activated in intestinal cells of Caenorhabditis elegans upon mitochondrial dysfunction protects neurons in a cell-non-autonomous fashion. Specifically, mitochondrial complex I dysfunction induced by rotenone activates the p38MAPK/CREB/ATF-7-dependent innate immune response pathway in intestinal cells of C. elegans. Activation of p38MAPK in the gut is neuroprotective. Enhancing the p38MAPK-mediated immune pathway in intestinal cells alone suppresses rotenone-induced dopaminergic neuron loss, while downregulating it in the intestine exacerbates neurodegeneration. The p38MAPK/ATF-7 immune pathway modulates autophagy and requires autophagy and the PTEN-induced putative kinase PINK-1 for conferring neuroprotection. Thus, mitochondrial damage induces the clearance of mitochondria by the immune pathway, protecting the organism from the toxic effects of mitochondrial dysfunction. We propose that mitochondria are subject to constant surveillance by innate immune mechanisms.
Graphical Abstract
In Brief
Chikka et al. find that mitochondrial complex I damage activates the p38MAPK/ATF-7 signaling pathway in the intestine of C. elegans. Activation of the p38MAPK/ATF-7 immune pathway in the intestine is neuroprotective and sufficient to prevent rotenone-induced degeneration of dopaminergic neurons.
INTRODUCTION
Although neurodegeneration has traditionally been viewed as a consequence of processes autonomous to the vulnerable neurons, it is becoming increasingly clear that cell-non-autonomous mechanisms can impact the process of degeneration. Activated microglia observed in a variety of degenerative neurological conditions, such as Parkinson’s disease (PD), Pick’s disease, amyotrophic lateral sclerosis (ALS), and Huntington’s disease, actively contribute to disease progression (Heneka et al., 2014). The presence of mutant superoxide dismutase (SOD) in neighboring non-neuronal supporting cells can cause cell-non-autonomous toxicity to motor neurons in mouse models of ALS (Ilieva et al., 2009). Similarly, in Caenorhabditis elegans models of ALS, the systemic activation of p38MAPK-mediated innate immunity is required for neurodegeneration of motor neurons (Vérièpe et al., 2015), and exposure of C. elegans to pathogenic bacteria such as Pseudomonas triggers neuronal changes that are hallmarks of neurodegeneration (Wu et al., 2015). In these examples, immunological mediators that originate both within and external to the nervous system exacerbate neuronal loss, suggesting roles for intercellular communication in the degenerative process.
However, despite multiple lines of evidence that the activation of the innate immune response is destructive, its precise role in neurodegenerative diseases remains unclear. This is because the innate immune response can also trigger the production of protective neurotrophic factors, promote repair and remyelination, and induce cytoprotective pathways such as autophagy (Heneka et al., 2014). Indeed, PD pathology is associated not only with the exacerbation of innate immunity upon chronic inflammation but also with the inhibition of the innate immune response, as in the case of Parkin mutations (Winklhofer, 2014). This dual nature of the immune response has been attributed in large part to the differences in the temporal and spatial aspects of immune activation and the specific immune pathways involved.
A widely used model to study mechanisms influencing neurodegeneration is the chronic exposure of organisms to the mitotoxin rotenone (Bové et al., 2005; Sherer et al., 2003). Mitochondrial dysfunction not only compromises the energy balance of cells to influence systemic physiology (Branicky et al., 2000; Ohsawa et al., 2012; Schmeisser et al., 2013) but also can elicit the degeneration of specific neurons (Perier and Vila, 2012; Przedborski and Jackson-Lewis, 1998) and activate the innate immune response (Estes et al., 2010; Kirienko et al., 2015; Lartigue and Faustin, 2013; Liu et al., 2014; Melo and Ruvkun, 2012; Pellegrino et al., 2014; Runkel et al., 2013; Weinberg et al., 2015; West et al., 2011). Therefore, to better understand the relationship between the activation of the innate immune response and neurodegeneration, we examined neurodegeneration in C. elegans caused by rotenone exposure.
RESULTS
Activation of p38MAPK and the ATF-7 Signaling Pathways upon Rotenone Exposure Is Neuroprotective
In C. elegans and other organisms, the dopaminergic neurons appear to be particularly susceptible to mitochondrial complex I dysfunction through as yet unknown mechanisms (Bové et al., 2005; Nass et al., 2002; Ray et al., 2014). While animals grown in the absence of rotenone retained all dopaminergic neurons as they matured into adults (% neurodegeneration = 0; n = 556), C. elegans that developed to adulthood in the presence of 3–5 μM rotenone from larval stage 1 (L1s) lost one or more of the six anterior dopaminergic neurons (two pairs of CEP and one pair of ADE neurons; Figures 1A and 1B). This could be readily visualized by expressing fluorescent mCherry under the dopaminergic neuron-specific dat-1 promoter (Figure 1A). Thermosensory neurons similarly exposed to rotenone did not degenerate (Figure S1A) (Nass et al., 2002). As seen in mammalian models, the ability of rotenone to induce neurodegeneration depended on its ability to interact with mitochondrial complex I (Johnson and Bobrovskaya, 2015; Richardson et al., 2007; Sherer et al., 2003), as downregulating complex I subunits nuo-1, nuo-2, nuo-3, nuo-4, nuo-6, nduf-6, nduf-b7, and nduf-s8 suppressed rotenone-induced dopaminergic neuron death, while downregulating complex III subunits cyc-1 and T02H6.11 did not (Figure 1C). Downregulating oxa-1, the more ubiquitous mitochondrial respiratory chain complex assembly factor (Maxwell et al., 2013), also suppressed neurodegeneration caused by rotenone (Figure 1C). However, while exposure to rotenone invariably caused dopaminergic neurons to degenerate, only an average of 7% of the animals (n > 3,000), ranging from as few as 2% to as many as 25% per experiment, lost dopaminergic neurons; the remaining animals showed no overt evidence of neurodegeneration (Figure 1B). This suggested that there may be innate protective mechanisms that are activated in the majority of animals to prevent rotenone-induced neurodegeneration.
Figure 1. Activation of the p38MAPK/ATF-7 Signaling Pathway in C. elegans upon Rotenone Exposure Is Neuroprotective.

(A) Representative maximum projection confocal images of heads of day 1 adults expressing mCherry in their dopaminergic (DA) neurons under the dat-1 promoter. (Top) Wild-type animals. (Bottom) Animals systemically exposed to 3–5 μM rotenone from larval stage 1 (L1). Scale bar, 20 μm.
(B) Percentage of animals that lost their dopaminergic neurons when exposed to 3–5 μM rotenone compared to control vehicle (DMSO)-treated animals. n > 3,000.
(C) Percentage of animals showing rotenone-dependent degeneration of dopaminergic neurons on control RNAi and upon RNAi-mediated downregulation of complex I, oxa-1, and complex III subunits. n = 300–400 animals per treatment.
(D) Relative mRNA levels determined by qRT-PCR of p38MAPK/ATF-7 innate immune genes (x axis) in wild-type animals exposed to 3–5 μM rotenone and in untreated controls.
(E) Relative mRNA levels measured by qRT-PCR of p38MAPK/ATF-7-dependent innate immune genes (x axis) in wild-type animals, pmk-1(km25) animals, and animals subject to atf-7 RNAi.
(D and E) mRNA levels of the transcripts (x axis) were normalized to wild-type control levels. Actin is the internal control. n ≥ 3 repeats of 20–25 animals foreach treatment.
(F) Percentage of wild-type animals and pmk-1(km25) animals that lost dopaminergic neurons following RNAi treatment (RNAi treatments listed on x axis). n = 200–1,300 animals per treatment.
(B–F) Bargraphs show mean ± SEM. Data from paired experiments and controls are compared. *p < 0.05, **p < 0.01, ***p < 0.001 (Student’s t test). Animals: day 1–2 adults. See also Figure S1.
Mitochondrial dysfunction upregulates multiple stress response pathways, including mitochondrial-repair pathways (Haynes and Ron, 2010; Melo and Ruvkun, 2012; Runkel et al., 2013) and oxidative stress response transcription factors such as the FOXO homolog DAF-16 (Lemire et al., 2009) and the Nrf2 homolog SKN-1 (Paek et al., 2012; Palikaras et al., 2015). Recently, it has been shown that in C. elegans mitochondrial stress activates the innate immune response, upregulating drug-detoxification and pathogen-response genes through the activity of the mitochondrial-stress-sensing transcription factor ATFS-1 and the bZIP transcription factor ZIP-2 (Estes et al., 2010; Kirienko et al., 2015; Lartigue and Faustin, 2013; Liu et al., 2014; Melo and Ruvkun, 2012; Pellegrino et al., 2014; Runkel et al., 2013; Weinberg et al., 2015; West et al., 2011). Accordingly, the DAF-16 target mtl-1, the SKN-1 target gst-4, and the ZIP-2 target irg-1 were all upregulated upon exposure to rotenone (Figures S1B–S1D). Besides ZIP-2 and DAF-16, in C. elegans, the p38MAPK signaling pathway also plays a major role in the innate immune response, upregulating numerous secreted proteins in response to pathogens such as Pseudomonas through the transcriptional activation of the CREB-like transcription factor ATF-7 (Ermolaeva and Schumacher, 2014; Ewbank and Pujol, 2016; Kim et al., 2002; Shivers et al., 2010). We found that systemic exposure of C. elegans to rotenone also upregulated the p38MAPK-dependent innate immune response pathway (Figure 1D). This was evidenced by the upregulation of numerous ATF-7 target genes in a p38MAPK (pmk-1) and atf-7 dependent manner (Figures 1D and 1E).
To investigate which, if any, of the multiple stress response pathways induced by rotenone treatment protected animals from rotenone-induced neurodegeneration, we downregulated each of these pathways by RNAi and scored effects on neurodegeneration (Figure 1F). Due to the variability in the range of rotenone-induced neurodegeneration in each experiment, these and all subsequent experiments were quantified as paired repeats. Downregulating SKN-1 through RNAi did not increase the numbers of animals that lost dopaminergic neurons, suggesting that the SKN-1 mediated transcriptional response, while upregulated upon rotenone exposure, did not protect dopaminergic neurons from degeneration under the conditions of our experiments (Figure 1F). Similarly, downregulating ZIP-2 or DAF-16 also did not enhance rotenone-induced neuron loss (Figure 1F). Instead, downregulating ZIP-2 decreased the fraction of animals that lost dopaminergic neurons, suggesting that the activation of ZIP-2 by rotenone may even be detrimental to neuronal health. In contrast, downregulating the p38MAPK signaling pathway exacerbated neuronal loss (Figure 1F). In pmk-1(km25) deletion mutants lacking p38MAPK activity, twice the number of animals lost dopaminergic neurons upon rotenone exposure when compared to wild-type animals. Similarly, RNAi-mediated knockdown of atf-7 increased the number of animals that lost dopaminergic neurons when exposed to rotenone (Figure 1F). Moreover, atf-7 RNAi did not further exacerbate dopaminergic neuron loss in pmk-1(km25) animals, suggesting that the activity of p38MAPK occurred through its regulation of ATF-7 (Figure 1F). None of these animals lost dopaminergic neurons in the absence of rotenone (% neurodegeneration = 0; n = 500–567). Downregulating another innate immune response pathway, the transforming growth factor-β (TGF-β)/SMA-3 pathway, had no effect (Figure 1F). Thus, it appeared that the p38MAPK/ATF-7-dependent innate immune response pathway that was induced by rotenone treatment protected animals from rotenone-induced dopaminergic neuron degeneration.
Activation of p38MAPK/ATF-7-Mediated Signaling in Intestinal Cells Is Sufficient to Confer Neuroprotection
To determine the site(s) of activation of the p38MAPK/ATF-7 innate immune pathway upon rotenone exposure, we conducted single-molecule fluorescent in situ hybridization (smFISH) in whole animals for the ATF-7 target gene C17H12.8 (Shivers et al., 2010) (Figures 2A, S1E, and S1F). Although animals were systemically exposed to rotenone and exhibited neurodegeneration, the activation of p38MAPK evidenced by C17H12.8 expression was restricted to intestinal nuclei and could not be detected in other cells of the animal (Figure S1E). In addition, it appeared that the induction of p38MAPK/ATF-7 in intestinal cells correlated with the susceptibility of animals to rotenone-induced dopaminergic neurodegeneration, as animals that did not lose their dopaminergic neurons despite exposure to rotenone displayed a larger ATF-7-dependent transcriptional response to rotenone and expressed more ATF-7 target gene C17H12.8 mRNA in a larger number of nuclei, as determined by smFISH (Figures 2B and 2C). Animals that had lost their dopaminergic neurons, on the other hand, showed little to no expression of C17H12.8 (Figures 2B and 2C).
Figure 2. Activation of p38MAPK/ATF-7 in Intestinal Cells Suppresses Rotenone-induced Degeneration of Dopaminergic Neurons.
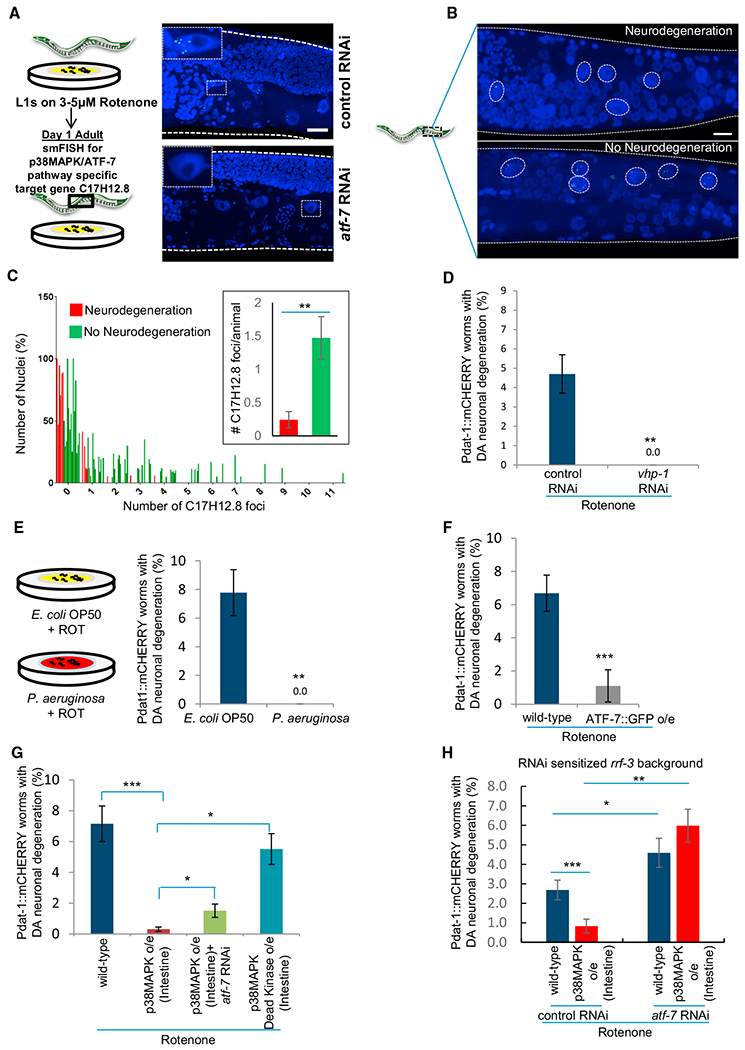
(A) Representative maximum projection confocal images of the ATF-7 target gene C17H12.8 mRNA in adult day 1, rotenone-treated animals on control and atf-7 RNAi. mRNA was detected by single-molecule RNA in situ hybridization (smFISH). Scale bar, 10 μm. n = 3 animals each treatment. Inset: single intestinal nucleus.
(B) Representative maximum projection confocal images of C17H12.8 mRNA in rotenone-treated animals that did, and did not, undergo neurodegeneration. Circles represent intestinal nuclei. Scale bar, 10 μm.
(C) Frequency distribution of percent of intestinal nuclei (y axis) that expressed 0–11 discrete C17H12.8 mRNA foci/nucleus (x axis) and mean number of C17H12.8 mRNA foci/animal counted in ~15 intestinal nuclei/animal (inset). Rotenone-treated wild-type animals that lost (red), and did not lose (green) dopaminergic neurons. n = 88–215 nuclei, from 6 to 16 animals.
(D) Percentage of animals subjected to vhp-1 RNAi that lost dopaminergic neurons on rotenone compared to control. n = 200–250 animals.
(E) Percentage of wild-type animals grown on Pseudomonas aeruginosa (PA14) that lost dopaminergic neurons on rotenone compared to control (OP50). n = 400 animals.
(F) Percentage of animals overexpressing ATF-7 that lost dopaminergic neurons on rotenone compared to wild-type. n = 300 animals.
(G) Percentage of wild-type animals, animals overexpressing p38MAPK in intestinal cells on control and atf-7 RNAi, and animals overexpressing kinase-dead p38MAPK in intestinal cells that lost dopaminergic neurons on rotenone treatment. n = 1,200 and 500 for wild-type animals and animals overexpressing active p38MAPK respectively and 127 for animals overexpressing intestinal kinase-dead p38MAPK.
(H) Percentage of rrf-3(mg373) animals and rrf-3(mg373) animals overexpressing p38MAPK in intestinal cells on control and atf-7 RNAi that lost dopaminergic neurons on rotenone. Data from paired experiments and controls are compared.
(C–H) Bar graphs show mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 (Student’s t test). Animals: day 1–2 adults. See also Figure S2.
We therefore examined whether activation of p38MAPK was sufficient to confer neuroprotection. RNAi-mediated downregulation of VHP-1 (Mizuno et al., 2004), the phosphatase that dephosphorylates p38MAPK leading to an increase in active p38MAPK and subsequent transcriptional activation of ATF-7 (Figure S2A), suppressed dopaminergic neurodegeneration in all animals (0% of the animals lost dopaminergic neurons; Figure 2D). Feeding C. elegans a diet of Pseudomonas aeruginosa, which induces the p38MAPK innate immune response in intestinal cells (Kim et al., 2002), also suppressed rotenone-induced neurodegeneration, and none of the animals lost dopaminergic neurons (Figures 2E and S2B). Overexpressing ATF-7 under the control of its own promoter (Shivers et al., 2010) was also sufficient to suppress rotenone-induced dopaminergic neuron loss (Figure 2F). Remarkably, overexpression of p38MAPK only in the gut under the control of a vha-6 promoter (Figure S2C) (Bolz et al., 2010) increased ATF-7 target gene expression upon rotenone exposure compared to controls (Figure S2E) and dramatically suppressed rotenone-induced dopaminergic neuron degeneration (Figure 2G). In contrast, animals that overexpressed inactive p38MAPK in intestinal cells due to a TGY→AGF substitution continued to display dopaminergic neuronal loss at similar rates as wild-type animals (Figures S2C and 2G). Overexpression of p38MAPK in muscle cells also suppressed dopaminergic neuronal degeneration, albeit to a lesser extent (Figures S2C, S2D, and S2F). The protection conferred by p38MAPK was ATF-7 dependent; RNAi-mediated knockdown of atf-7 partially suppressed the rescue observed upon intestinal overexpression of p38MAPK in a wild-type background (Figure 2G) and completely suppressed the rescue in an rrf-3 mutant background (Figure 2H), which undergoes enhanced RNAi (Simmer et al., 2002), although for unknown reasons, the extent of rotenone-induced neurodegeneration itself varied between the rrf-3 and wild-type strains. Consistent with this, and as expected, the levels of atf-7 mRNA were reduced more in the rrf-3 background upon atf-7 RNAi than in wild-type (Figure S2H). RNAi-mediated knockdown of atf-7 also suppressed the modest rescue observed upon overexpression of p38MAPK in muscle cells (Figure S2F). On the other hand, RNAi-mediated knockdown of SKN-1 or DAF-16 did not reverse p38MAPK-mediated neuroprotection (Figure S2G). These data, taken together, indicate that p38MAPK-mediated activation of ATF-7 in intestinal cells, as occurred upon rotenone treatment, was sufficient to protect dopaminergic neurons from the degenerative effects of rotenone. Experimentally inducing p38MAPK-mediated activation of ATF-7 in muscle cells by overexpressing p38MAPK was also neuroprotective, albeit to a lesser extent.
Activation of p38MAPK-Dependent ATF-7 Transcription Is Induced by Mitochondrial Complex I Dysfunction
Rotenone inhibits mitochondrial complex I. Dysfunctional mitochondria activate mitochondrial retrograde signaling pathways that induce adaptive changes in the cell upon the loss of mitochondrial function (Kleine and Leister, 2016). We therefore examined whether the activation of p38MAPK and ATF-7 was triggered by downregulating complex I, rather than being a more general consequence of rotenone toxicity. To this end, we used RNAi to downregulate complex I subunits and assessed the levels of phosphorylated p38MAPK- and ATF-7-dependent target genes. Downregulating complex I genesnuo-2 and nduf-6 caused a significant increase in p38MAPK phosphorylation (Figure 3A). The increase in active p38MAPK levels was similar if not greater than that seen upon rotenone exposure (Figure S3A), perhaps because rotenone has more pleiotropic effects than only complex I inhibition. Downregulation of mitochondrial complex I also upregulated four of the seven ATF-7-dependent genes induced by rotenone: C17H12.8, F56D6.2. and M02F4.7 (C-type lectins) and F49F1.6 (mul-1, a mucin-like protein). These genes were upregulated upon RNAi-induced downregulation of at least five of the six tested complex I genes to a comparable (F49F1.6), lower (F56D6.2), or higher (M02F4.7) extent (Figure 3C). As previously shown (Shivers et al., 2010), the expression of these four ATF-7 targets was p38MAPK dependent under basal conditions, and this was also true upon RNAi-mediated knockdown of complex I genes (Figure S3B). In support of the possibility that phosphorylation of p38MAPK and induction of ATF-7-dependent gene expression was a specific retrograde signaling pathway activated by complex I dysfunction, RNAi-mediated downregulation of the respiratory chain complex assembly factor oxa-1, complex III, complex IV, and complex V ATP synthase subunits did not increase the levels of phosphorylated p38MAPK (Figure 3B). This was the case despite the fact that both nuo-6 and oxa-1 RNAi caused similar reactive oxygen species (ROS) accumulation, as measured by the levels of dihydroethidium (DHE) fluorescence (Figure 3D). As in wild-type animals, downregulation of complex I subunits also inhibited rotenone-induced degeneration in pmk-1(km25) animals (Figure 3E). These data, taken together, suggest that mitochondrial complex I dysfunction acted upstream of p38MAPK signaling and through unknown mechanisms activated p38MAPK/ATF-7-dependent gene expression. (Figure 3F).
Figure 3. Mitochondrial Complex I Dysfunction Activates p38MAPK/ATF-7.

(A and B) Levels of phosphorylated p38MAPK following RNAi-mediated downregulation of complex I (C-I) subunits (A) and complex V (C-V), oxa-1, complex III (C-III), and complex IV (C-IV) subunits (B) relative to control RNAi-treated animals. Levels of phospho-p38MAPK (top lanes) were quantified relative to alpha-tubulin (bottom lanes). n ≥ 3 repeats of 20–25 animals per treatment.
(C) mRNA levels of ATF-7-dependent innate immune genes in control animals and animals on complex I RNAi, determined by qRT-PCR. mRNA levels of the transcripts (x axis) were normalized to wild-type control levels. Actin is the internal control. n ≥ 3 repeats of 20–25 animals per treatment.
(D) Levels of dihydroethidium (DHE) fluorescence quantitated in proximal intestinal cells of animals on control RNAi, complex I subunit nuo-6 RNAi, and mitochondrial assembly factor oxa-1 RNAi. Representative micrographs are of the anterior regions of animals. R.F.U., relative fluorescence units. Scale bar, 30 μm. n > 20 animals for each treatment.
(E) Percent animals showing rotenone-dependent dopaminergic neuron loss in wild-type and pmk-1(km25) animals on control RNAi and upon RNAi-mediated downregulation of complex I. n = 200–600 animals per treatment.
(F) Model for activation of p38MAPK upon mitochondrial dysfunction.
(A–E) Bar graphs show mean ± SEM. Data from paired experiments and controls are compared. *p < 0.05, **p < 0.01, ***p < 0.001 (Student’s t test). Animals: day 1–2 adults. See also Figure S3.
Overexpression of p38MAPK in Intestinal Cells Causes a Decrease in Mitochondrial Content
How could the activation of p38MAPK and ATF-7 in intestinal cells upon mitochondrial dysfunction protect dopaminergic neurons? Since rotenone affects the electron transport chain (ETC) and mitochondrial oxidative phosphorylation (OXPHOS), we examined whether the p38MAPK/ATF-7-signaling pathway conferred neuroprotection by preserving systemic ATP levels and/or the biogenesis of mitochondrial ETC components. This did not appear to be the case, as (1) ATP levels did not decrease in wild-type animals on rotenone treatment, nor were there significant differences among wild-type animals, animals on atf-7 RNAi, and animals overexpressing p38MAPK (Figure S4A); and (2) steady-state mRNA levels of mitochondrial ETC components and protein levels of the ATPB subunit of the ATP synthase did not consistently or significantly change in animals overexpressing p38MAPK in intestinal cells or in animals where atf-7 was downregulated (Figures S4B and S4C). For instance, while steady-state mRNA levels of atp-5, the ATP synthase subunit, were higher in p38MAPK-overexpressing animals and lower upon atf-7 RNAi (Figure S4B), protein levels of the ATP synthase ATPB subunit did change accordingly (Figure S4C).
One of the described causes for neuronal death upon rotenone poisoning is the increase in oxidative damage (Sherer et al., 2003). Assessing the fluorescence levels of DHE in the first two intestinal cells showed that rotenone increased ROS levels in wild-type animals and upon atf-7 RNAi (Figure 4A). However, in p38MAPK-overexpressing animals, the DHE fluorescence remained low despite rotenone exposure, even though these animals did not show obvious increases in mRNA levels of genes required to eliminate ROS (e.g., mitochondrial SOD levels sod-2, or gst-4, etc.; Figure S4D; data not shown). RNAi-mediated knockdown of atf-7 moderately yet significantly increased ROS levels in p38MAPK-overexpressing animals (Figure 4A). These data suggested that p38MAPK/ATF-7-dependent decrease in oxidative damage could be responsible for the observed neuroprotection.
Figure 4. Activation of p38MAPK/ATF-7 in Intestinal Cells Decreases Mitochondrial Content.
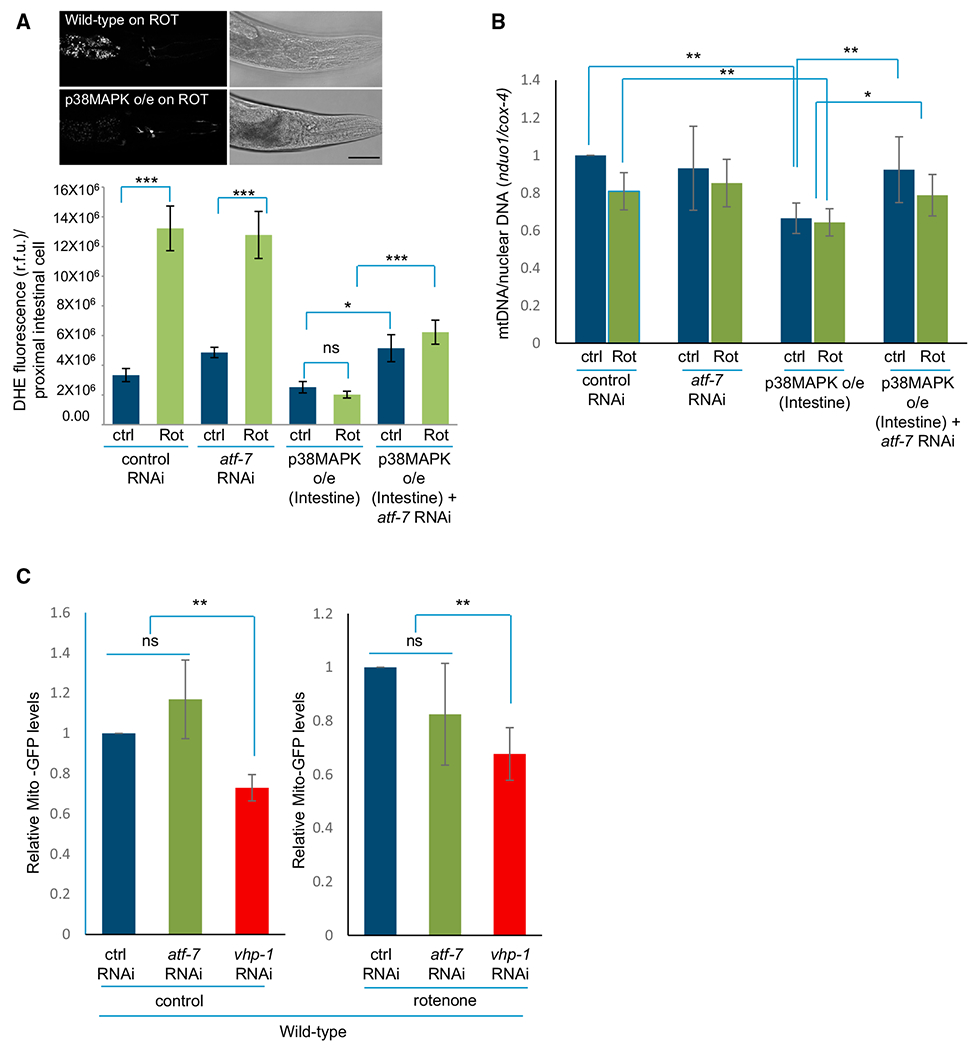
(A) (Top) Micrographs of dihydroethidium (DHE) fluorescence in the proximal intestinal cells of wild-type animals and animals overexpressing p38MAPK following rotenone treatment. Scale bar, 30 mm. (Bottom) Quantitation of DHE fluorescence in control and rotenone treated wild-type and p38MAPK-overexpressing animals on control RNAi and atf-7 RNAi. R.F.U., relative fluorescence units. n > 100–150 animals for each treatment.
(B) Ratio of mtDNA gene nduo-1 to the nuclear gene cox-4 as determined by qPCR. Samples are wild-type animals and animals overexpressing p38MAPK on control RNAi and atf-7 RNAi.
(C) Quantitation of GFP protein levels in ges-1pr::gfpmt transgenic worms: wild-type animals on control RNAi, animals on atf-7 RNAi, and animals where p38MAPK was activated by vhp-1 RNAi under control conditions and upon rotenone treatment. GFP levels are quantified relative to alpha-tubulin and normalized to the respective control RNAi-treated animals. n ≥ 3 repeats of 20–25 animals per treatment.
(A–C) Bar graphs show mean ± SEM. Data from paired experiments and controls are compared. *p < 0.05, **p < 0.01, ***p < 0.001 (Student’s t test). Animals: day 1–2 adults. See also Figure S4.
A mechanism that would limit ROS accumulation and oxidative damage is mitochondrial repair through the activation of the unfolded protein response (UPRmito) (Runkel et al., 2013). Since, in C. elegans, activation of UPRmito in neuronal cells affects UPRmito in the intestine (Durieux et al., 2011), we examined whether UPRmito-mediated signaling could allow for the observed cell-non-autonomous neuronal protection. However, animals deficient in p38MAPK or ATF-7 were not deficient in inducing UPRmito upon rotenone treatment and expressed the UPRmito hsp-6p::GFP reporter, upregulated hsp-6 and hsp-60 mRNA to wild-type levels, and expressed wild-type levels of HSP-60 protein (Figures S4E–S4G). Moreover, RNAi-mediated downregulation of the transcription factor ATFS-1 (Nargund et al., 2012), responsible for UPRmito induction, retarded growth upon rotenone exposure but suppressed, rather than exacerbated, dopaminergic neuron loss in wild-type and pmk-1(km25) animals (Figure S4H). Thus, UPRmito did not explain the observed decrease in ROS levels in the p38MAPK-overexpressing animals.
Another mechanism that could limit ROS and oxidative damage would be the removal of dysfunctional mitochondria by mitochondrial autophagy (mitophagy). We therefore assessed whether overexpressing p38MAPK affected mitochondrial content by measuring the ratio of mitochondrial to nuclear genomes by qPCR (Figure 4B). As previously reported (Luz et al., 2015), rotenone poisoning did not significantly change mitochondrial copy number in wild-type animals; however, animals overexpressing p38MAPK in intestinal cells had significantly lower numbers of mitochondria, both under control conditions and upon rotenone exposure (Figure 4B), consistent with a lowering of mitochondrial numbers and increased mitophagy. The decrease in relative mitochondrial genome copy number upon intestinal overexpression of p38MAPK was also ATF-7 dependent, as mitochondrial copy numbers were restored to wild-type levels upon RNAi-mediated knockdown of atf-7 (Figure 4B). To directly examine whether mitochondrial content was reduced in animals overexpressing p38MAPK, we utilized a transgenic strain expressing GFP fused to a mitochondrial signal sequence in intestinal cells (ges-1pr::gfpmt) and assessed GFP levels using western blot analysis in wild-type animals, in animals where p38MAPK was activated by vhp-1 RNAi, and in animals where atf-7 was downregulated (Figure 4C). Consistent with the decrease in mitochondrial genome copy number in p38MAPK overexpressing animals, activation of the innate immune response by vhp-1 RNAi significantly decreased GFPmt levels in the intestine of animals both under control conditions and upon rotenone treatment (Figure 4C). Mitochondrial content upon atf-7 downregulation did not increase, but this could be, in part, due to the variability inherent in RNAi and the resolution of our methods. Nevertheless, these data, taken together, suggested that p38MAPK signaling in intestinal cells may be promoting a decrease in mitochondrial number through an ATF-7-dependent mechanism, protecting animals from the detrimental consequences of OXPHOS dysregulation upon rotenone exposure.
Neuroprotection by p38MAPK-Mediated Immune Pathway Occurs through Mitophagy
In C. elegans, mitophagy has been shown to occur through the core autophagy machinery that includes the Atg6/Vps30/Beclin 1 ortholog BEC-1, the mammalian LC3/GABARAP ortholog LGG-1, and the mitochondrial PTEN-induced putative kinase 1 (PINK-1) (Kirienko et al., 2015; Palikaras et al., 2015). To determine whether mitophagy was indeed responsible for neuroprotection conferred by p38MAPK-mediated immunity, we downregulated these genes by RNAi in an rrf-3 background, in the absence and presence of p38MAPK intestinal overexpression. Downregulation of lgg-1 increased rotenone-induced dopaminergic neuron degeneration in animals overexpressing p38MAPK from 0.7% to 5.7%, similar to that seen upon the knockdown of atf-7, representing almost an 8-fold increase in the incidence of neurodegeneration (Figure 5A). Similarly, downregulation of bec-1 and pink-1 reversed the protection conferred by p38MAPK overexpression (Figure 5A). Knockdown of atg-18, a WD40 repeat containing protein required for autophagosome formation, also reversed the neuroprotection conferred by p38MAPK overexpression (Figure 5A). Downregulation of these autophagy genes did not induce neurodegeneration in the absence of rotenone (% neurodegeneration = 0; n = 300–823 animals/RNAi). To test whether the suppression of p38MAPK-mediated protection could be attributed to an increase in mitochondrial content as a result of downregulating autophagy genes, we once again assessed mitochondrial content in ges-1pr::gfpmt animals upon p38MAPK activation, but this time in pink-1 partial deletion mutants (Figure 5B). In contrast to what was seen in animals with functional PINK-1, in pink-1(ok3538) mutants, GFP levels did not decrease upon activation of p38MAPK either under control or rotenone treatment (Figure 5B; compare to Figure 4C; samples prepared in parallel). These data indicate that the neuroprotection conferred by activating p38MAPK-mediated signaling in intestinal cells occurred through increased mitophagy, as knockdown of genes involved in this process completely reversed the p38MAPK-mediated neuroprotection and prevented the decrease of mitochondrial content that occurred upon p38MAPK activation.
Figure 5. Neuroprotection by p38MAPK/ATF-7 Requires Autophagy Genes and PINK-1.

(A) Percentage of rrf-3(mg373) animals and rrf-3(mg373) animals overexpressing p38MAPK in intestinal cells subjected to RNAi treatment (RNAi treatments on x axis) that lost dopaminergic neurons on rotenone.
(B) Quantitation of GFP protein levels in ges-1pr::gfpmt animals in a pink-1(ok3538) mutant background: wild-type animals on control RNAi, atf-7 RNAi, and vhp-1 RNAi under control conditions and upon rotenone treatment. GFP levels are quantified relative to alpha-tubulin, and normalized to the respective control RNAi treated animals. n ≥ 3 repeats of 20–25 animals per treatment. Compare to Figure 4C.
(C) Percentage of rotenone-induced neurodegeneration upon intestine-specific RNAi: rde-1(ne219) mutants expressing RDE-1 under the intestinal nhx-2 promoter were exposed to RNAi (RNAi treatments on x axis).
(D) Percentage of rotenone-induced neurodegeneration upon neuron-specific RNAi: sid-1(pk3321) animals expressing SID-1 under the neuronal unc-119 promoter were exposed to RNAi (RNAi treatments on x axis).
(A–D) Graphs show mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 (Student’s t test). Animals: day 1–2 adults. See also Figure S5.
We further examined whether the innate immune response and autophagy both had to be activated in intestinal cells to confer protection from rotenone-induced neurodegeneration or whether autophagy was required in neurons for its neuroprotective effects. To do this, we knocked down genes involved in mitophagy specifically either in intestinal cells or in neurons, using strains sensitized to undergo RNAi only in the intestine or neurons: rde-1(ne219) mutants expressing RDE-1 under the intestinal nhx-2 promoter (Kumsta and Hansen, 2012; Qadota et al., 2007) and sid-1(pk3321) expressing SID-1 under the neuronal unc-119 promoter (Calixto et al., 2010), respectively. Once again, although, for unknown reasons, the basal extent of rotenone-toxicity differed in these strains, RNAi-mediated downregulation of atf-7 in intestinal tissue enhanced neurodegeneration, while knockdown only in neuronal tissue had no effect (Figures 5C and 5D). Thus, activation of p38MAPK/ATF-7 in intestinal cells was not only sufficient but also necessary for neuroprotection. Downregulation of PINK-1 and LGG-1 in intestinal cells, but not in neurons, also exacerbated neurodegeneration, suggesting that their activity was also required in the intestine to limit neurodegeneration (Figures 5C and 5D). Neurodegeneration in the absence of rotenone for all RNAi was 0% (n = 315–579/RNAi). However, RNAi-mediated knockdown of bec-1 in both intestine and neurons increased neurodegeneration (Figures 5C and 5D), and knockdown of bec-1 in neurons increased neurodegeneration even in the absence of rotenone (neurodegeneration in the absence of rotenone = 6% ± 3.2%; n = 467), suggesting a more complex role for autophagy in the protection of neuronal cells. Similarly, knockdown of mboa-1 a putative acyl-coenzyme A:cholesterol (“sterol”) O-acyltransferase previously shown to be required for mitophagy in C. elegans (Kirienko et al., 2015) had no effect when downregulated in intestinal cells but decreased rather than exacerbated neurodegeneration when targeted to neuronal cells alone (Figures S5A and S5B). These data, taken together, showed that activation of p38MAPK/ATF-7, as well as PINK-1 and LGG-1, in the gut was necessary to confer neuroprotection. The tissue requirements of autophagy, however, appeared more complex, and our data cannot distinguish whether autophagy occurred only in intestinal cells or also in neurons as a neuroprotective consequence of p38MAPK signaling in the intestine, and further studies will be required.
The p38MAPK/ATF-7 Pathway Regulates Autophagy
Since neuroprotection through p38MAPK-dependent signaling pathways required autophagy, we tested whether the p38MAPK/ATF-7 immune pathway regulated the autophagy process. To address this, we assessed whether the transcription and/or translation of autophagy-pathway associated genes and pink-1 were altered upon p38MAPK overexpression and atf-7 downregulation. Examination of a panel of autophagy-related genes that had previously been shown to be required for longevity assurance, including of clk-1 mitochondrial respiration mutants (Hansen et al., 2008; Lapierre et al., 2011), showed that in wild-type animals, rotenone exposure caused a slight but significant increase in mRNA levels of some, but not all, of these autophagy genes (Figure 6A). Intestinal overexpression of p38MAPK further increased the steady-state mRNA levels of many of these genes above wild-type levels, in some cases under control conditions and in some cases upon rotenone exposure (Figure 6A). These genes included atg-18 and genes involved in the acidification and fusion of autophagosomes (Figure 6A). Downregulation of atf-7 through RNAi had a more dramatic and consistent effect, decreasing steady-state mRNA levels of eight of the nine genes tested (Figure 6A). This was the case for both basal levels of autophagy-pathway associated genes, as well as levels in the presence of rotenone. Steady-state protein levels of LGG-1 tagged to GFP, GFP:: LGG-1 (Meléndez and Levine, 2009; Zhang et al., 2015), a widely used monitor of autophagy in C. elegans, also significantly increased with rotenone treatment in animals where p38MAPK was activated (Figure S6A) and significantly decreased upon RNAi-mediated knockdown of atf-7 (Figure S6B). Deletion of p38MAPK in pmk-1(km25) animals resulted in a decrease in GFP::LGG-1 protein levels, although this decrease did not reach significance, as there was more variability in GFP::LGG-1 expression across numerous repeated trials (Figure S6C). The number of autophagosomes measured as GFP::LGG-1 puncta (Meléndez and Levine, 2009; Zhang et al., 2015) increased in intestinal cells of wild-type animals when exposed to rotenone. This number was once again significantly lower in rotenone-treated pmk-1(km25) animals lacking p38MAPK and rotenone-treated animals on atf-7 RNAi and higher in rotenone-treated animals where p38MAPK was activated using vhp-1 RNAi (Figures 6B and 6C). Animals lacking p38MAPK, pmk-1(km25) animals, had lower numbers of GFP::LGG-1 puncta even in the absence of rotenone treatment (Figures 6B and 6C). However, the number of autophagosomes did increase above control levels upon rotenone treatment in all animals except those on atf-7 RNAi (Figures 6B and 6C) consistent with the presence of other signaling pathways besides p38MAPK that regulate autophagy. In contrast to the clear increase in GFP::LGG-1 puncta in intestinal cells upon rotenone treatment, the number of GFP::LGG-1 puncta in the pair of ADE dopaminergic neurons susceptible to degeneration was not significantly changed upon rotenone treatment (Figures S6D and S6E). In addition, GFP::LGG-1 puncta in ADE neurons of animals where p38MAPK immunity was activated or downregulated remained comparable, although the basal numbers of GFP::LGG-1 puncta in these neurons differed (Figures S6D and S6E).
Figure 6. p38MAPK/ATF-7 Modulates the Expression and Activity of Autophagy Genes.
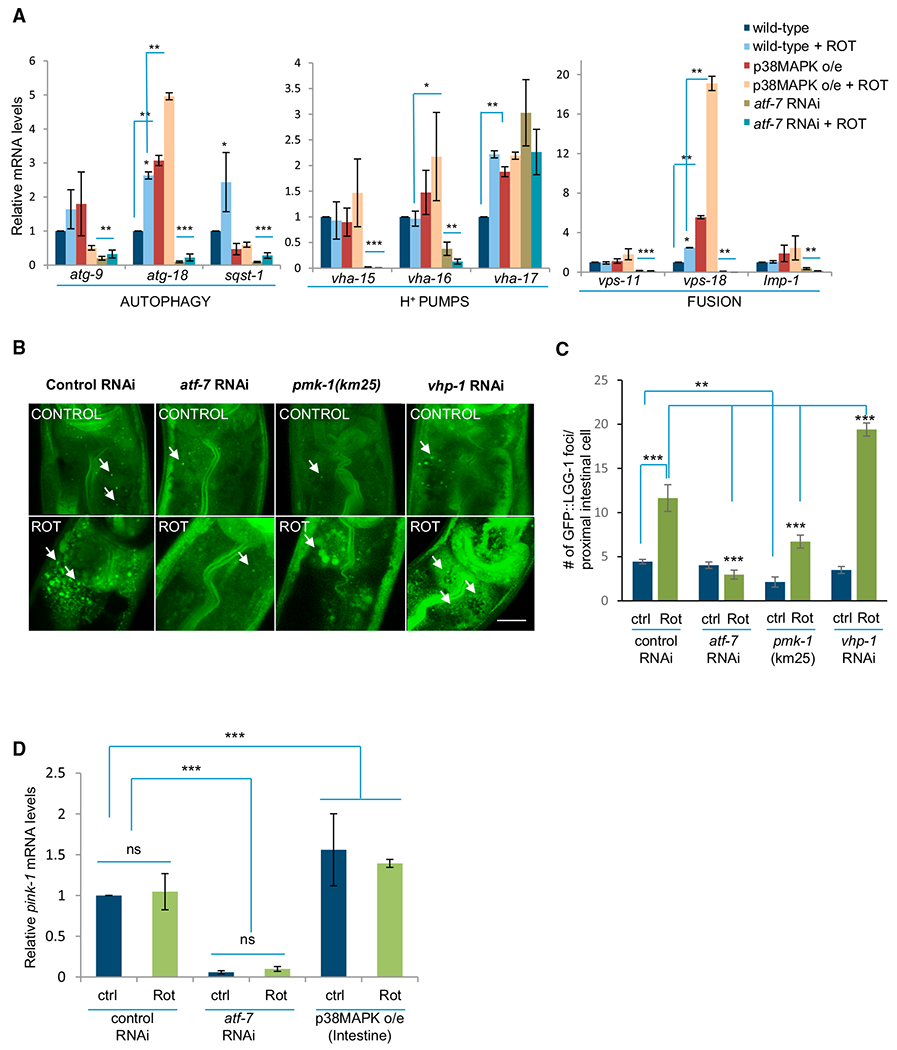
(A) qRT-PCR measurements of mRNA levels of autophagy-related genes in wild-type animals on control RNAi, atf-7 RNAi, and upon p38MAPK intestinal overexpression under control conditions and upon rotenone treatment. n ≥ 3 repeats of 20 animals for each treatment. Actin is the internal control, and values were normalized to control untreated levels. n ≥ 3 repeats of 20 animals for each treatment.
(B) Representative maximum projection confocal images showing GFP::LGG-1 puncta in the proximal intestinal nuclei of wild-type animals on control RNAi, atf-7 RNAi, pmk-1(km25) animals and animals on vhp-1 RNAi under control conditions and upon rotenone treatment. Arrows mark representative GFP::LGG-1 puncta. Scale bar, 10 μm.
(C) Quantitation of GFP::LGG-1 puncta averaged from counts in the first four intestinal cells of n = 20–25 animals per treatment.
(D) mRNA levels of pink-1 as determined by qRT-PCR in controls and upon rotenone treatment of animals on control RNAi, atf-7 RNAi, and animals overexpressing p38MAPK in intestinal cells. mRNA levels of the transcripts are normalized to wild-type control levels. Actin is the internal control. n ≥ 3 repeats of 20 animals for each treatment.
(A, C, and D) Bar graphs show mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 (Student’s t test). (A–D) Animals: day 1–2 adults. See also Figure S6.
Steady-state pink-1 mRNA levels also significantly increased in animals overexpressing p38MAPK in intestinal cells (Figure 6D). Conversely, pink-1 mRNA levels were dramatically reduced both under control conditions and upon rotenone exposure when atf-7 was downregulated (Figure 6D). As the mechanism of action of PINK-1 is post-translational and not necessarily through the modulation of mRNA levels, we also examined PINK-1::GFP protein levels in transgenic animals expressing PINK-1::GFP under the pink-1 promoter (Sämann et al., 2009). In these animals, PINK-1::GFP is consistently visible in CAN neurons but not apparent in the intestine. Nonetheless, in accordance with the mRNA data, PINK-1::GFP levels assessed by fluorescence intensity in CAN neurons increased upon p38MAPK overexpression (Figure S6F). These data suggest that p38MAPK/ATF-7 signaling pathway regulated autophagy at the transcriptional and translational level, as well as its activity, supporting the role of autophagy in the neuroprotection conferred by intestinal overexpression of p38MAPK. In addition, it appeared that the activation of p38MAPK in intestinal cells could not only affect autophagy in the intestinal cells but to some extent also modified autophagy in neurons.
DISCUSSION
In summary, our studies uncover a surprising neuroprotective role for the triggering of the innate immune response pathway in the gut of C. elegans by mitochondrial damage (Figure 7). Specifically, we show that the activating the p38MAPK/ATF-7 pathway in C. elegans intestinal cells by mitochondrial dysfunction non-autonomously protects dopaminergic neurons from rotenone-induced degeneration, while downregulating this pathway exacerbates neurodegeneration. Neuroprotection by p38MAPK/ATF-7 requires the core autophagy machinery and PINK-1, which, in turn, are regulated by p38MAPK and ATF-7, leading us to propose that p38MAPK-mediated immunity activates mitophagy to confer neuroprotection.
Figure 7. Model for Neuroprotection Conferred by Innate Immune Activation.

p38MAPK/ATF-7 pathway of C. elegans is activated in intestinal cells by mitochondrial complex I dysfunction induced by rotenone treatment. Activation of p38MAPK in intestinal cells activates the core autophagy machinery and PTEN-induced putative kinase 1 (PINK-1) to remove dysfunctional mitochondria in intestinal cells, conferring neuroprotection by unknown means.
While our data show that activation of p38MAPK in intestinal cells is sufficient to confer neuroprotection, it is arguable that activating p38MAPK in any tissue could be neuroprotective. Thus, overexpression of p38MAPK in muscle cells also conferred significant protection. However, rotenone appears to activate p38MAPK/ATF-7 signaling predominantly in the intestine and not in other tissues, supporting a role for signaling between the intestine and neurons in the normal course of neuroprotection. Moreover, activating p38MAPK pathways in neuronal tissue has been shown to exacerbate rather than protect against neuron degeneration (Kumar et al., 2003; Tortarolo et al., 2003; Vérièpe et al., 2015; Wu et al., 2015). These data together suggest that the location, mechanisms that initiate neurodegeneration, and targets of the p38MAPK signaling cascade, which include the mediators of innate immunity, may all collude to determine whether its activation is protective or detrimental.
It is also unclear whether the protection conferred by the p38MAPK/ATF-7 pathway is specific to dopaminergic neurons. Neural circuits are able to cell-non-autonomously modulate somatic cellular stress responses, including the innate immune response (Kumsta et al., 2014; Maman et al., 2013; Prahlad and Morimoto, 2011; Styer et al., 2008; Sun et al., 2011; Tracey, 2014). Interestingly, dopaminergic neurons are required for some aspects of p38MAPK-mediated immunity in C. elegans and mammals (Shi et al., 2013; Torres-Rosas et al., 2014). That the activation of p38MAPK-mediated immunity in the gut in turn protects dopaminergic neurons may suggest the existence of a feedback loop in this regulatory mechanism.
How p38MAPK and/or ATF-7 regulate autophagy in C. elegans remains to be understood. In other model systems, the p38MAPK-mediated innate immune response also upregulates autophagy (Hirota et al., 2015; Prick et al., 2006; Wei et al., 2015) , and in C. elegans, mitophagy is also activated by p38MAPK-independent innate immune responses (Kirienko et al., 2015). Animals treated with atf-7 RNAi are wild-type with respect to number of GFP::LGG-1 puncta under control conditions (but express less GFP::LGG-1 protein and have lower autophagy gene-mRNA levels) but have significantly fewer puncta upon rotenone exposure. Animals deficient in p38MAPK, pmk-1(km25) mutants, on the other hand have significantly fewer GFP::LGG-1 puncta both on rotenone treatment as well as under control conditions. This could mean that both p38MAPK and ATF-7 are necessary and sufficient to upregulate autophagy upon rotenone exposure when p38MAPK is activated, and indeed activating p38MAPK by vhp-1 RNAi significantly increases GFP::LGG-1 autophagosomes above control levels. However, only p38MAPK appears to be required for normal autophagosome numbers under control conditions.
It is also interesting to speculate how p38MAPK-dependent immunity and autophagy confer cell-non-autonomous protection. p38MAPK activity is critical for normal immune and inflammatory responses in all animal models (Kumar et al., 2003), and autophagy is increasingly being considered a bona fide immunological process (Deretic, 2012). One likely explanation for the observed protection is that mitophagy induced by p38MAPK reduces the detrimental consequences of OXPHOS dysregulation by limiting the numbers of damaged mitochondria. In support of this hypothesis, ZIP-2 and ATFS-1 activity, known to activate the UPRmito and maintain, rather than clear, dysfunctional mitochondria, increase neurodegeneration (Lin et al., 2016). Besides its role in preventing OXPHOS dysregulation, mitophagy plays a central role in preventing the exacerbation of the innate immune response itself (Lazarou, 2015; Nakahira et al., 2011; West et al., 2011). It is therefore possible that activation of p38MAPK-mediated immunity in remote tissues like the gut may ultimately prevent neuroinflammation through inter-tissue signaling mechanisms. Alternatively, an increase in autophagy in the intestine may facilitate more general cytoprotective mechanisms, such as changes in energy balance or alterations in ROS signaling (Barone et al., 2011; D’Souza et al., 2013; Schroeder and Shadel, 2012), making neurons less susceptible to rotenone-induced damage of Complex I.
It is also possible that p38MAPK/ATF-7 controls autophagy independent of its role in regulating the innate immune response. Indeed, other p38MAPK targets, such as the transcription factor Nrf2, the SKN-1 homolog, are known to also play neuroprotective roles (Barone et al., 2011). While ATF-7, the ortholog of mammalian ATF2/ATF7, has been best characterized for its role in the upregulation of antibacterial genes like mammalian ATF2, it may play more general roles in other stress-response pathways. In addition, although activation of p38MAPK in intestinal cells is one prominent neuroprotective pathway activated by rotenone, there also appear to be other additional pathways, not revealed by our studies, that protect against rotenone-induced dopaminergic neuron loss. This is supported by the fact that while almost all wild-type animals with intact dopaminergic neurons display an increase in the ATF-7 target gene C17H12.8 as determined by smFISH, only ~10% lose dopaminergic neurons upon atf-7 RNAi.
Our data also implicate the activation of p38MAPK/ATF-7 in retrograde signaling by mitochondria upon complex I dysfunction, even in the absence of rotenone (Munkacsy et al., 2016). It is intriguing that p38MAPK-dependent innate immunity activated by mitochondrial dysfunction can mobilize mitophagy. Cells unleash complex and nuanced immune responses to pathogens through the ability of their innate immune response to differentiate between self and non-self, and autophagy is increasingly being regarded as an intrinsic part of this immune process. Thus, just as activation of the innate immune response can clear the cell of pathogens and protect against infection, the activation of the innate immune response upon mitochondrial dysfunction may clear the cell of mitochondria and protect the organism from defective mitochondria (Lazarou, 2015; Randow and Youle, 2014). Mitochondrial DNA and chaperones are potent antigens and elicit inflammatory responses through the activation of p38MAPK and other pathways (Kirienko et al., 2015; Lazarou, 2015; Randow and Youle, 2014; Weinberg et al., 2015; West et al., 2011). Given the origins of mitochondria, the recognition of mitochondria as integral components of the cell may be ambiguous and subject to constant surveillance by innate immune mechanisms. Here, we propose that one such surveillance pathway acting through p38MAPK/ATF-7-mediated signaling can prove protective against neurodegeneration.
EXPERIMENTAL PROCEDURES
C. elegans Strains
A detailed strain list is provided in Supplemental Experimental Procedures
Growth Conditions
All strains were grown at low densities at 20°C as described in Supplemental Experimental Procedures.
RNAi Experiments and Constructs
All RNAi clones were sequenced. atf-7 RNAi plasmids were a gift from Dr. Dennis Kim (MIT) and also obtained from Open Biosystems (GE Dharmacon, Fisher Scientific).
Dopaminergic Neuronal Loss Assay
L1s were exposed to 3–5 μM rotenone as detailed in Supplemental Experimental Procedures and scored on days 1–2 of adulthood.
Single-Molecule Fluorescent In Situ Hybridization
smFISH probes were designed against C17H12.8 by utilizing the Stellaris FISH Probe Designer (Biosearch Technologies) as detailed in Supplemental Experimental Procedures.
mtDNA/Nuclear DNA Ratio
For quantification of mtDNA/nuclear genome, relative values of nduo-1 and cox-4 were compared as detailed in Supplemental Experimental Procedures.
Autophagy Analysis by GFP::LGG-1 Reporter
MAH236 line obtained from the CGC was used for the analysis of GFP::LGG-1 puncta as detailed in Supplemental Experimental Procedures.
Statistical Methods
In all cases, data from paired experiments and controls were compared. N for experiments is indicated in the legend, and data are presented as mean ± SEM. Significance was calculated using a paired Student’s t test, and p values < 0.05 were considered significant (*p < 0.05, **p < 0.01, ***p < 0.001).
Supplementary Material
Highlights.
Rotenone exposure activates the p38MAPK/ATF-7 immune pathway in the C. elegans gut
The p38MAPK/ATF-7 immune pathway is triggered by mitochondrial complex I dysfunction
p38MAPK/ATF-7 activity in the gut protects from rotenone-induced neurodegeneration
Neuroprotection through p38MAPK/ATF-7 activation occurs through mitophagy
ACKNOWLEDGMENTS
We apologize to all colleagues whose work could not be cited due to space limitations. We thank Dr. Dennis Kim (Department of Biology, MIT) for sharing the atf-7 plasmid, Dr. Anton Gartner (University of Dundee, U.K.) for C. elegans strains expressing pdat-1::mcherry, and Dr. Bryan Phillips (University of Iowa) for various reporter plasmids. We thank the Ellison Medical Foundation (AG-NS-1056-13) for financial support.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures and six figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.07.077.
REFERENCES
- Barone MC, Sykiotis GP, and Bohmann D (2011). Genetic activation of Nrf2 signaling is sufficient to ameliorate neurodegenerative phenotypes in a Drosophila model of Parkinson’s disease. Dis. Model. Mech 4, 701–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolz DD, Tenor JL, and Aballay A (2010).A conserved PMK-1/p38 MAPK is required in Caenorhabditis elegans tissue-specific immune response to Yersinia pestis infection. J. Biol. Chem 285, 10832–10840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bové J, Prou D, Perier C, and Przedborski S (2005). Toxin-induced models of Parkinson’s disease. NeuroRx 2, 484–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branicky R, Bénard C, and Hekimi S (2000). clk-1, mitochondria, and physiological rates. BioEssays 22, 48–56. [DOI] [PubMed] [Google Scholar]
- Calixto A, Chelur D, Topalidou I, Chen X, and Chalfie M (2010). Enhanced neuronal RNAi in C. elegans using SID-1. Nat. Methods 7,554–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza AD, Parish IA, Krause DS, Kaech SM, and Shadel GS (2013). Reducing mitochondrial ROS improves disease-related pathology in a mouse model of ataxia-telangiectasia. Mol. Ther 21, 42–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deretic V (2012). Autophagy: an emerging immunological paradigm. J. Immunol 189,15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durieux J, Wolff S, and Dillin A (2011). The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144, 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermolaeva MA, and Schumacher B (2014). Insights from the worm: the C. elegans model for innate immunity. Semin. Immunol 26, 303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estes KA, Dunbar TL, Powell JR, Ausubel FM, and Troemel ER (2010). bZIP transcription factor zip-2 mediates an early response to Pseudomonas aeruginosa infection in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 107,2153–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewbank JJ, and Pujol N (2016). Local and long-range activation of innate immunity by infection and damage in C. elegans. Curr. Opin. Immunol 88,1–7. [DOI] [PubMed] [Google Scholar]
- Hansen M, Chandra A, Mitic LL, Onken B, Driscoll M, and Kenyon C (2008). A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet. 4, e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes CM, and Ron D (2010). The mitochondrial UPR - protecting organelle protein homeostasis. J. Cell Sci 123, 3849–3855. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, and Latz E (2014). Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol 14, 463–477. [DOI] [PubMed] [Google Scholar]
- Hirota Y, Yamashita S, Kurihara Y, Jin X,Aihara M, Saigusa T, Kang D, and Kanki T (2015). Mitophagy is primarily due to alternative autophagy and requires the MAPK1 and MAPK14 signaling pathways. Autophagy 11, 332–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilieva H, Polymenidou M, and Cleveland DW (2009). Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol 187, 761–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ME, and Bobrovskaya L (2015). An update on the rotenone models of Parkinson’s disease: their ability to reproduce the features of clinical disease and model gene-environment interactions. Neurotoxicology 46, 101–116. [DOI] [PubMed] [Google Scholar]
- Kim DH, Feinbaum R, Alloing G, Emerson FE, Garsin DA, Inoue H, Tanaka-Hino M, Hisamoto N, Matsumoto K, Tan MW, and Ausubel FM (2002). A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science 297, 623–626. [DOI] [PubMed] [Google Scholar]
- Kirienko NV, Ausubel FM, and Ruvkun G (2015). Mitophagy confers resistance to siderophore-mediated killing by Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 112, 1821–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleine T, and Leister D (2016). Retrograde signaling: Organelles go networking. Biochim. Biophys. Acta 1857, 1313–1325. [DOI] [PubMed] [Google Scholar]
- Kumar S, Boehm J, and Lee JC (2003). p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat. Rev. Drug Discov 2, 717–726. [DOI] [PubMed] [Google Scholar]
- Kumsta C, and Hansen M (2012). C. elegans rrf-1 mutations maintain RNAi efficiency in the soma in addition to the germline. PLoS ONE 7, e35428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumsta C, Ching TT, Nishimura M, Davis AE, Gelino S, Catan HH,Yu X, Chu CC, Ong B, Panowski SH, et al. (2014). Integrin-linked kinase modulates longevity and thermotolerance in C. elegans through neuronal control of HSF-1. Aging Cell 13, 419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapierre LR, Gelino S, Meléndez A, and Hansen M (2011). Autophagy and lipid metabolism coordinately modulate life span in germline-less C. elegans. Curr. Biol 21, 1507–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lartigue L, and Faustin B (2013). Mitochondria: metabolic regulators of innate immune responses to pathogens and cell stress. Int. J. Biochem. Cell Biol 45, 2052–2056. [DOI] [PubMed] [Google Scholar]
- Lazarou M (2015). Keeping the immune system in check: a role for mitophagy. Immunol. Cell Biol 93, 3–10. [DOI] [PubMed] [Google Scholar]
- Lemire BD, Behrendt M, DeCorby A, and Gásková D (2009). C. elegans longevity pathways converge to decrease mitochondrial membrane potential. Mech. Ageing Dev 130, 461–465. [DOI] [PubMed] [Google Scholar]
- Lin YF, Schulz AM, Pellegrino MW, Lu Y, Shaham S, and Haynes CM (2016). Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. Nature 533, 416–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Samuel BS, Breen PC, and Ruvkun G (2014). Caenorhabditis elegans pathways that surveil and defend mitochondria. Nature 508, 406–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luz AL, Rooney JP, Kubik LL, Gonzalez CP, Song DH, and Meyer JN (2015). Mitochondrial morphology and fundamental parameters of the mitochondrial respiratory chain are altered in Caenorhabditis elegans Strains deficient in mitochondrial dynamics and homeostasis processes. PLoS ONE 10, e0130940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maman M, Carvalhal Marques F, Volovik Y, Dubnikov T, Bejerano-Sagie M, and Cohen E (2013). A neuronal GPCR is critical for the induction of the heat shock response in the nematode C. elegans. J. Neurosci 33,6102–6111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell S, Harding J, Brabin C, Appleford PJ, Brown R, Delaney C, Brown G, and Woollard A (2013). The SFT-1 and OXA-1 respiratory chain complex assembly factors influence lifespan by distinct mechanisms in C. elegans. Longev. Healthspan 2, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meléndez A, and Levine B (2009). Autophagy in C. elegans. WormBook, 1–26. [DOI] [PubMed] [Google Scholar]
- Melo JA, and Ruvkun G (2012). Inactivation of conserved C. elegans genes engages pathogen- and xenobiotic-associated defenses. Cell 149, 452–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno T, Hisamoto N, Terada T, Kondo T, Adachi M, Nishida E, Kim DH, Ausubel FM, and Matsumoto K (2004). The Caenorhabditis elegans MAPK phosphatase VHP-1 mediates a novel JNK-like signaling pathway in stress response. EMBO J. 23, 2226–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munkacsy E, Khan MH, Lane RK, Borror MB, Park JH, Bokov AF, Fisher AL, Link CD, and Rea SL (2016). DLK-1, SEK-3 and PMK-3 are required for the life extension induced by mitochondrial bioenergetic disruption in C. elegans. PLoS Genet. 12, e1006133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, et al. (2011). Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol 12, 222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, and Haynes CM (2012). Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 337, 587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nass R, Hall DH, Miller DM 3rd, and Blakely RD (2002). Neurotoxin-induced degeneration of dopamine neurons in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 99, 3264–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsawa S, Sato Y, Enomoto M, Nakamura M, Betsumiya A, and Igaki T (2012). Mitochondrial defect drives non-autonomous tumour progression through Hippo signalling in Drosophila. Nature 490, 547–551. [DOI] [PubMed] [Google Scholar]
- Paek J, Lo JY, Narasimhan SD, Nguyen TN, Glover-Cutter K, Robida-Stubbs S, Suzuki T, Yamamoto M, Blackwell TK, and Curran SP (2012). Mitochondrial SKN-1/Nrf mediates a conserved starvation response. Cell Metab. 16, 526–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palikaras K, Lionaki E, and Tavernarakis N (2015). Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521, 525–528. [DOI] [PubMed] [Google Scholar]
- Pellegrino MW, Nargund AM, Kirienko NV, Gillis R, Fiorese CJ, and Haynes CM (2014). Mitochondrial UPR-regulated innate immunity provides resistance to pathogen infection. Nature 516, 414–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perier C, and Vila M (2012). Mitochondrial biology and Parkinson’s disease. Cold Spring Harb. Perspect. Med 2, a009332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prahlad V, and Morimoto RI (2011). Neuronal circuitry regulates the response of Caenorhabditis elegans to misfolded proteins. Proc. Natl. Acad. Sci. USA 108, 14204–14209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prick T, Thumm M, Köhrer K, Häussinger D, and Vom Dahl S (2006). In yeast, loss of Hog1 leads to osmosensitivity of autophagy. Biochem. J 394, 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przedborski S, and Jackson-Lewis V (1998). Mechanisms of MPTP toxicity. Mov. Disord 13 (Suppl 1), 35–38. [PubMed] [Google Scholar]
- Qadota H, Inoue M, Hikita T, Köppen M, Hardin JD, Amano M, Moerman DG, and Kaibuchi K (2007). Establishment of a tissue-specific RNAi system in C. elegans. Gene 400, 166–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randow F, and Youle RJ (2014). Self and nonself: how autophagy targets mitochondria and bacteria. Cell Host Microbe 15, 403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray A, Martinez BA, Berkowitz LA, Caldwell GA, and Caldwell KA (2014). Mitochondrial dysfunction, oxidative stress, and neurodegeneration elicited by a bacterial metabolite in a C. elegans Parkinson’s model. Cell Death Dis. 5, e984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson JR, Caudle WM, Guillot TS, Watson JL, Nakamaru-Ogiso E, Seo BB, Sherer TB, Greenamyre JT, Yagi T, Matsuno-Yagi A, and Miller GW (2007). Obligatory role for complex I inhibition in the dopaminergic neurotoxicity of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Toxicol. Sci 95, 196–204. [DOI] [PubMed] [Google Scholar]
- Runkel ED, Liu S, Baumeister R, and Schulze E (2013). Surveillance-activated defenses block the ROS-induced mitochondrial unfolded protein response. PLoS Genet. 9, e1003346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sämann J, Hegermann J, von Gromoff E, Eimer S, Baumeister R, and Schmidt E (2009). Caenorhabditits elegans LRK-1 and PINK-1 act antagonistically in stress response and neurite outgrowth. J. Biol. Chem 284, 16482–16491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeisser S, Priebe S, Groth M, Monajembashi S, Hemmerich P, Guthke R, Platzer M, and Ristow M (2013). Neuronal ROS signaling rather than AMPK/sirtuin-mediated energy sensing links dietary restriction to lifespan extension. Mol. Metab 2, 92–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder EA, and Shadel GS (2012). Alternative mitochondrial fuel extends life span. Cell Metab. 15, 417–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, Miller GW, Yagi T, Matsuno-Yagi A, and Greenamyre JT (2003). Mechanism of toxicity in rotenone models of Parkinson’s disease. J. Neurosci 23, 10756–10764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z, Lu Z, Zhao Y, Wang Y, Zhao-Wilson X, Guan P, Duan X, Chang YZ, and Zhao B (2013). Neuroprotective effects of aqueous extracts of Uncaria tomentosa: Insights from 6-OHDA induced cell damage and transgenic Caenorhabditis elegans model. Neurochem. Int 62, 940–947. [DOI] [PubMed] [Google Scholar]
- Shivers RP, Pagano DJ, Kooistra T, Richardson CE, Reddy KC, Whitney JK, Kamanzi O, Matsumoto K, Hisamoto N, and Kim DH (2010). Phosphorylation of the conserved transcription factor ATF-7 by PMK-1 p38 MAPK regulates innate immunity in Caenorhabditis elegans. PLoS Genet. 6, e1000892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmer F, Tijsterman M, Parrish S, Koushika SP, Nonet ML, Fire A, Ahringer J, and Plasterk RH (2002). Loss of the putative RNA-directed RNA polymerase RRF-3 makes C. elegans hypersensitive to RNAi. Curr. Biol 12, 1317–1319. [DOI] [PubMed] [Google Scholar]
- Styer KL, Singh V, Macosko E, Steele SE, Bargmann CI, and Aballay A (2008). Innate immunity in Caenorhabditis elegans is regulated by neurons expressing NPR-1/GPCR. Science 322, 460–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Singh V, Kajino-Sakamoto R, and Aballay A (2011). Neuronal GPCR controls innate immunity by regulating noncanonical unfolded protein response genes. Science 332, 729–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Rosas R, Yehia G, Peña G, Mishra P, del Rocio Thompson-Bonilla M, Moreno-Eutimio MA, Arriaga-Pizano LA, Isibasi A, and Ulloa L (2014). Dopamine mediates vagal modulation of the immune system by electroacupuncture. Nat. Med 20, 291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortarolo M, Veglianese P, Calvaresi N, Botturi A, Rossi C, Giorgini A, Migheli A, and Bendotti C (2003). Persistent activation of p38 mitogen-activated protein kinase in a mouse model of familial amyotrophic lateral sclerosis correlates with disease progression. Mol. Cell. Neurosci 23, 180–192. [DOI] [PubMed] [Google Scholar]
- Tracey KJ (2014). Approaching the next revolution? Evolutionary integration of neural and immune pathogen sensing and response. Cold Spring Harb. Perspect. Biol 7, a016360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vérièpe J, Fossouo L, and Parker JA (2015). Neurodegeneration in C. elegans models of ALS requires TIR-1/Sarm1 immune pathway activation in neurons. Nat. Commun 6, 7319. [DOI] [PubMed] [Google Scholar]
- Wei Y, An Z, Zou Z, Sumpter R, Su M, Zang X, Sinha S, Gaestel M, and Levine B (2015). The stress-responsive kinases MAPKAPK2/MAPKAPK3 activate starvation-induced autophagy through Beclin 1 phosphorylation. eLife 4, e05289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg SE, Sena LA, and Chandel NS (2015). Mitochondria in the regulation of innate and adaptive immunity. Immunity 42, 406–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Shadel GS, and Ghosh S (2011). Mitochondria in innate immune responses. Nat. Rev. Immunol 11, 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winklhofer KF (2014). Parkin and mitochondrial quality control: toward assembling the puzzle. Trends Cell Biol. 24, 332–341. [DOI] [PubMed] [Google Scholar]
- Wu Q, Cao X, Yan D, Wang D, and Aballay A (2015). Genetic screen reveals link between the Maternal effect sterile gene mes-1 and Pseudomonas aeruginosa-induced neurodegeneration in Caenorhabditis elegans. J. Biol. Chem 290, 29231–29239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Chang JT, Guo B, Hansen M, Jia K, Kovács AL, Kumsta C, Lapierre LR, Legouis R, Lin L, et al. (2015). Guidelines for monitoring autophagy in Caenorhabditis elegans. Autophagy 11, 9–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.